
Synthesis of metal oxide nanoparticles via a robust “solvent-deficient” method†
Stacey J.
Smith
,
Baiyu
Huang
,
Shengfeng
Liu
,
Qingyuan
Liu‡
,
Rebecca E.
Olsen
,
Juliana
Boerio-Goates
and
Brian F.
Woodfield
*
Department of Chemistry and Biochemistry, Brigham Young University, Provo, UT 84602, USA. E-mail: Brian_Woodfield@byu.edu
First published on 5th November 2014
Abstract
We report an efficient, general methodology for producing high-surface area metal oxide nanomaterials for a vast range of metal oxides, including at least one metal oxide nanomaterial from nearly every transition metal and semi-metal group in the periodic table (groups 3–4 and 6–15) as well as several from the lanthanide group (see Table 1). The method requires only 2–3 simple steps; a hydrated metal salt (usually a nitrate or chloride salt) is ground with bicarbonate (usually NH4HCO3) for 10–30 minutes to form a precursor that is then either untreated or rinsed before being calcined at relatively low temperatures (220–550 °C) for 1–3 hours. The method is thus similar to surfactant-free aqueous methods such as co-precipitation but is unique in that no solvents are added. The resulting “solvent-deficient” environment has interesting and unique consequences, including increased crystallinity of the products over other aqueous methods and a mesoporous nature in the inevitable agglomerates. The products are chemically pure and phase pure with crystallites generally 3–30 nm in average size that aggregate into high surface area, mesoporous agglomerates 50–300 nm in size that would be useful for catalyst and gas sensing applications. The versatility of products and efficiency of the method lend its unique potential for improving the industrial viability of a broad family of useful metal oxide nanomaterials. In this paper, we outline the methodology of the solvent-deficient method using our understanding of its mechanism, and we describe the range and quality of nanomaterials it has produced thus far.
A. Introduction
Nanomaterials have attracted numerous research efforts in the past two decades.1 Due to their unique chemical and physical properties, nanomaterials, especially metal oxide nanoparticles, have been used in a wide range of applications, including catalysis,2–4 sensing,5 optical,1 semiconducts,6,7 solar cells,8 batteries,9 ultrafiltration membranes,10 and biotech industries.11,12 Because most of these applications require particles below 100 nm in size2,11,13,14 with many requiring sizes below 20 nm,3,4,15–17 the key to developing and utilizing these burgeoning industrial nanotechnologies lies in being able to efficiently produce the nanomaterials they use.Numerous and varied synthetic techniques have been developed for producing metal oxide nanoparticles. These can be generally grouped into three categories: solid-state (milling, sonication), solution-phase (co-precipitation, sol–gel, microemulsion, solvothermal/hydrothermal, non-aqueous), and vapor-phase methods (spray pyrolysis, inert gas condensation, plasma or flame based methods).12 Each technique has its own advantages and disadvantages, as several excellent reviews of these methods reveal in greater detail.1,12,14,18–30 To summarize, solid-state methods are typically fast, inexpensive, and easily scaled to produce industrial quantities, but the particles produced are usually >30 nm (if not >100 nm) in size and thus tend to have lower surface areas.12 The milling or sonication process can also cause stress/strain on the crystal structure as well as introduce impurities.13,18 Vapor-phase methods are highly successful in synthesizing nanoparticles, particularly one-dimensional nanostructures (nanowires, nanobelts, etc.),21 but the equipment and energy required to vaporize the reagents tend to be costly, and with a few exceptions (e.g. TiO2), these methods are more widely used in the production of metal rather than metal oxide nanomaterials.12 Solution-phase methods, particularly those which employ either surfactants or non-aqueous solvents, can produce uniform metal oxide nanoparticles with excellent control over size, size distribution, morphology, and dispersability. However, organic impurities can be an issue,12,13 and for certain applications (e.g. catalysis and gas sensing), the capping agents are an obstacle that must be removed in order to access the oxide surface.12,13,21 Surfactant-free aqueous processes introduce fewer impurities and are readily scaled up for industrial production, but low crystallinity, larger size distributions, and agglomeration can be issues.21,31
In essence, no single technique is ideal in every way or for every application.21 Perhaps as importantly, however, most methods are only successful for certain nanomaterials as well as certain applications. The variation in equipment and procedures thus necessary for the synthesis of the various useful metal oxide nanomaterials represents one of the challenges to their widespread industrial production.
