
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interactions between lipid-free apolipoprotein-AI and a lipopeptide incorporating the RGDS cell adhesion motif
V.
Castelletto†
*a,
I. W.
Hamley
a,
M.
Reza
b and
J.
Ruokolainen
b
aDepartment of Chemistry, University of Reading, Whiteknights, Reading, RG6 6AD, UK. E-mail: I.W.Hamley@reading.ac.uk
bDepartment of Applied Physics, Aalto University School of Science, P.O. Box 15100 FI-00076 Aalto, Finland
First published on 23rd October 2014
Abstract
The interaction of a designed bioactive lipopeptide C16-GGGRGDS, comprising a hexadecyl lipid chain attached to a functional heptapeptide, with the lipid-free apoliprotein, Apo-AI, is examined. This apolipoprotein is a major component of high density lipoprotein and it is involved in lipid metabolism and may serve as a biomarker for cardiovascular disease and Alzheimers’ disease. We find via isothermal titration calorimetry that binding between the lipopeptide and Apo-AI occurs up to a saturation condition, just above equimolar for a 10.7 μM concentration of Apo-AI. A similar value is obtained from circular dichroism spectroscopy, which probes the reduction in α-helical secondary structure of Apo-AI upon addition of C16-GGGRGDS. Electron microscopy images show a persistence of fibrillar structures due to self-assembly of C16-GGGRGDS in mixtures with Apo-AI above the saturation binding condition. A small fraction of spheroidal or possibly “nanodisc” structures was observed. Small-angle X-ray scattering (SAXS) data for Apo-AI can be fitted using a published crystal structure of the Apo-AI dimer. The SAXS data for the lipopeptide/Apo-AI mixtures above the saturation binding conditions can be fitted to the contribution from fibrillar structures coexisting with flat discs corresponding to Apo-AI/lipopeptide aggregates.
Introduction
Apolipoproteins are proteins that bind lipids, and are involved in lipid transport in vivo. There are six classes of apolipoprotein with different structures and biological activities. The apoliprotein A family is a major component of high density lipoprotein (HDL). HDL removes fat and cholesterol from cells and therefore there is significant research interest in the interaction of Apo-A apolipoproteins with lipids due to biomedical relevance, in particular to cardiovascular disease. Among this class, Apo-AI is the major component of HDL, comprising 70% of total protein.1 Polymorphism in the gene associated with Apo-AI has been associated with early onset Alzheimer's disease.2The structure of Apo-AI comprises two domains – the N terminal (residue 1–186) domain comprises a four-helix bundle of amphipathic α-helices that result from 11- and 12-residue repeats, whilst the C terminal domain comprises two further helices, when bound to lipid although less ordered in the absence of lipid.3–5 Apolipoproteins such as Apo-AI may adopt a necklace-like structure6 (Fig. 1) and these are able to wrap around bundles of lipid molecules to form so-called “nanodiscs”.7,8
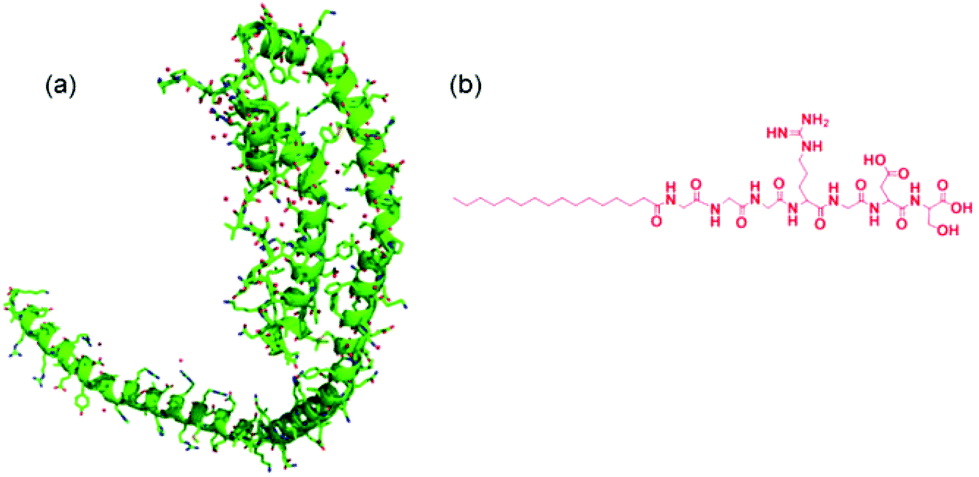
The interaction of Apo-AI with various lipids has been the subject of several studies which indicate that binding between the apolipoprotein and lipids leads to enhanced α-helix content.9–11 The binding is believed to be achieved via the C-terminal α-helices in Apo-AI5,10 which insert into lipid membranes. There is a concomitant increase in α-helical content in the C terminal domain along with a second step conformational transition in the N terminal helix bundle exposing the helices to lipid.12 Binding leads to the formation of HDL-like aggregates including nascent discoidal particles and circulating spherical particles.12–14
Here, we investigate for the first time to our knowledge, the interaction of Apo-AI with a designed lipopeptide. We selected the lipopeptide C16-GGGRGDS (G: Glycine, R: Arginine, D: Aspartic Acid and S: Serine) because firstly it contains a bioactive cell adhesion motif (RGDS) which has been exploited by us in applications in cell culture supports for tissue engineering.15,16 Similar lipopeptides have been used by others in materials for tissue engineering and other cell-based therapeutic materials.17,18 Second, its self-assembly has been extensively characterized by ourselves19 and that of similar peptide amphiphiles has been extensively examined by others.20
Experimental
Materials
Apolipoprotein A-I (Apo-AI), a high density lipoprotein extracted from human plasma, was purchased from Calbiochem (US) and received as a stock solution containing 53.6 μM Apo-AI in 10 mM NH4HCO3 (pH 7.4). The Apo-I molecular weight (Mw) is 28 kDa. Lipopeptide C16-GGGRGDS was custom synthesized by CS Bio (Menlo Park, USA) as a TFA salt. The purity was 95.46% (by analytical HPLC in a TFA water/acetonitrile gradient). MW was obtained by electrospray-mass spectrometry. Mw,Found = 842.99 and Mw,Expected = 842.40.Sample preparation
