
RNA-controlled assembly of tobacco mosaic virus-derived complex structures: from nanoboomerangs to tetrapods
Fabian J.
Eber
,
Sabine
Eiben
,
Holger
Jeske
and
Christina
Wege
*
Department of Molecular Biology and Plant Virology, Institute of Biomaterials and Biomolecular Systems, University of Stuttgart, Pfaffenwaldring 57, D-70569 Stuttgart, Germany. E-mail: christina.wege@bio.uni-stuttgart.de; Tel: +49-711-685-65073
First published on 10th November 2014
Abstract
The in vitro assembly of artificial nanotubular nucleoprotein shapes based on tobacco mosaic virus-(TMV-)-derived building blocks yielded different spatial organizations of viral coat protein subunits on genetically engineered RNA molecules, containing two or multiple TMV origins of assembly (OAs). The growth of kinked nanoboomerangs as well as of branched multipods was determined by the encapsidated RNAs. A largely simultaneous initiation at two origins and subsequent bidirectional tube elongation could be visualized by transmission electron microscopy of intermediates and final products. Collision of the nascent tubes’ ends produced angular particles with well-defined arm lengths. RNAs with three to five OAs generated branched multipods with a maximum of four arms. The potential of such an RNA-directed self-assembly of uncommon nanotubular architectures for the fabrication of complex multivalent nanotemplates used in functional hybrid materials is discussed.
Introduction
Tobacco mosaic virus (TMV)1 has been used increasingly for nanobiotechnology applications throughout the last decade.2–13 It contains one positive-sense single-stranded RNA of ∼6395 nucleotides carrying the genetic information,14 embedded within a protein coat of ∼2130 identical, helically arranged monomers. This rigid rod-like nucleoprotein structure – the virion – of about 300 × 18 nm encloses a longitudinal channel 4 nm in diameter.15–17 Virion-like particles can be generated also in vitro, by self-assembly of coat protein (CP) intermediate aggregates, mainly ring-shaped multimers (20S disks), with RNA.18 In addition, pure protein nanotubes can be formed in the absence of RNA, depending on pH, protein and ion concentration.19 According to the prevalent model of TMV assembly (ref. 20–22 and references therein), an origin of assembly (OAs) is located within the 3′ region of the viral RNA, which folds into a stem-loop structure with high affinity to the 20S aggregates, i.e. particularly double-layered 20S disks composed of 34 CP subunits.23–26Upon binding of such disk to the OAs nucleic acid loop with a characteristic nonanucleotide sequence, the disk undergoes a conformational change into a helical “lockwasher” structure.23 Concomitantly, a portion of the RNA becomes sandwiched between the protein layers while both RNA ends protrude from one side (the ‘tail’ side) of this initial complex.27–29 Further disks then serially attach to the ‘front’ side of the nascent tube in a cooperative manner, each rearranging and thereby pulling the 5′-terminus of the RNA through the internal channel to elongate the nucleoprotein helix.14,23,30 The 3′ portion of the RNA is packaged by small oligomeric CPs (“A-Protein”) in the opposite direction and at a slower rate.20,31–33 This mechanistic principle is illustrated in Fig. 1 (for an artificial RNA scaffold containing two TMV OAs sites, as introduced below). The efficiency of the assembly is affected by the availability of CP 20S aggregates for rod nucleation at the OAs sequences, with higher protein concentrations accelerating completion of these initiation reactions (and kinetics corresponding to bimolecular reactions).29 Subsequent elongation contributes to the overall assembly rate with a CP saturation concentration, determined by the incorporation velocity of CP 20S aggregates cooperatively in 5′ direction, and “A-Protein” in 3′ direction, respectively.20,29 Apart from the CP-to-RNA ratio, also the overall concentration of the reaction partners drives nanotube formation. For temperature, ionic and buffer strength as well as type, optimum conditions have to be considered (as reviewed extensively by Butler20,34).
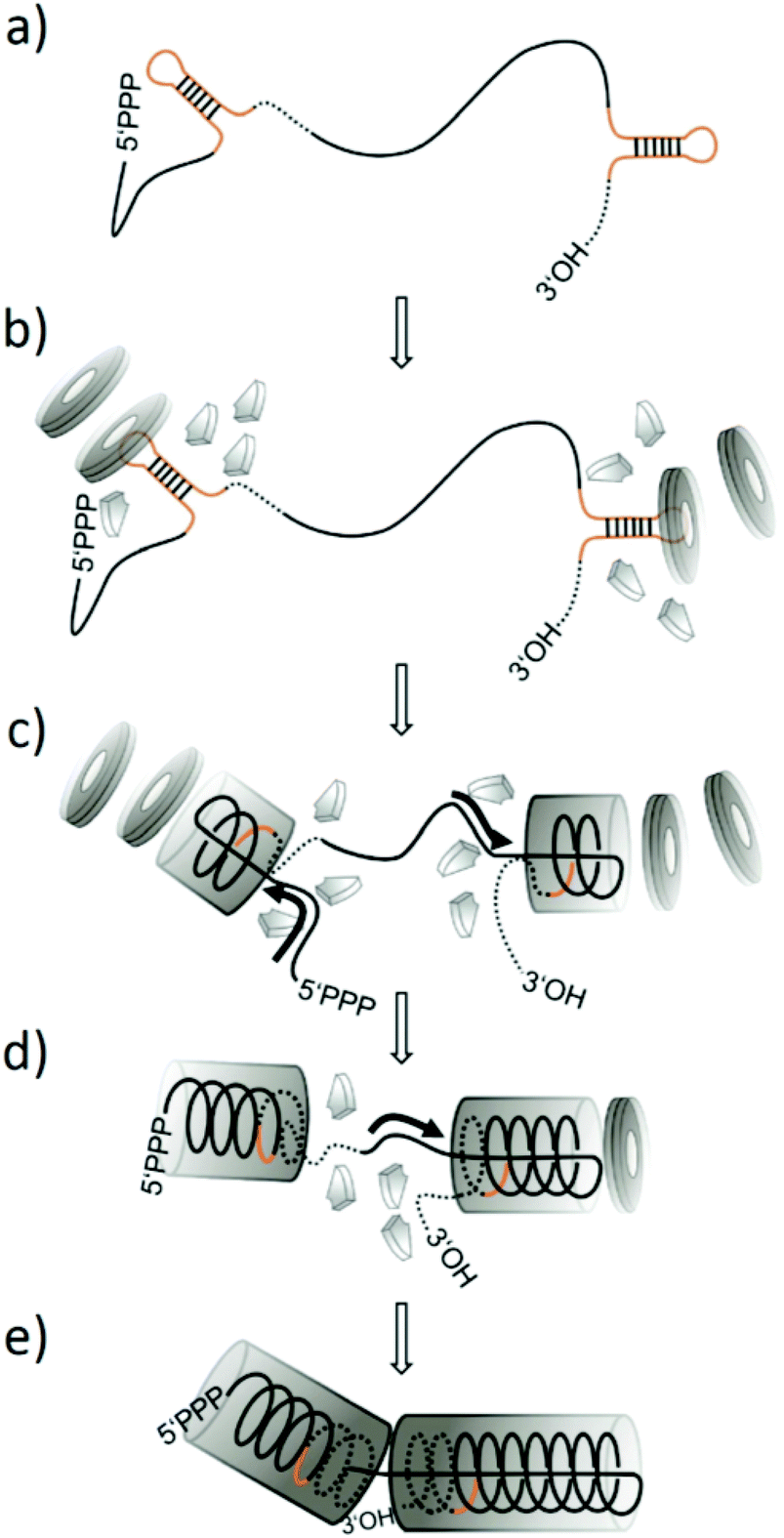 | ||
| Fig. 1 Scheme of the prevalent model of TMV nanotube assembly in vitro, illustrating the process predicted for an artificial RNA scaffold with two tandemly repeated OAs-containing segments which nucleate growth of two nucleoprotein domains simultaneously, with their final collision resulting in a nanoboomerang shape. (a–e) visualize successive stages. (a) RNA construct with two OAs structures depicted in brown colour. (b) Assembly initiation on both OAs sequences, induced by their interaction with two-layered ring-shaped 20S CP aggregates (grey “disks”; in the presence of further smaller CP “A-protein” aggregates). (c–d) Assembly of two helical RNA-CP tube domains, elongating every initiation complex bidirectionally. Growth in 5′-direction (left and right: tube fronts with RNA “traveling loops”) occurs via serial addition of further disks, concomitantly sandwiching an RNA helix between CP layers and pulling the 5′-portions of every RNA segment [continuous lines] into the inner tube channel (black arrows: direction of RNA movement). 3′-elongation is realized by the addition of A-protein, packaging the corresponding RNA portions [dotted lines] into the nascent CP helix. (e) The RNA strand between the two OAs is consumed so that the two 3′-directionally growing arms collide to form a kinked structure. For further details, refer to the text. | ||