Here, we report a new method32 that is successful for a vast range of nanomaterials, including at least one metal oxide nanomaterial from nearly every transition metal group (groups 3–4 and 6–12: Y2O3, TiO2,33 ZrO2, MoO3, Mn2O3, Fe2O3, Fe3O4, CoO, Co3O4, NiO, PdO, CuO, Ag2O, ZnO) and semi-metal group (groups 13–15: Al2O3,34–36 In2O3, SnO2, Bi2O3) in the periodic table as well as several from the lanthanide group (Ce4+, Pr3+, Nd3+). Simply stated, the method involves grinding a hydrated metal salt (usually a nitrate or chloride salt) with bicarbonate (usually NH4HCO3) for 10–30 minutes to form a precursor that is then either untreated or rinsed before being calcined at relatively low temperatures (220–550 °C) for 1–3 hours. The method is thus similar to surfactant-free aqueous methods such as co-precipitation,13,37 but is unique in that no solvents are added. The resulting “solvent-deficient” environment has interesting and unique consequences, including increased crystallinity of the products over other aqueous methods and a useful mesoporous nature in the inevitable agglomerates.
Requiring only 2–3 steps and a total of ≤5 hours, the technique provides an efficient, general methodology for producing a wide range of high-surface area metal oxide nanomaterials. As with all other methods, the products are suited for particular applications such as catalysis and gas sensing, but the versatility of products and efficiency of the method lend its unique potential for improving the industrial viability of a broad family of metal oxide nanomaterials. In this paper, we outline the methodology of the solvent-deficient method using our understanding of its mechanism, and we describe the range and quality of nanomaterials it has produced thus far.
B. Methods
I. Synthetic method
The process consists of two main steps with a third intermediate step (in gray) incorporated for some samples: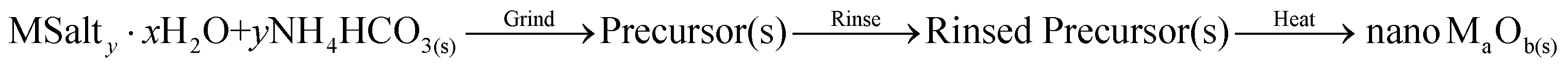 | (1) |
In the first step, a mortar and pestle is used to grind a hydrated metal salt (MSalty·xH2O, where Salt = NO3− or Cl− and x = 6–9) with bicarbonate (usually NH4HCO3 but NaHCO3 can also be used). The moles of bicarbonate should equal the moles of anion from the metal salt, as eqn (1) shows (or, equivalently, the moles of bicarbonate should equal the oxidation number of the metal cation). If the metal salt is anhydrous or has <6 moles of water per mole of salt, enough water should be added as the reagents are mixed to mimic the 6–9 moles of hydration present in most metal salts. For chloride metal salt reagents which have very exothermic reactions with water in which HCl gas is released, the water should be added slowly or dropwise while the reagents are ground together under a ventilated hood.
As Fig. 1a–d shows, even though no true solvent is added, when the (usually solid) reagents are ground together, the mixture liquefies and bubbles vigorously, producing faint popping noises. The reagents should be ground until this bubbling/popping subsides (see Fig. 1e), which is typically after 5–30 min of grinding. The grinding time decreases with increasing levels of hydration, and we have found that adding enough water to make the metal salt a nonahydrate decreases the grinding time ubiquitously to 5–15 minutes. We therefore typically add enough water to make the metal salt a nona-hydrate.
 | ||
| Fig. 1 Visual images of the first step (grinding) in the solvent-deficient synthetic method. Image (a) shows the two reagents (Fe(NO3)3·9H2O and NH4HCO3) before they are mixed. Images (b) through (e) show the progression of the reaction during 15 minutes of grinding; in (b) the reagents are mixed, in (c) bubbling begins as the reagents are ground together, in (d) the bubbling wanes, and in (e) the bubbling has ceased. The precursor, which in (e) is a slurry, can be dried and ground into the powder shown in (f). | ||
The grinding reaction produces a slurry/paste of solid precipitate particles (see Fig. 1e), which we call the precursor. For several materials (see Table 1), the precursor slurry should then be rinsed using a polar solvent; we typically use distilled H2O and a vacuum filter flask to wash the precursor 2–5 times. The precursor (either rinsed or untreated) can be dried at 50–100 °C for several hours to form a powder (see Fig. 1f) that can be stored indefinitely, but in general, we do not dry the precursor before the final calcination step.