53.6 μM Apo-AI stock solution was diluted in 10 mM Tris, 0.1 mM NaCl (pH 7.4), while aqueous C16-GGGRGDS solutions were prepared at pH 8, by titrating 0.1 M NaOH. Binary samples were made by mixing weighed solutions of Apo-AI and C16-GGGRGDS at different molar ratios Mr = [C16-GGGRGDS]/[Apo-AI].Circular dichroism (CD)
Spectra were recorded using a Chirascan spectropolarimeter (Applied Photophysics, UK). The sample was placed in a cover slip cuvette (0.01 cm thick). Spectra are presented with absorbance A < 2 at any measured point with a 0.5 nm step, 1 nm bandwidth, and 1 second collection time per step at 20 °C. The post-acquisition smoothing tool from Chirascan software was used to remove random noise elements from the averaged spectra. A residual plot was generated for each curve in order to verify whether or not the spectrum had been distorted during the smoothing process. The CD signal from the water was subtracted from the CD data of the peptide solutions. The spectra were collected as ellipticity θ as a function of wavelength and converted to mean residue molar ellipticity [θ] = θ/(l10c) using the sample molar concentration c = 243 × c1 + 7 × c2 (c1: molar concentration of Apo-AI, 243: number of residues on Apo-AI, c2: molar concentration of C16-GGGRGDS, 7: number of residues on C16-GGGRGDS).Small-angle X-ray scattering (SAXS)
Experiments were performed on beamline BM29 at the ESRF (Grenoble, France). A few microlitres of samples were injected via an automated sample exchanger at a slow and very reproducible flux into a quartz capillary (1.8 mm internal diameter), which was then placed in front of the X-ray beam. The quartz capillary was enclosed in a vacuum chamber, in order to avoid parasitic scattering. After the sample was injected in the capillary and reached the X-ray beam, the flow was stopped during the SAXS data acquisition. The sample was thermostated throughout its entire travel from the injector to the quartz capillary. SAXS experiments were performed at 20 °C. The q = 4πsinθ/λ range was set to 0.04–4 nm−1, with λ = 0.1 nm (12 keV). The images were captured using a PILATUS 1M detector. Data processing (background subtraction, radial averaging) was performed using dedicated beamline software ISPYB.SAXS theory
The SAXS data for the 10 μM Apo-AI (Mr = 0) was modelled using the software Crysol (Version 2.8 ©ATSAS team 1995–2011).21,22 Crysol evaluates the solution scattering from macromolecules with known atomic structure. In this work, we used the atomic coordinates for the structure of C Terminal Truncated Human Apolipoprotein A-I listed in the Protein Databank File pdb file 3R2P.23 The crystal structure is displayed in Fig. 1a. SAXS data for samples with Mr = 2.2–14.9 was modelled according to a co-existence of flat cylinders, with diameter D and length L, with bilayer tapes described as Gaussian bilayers interacting through the Caillé structure factor for multi-layer systems. The details of the model used to describe the bilayers are provided elsewhere.19,24–26 The model assumes an electron density profile comprising one Gaussian function for each headgroup on either side of the bilayer electron density (ρH) profile, and one Gaussian function for the chains in the core of the bilayer electron density (ρC) profile. The position of the Gaussian peaks is at zH and zC for ρH and ρC respectively. The model assumes a standard deviation σH and σC for zH and zC respectively, while the bilayer is centred at z = zC = 0. We used a Gaussian distribution of zH, with associated degree of polydispersity ΔZH. The background was taken to be a constant C0. The fitting parameters of the model are zH, ρH, σH, ρC, σC and C0. The modified Caillé theory appropriate for lamellar systems corresponds to a multilayer structure influenced by thermal fluctuations. It is described by the total number of layers N, the layer spacing d and the Caillé parameter η.25,26Cryo-transmission electron microscopy (cryo-TEM)
Experiments were carried out using a field emission cryo-electron microscope (JEOL JEM-3200FSC) operating at 200 kV. Images were taken using bright-field mode and zero loss energy filtering (omega type) with a slit with 20 eV. Micrographs were recorded using a Gatan Ultrascan 4000 CCD camera. The specimen temperature was maintained at −187 °C during the imaging. Vitrified specimens were prepared using an automated FEI Vitrobot device using Quantifoil 3.5/1 holey carbon copper grids with 3.5 μm hole sizes. Grids were cleaned using a Gatan Solarus 9500 plasma cleaner just prior to use and then transferred into an environmental chamber of an FEI Vitrobot at room temperature and 100% humidity. Thereafter, 3 μl of sample solution at 2 wt% concentration was applied on the grid, blotted once for 1 second and then vitrified in a 1/1 mixture of liquid ethane and propane at −180 °C. Grids with vitrified sample solutions were maintained in a liquid nitrogen atmosphere and then cryo-transferred into the microscope.Transmission electron microscopy (TEM)