The stability of TMV and the capability of its building blocks to self-assemble into different complexes, in conjunction with the comprehensive knowledge of their physical and chemical properties, has led to the fabrication of particular TMV-derived biotemplates with tailored nanometric dimensions or internal sub-structures. These offer specific opportunities for applications relying on a well-ordered arrangement of functional units, to form e.g. linear35 or ring-shaped36 plasmonic nanoparticle arrays in suspension, energy transfer and light-harvesting systems,37–39 spatially controlled supramolecular high-relaxivity gadolinium(III) chelate assemblies as contrast agents for magnetic resonance imaging,40 photovoltaic devices,41 multivalent nanotubes in distinct length classes stabilizing and regulating the magnetoviscosity of ferrofluids42 also allowing covalent installation of functional molecules,43 high-precision positioning tools for collaborating amino acids gaining artificial enzyme activity,44 arrays of carrier rods with programmable length grown bottom-up on technical substrates45 or on metal cores yielding nanostar colloids with ultrahigh soft-matter surface,46 support sticks for the oriented immobilization e.g. of bioactive antigens,47 rods with selectively addressable sub-domains enabling a highly ordered presentation of distinct molecule types,48 or particles interlinked by host–guest molecular interactions permitting convenient assembly of multifunctional ensembles.49 These and other studies illustrate a growing body of evidence for the versatility of well-defined TMV-deduced 3D architectures for specific fields of applications. The availability of engineered TMV CP variants promoting certain aggregation states such as stabilized rods or disks,50–54 and refined procedures to even transform TMV into spherical vaccine carrier particles in a controlled manner,40,55,56 have further expanded research on TMV-based novel nanodevices and functional hybrids.
So far, however, only few approaches have exploited the potential of genetically modified RNA molecules to govern length and stability of TMV-like nanotubes prepared for technical uses.42,43,45,46,48,51,57,58 On the contrary, earlier findings hint at immense additional degrees of freedom during the assembly of TMV-derived particles with atypical RNA, by which even their overall shape might be converted into non-linear kinked or branched products.
While TMV in vitro assembly usually starts at the primary OAs as described above, a certain mutation of the sequence slowed down initiation rates at elevated temperatures59,60 and allowed an alternative RNA site closer to the 3′ terminus (within the CP gene) to act as secondary (cryptic) OAs simultaneously. Such twofold initiation on single RNA molecules led to the growth of nucleoprotein tube pairs, interlinked by a stretch of free RNA. Kinked and branched particles of further increased structural complexity could be assembled with RNA containing concatemeric OAs-comprising repeats.61 Those authors used mixed populations of RNA molecules with variable OAs numbers, obtained by in vitro rolling circle transcription of supercoiled DNA plasmids with a single cDNA copy of the TMV OAs region. Largely simultaneous induction at separate OAs sites of individual transcripts led to the bidirectional growth of discrete tubular domains on single RNAs. Collisions of their approaching nucleoprotein ends generated various types of non-linear structures.
Against the background of these findings, we sought to investigate the feasibility of a predictable formation of kinked or branched particles with defined complex shapes, with regard to reaction efficiencies, mechanistic details of the viral self-assembly and potential uses of such biotemplates. RNA molecules containing two to five OAs modules at defined positions were engineered, and their assembly with TMV CP and the resulting products characterized systematically. Our data point at intriguing possibilities to modify and pre-determine the shape of tobamovirus-derived nanotubular architectures.
Results and discussion
In vitro assembly products of RNA molecules with two OAs sequences
To test which nanostructures typically arise if bidirectionally self-assembling TMV nucleoprotein domains collide with each other, RNA molecules containing two viral OAs at defined sequence positions were created: A truncated TMV cDNA portion (“TMV_C”) including the OAs was duplicated and cloned into a bacterial plasmid in two different designs allowing in vitro transcription (pGEM-T/dupTMV_C+RZ and pGEM-T/dupTMV_C−RZ, Fig. 2). Both constructs harbor the same direct repeat of the target sequence but differ in the presence of a ribozyme at the 3′ end (+RZ or −RZ; Fig. 2a and c), which trims the terminus autocatalytically and therefore shortens the 3′ portion of the corresponding RNA. In vitro transcription of the plasmids linearized by the restriction enzyme NdeI (for +RZ) or ScaI (for −RZ) thus yielded RNA molecules of 3958 nts (plus the 3′ ribozyme fragment) or 5638 nts, named dupOAs3958 and dupOAs5638, respectively (Fig. 2b and d). Both are expected to establish two hairpins with nonanucleotide-loops each, which serve as initiation sites for assembly according to virtual secondary structure predictions (Fig. 2e and f). After mixing these RNA molecules with assembly-competent TMV CP under appropriate conditions,50 elongated TMV-like particles (TLPs) were formed (Fig. 2g, i and k), with the typical diameter of 18 nm. Many of them exhibited a boomerang-like shape with two arms separated by a kink. | ||
| Fig. 2 Kinked nanostructures scaffolded by two RNA constructs each with two OAs sites but different length (dupOAs3958 and dupOAs5638). Plasmids pGEM-T/dupTMV_C+RZ (a) and pGEM-T/dupTMV_C−RZ (c) were used for in vitro transcription of RNA (red lines) after linearization with NdeI or ScaI, respectively. The position of the ribozyme sequence (RZ) is indicated (a). The size of the transcripts was determined in denaturing 1% agarose gels (b, d) in comparison to marker RNA (M) with the indicated sizes. The predicted secondary structures of the transcripts (e, f) with calculated ΔG values at 37 °C of −1171 kcal mol−1 for dupOAs3958 and −1699 kcal mol−1 for dupOAs5638 are based on RNA folding software.62 The OAs hairpins with nonanucleotide loops are marked by a black frame. Assembly products of TMV CP with both scaffold RNAs (g, h) in TEM images are shown after negative staining with uranyl acetate, with close-up of single particles (i, k) directly or after RNase treatment (j, l) which destroys protein-free RNA linkers. Kinked structures are labeled with arrowheads. | ||
The two arms of individual particles were anticipated to be connected by non-packaged RNA, as inferred from the assembly mechanism.20–22,61 Correspondingly, they were separated by RNase treatment (Fig. 2j and l). With both RNA constructs, one arm was shorter (about 70 nm in length) than the other (in dupOAs3958 about 110 nm; in dupOAs5638 about 120 nm). As specified in the subsequent section, their length distributions could be explained well on the basis of former studies on the differential velocities of the TMV coat assembly in 3′ vs. 5′ direction.