| Group | Metal salt reagent(s) | Mole ratio (Salt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BC) :BC) |
Rinse step | Calc. temp. (°C) | Calc. time (hrs) | Avg. yield | Product |
|---|---|---|---|---|---|---|---|
| a NaHCO3 must be used instead of NH4HCO3 to avoid NH3 complexation b These precursors must be calcined in an inert environment to maintain the correct metal oxidation state. | |||||||
| Transition metal oxides | |||||||
| 3 | Y(NO3)3·6H2O | 1:3 |
No | 475 | 1 | >95% | Y2O3 |
| 4 | TiCl4 | 1:4 |
YesP | 400 | 3 | 60% | TiO2anatase |
| 4 | ZrCl4 | 1:4 |
YesP | 350 | 2 | 80% | ZrO2 |
| 6 | MoCl5 | 1:5 |
YesP,O | 350 | 1 | 60% | MoO3 |
| 7 | MnCl2·4H2O | 1:2 |
YesP,O | 500 | 2 | 60% | α-Mn2O3 |
| 8 | Fe(NO3)3·9H2O | 1:3 |
YesS | 350 | 1 | >95% | Fe2O3 |
| 8 | FeCl3 + FeCl2 | 1:1.5:11 |
YesP | 550b | 3.5 | Fe3O4 | |
| 9 | Co(NO3)2·6H2O | 1:2 |
YesO,S | 300b | 1 | >95% | CoO |
| 9 | Co(NO3)2·6H2O | 1:2 |
YesS | 300 | 2 | >95% | Co3O4 |
| 10 | Ni(NO3)2·6H2O | 1:2 |
YesS | 300 | 2 | 60% | NiO |
| 10 | Pd(NO3)2·xH2O | 1:2a |
YesP | 375 | 1 | >95% | PdO |
| 11 | Cu(NO3)2·6H2O | 1:2 |
YesS | 250 | 1 | 70% | CuO |
| 11 | AgNO3 | 1:1a |
YesP | 220 | 1 | >95% | Ag2O |
| 12 | Zn(NO3)2·6H2O | 1:2 |
YesP,S | 250 | 1 | >95% | ZnOwhite |
| Zn(NO3)2·6H2O | 1:2 |
No | 300 | 2 | >95% | ZnOpink | |
| Semi-metal oxide | |||||||
| 13 | Al(NO3)3·9H2O | 1:3 |
No | 700 | 2 | >95% | γ-Al2O3 |
| 13 | Al(NO3)3·9H2O | 1:3 |
No | 1100 | 2 | >95% | α-Al2O3 |
| 13 | In(NO3)3·xH2O | 1:3 |
No | 300 | 2 | 60% | In2O3 |
| 14 | SnCl4 | 1:4 |
YesP,O | 340 | 1 | >95% | SnO2 |
| 15 | Bi(NO3)3·5H2O | 1:3 |
YesO | 400 | 2 | >95% | Bi2O3 |
| Lanthanide metal oxide | |||||||
| Ce(NO3)4·6H2O | 1:4 |
No | 300 | 2 | >95% | CeO2 | |
| Pr(NO3)3·6H2O | 1:4 |
No | 550 | 3 | >95% | PrO2 | |
| Nd(NO3)3·6H2O | 1:3 |
No | 550 | 3 | >95% | Nd2O3 | |
In the final calcination step, which is either the second or third step depending on whether or not rinsing is performed, the precursor (in the slurry, rinsed, or dried form) is transferred to a crucible and calcined at relatively low temperatures (220–550 °C) in air (unless otherwise specified) for 1–3 hours to produce the desired nanomaterial. For each material, a heating rate of 5 °C min−1 is generally used to reach the calcination temperature. If large quantities (≥5 g) of nanomaterial are being calcined, the effluent gases should be directed through either a filtration system or a hood.
These steps form the basis of the method. Table 1 provides a list of the specific reaction parameters used for each material produced thus far. The methodology for setting the various parameters will be discussed with the mechanism of the method.