TEM imaging was performed using a Philips CM20 TEM microscope operated at 200 kV. Droplets of solutions were placed on Cu grids coated with a carbon film (Agar Scientific, UK), stained with 1 wt% uranyl acetate, and air-dried.Isothermal titration calorimetry (ITC)
ITC experiments were carried out using an iTC200 microcalorimeter. The Apo-AI stock solution (53.6 μM Apo-AI diluted in 10 mM NH4HCO3, pH 7.4) was dissolved to give a 10.7 μM solution of Apo-AI using 10 mM Tris, 0.1 mM NaCl, pH 7.4. The working cell was filled with 200 μL of the 10.7 μM Apo-AI solution and the reference cell was filled with deionized water. The titrant syringe was filled with a solution of 1.54 mM C16-GGGRGDS (dissolved in the same buffer used for the Apo-AI solution in the working cell). The ITC experiment was programmed to run 18 injections of 2 μL volume of the titrant solution (1.54 mM C16-GGGRGDS) into the working cell (10.4 μM of Apo-AI) with 180 s lag between each injection to ensure return to the baseline. The syringe was stirred throughout the experiment at 750 rpm and the working cell was set at 25 °C. The data was analysed using Origin 7 (MicroCal) by fitting the curve using the one set of sites model.Results
To first probe the possible interactions between Apo-AI and C16-GGGRGDS (Fig. 1 shows, respectively the structure of Apo-AI from X-ray crystallography and a molecular model of the PA) we employed isothermal titration calorimetry (ITC), a well-known technique to enable investigation of the molecular interactions of biological samples, in this case a protein-lipopeptide interaction. The experiment was designed to inject 1.54 mM C16-GGGRGDS into 53.6 μM Apo-AI, i.e. C16-GGGRGDS is the ligand and Apo-AI is the receptor. The relatively low Apo-AI concentration selected for ITC reduces the total number of binding sites to ensure saturation of C16-GGGRGDS binding to Apo-AI is reached. The ITC profile for the titration of C16-GGGRGDS into Apo-AI (Fig. 2a) exhibits a series of positive peaks, indicating an endothermic reaction. The integration and normalization of the endothermic peaks produced a sigmoidal curve relative to the moles of ligand added, which was then appropriately fitted to a one set of sites model. The one set of sites model was used for the peptide/lipopetide system as it is assumed that each binding site has the same binding affinity. A binding constant K = (3.21 ± 1.7) × 106 M−1 was determined from the fitting parameters (Fig. 2b). Other parameters including the number of binding sites N = (0.59 ± 0.04), enthalpy ΔH = (6.6 ± 0.6) kcal mol−1 and entropy ΔS = 0.05 Kcal mol−1 °C−1 were also obtained from the fit.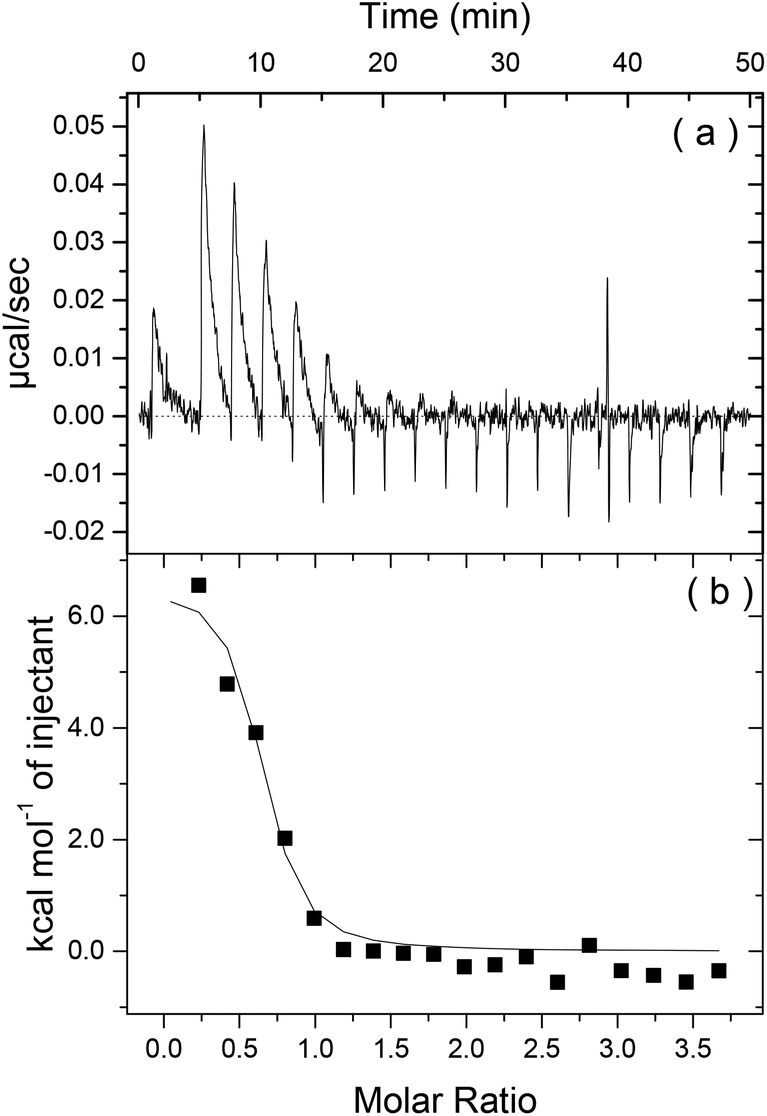 | ||
| Fig. 2 (a) Heat release as a function of time and (b) energy released as a function of the molar ratio for Apo-I titrated with C16-GGGRGDS. | ||
Fig. 2b shows that the system reaches saturation at Mr = 1.2, because only heat of dilution is observed for Mr > 1.2. ΔH reflects the strength of the ligand/target interaction enthalphy relative to that with solvent, primarily due to hydrogen bond formation and van der Waals interactions. It is highly unlikely that an endothermic reaction (ΔH > 0), is dominated by hydrogen bonding or van der Waals interactions. The isotherm corresponds to a favourable change in entropy (−TΔS < 0). Therefore it is possible that Apo-AI/C16-GGGRGDS binding is driven by hydrophobic interactions. The ITC signal is a result of many interactions – not just the binding process of Apo-AI to the lipopeptide but also potential protein conformational changes upon dilution, lipopeptide/lipopeptide and hydration effects. However, an unfavourable ΔH associated with an endothermic process is possibly due to conformational constraints of the Apo-AI.27
To our knowledge, there are no prior reports on binding of Apo-AI to lipopeptides. ITC experiments have been mostly performed to study the binding of lipids to Apo-AI. Although these experiments show that lipids bind to Apo-AI at room temperature through an exothermic reaction,10,28–30 one report shows an endothermic reaction for the titration of Apo-AI into 1-palmitoyl,2-oleoyl phosphatidylcholine/sphingomyelin small unilamellar vesicles at 37 °C.29
Having determined Mr = 1.2 as the saturation limit for Apo-AI/C16-GGGRGDS binding at 10.7 μM protein (Fig. 2), further studies were performed for a fixed concentration of 10.7 μM Apo-AI, to allow for comparison with results from ITC experiments.