Quantitative analysis of at least 100 randomly selected particles for each RNA construct per experiment (in three independent experiments) revealed that about 70% showed bending in the TEM images (mean ± standard deviation: 75 ± 12% for dupOAS3958; 68 ± 5% for dupOAs5638). This indicates that assembly was initiated regularly if not generally on both OAs sequences of the same RNA molecule almost simultaneously, and that the subsequent bidirectional elongation of the stiff protein tubes resulted in their collision, to form a kink in-between the two initiation sites.
In 30% of the assembly products, no kink was detectable. These particles were, however, only 70 to 140 nm in length for both constructs, with no prominent peak in the size histograms (not shown). Since complete encapsidation should result in substantially longer particles (186 nm for dupOAs3958, 268 nm for dupOAs5638), most straight products likely represent incompletely assembled tubes, initiated by only one OAs. Such failure of initiation of the second OAs might rely on some degree of aberrant folding of the RNA secondary structures. Furthermore, a fraction of kinked particles may also appear linear (and shortened) due to their position on the TEM support, while a significant contribution of truncated RNA scaffolds can be largely excluded (see Fig. 2b and d). Taken together, simultaneously growing nanotube domains were generated with at least 70% efficiency via the combination of two OAs separated by about 1900 nts on a single RNA template, and their collision resulted in a kink on an almost regular basis.
Length distribution of nanoboomerang arms
Comparing the combined tube lengths of fully assembled nanoboomerangs to those of the individual arms for the two different RNA scaffolds allowed inferring which RNA domain had determined the length of which arm. To this aim, the arm lengths of individual particles were registered pairwise, together with the total length of the tube system. As a result, the size distributions of the shorter and the longer arms could be determined separately. The median values for the long versus the short arms were established for every experiment, and the respective averages calculated from different experiments (Fig. 3a). When we compared the median lengths of the particles’ arms of either construct, we found that the short arms were very similar in length (for dupOAs3958: 71.3 ± 1.7 nm SD; for dupOAs5638: 72.5 nm ± 0.5 nm SD), while the median length of the long arms differed significantly from each other (dupOAs3958: 106.4 nm ± 8.4 nm; dupOAs5638: 123.1 ± 0.9 nm). Since dupOAs5638 has an extended 3′ end after the second (3′) OAs but is otherwise identical to dupOAs3958, the longer arms originated from initiation at the second (3′) OAs (Fig. 3b and c). | ||
| Fig. 3 Median lengths (a) of short and long arms and total tube system of nanoboomerangs after in vitro assembly of CP, directed by two distinct RNA molecules with 3958 nts (empty bars) or 5638 nts (filled bars), each containing two OAs. Averages of the median values of the length distributions of three independent experiments are depicted with standard deviations. Schematic drawings (b, c) show the free RNA constructs and the configurations in the kinked particles, inferred from the median lengths of the particle arms and the corresponding RNA. Grey: Origin of assembly; RNA stretches expected to be encapsidated by the slower 3′ assembly (dotted line) or by the fast 5′ assembly (black line) mechanism.33 Data were obtained from TEM analyses of negatively stained specimens from three independent experiments, each with at least 100 structures. Selected images of particles arisen from assembly with dupTMV5638 after spreading and shadowing, showing a long RNA tail (d) or a third particle arm (e) protruding from the collision zone. | ||
Using the predicted relation of 0.0469 nm particle length per nucleotide of the RNA in assembled straight TMV,14 the 3958 nts RNA should yield 186 nm rods as a sum of both arms which is only slightly above the observed length of about 178 nm and fits within the limits of determination. In contrast, however, dupOAs5638 with an expected particle length of 264 nm was about 67 nm shorter. This deviation may result from a protruding RNA 3′ tail which was impaired in assembly in the collision zone, as drawn schematically in Fig. 2c. To test this hypothesis, particles were spread on a water hypophase,63 attached to filmed grids and metal-shadowed to visualize free RNA stretches. Indeed, TMV-derived structures with free RNA extensions or, less frequently, with a short third arm protruding from the collision zone were observed (Fig. 3d and e). The formation of the third arm could have been initiated at a secondary OAs site formed within the CP gene at a low frequency,23,34,60 or by 20S disk-independent packaging with small CP aggregates (A-protein). The relative positions of OAs nucleation sites to each other, if combined on single RNA templates, thus may affect structural homogeneity and completeness of the resulting particles, with steric hindrance at the collision sites introducing discontinuities of both shape and dimension.
Partial assembly
Early forms of the assembly directed by dupOAs3958 were trapped by adding less than one equivalent (eq.; the amount of CP needed to encapsidate the complete RNA) of CP to the RNA scaffold. After 16 hours of assembly, the structures were spread and shadowed with platinum in order to visualize the RNA. Complexes with two particle arms connected by free RNA were resolved, with two RNA tails protruding from each nascent arm on its same pole (Fig. 4a), thus visualizing two initiation events occurring simultaneously on one RNA molecule. The two nascent arms were usually similar in length. In negatively stained specimens prepared five minutes after induction of the assembly, distinct structures were found with two disks in spatial proximity, presumably representing initial assembly stages (Fig. 4b, boxes). | ||
| Fig. 4 Early assembly of VLPs. TEM images of selected structures after partial assembly of TMV CP directed by a 3958 nts RNA with 0.2 eq. of CP after 16 h (a), spread and rotary-shadowed to visualize free RNA, or with 1.3 eq. of CP after 5 min (b), stained negatively with uranyl acetate. Boxed insets with pairs of disks in spatial proximity are shown at higher magnification. Disks are likely interconnected by RNA (not visible). Bars: 100 nm. | ||
Quantitative analysis of the tube lengths (Fig. 5) of these incompletely assembled TMV-deduced structures revealed that CP domains distributed unevenly on RNA molecules under limiting CP concentrations. For example, 0.2 eq. of CP yielded total particle tube lengths decreased only to about 60% of the size of fully encapsidated RNAs for both kinked and straight structures, which is three times the length expected if all RNAs would have been targeted in parallel (Fig. 5a; black and white columns). This suggests that no more than one third of the RNA molecules participated in the assembly, and is in accordance with earlier observations by others at similar substoichiometric CP concentrations.29 With 0.5 eq. of CP, the median length of the tubes reached about 80% of the maximum length, corresponding to 62% of the OAs used. Accordingly, less kinked particles were observed after applying reduced CP to RNA ratios (51% with 0.5 eq. CP, and 33% with 0.2 eq. CP, respectively).