The influence of interactions between Apo-AI and the lipopeptide C16-GGGRGDS on the secondary structure of the lipopeptide were examined by CD spectroscopy. CD spectra for mixtures with Mr = 2.2–14.4 are shown in Fig. 3a. Fig. 3b shows the dependence with Mr of the ellipticity at the minima at 209 and 221 nm. Again, saturation is observed above equimolar conditions. The results in Fig. 3 show that the amount of alpha-helix secondary structure decreases upon increasing Mr. Previous studies have shown that human Apo-AI has high α-helical content, values between 43%–68% having been reported,1,9–11,31 in good agreement with the CD for pure Apo-AI in Fig. 3a. To further evaluate whether interactions between Apo-AI and C16-GGGRGDS modify the secondary structure, we compared measured CD spectrum with those calculated by the addition of the spectra for the individual components. Fig. 4 is a representative comparison showing a large difference between the measured and calculated spectra for one of the mixtures studied (Mr = 4.5). In this case, the calculated spectra corresponds to the one-to-one addition of the CD spectrum measured for 10.7 μM Apo-AI to the CD measured for 48 μM C16-GGGRGDS (Mr = 48/10.7 = 4.5). The calculated CD spectra for Mr = 4.5 (Fig. 4) presents a maxima at 193 nm and a smaller minimum at 225 nm. This spectra is similar to that reported by us for C16-KTTKS32 and 154 μM C16-GGGRGDS.19 Our interpretation for the features in the calculated CD spectra for Mr = 4.5 (Fig. 4) is that they result from red-shifted β-sheet peaks associated with the lipopeptide fibrils,33 the expected maximum for this structure being around 195–200 nm and the minimum being near 216 nm. In contrast, the CD spectra measured for Mr = 4.5 (Fig. 3–4) is dominated by an α-helical structure. Fig. 3 and Fig. 4 together indicate that there are interactions between Apo-AI and C16-GGGRGDS which modify the secondary structure of each component, specifically there appears to be a decrease in α-helix content of Apo-AI and a reduction of β-sheet content of C16-GGGRGDS.
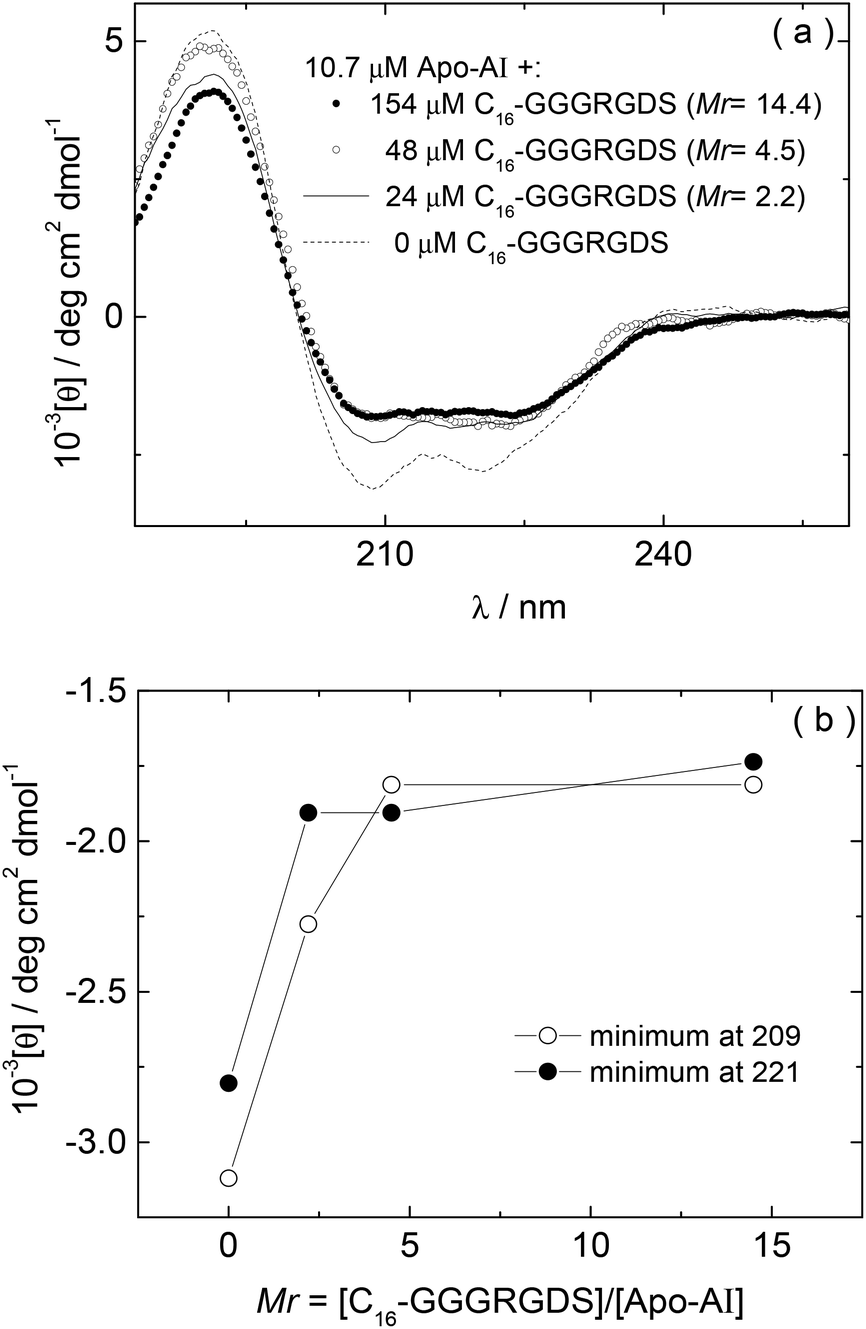 | ||
| Fig. 3 (a) CD for Apo-AI/C16-GGGRGDS mixtures measured as a function of Mr, (b) Dependence on Mr of the CD minima at 209 nm and 221 nm measured in (a). | ||
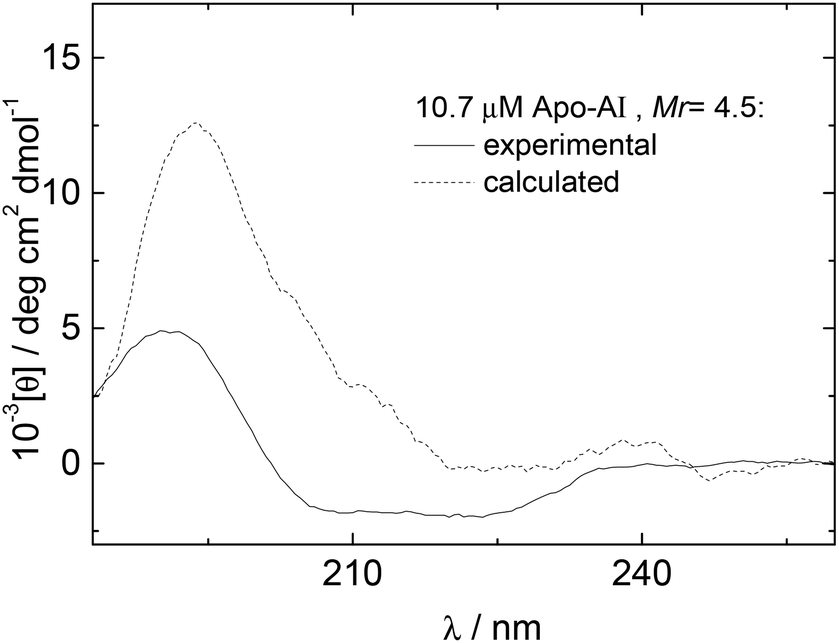 | ||
| Fig. 4 (- - -) Experimental and (–––) calculated CD data for Mr = 4.5. | ||
According to the results from CD, the α-helical content of Apo-AI progressively reduces upon addition of C16-GGGRGDS. In this way, the effect of lipopeptide titration into the Apo-AI solution is opposite to the interaction of Apo-AI with various lipids, in which case binding between the apolipoprotein and lipids leads to enhanced α-helix content.9–11 However, the results from CD are in good agreement with ITC (Fig. 2), because it is possible that Apo-AI forms entropically favoured structures (such as aggregates) that might lead to the endothermic reaction measured by ITC experiments.
Cryo-TEM was used to image self-assembled structures in mixtures of Apo-AI and C16-GGGRGDS above the saturation condition (Mr> 1). Fig. 5 shows cryo-TEM images measured for samples with 10.4 μM of Apo-AI and Mr = 2.2, 4.4 and 14.4. For the sample with Mr = 2.2, thin fibrils can be seen (Fig. 5a) with a diameter of approximately 10 nm. The fibrils are highly extended, with lengths up to tens of microns. Upon increasing the content of C16-GGGRGDS in the solutions, the fibrils become more twisted as evident in Fig. 5b for Mr = 4.4. Of particular note, cryo-TEM shows a co-existence of fibrils and spheroidal aggregates for Mr = 2.2–14.4 (Fig. 5). The average size of these aggregates was (17.8 ± 4.2) nm, and did not show a particular dependence with Mr.
 | ||
| Fig. 5 Cryo-TEM images measured for samples with 10.4 μM of Apo-AI and Mr = (a) 2.2, (b) 4.4 and (c) 14.4. The insets in (a) and (c) show details of the aggregates observed for Mr = 2.2 and 14.4 respectively. | ||
The aggregates shown in Fig. 5a–b have similar sizes to the 13 nm and ∼(11.1–13) nm diameter nanodiscs formed through complexation of Apo-E with dipalmitoylphosphatidylcholine34 and Human Apo-AI with dimyristoylphosphatidylcholine respectively.7,34 Therefore, it is likely that the aggregates displayed in Fig. 5a–c arise from the complexation of the Apo-AI with C16-GGGRGDS to form nanodiscs. This was further probed by SAXS (vide infra).