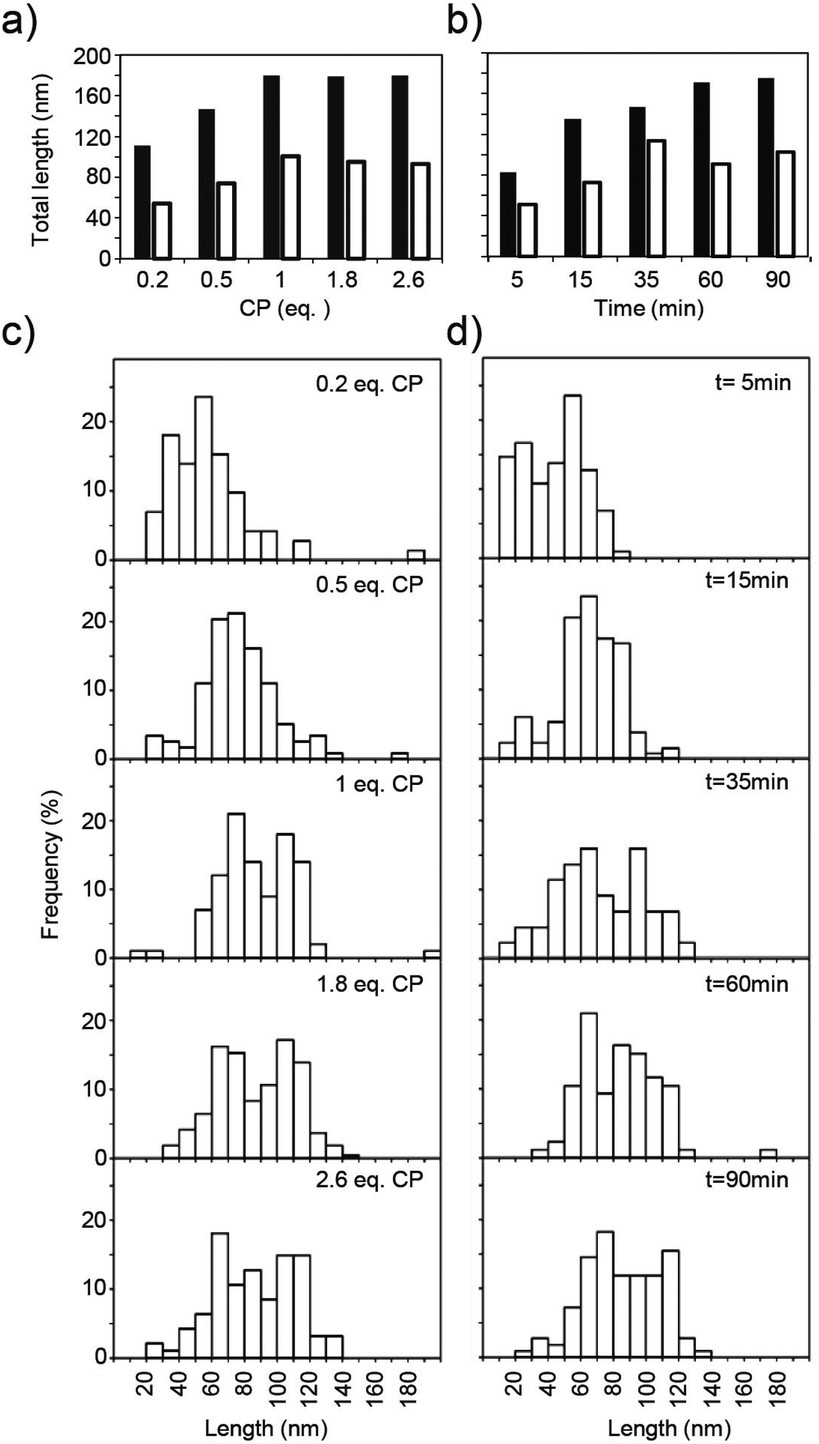 | ||
| Fig. 5 Partial assembly of TMV CP directed by the 3958 nts long RNA achieved with different equivalents (eq.) of CP after assembly for 16 h (a), or in a time course during assembly with 1.3 eq. of CP (b). The median values of the total nanotubular domain lengths of kinked (black bars) and straight (white bars) particles are depicted (a, b). The individual arm lengths of the nanoboomerangs are indicated by histograms (c, d). For data collection, TEM specimens were stained negatively, and for each preparation at least 100 particles were evaluated. | ||
To investigate nanostructure growth under saturation conditions, specimens with 1.3 eq. TMV CP were prepared at different time points after combining RNA and CP educts (Fig. 5b). For this protein concentration and 1.0 protein eq., nucleation had been determined to occur on 90% of the OAs within 30 s, and to reach 99% initiation efficiency within 60 s.29 With construct dupOAs3958, the total lengths of kinked and straight particles rose over time and reached their final values (180 or 100 nm) after 60 min (Fig. 5b). This time course was in good agreement with data obtained for the reassembly with native TMV RNA,64 taking into account the distinct velocity of 3′- and 5′-directed assembly as follows.
The typical bimodal arm length distribution of kinked particles was observed if more than 1 CP eq. was applied (Fig. 5c), and at 1.3 eq. of CP it took 35 min for appearance (Fig. 5d). Hence, an elongated arm was only formed if sufficient CP was supplied. For the smaller arm, the 5′ terminus-directed assembly can contribute maximally 54 nm to the total length (as calculated from the distance of OAs to the 5′ end of the RNA). With the 73 nm observed (Fig. 3c), 19 nm remain for the 3′-directed assembly of this arm, which is probably terminated by its collision with the longer nascent arm (Fig. 4c).
Given that upon bidirectional assembly, 5′-directed tube elongation is about 3 to 5 times faster than 3′-directed growth,33,64 the regular composition of boomerangs with unequal arms supports the conclusion that the longer arm is initiated at the second OAs in the construct (Fig. 3c).
Multiple origins of assembly
To find out if synchronous growth of interconnected arms is also possible for more complex RNA scaffolds, constructs containing more than two OAs sequences were generated. A circularized 640 bp PCR product of an OAs-containing cDNA portion was used as a template for rolling circle amplification (RCA65) to yield directly repeated sequences of this fragment. Partial enzymatic hydrolysis,66 electrophoretic separation and cloning of distinct products containing three, four and five repeats yielded plasmids which were used for in vitro transcription. For comparison, the linear monomeric PCR product was directly cloned in parallel. Thus, four RNA species were generated that differed in length and number of OAs-containing direct repeats (Table 1 and Fig. 6a). | ||
| Fig. 6 Transcripts with single or concatemeric OAs modules. Gel electrophoretic separation (a) and secondary structure prediction (b, c) of engineered RNA molecules obtained by in vitro transcription (a: denaturing 1% agarose gel). The secondary structures for four RNAs with 1 to 4 complete OAs [x] and zero or one split OAs [y] were predicted using mfold software62 with ΔG values of −196; −549; −728 and −895 kcal mol−1 at 37 °C, respectively (b). Complete OAs hairpins are framed with continuous lines, the splitOAs structures with dotted lines, and structure predictions shown in detail in (c). nx indicates numbers of complete OAs structures, ny numbers of split OAs structures, and ∑n the sum of both. In (c), compact arcs depict the nonanucleotide loop sequences; interrupted or dashed lines RNA strands stemming from 5′ or 3′ transcript portions, respectively. | ||
| Number of OAs | n x = | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| n y = | 0 | 1 | 1 | 1 | |
| ∑n = | 1 | 3 | 4 | 5 | |
| a n x – number of complete OAs structures; ny – number of split-OAs structures; ∑n = sum of both. b The expected nanotube lengths were calculated with the factor 0.0469 nm nt−1. c Maximal tube lengths were calculated for a single initiation event to encapsidate the whole RNA. d Minimal tube lengths were calculated for the case that all OAs modules were packaged simultaneously, thereby burying part of the RNA within the channel of the tube using the estimation of 0.33 nm nt−1 for the RNA length.67 | |||||
| RNA length (nts) | 726 | 2004 | 2644 | 3284 | |
| Tube length maximal (nm)c | 34 | 94 | 124 | 154 | |
| Tube length minimal (nm)d | 34 | 82 | 109 | 135 | |
Secondary structure prediction by in silico folding of the distinct molecules indicated that the OAs hairpins are exposed in the context of the repeats (Fig. 6b). Since the RCA products employed for construction of the plasmids were partially cleaved within one OAs sequence, this was split. Its 5′ portion yielded the 3′ terminus of the transcripts, and its 3′ portion with the OAs hairpin-nonanucleotide loop the very 5′ end of the corresponding RNA molecules (Fig. 6c). Both RNA portions are predicted to pair by hybridization, thereby attaining a conformation with a series of stem-loop and bulge structures similar to a complete OAs.