TEM experiments were performed for control solutions of C16-GGGRGDS. The images displayed in Fig. 6 are for the lipopeptide solutions, with the same concentrations used to prepare solutions with Mr = 2.2, 4.4 and 14.4. Extended fibrils can be observed for all three concentrations, without amorphous aggregates in contrast to the mixtures with Apo-AI (Fig. 5). The formation of fibres is consistent with the low critical aggregation concentration for C16-GGGRGDS, determined to be ∼59 μM (in water).19 Control experiments with Apo-AI only revealed no observable structures in TEM images at 10.7 μM concentration. It is evident that the irregular circular aggregates observed in Fig. 5 originate from the peptide/protein binding, since they are absent in Fig. 6.
 | ||
| Fig. 6 TEM images measured for (a) 24, (b) 47 and (c) 154 μM C16-GGGRGDS. (a), (b) and (c) are the control peptide solutions for the mixture samples with Mr = 2.2, 4.4 and 14.4 respectively. | ||
Consequently, the circular objects measured in Fig. 5 might be closely related to the nanodisc structures observed in previous lipid/Apo-AI binding studies.7,8
Electron microscopy imaging was complemented with SAXS experiments. The SAXS curves measured for 10 μM Apo-AI and Apo-AI/C16-GGGRGDS mixtures above the saturation condition (Mr> 1) are displayed in Fig. 7. The resulting intensity profiles provide information on sample-averaged solution nanostructures, via analysis of the form factor. The form factor models are described in the Experimental section.
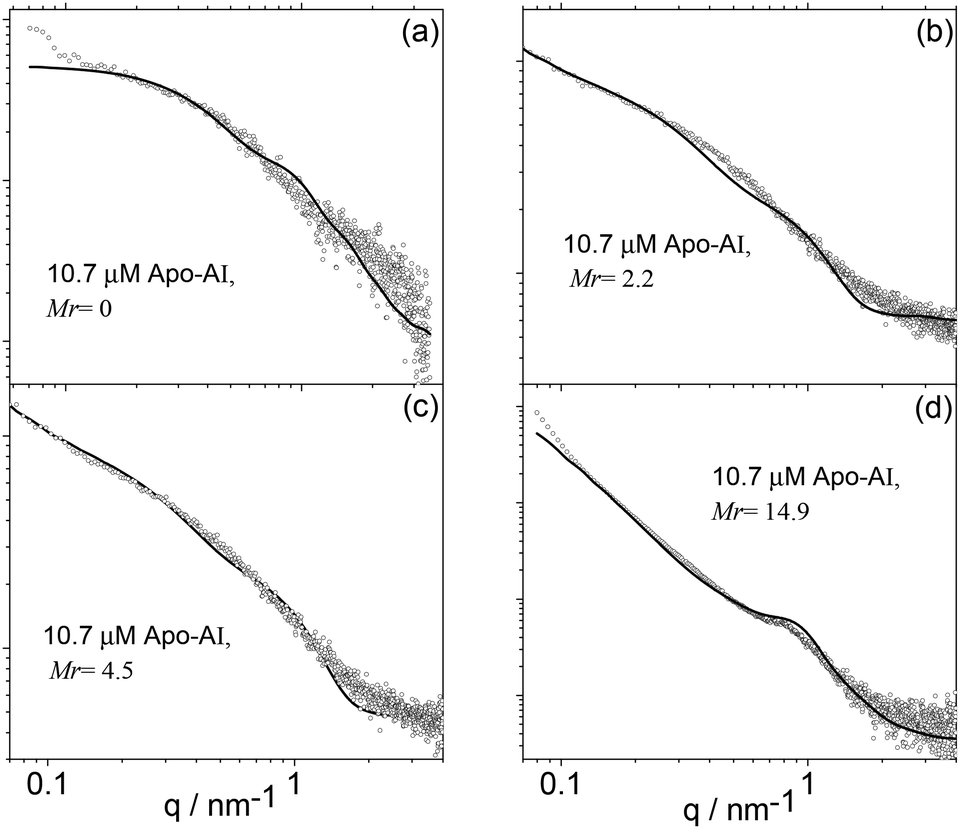 | ||
| Fig. 7 SAXS data (symbols) along with calculated form factor profiles (full lines) according to the models described in the text. (a) Mr = 0, (b) Mr = 2.2, (c) Mr = 4.5 and (d) Mr = 14.9 (Table 1). | ||
The form factor of Apo-AI (Fig. 6a) was computed from the published crystal structure as described in the SAXS Theory section and displayed in Fig. 1a. It agrees very well with the data. It has been shown that the Apo-AI is not monomeric for concentrations higher than 0.1 μM.35 The crystallographic structure used to fit the experimental data corresponds to dimers of the C-terminal truncated apolipoprotein.23 Indeed, attempts to model the observed SAXS data using published crystal structures for several monomeric forms of Apo-AI6,36 were not successful. The presence of other oligomeric structures in the sample, as reported on the basis of analytical ultracentriguation experiments,35 is highly probable, but a dimer model proved sufficient to fit the SAXS data.
Apparently, the incomplete loop structure of the Apo-AI dimer shown in Fig. 1a is, in the main, not able to wrap around the lipopeptide molecules to produce regular nanodiscs. However, cryo-TEM results show a few “nanodisc” structures in equilibrium with peptide nanofibres, suggesting that there is a dynamic competition between lipopeptide self-assembly and lipopeptide binding to Apo-AI.