After incubating TMV CP with preparations of the RNAs with repetitive OAs modules, the majority of the resulting assemblies were mono-, di- and tripod shapes (Fig. 7). Even for RNA with only two complete and one split OAs (Fig. 7; nx = 2; ny = 1), about 40% of the particles were tripods (Fig. 7a and b; number of arms na = 3). Their total tube length corresponded to fully encapsidated RNA (Fig. 7b), indicating that the split OAs was able to initiate assembly at least to a certain extent. This conclusion was confirmed further by an RNA species containing only the incomplete split OAs, which induced nanotube formation at a significant frequency as well (data not shown).
 | ||
| Fig. 7 Structures and lengths of RNA-scaffolded mono- to tetrapods. The assembly of TMV CP with RNA molecules containing in total one, three, four and five copies of the OAs (∑n total number of OAs sequences; thereof: nx number of complete OAs; ny number of split OAs). TEM images (a) of specimens after negative staining, scale bars: 100 nm. Size distributions (b) represented in histogram (top) and the corresponding median total tube system lengths (bottom). na number of arms. Box plots as described in Fig. 3. At least 100 structures were examined with each specimen. Dotted lines mark the minimal and maximal tube length expected for complete RNA encapsidation (see Table 1). | ||
With more OAs modules (i.e. a sum ∑n of x + y ≥ 4), also structures with four arms (tetrapods) were formed (7% for 4 OAs, 10% for 5 OAs), but not a single five-armed one was found. Sterical hindrance, the multiplicity of aberrant folding events in five independent OAs sequences, or an inability to initiate or elongate five tubes on the specific construct may be the reasons.
The formation of TMV-derived dipod and tripod structures has been observed previously, applying scaffold populations with variable numbers of OAs sequences in RNA concatemers of more than 3 kb each. Those had been obtained by run-off transcription of covalently closed circular plasmids.61 In the current study, much shorter repetitive OAs modules of defined frequency allowed delimiting initiation and encapsidation efficiencies of multiple OAs-templates for the first time, as well as initial tests with four to five nucleation sites on single transcripts. For all scaffolds with more than two OAs sequences, complete encapsidation did only occur if at least the number of complete OAs sequences (x) was used for initiation (Fig. 7b; total lengths: stippled lines, and Table 1), e.g. the construct carrying three complete and one split OAs (nx = 3; ny = 1; ∑n = 4) was fully packaged only in tri- or tetrapods. In the case of both ∑n = 3 or 4, an additional nucleation event did not lead to a further elongated complete tube system, which might be attributed to the terminal positions of the split OAs segments. In turn, when initiation failed to take place in one or more complete OAs, RNA encapsidation was only partial and obviously impeded by conformational hindrance exerted by the non-utilized OAs sites. This is in agreement with our findings on dupOAs constructs carrying two native OAs sequences: with these templates, single initiation events resulted in continuous straight tubes (na = 1) of incomplete total length on a regular basis.
For future applications it might be possible to increase the efficiency of OAs-dependent initiation and thereby the proportion of higher-order branched systems growing on scaffolds with multiple OAs sites. It is worth to investigate if extended RNA segments between the individual OAs sequences and higher initial CP concentrations29 can raise the nucleation frequency of nanotubes which elongate prior to their collision with adjacently assembling domains. An interesting question is whether shortening the OAs sequence to its functionally important loop68 would help to reduce RNA misfolding in complex constructs and thus promote formation of more homogeneous products. Besides improving the reliability of the assembly reactions, also efficient sorting of distinct nanoarchitectures e.g. according to their sedimentation coefficients or diameters seems feasible by routine procedures.
Conclusion
A novel scaffolding set comprising RNA templates with two to five repetitive TMV assembly-nucleating sequence modules (OAs), combined at distinct positions on single RNA strands, allowed the RNA-guided assembly of complex kinked and branched proteinaceous nanotube systems in a predictable manner. Twofold initiation of TMV CP assembly on RNA molecules containing two OAs in a sterically favorable arrangement produced about 75% kinked particles with arms of the expected lengths, with a largely constant amount of straight and shortened particles formed in addition. Under conditions of educt saturation, their individual tube domains assembled simultaneously and induced kinking upon collision with each other. If the collision site's position could lead to pre-mature termination of tube growth, product heterogeneity increased. With multiple OAs modules on single template RNAs, up to four arms could be generated, with tripod yields of 30 to 40%. Fivefold initiation was not observed at all. Kinked69–71 and branched71–78 nanoarchitectures have gained increasing interest during recent years because they may exhibit exceptional electric,79 magnetic,80 plasmonic81 or placement82 properties. In combination with the ultrahigh density of selectively addressable surface-exposed reactive groups on the multivalent TMV coat, the fabrication of nanometric TMV-derived kinked and branched structures, as well as detailed insights into their assembly processes, might prove useful for the next generation of multi-purpose biotemplates.Experimental
Materials
Restriction enzymes and T4 DNA ligase were from NEB (Ipswich, USA), Taq polymerase from Qiagen (Hilden, Germany). In vitro transcription was performed with the MEGAScript® T7 High Yield Transcription kit (Ambion, Austin, USA). All enzymes were used according to the manufacturer's protocols unless otherwise stated. Primers were supplied by Metabion (Martinsried, Germany) and chemicals by Roth (Karlsruhe, Germany). T/A-cloning was performed with the pGEM-T vector system (Promega, Madison, USA). Plasmids were purified with a Plasmid Mini Kit or by elution from agarose gels with a QIAquick Gel Extraction Kit (both from Qiagen, Hilden, Germany). Sequencing made use of a CEQ™ 8000 Genetic Analysis System (Beckmann Coulter, Krefeld, Germany). For electron microscopy, copper and nickel grids (400 mesh, Science Services, Munich, Germany) were coated with Formvar©-carbon or collodium, respectively.83 A Tecnai G2 Sphera electron microscope (FEI, Hillsboro, USA) with a Tietz F214 (TVIPS, München, Germany) camera was used. Absorption spectra of protein, nucleic acid or virus preparations were determined with Nanodrop© instrumentation (Peqlab, Erlangen, Germany). Images were edited with Image J84 and statistic evaluation was performed with SigmaStat software (Systat, Erkrath, Germany).Cloning and in vitro transcription
pTMV-C (also named p843peTMVΔ9353-4411), a 4941 bp plasmid containing a truncated cDNA copy of TMV U1 genomic RNA corresponding to TMV RNA residues 4412–6395, was prepared by restriction hydrolysis of p843pe35TMVr185 with SnaBI and AatII followed by religation. pTMV-C served as template for polymerase chain reactions (PCR) amplifying cDNA portions containing the OAs, using Taq DNA polymerase (Qiagen, Hilden, Germany) and two pairs of primers (Table 2, #1 and #2; #3 and #4).| # | Name | Sequence 5′ to 3′ |
|---|---|---|
| 1 | FwA | AATTAACCCTCACTAAAGGG |
| 2 | RevA | CTCGAGTCATGCTAGCGCACCACGTGTGATTACG |
| 3 | FwB | GCTAGCATGACTCGAGGGGAGAGACGAATTCGAG |
| 4 | RevB | TAGCGGATCCCTTGCATGCCTGCAGGAATTCG |
| 5 | pGEMfw | CGCCAGGGTTTTCCCAGTCAC |
| 6 | pGEMrev | AGCGGATAACAATTTCACACA |
| 7 | AKOAsTMV5′BamHI5101 | TAGCGGATCCGTCTGTTTAGCCGGTTTGGTCG |
| 8 | SEI_CP_pTMV_QC_XhoIR | GATACTATAAGACATCTCGAGTTAAAACGAATCC |
The temperature profile was set to: 94 °C, 4 min; 94 °C, 30 s; 50 °C, 45 s; 72 °C, 2 min; 30 repeats of step 2–4; 72 °C, 10 min. The resulting PCR products (1893 or 2138 base pairs [bps]) were cut with XhoI, and, after purification, ligated with each other using T4 DNA ligase. The product was gel-purified, ligated into pGEM-T and propagated in Escherichia coli DH5α.86 Correct insertion of the target sequence was verified by sequencing with primers #5 and #6 (Table 2). The resulting recombinant plasmid contained a duplicated cDNA portion with two complete OAs regions, corresponding to residues 4412–6117 (plus an extension encompassing residues 6118–6395 of the 3′ module; see Fig. 2) of the total TMV RNA sequence, fused in addition to a 3′-terminal hammerhead ribozyme sequence. It was named pGEM/dupTMV_C+RZ. In order to extend the transcript length, the ribozyme-coding region at the 3′ end of the insert in pTMV-C was removed using BamHI and BsiWI digestion and religation, yielding the recombinant pGEM/dupTMV_C−RZ. The linearized plasmids (with NdeI for pGEM/dupTMV_C+RZ for RNA dupOAs3958; with ScaI for pGEM/dupTMV_C−RZ for RNA dupOAs5638) were transcribed in vitro with T7 RNA polymerase. Transcription of the linearized plasmid pGEM/dupTMV_C−RZ was performed at 30 °C to avoid pre-mature termination events which occurred at 37 °C, probably due to polyA-stretches at positions 4870 and 4903 in the pGEM vector backbone (data not shown).
To generate RNA molecules with multiple OAs copies, a fragment containing TMV cDNA residues 5101–5726 with both core (position 5420–5546) and extended (position 5313–5546) regions of the OAs26 was amplified by PCR using p843pe35TMVr.185 as template and primers #7 and #8 (Table 2), with 0.2 ng μl−1 template DNA, 0.5 pmol μl−1 of each primer, 50 mU μl−1 Pfu DNA polymerase (Fermentas, St. Leon-Rot, Germany), 0.8 mM dNTP mix, and a temperature profile of 94 °C, 5 min; 94 °C, 30 s; 55 °C, 30 s with a touchdown of 0.25 °C per cycle; 68 °C, 2 min; 25 repeats of step 2–4; 68 °C, 5 min. The 640 bps fragment (25 ng gel-purified in 10 μl) was circularized with T4 DNA ligase. Subsequent RCA was performed using 4 μl of the ligation product in 40 μl as template and a TempliPhi kit (GE Healthcare, Freiburg, Germany). Partial ApaI restriction (35 U; 20 μl RCA product in 100 μl; 15 min at room temperature and 90 s at 37 °C), inactivation of the enzyme and gel purification yielded fragments with 3, 4 or 5 repeats of the OAs module, which were ligated into ApaI-linearized pGEM-T using T4 DNA ligase and a 3-fold molar excess of the insert. For comparison, the PCR product was cloned into pGEM-T directly after dA-tailing with Taq DNA polymerase and dATP as described in the manufacturer's protocol. After chemically transforming E. coli DH 5α cells, plasmids were isolated and used for in vitro transcription after linearization with NdeI. It is worth to mention that recombination events in E. coli led to the deletion of copies of repeated fragments and shortening of the plasmids, but to an overall very low extent. No transcripts were detectable for these shortened plasmids. Full-length transcripts were precipitated with LiCl solution according to standard procedures,86 dissolved in RNase-free water and stored at a concentration of 1 mg ml−1 at −80 °C. The RNA concentration was determined spectrophotometrically using the extinction coefficient of 0.025 ml μg−1 cm−1 at 260 nm and 25 °C,87 and its integrity was checked on denaturing agarose gels.86,88
TMV CP preparation
TMV CP was prepared from infected plants. Nicotiana tabacum ‘Samsun’ nn plants were inoculated mechanically with TMV (strain vulgare; PV0107, DSMZ, Germany) and leaves harvested 30 days post inoculation. Virus particles were purified as described.89 CP from purified virions was obtained via acetic acid–based particle decomposition.90 In order to induce 20S disk formation,28 2 to 20 μg μl−1 CP was incubated in 50 mM sodium potassium phosphate (SPP) buffer at pH 7.2 for at least 48 hours at room temperature (RT). Before use for in vitro assembly experiments, the suspension was centrifuged for 20 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and RT to remove gross material. The purity and concentration of TMV CP was determined spectrophotometrically.18,91
000g and RT to remove gross material. The purity and concentration of TMV CP was determined spectrophotometrically.18,91
In vitro assembly reactions and electron microscopy
Typically 1.3 μg μl−1 CP (final concentration, f.c.) was mixed with 50 ng μl−1 RNA (f.c.) in 50 mM SPP buffer pH 7.2. After incubation for 16 h at 22 °C, assembly products were adsorbed onto Formvar©/carbon-coated copper grids and stained with 1% (w/v) uranyl acetate in the presence of 0.25 mg ml−1 bacitracin. For spreading of partially assembled structures to visualize free RNA stretches, samples from in vitro assembly reactions were diluted 25-fold in the spreading solution containing cytochrome c (0.1 mg ml−1 f.c.) and 60% (v/v) dimethyl sulfoxide (DMSO),92 and 1–5 μl drops were spread onto a water hypophase.63 The film developing at the water–air interface was adsorbed onto parlodium-coated nickel grids (300 mesh)83 and rotary-shadowed first with 2 nm platinum at an angle of 7.5° for contrasting, and subsequently with 5 to 10 nm carbon at an angle of 90° to stabilize the film.To evaluate length and shape of the particles assembled with RNA containing two OAs, about 100 randomly selected particles per experiment were measured using ImageJ software,84 and data displayed in histograms using Microsoft Excel. The length distributions were characterized regarding median values, quartiles, and maximum and minimum values. The median values of independent experiments were used to calculate an average with a standard deviation. Particles with multiple OAs modules were only evaluated with respect to the total lengths of their tube systems.
Acknowledgements
This work was supported by the Baden-Wuerttemberg-Stiftung, Network of Competence “Functional Nanostructures”, and the DFG (SPP1569). We thank Sigrid Kober for the preparation of TMV and our gardeners Annika Allinger and Diether Gotthardt for taking great care of the plants. We also like to thank Enrico DiPoto for creating first multiple OAs clones and Cornelia Kocher for her great support with knowledge and experience on nucleic acid spreading and shadowing. For help with the statistical analysis, we are grateful to Kathrin Richter. Discussions with Fania Geiger and Sven Degenhard have always been of a great help. We are also indebted to Prof. Stephan Nussberger and PD Dr Michael Schweikert for providing access to and taking care of the TEM facility.References
-
M. J. Adams, C. Heinze, A. O. Jackson, J. F. Kreuze, S. A. MacFarlane and L. Torrance, in Virus taxonomy. Classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses, ed. A. M. Q. King, M. J. Adams, E. B. Carstens and E. J. Lefkowitz, Elsevier/Academic Press, Amsterdam, Boston, Heidelberg, London, New York, Oxford, Paris, San Diego, San Francisco, Singapore, Sydney, Tokyo, 2012, pp. 1139–1162 Search PubMed
.