Therefore, the SAXS data for Mr = 2.2–14.9 was modelled as a co-existence of flat cylinders with nanotape-like structures, which accounts for the mixture of extended fibrils and disk-like objects imaged/revealed by cryo-TEM (Fig. 5). The form factor for flat discs represents the Apo-AI/lipopetide aggregates, while the nanotape-like structure represents the C16-GGGRGDS fibrils. The nanotape-like structure can be described using a lipid-like bilayer form factor in which the bilayer is represented by three Gaussian functions (one for the electron-depleted alkyl chain region and two for the headgroups at the surface of each bilayer) and has been previously used by us to describe C16-GGGRGDS and related self-assembling peptide amphiphiles (PAs).19 A structure factor for lamellar systems is used to account for the stacking of the bilayers within the PA tapes. The details of the form factors are given in the Experimental section. The SAXS data is displayed in Fig. 7b–d, together with the fitted curves using the corresponding form factors. The parameters obtained from the SAXS fitting are listed in Table 1.
| M r | D [Å] | L [Å] | 2lT [Å] | 104 × ρH [rel. u.] | σ H [Å] | 103 × ρC [rel. u.] | σ C [Å] | Co [a. u.] |
|---|---|---|---|---|---|---|---|---|
| a N = 2, d = 65 Å and η = 0.4 in structure factor used only for Mr = 14.9. | ||||||||
| 2.2 | 106 ± 34 | 11.8 | 65 ± 5 | 1.9 | 2.5 | −2.6 | 5 | 6 |
| 4.5 | 108 ± 34 | 11.8 | 65 ± 5 | 1.8 | 2.5 | −2.7 | 5 | 4.5 |
| 14.9 | 108 ± 34 | 11.2 | 70 ± 5 | 0.09 | 5 | −1.5 | 5 | 0.4 |
The diameter of the flat discs used to fit data in Fig. 7b–d agrees with the size of the circular objects measured from the cryo-TEM images in Fig. 5. The estimated length of the extended C16-GGGRGDS molecule is le = 42 Å, calculated by adding the length of a C16 chain (18 Å) to the total length of the peptide block (individual residue period of 3.4 Å).37 The thickness of the bilayer lT ∼ 2zH + 2σH with Gaussian polydispersity ΔZH for zH (Table 1), is shorter than two extended lipopeptides molecules lT< 2 × le for Mr = 2.2–14.4, indicating that the C16 chain probably overlaps giving an interdigitated structure. It can also be noted that according to previous SAXS studies, pure C16-GGGRGDS nanotapes are present in solution as a co-existence of two populations with d = 55 and 70 Å.19 Binding of Apo-AI to C16-GGGRGDS resulted in only one population of nanotapes with d = 65 Å (Table 1).
Conclusions
The original aim of our work was to prepare lipopeptide/apoliproprotein nanodiscs, analogous to those reported for lipids but containing peptide-functionalised lipopeptide molecules wrapped with the Apo-AI. We have indeed observed a fraction of nanodiscs above the saturation binding condition, although these coexist with fibrillar structures from the lipopeptide self-assembly process.We have shown that the lipopeptide C16-GGGRGDS binds to Apo-AI with a saturation-binding type behaviour. The interaction is endothermic, which indicates that the binding process is driven by entropic processes which may correspond to changes in the conformation of the lipopeptide and/or apolipoprotein substrate. Circular dichroism spectroscopy provides a similar saturation binding concentration by monitoring the reduction in α-helical content of Apo-AI upon addition of C16-GGGRGDS. There also seems to be a reduction in the β-sheet structure of C16-GGGRGDS. In other words, CD indicates conformational changes of both Apo-AI and lipopeptide. This points to the origin of the entropically driven process which leads to a loss of secondary structure as the two molecules interact.
Electron microscopy imaging reveals the presence of a mixture of fibrils and nanodiscs above the saturation binding condition. SAXS provided more insight into the nature of the self-assembly process and binding interaction. First, it confirms the findings from TEM of the presence of a mixture of fibrils and nanodiscs, also providing detailed (internal) structural parameters for these assemblies. Secondly, and most importantly, it shows that the Apo-AI itself is present in solution in the form of multimers, specifically dimers – although smaller amounts of other species may also be present. The fact that the Apo-AI was not present in monomeric form may be one reason why nanodiscs comprising Apo-AI wrapped around the lipopeptide were not able to assemble as the main morphology. A fibrillar structure was predominant, although some nanodiscs were seen reflecting the nature of the Apo-AI monomer/oligomer equilibrium as well as the lipopeptide self-assembly process and the possible interplay between these two equilibria in the mixed system.
This work forms part of a continuing effort to examine the bioactivity of biofunctional lipopeptides and further efforts to create lipopeptide nanodiscs are ongoing. Our work suggests that preparing Apo-AI in monomeric form may be critical to the synthesis of (entirely) nanodisc assemblies resulting from lipopeptide/apoliprotein complexation.
Acknowledgements
This work was supported by EPSRC grant EP/G026203/1. We would like to acknowledge A. Round and Barbara Calisto for support during beamtime sessions at the ESRF (Grenoble, France) beamline BM29 (Projects Number MX1511 and MX1620).Notes and references
- D. P. Rogers, L. M. Roberts, J. Lebowitz, J. A. Engler and C. G. Brouillette, Biochemistry, 1998, 37, 945 CrossRef CAS PubMed.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147 CrossRef CAS PubMed.
- K. Nagao, M. Hata, K. Tanaka, Y. Takechi, D. Nguyen and P. Dhanasekaran, et al. , Biochim. Biophys. Acta, Bioenerg., 2014, 1841, 80 CrossRef CAS PubMed.
- A. A. Ajees, G. M. Anantharamaiah, V. K. Mishra, M. M. Hussain and H. M. K. Murthy, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2126 CrossRef CAS PubMed.
- W. S. Davidson, T. Hazlett, W. W. Mantulin and A. Jonas, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13605 CrossRef CAS.
- D. W. Borhani, D. P. Roger, J. A. Engler and C. G. Brouillette, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12291 CrossRef CAS.
- Y. Fang, O. Gursky and D. Atkinson, Biochemistry, 2003, 42, 13260 CrossRef CAS PubMed.
- A. Nath, W. M. Atkins and S. G. Sligar, Biochemistry, 2007, 46, 2059 CrossRef CAS PubMed.
- G. Franceschini, G. Vecchio, G. Gianfranceschi, D. Magani and C. R. Sirtori, J. Biol. Chem., 1985, 260, 6321 Search PubMed.
- H. Saito, P. Dhanasekaran, D. Nguyen, E. Deridder, P. Holvoet and S. Lund-Katz, et al. , J. Biol. Chem., 2004, 279, 20974 CrossRef CAS PubMed.
- S. E. Lux, R. I. Shrager, R. Hirz and A. M. Gotto, J. Biol. Chem., 1972, 247, 2598 CAS.
- H. Saito, S. Lund-Katz and M. C. Phillips, Progr. Lipid Res., 2004, 43, 350 CrossRef CAS PubMed.
- A. Jonas, J. H. Wald, K. L. H. Toohill, E. S. Krul and K. E. Kezdy, J. Biol. Chem., 1990, 265, 22123 CAS.
- J. P. Segrest, M. K. Jones, A. E. Klon, C. J. Sheldahl, M. Hellinger and H. De Loof, et al. , J. Biol. Chem., 1999, 274, 31755 CrossRef CAS PubMed.
- V. Castelletto, R. M. Gouveia, C. J. Connon, I. W. Hamley, J. Seitsonen and A. Nykanen, et al. , Biomat. Sci., 2014, 2, 362 RSC.
- R. M. Gouveia, V. Castelletto, I. W. Hamley and C. J. Connon, Adv. Healthcare Mater., 2014 Search PubMed , Submitted.
- M. J. Webber, J. Tongers, M.-A. Renault, J. G. Roncalli, D. W. Losordo and S. I. Stupp, Acta Biomater., 2010, 6, 3 CrossRef CAS PubMed.
- D. A. Harrington, E. Y. Cheng, M. O. Guler, L. K. Lee, J. L. Donovan and R. C. Claussen, et al. , J. Biomed. Mat. Res., Part A, 2006, 78A, 157 CrossRef CAS PubMed.
- V. Castelletto, R. Gouveia, C. J. Connon and I. W. Hamley, Faraday Discuss., 2013, 166, 381 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133 CrossRef CAS PubMed.
- ATSAS. Crysol - Version 2.8, 1995–2011 Search PubMed.
- D. Svergun, C. Barberato and M. H. J. Koch, J. Appl. Crystallogr., 1995, 28, 768 CrossRef CAS.
- X. Mei and D. Atkinson, J. Biol. Chem., 2011, 286, 38570 CrossRef CAS PubMed.
- G. Pabst, M. Rappolt, H. Amenitsch and P. Laggner, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 4000 CrossRef CAS.
- A. Caillé, C. R. Acad. Sci. Paris, 1972, 274, 891 Search PubMed.
- R. Zhang, R. M. Suter and J. F. Nagle, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50, 5047 CrossRef CAS.
- R. O'Brien and I. Haq. Applications of Biocalorimetry: Binding, Stability and Enzyme Kinetics, in Biocalorimetry 2, ed. J. E. Ladbury and M. Doyle, Wiley & Sons, London, 2004. pp. 3–34 Search PubMed.
- M. Kono, Y. Okumura, M. Tanaka, D. Nguyen, P. Dhanasekaran and S. Lund-Katz, et al. , Biochemistry, 2008, 47, 11340 CrossRef CAS PubMed.
- C. Arnulphi, S. A. Sanchez, M. A. Tricerri, E. Gratton and A. Jonas, Biophys. J., 2005, 89, 285 CrossRef CAS PubMed.
- A. Derksen, D. Gantz and D. M. Small, Biophys. J., 1996, 70, 330 CrossRef CAS.
- L. Calabresi, G. Vecchio, R. Longhi, E. Gianazza, G. Palm and H. Wadensten, et al. , J. Biol. Chem., 1994, 269, 32168 CAS.
- V. Castelletto, I. W. Hamley, J. Perez, L. Abezgauz and D. Danino, Chem. Commun., 2010, 46, 9185 RSC.
- I. W. Hamley, D. R. Nutt, G. D. Brown, J. F. Miravet, B. Escuder and F. Rodriguez-Llansola, J. Phys. Chem. B, 2010, 114, 940 CrossRef CAS PubMed.
- C. A. Peters-Libeu, Y. Newhouse, S. C. Hall, H. E. Witkowska and K. H. Weisgraber, J. Lipid Res., 2007, 48, 1035 CrossRef CAS PubMed.
- L. B. Vitello and A. M. Scanu, J. Biol. Chem., 1976, 251, 1131 CAS.
- A. A. Ajees, G. M. Anantharamaiah, V. K. Mishra, M. M. Hussain and K. H. M. Murthy, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2126 CrossRef CAS PubMed.
- T. E. Creighton, Protein Folding, W W. H. Freeman, New York, 1992 Search PubMed.
Footnote |
| † Current address: National Physical Laboratory, Hampton Rd, Teddington, Middlesex TW11 0LW, UK. |
| This journal is © The Royal Society of Chemistry 2015 |