- D. J. Evans, Biochem. Soc. Trans., 2009, 37, 665–670 CrossRef CAS PubMed
- M. Fischlechner and E. Donath, Angew. Chem., Int. Ed., 2007, 46, 3184–3193 CrossRef CAS PubMed
- M. Young, D. Willits, M. Uchida and T. Douglas, Annu. Rev. Phytopathol., 2008, 46, 361–384 CrossRef CAS PubMed
- L. A. Lee, Z. W. Niu and Q. Wang, Nano Res., 2009, 2, 349–364 CrossRef CAS PubMed
- L. A. Lee, H. G. Nguyen and Q. Wang, Org. Biomol. Chem., 2011, 9, 6189–6195 CAS
- S. Y. Lee, J. S. Lim and M. T. Harris, Biotechnol. Bioeng., 2012, 109, 16–30 CrossRef CAS PubMed
- C. M. Soto and B. R. Ratna, Curr. Opin. Biotechnol., 2010, 21, 426–438 CrossRef CAS PubMed
- C. Mao, A. Liu and B. Cao, Angew. Chem., Int. Ed., 2009, 48, 6790–6810 CrossRef CAS PubMed
- J. K. Pokorski and N. F. Steinmetz, Mol. Pharm., 2011, 8, 29–43 CrossRef CAS PubMed
-
A. M. Bittner, J. M. Alonso, M. L. Górzny and C. Wege, in Structure and physics of viruses: an integrated textbook, ed. M. G. Mateu, Springer Science+Business Media, Dordrecht, 2013, vol. 68, pp. 667–702 Search PubMed
- F. Li and Q. Wang, Small, 2014, 10, 230–245 CrossRef CAS PubMed
- J. M. Alonso, M. Ł. Górzny and A. M. Bittner, Trends Biotechnol., 2013, 31, 530–538 CrossRef CAS PubMed
- P. Goelet, G. P. Lomonossoff, P. J. G. Butler, M. E. Akam, M. J. Gait and J. Karn, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 5818–5822 CrossRef CAS
- K. Namba, R. Pattanayek and G. Stubbs, J. Mol. Biol., 1989, 208, 307–325 CrossRef CAS
- D. K. Clare and E. V. Orlova, J. Struct. Biol., 2010, 171, 303–308 CrossRef CAS PubMed
- P. Ge and Z. H. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9637–9642 CrossRef CAS PubMed
- H. Fraenkel-Conrat and R. C. Williams, Proc. Natl. Acad. Sci. U. S. A., 1955, 41, 690–698 CrossRef CAS
- A. C. H. Durham and A. Klug, Nat. New Biol., 1971, 229, 42–46 CrossRef CAS
- P. J. G. Butler, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 537–550 CrossRef CAS PubMed
- D. L. D. Caspar and K. Namba, Adv. Biophys., 1990, 26, 157–185 CrossRef CAS
- Y. Okada, Adv. Biophys., 1986, 22, 95–149 CrossRef CAS
- D. Zimmern, Cell, 1977, 11, 463–482 CrossRef CAS
- P. J. G. Butler and A. C. Durham, J. Mol. Biol., 1972, 72, 19–24 CrossRef CAS
- P. J. G. Butler, A. C. Durham and A. Klug, J. Mol. Biol., 1972, 72, 1–18 CrossRef CAS
- D. Zimmern, EMBO J., 1983, 2, 1901–1907 CAS
- O. Jardetzky, K. Akasaka, D. Vogel, S. Morris and K. C. Holmes, Nature, 1978, 273, 564–566 CrossRef CAS
- P. J. G. Butler, J. Mol. Biol., 1972, 72, 25–28 CrossRef CAS
- P. J. G. Butler, J. Mol. Biol., 1974, 82, 343–353 CrossRef CAS
- H. Guilley, G. Jonard, B. Kukla and K. E. Richards, Nucl. Acids Res., 1979, 6, 1287–1308 CrossRef CAS PubMed
- D. R. Turner, C. J. McGuigan and P. J. G. Butler, J. Mol. Biol., 1989, 209, 407–422 CrossRef CAS
- Y. Otsuki, I. Takebe, T. Ohno, M. Fukuda and Y. Okada, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 1913–1917 CrossRef CAS
- P. J. G. Butler and G. P. Lomonossoff, Biophys. J., 1980, 32, 295–312 CrossRef CAS
- P. J. G. Butler, J. Gen. Virol., 1984, 65, 253–279 CrossRef CAS
- E. Dujardin, C. Peet, G. Stubbs, J. N. Culver and S. Mann, Nano Lett., 2003, 3, 413–417 CrossRef CAS
- O. K. Zahr and A. S. Blum, Nano Lett., 2012, 12, 629–633 CrossRef CAS PubMed
- R. A. Miller, N. Stephanopoulos, J. M. McFarland, A. S. Rosko, P. L. Geissler and M. B. Francis, J. Am. Chem. Soc., 2010, 132, 6068–6074 CrossRef CAS PubMed
- R. A. Miller, A. D. Presley and M. B. Francis, J. Am. Chem. Soc., 2007, 129, 3104–3109 CrossRef CAS PubMed
- M. Endo, M. Fujitsuka and T. Majima, Chemistry, 2007, 13, 8660–8666 CrossRef CAS PubMed
- M. A. Bruckman, S. Hern, K. Jiang, C. A. Flask, X. Yu and N. F. Steinmetz, J. Mater. Chem. B, 2013, 1, 1482–1490 RSC
- C. Y. Chiang, J. Epstein, A. Brown, J. N. Munday, J. N. Culver and S. Ehrman, Nano Lett., 2012, 12, 6005–6011 CrossRef CAS PubMed
- Z. Wu, A. Mueller, S. Degenhard, S. E. Ruff, F. Geiger, A. Bittner, C. Wege and C. Krill III, ACS Nano, 2010, 4, 4531–4538 CrossRef CAS PubMed
- J. M. Rego, J. H. Lee, D. H. Lee and H. Yi, Biotechnol. J., 2013, 8, 237–246 CrossRef CAS PubMed
- C. Hou, Q. Luo, J. Liu, L. Miao, C. Zhang, Y. Gao, X. Zhang, J. Xu, Z. Dong and J. Liu, ACS Nano, 2012, 6, 8692–8701 CrossRef CAS PubMed
- A. Mueller, F. J. Eber, C. Azucena, A. Petershans, A. M. Bittner, H. Gliemann, H. Jeske and C. Wege, ACS Nano, 2011, 5, 4512–4520 CrossRef CAS PubMed
- F. J. Eber, S. Eiben, H. Jeske and C. Wege, Angew. Chem., Int. Ed., 2013, 52, 7203–7207 CrossRef CAS PubMed
- Z. Yin, H. G. Nguyen, S. Chowdhury, P. Bentley, M. A. Bruckman, A. Miermont, J. C. Gildersleeve, Q. Wang and X. Huang, Bioconjugate Chem., 2012, 23, 1694–1703 CrossRef CAS PubMed
- F. C. Geiger, F. J. Eber, S. Eiben, A. Mueller, H. Jeske, J. P. Spatz and C. Wege, Nanoscale, 2013, 5, 3808–3816 RSC
- L. Chen, X. Zhao, Y. Lin, Y. Huang and Q. Wang, Chem. Commun., 2013, 49, 9678–9680 RSC
- A. Mueller, A. Kadri, H. Jeske and C. Wege, J. Virol. Methods, 2010, 166, 77–85 CrossRef CAS PubMed
- S. Eiben, N. Stitz, F. Eber, J. Wagner, P. Atanasova, J. Bill, C. Wege and H. Jeske, Virus Res., 2014, 180, 92–96 CrossRef CAS PubMed
- M. T. Dedeo, K. E. Duderstadt, J. M. Berger and M. B. Francis, Nano Lett., 2010, 10, 181–186 CrossRef CAS PubMed
- M. A. Bruckman, C. M. Soto, H. McDowell, J. L. Liu, B. R. Ratna, K. V. Korpany, O. K. Zahr and A. S. Blum, ACS Nano, 2011, 5, 1606–1616 CrossRef CAS PubMed
- A. D. Brown, L. Naves, X. Wang, R. Ghodssi and J. N. Culver, Biomacromolecules, 2013, 14, 3123–3129 CrossRef CAS PubMed
- J. Atabekov, N. Nikitin, M. Arkhipenko, S. Chirkov and O. Karpova, J. Gen. Virol., 2011, 92, 453–456 CrossRef CAS PubMed
- O. Karpova, N. Nikitin, S. Chirkov, E. Trifonova, A. Sheveleva, E. Lazareva and J. Atabekov, J. Gen. Virol., 2012, 93, 400–407 CrossRef CAS PubMed
- C. Azucena, F. J. Eber, V. Trouillet, M. Hirtz, S. Heissler, M. Franzreb, H. Fuchs, C. Wege and H. Gliemann, Langmuir, 2012, 28, 14867–14877 CrossRef CAS PubMed
- A. Kadri, C. Wege and H. Jeske, J. Virol. Methods, 2013, 189, 328–340 CrossRef CAS PubMed
- I. B. Kaplan, Y. V. Kozlov, E. S. Pshennikova, M. E. Taliansky and J. G. Atabekov, Virology, 1982, 118, 317–323 CrossRef CAS
- M. E. Taliansky, I. B. Kaplan, L. V. Yarvekulg, T. I. Atabekova, A. A. Agranovsky and J. G. Atabekov, Virology, 1982, 118, 309–316 CrossRef CAS
- D. R. Gallie, K. A. Plaskitt and T. M. A. Wilson, Virology, 1987, 158, 473–476 CrossRef CAS
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed
- A. K. Kleinschmidt, D. Lang, D. Jacherts and R. K. Zahn, Biochim. Biophys. Acta, 1962, 61, 857–864 CAS
- M. Fukuda, T. Ohno, Y. Okada, Y. Otsuki and I. Takebe, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 1727–1730 CrossRef CAS
- D. Haible, S. Kober and H. Jeske, J. Virol. Methods, 2006, 135, 9–16 CrossRef CAS PubMed
- P. S. Wyant, S. Kober, A. Schwierzok, C. Kocher, B. Schäfer, H. Jeske and C. Wege, Virus Res., 2012, 167, 397–403 CrossRef CAS PubMed
- A. Nicolaieff, L. Pinck, A. M. Koenig-Nikes and L. Hirth, J. Gen. Virol., 1972, 16, 47–59 CrossRef CAS PubMed
- D. R. Turner and P. J. G. Butler, Nucleic Acids Res., 1986, 14, 9229–9242 CrossRef CAS PubMed
- M. G. Ryadnov and D. N. Woolfson, Nat. Mater., 2003, 2, 329–332 CrossRef CAS PubMed
- Y. J. Hyun, A. Lugstein, M. Steinmair, E. Bertagnolli and P. Pongratz, Nanotechnology, 2009, 20, 125606 CrossRef PubMed
- W. T. Yao, S. H. Yu, S. J. Liu, J. P. Chen, X. M. Liu and F. Q. Li, J. Phys. Chem. B, 2006, 110, 11704–11710 CrossRef CAS PubMed
- M. G. Ryadnov and D. N. Woolfson, Angew. Chem., Int. Ed., 2003, 42, 3021–3023 CrossRef CAS PubMed
- K. A. Dick, K. Deppert, M. W. Larsson, T. Martensson, W. Seifert, L. R. Wallenberg and L. Samuelson, Nat. Mater., 2004, 3, 380–384 CrossRef CAS PubMed
- K. A. Dick, K. Deppert, L. S. Karlsson, W. Seifert, L. R. Wallenberg and L. Samuelson, Nano Lett., 2006, 6, 2842–2847 CrossRef CAS PubMed
- K. A. Dick, S. Kodambaka, M. C. Reuter, K. Deppert, L. Samuelson, W. Seifert, L. R. Wallenberg and F. M. Ross, Nano Lett., 2007, 7, 1817–1822 CrossRef CAS PubMed
- S. Kar, B. N. Pal, S. Chaudhuri and D. Chakravorty, J. Phys. Chem. B, 2006, 110, 4605–4611 CrossRef CAS PubMed
- C. Lai, Q. Wu, J. Chen, L. Wen and S. Ren, Nanotechnology, 2010, 21, 215602 CrossRef PubMed
- F. Oehler, P. Gentile, T. Baron, M. D. Hertog, J. Rouviere and P. Ferret, Nanotechnology, 2009, 20, 245602 CrossRef CAS PubMed
- T. Markussen, A. P. Jauho and M. Brandbyge, Phys. Rev. Lett., 2009, 103, 055502 CrossRef
- Y. Chu, J. Hu, W. Yang, C. Wang and J. Z. Zhang, J. Phys. Chem. B, 2006, 110, 3135–3139 CrossRef CAS PubMed
- A. Guerrero-Martinez, S. Barbosa, I. Pastoriza-Santos and L. M. Liz-Marzan, Curr. Opin. Colloid Interface Sci., 2011, 16, 118–127 CrossRef CAS PubMed
- K. Risveden, K. A. Dick, S. Bhand, P. Rydberg, L. Samuelson and B. Danielsson, Nanotechnology, 2010, 21, 055102 CrossRef PubMed
-
J. Sommerville and U. Scheer, Electron microscopy in molecular biology a practical approach, IRL Press, Oxford, 1987 Search PubMed
-
W. S. Rasband, ImageJ, Program for image processing, US National Institutes of Health, Bethesda, Maryland, USA, 1.43 edn, 1997–2010, http://rsb.info.nih.gov/ij/ Search PubMed
- A. Kadri, E. Maiss, N. Amsharov, A. M. Bittner, S. Balci, K. Kern, H. Jeske and C. Wege, Virus Res., 2011, 157, 35–46 CrossRef CAS PubMed
-
J. Sambrook and D. W. Russell, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 3rd edn, 2001 Search PubMed
-
F. Lottspeich, Bioanalytik, Elsevier, München, 2nd edn, 2006 Search PubMed
- Y. C. Liu and Y. C. Chou, Biotechniques, 1990, 9, 558 CAS
- G. V. Gooding and T. T. Hebert, Phytopathology, 1967, 1285 Search PubMed
- H. Fraenkel-Conrat, Virology, 1957, 4, 1–4 CrossRef CAS
- R. Jaenicke and M. A. Lauffer, Biochemistry, 1969, 8, 3077–3082 CrossRef CAS
- G. Lebeurier, A. Nicolaieff and K. E. Richards, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 149–153 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2015 |