II. Characterization methods
All nanomaterials produced and their crystallographic phases/phase-purities were identified using powder X-ray diffraction (XRD). A PANalytical X'Pert Pro diffractometer with a Cu source and a Ge monochromator tuned to the Cu-Kα1 wavelength (λ = 1.540598 Å) was used to scan each nanomaterial from 10–90° 2θ with a step size of 0.016° at rates between 100–400 s per step. Each pattern was matched to a standard pattern in the ICDD (International Center for Diffraction Data) database. Crystallite sizes were estimated using the Scherrer formula for size-related peak broadening.38 Transmission electron microscopy (TEM) images were used to confirm/correct these initial estimates.TEM images of each nanomaterial were recorded using either a FEI Philips Technai F30 operating at 300 kV or a FEI Philips Technai F20 Analytical STEM operating at 200 kV. Specimens were prepared by dispersing the nanoparticles in ethanol without sonication and placing a drop of the dilute solution on a formvar/carbon film supported by a 200 mesh Cu or Ni grid (Ted-Pella Inc.). The solvent was allowed to evaporate, and images were recorded in standard high-resolution mode. The images were used to assess the crystallinity, size, and morphology of both the particles and agglomerates. The d-spacings of the visible lattice fringes were measured and compared with those of the standard crystal structures from the XRD analyses to assign crystal plane indices.
Chemical purity was assessed for a representative subgroup of samples including at least 1 sample from each group in the periodic table from which a metal oxide nanomaterial has been produced (groups 3, 4, 6, 7, 8, 9, 10, 11, 12, 13, 14, and 15 as well as the Lanthanides). Purities were ascertained by quantifying the contents of the possible impurities (C, H, N, and Cl) via combustion and nonaqueous chloride analyses performed by Galbraith Laboratories.
Agglomerate sizes of the same subgroup of samples were analyzed using a Nanosight LM20 microscope. Using a small drop of a very dilute aqueous suspension, the agglomerate sizes under which 50% (d50) and 90% (d90) of the agglomerates fall were recorded. The agglomerates of a few samples (Al2O3, CeO2) were milled using a Netzsch MiniCer mill to investigate the feasibility of breaking up the nanoparticle agglomerates. For the milling, the nanomaterials were suspended in distilled water, forming a 30% solution by mass.
Several experiments were performed to elucidate the reactions occurring. The gases released during bubbling in the initial grinding step were identified using a Jeol JMS-SX102A electron impact (EI) mass spectrometer. In this experiment, Fe(NO3)3·6H2O(s) and NH4HCO3(s) in the typical 1:3 mole ratio were mixed in an air-tight reaction vessel filled with nitrogen. Vigorous stirring was maintained (to mimic grinding) using a mechanical stirrer. Nitrogen gas was flowed through the reaction vessel, carrying the gases released from the bubbling through a tube into the spectrometer.
The compositions of the precursors of most materials were identified using the same PANalytical XRD instrument and measurements described previously. For these analyses, precursors were produced and split into either two or three portions: the first portion was simply dried; the second portion was dried, rinsed, and then dried again; and the occasional third portion was rinsed before being dried. Samples were dried by heating in a drying oven at 80–100 °C in air until dry (typically ∼24 hours). Samples were rinsed by washing the precipitate 3–5 times on a filter flask with enough distilled water to cover the solid. The XRD patterns of all portions were matched to standard patterns in the ICDD database to identify the compound(s) in each precursor.
The gases released during calcination of each precursor were identified though tandem TG/DTA-MS analyses. The thermogravimetric and differential-temperature analyses (TG/DTA) were performed using a Netzsch STA 409PC instrument. The mass spectrometry (MS) measurements were collected in tandem with the TG/DTA measurements by attaching a miniature quadrupole MS unit built in-house39 to the out-gas line of the Netzsch TG/DTA instrument. For these experiments, roughly 30 mg of each precursor material were heated in an alumina crucible to either 300 °C (for most un-rinsed precursors made with nitrate metal salts), 350 °C (for un-rinsed precursors made with chloride metal salts), or 650 °C (for rinsed precursors and some un-rinsed nitrate precursors) at a rate of 5 °C min−1 under He gas flow. The precursors heated to 300 °C and 350 °C were held at these temperatures for a minimum of 2 hours.
Nitrogen adsorption analysis was carried out using a Micromeritics Tristar 3020 apparatus at 77 K. Samples were degassed at 200 °C with nitrogen flow over night prior to the measurements. Specific surface area was calculated by the Brunauner–Emmett–Teller (BET) method, using a P/P0 range between 0.05 and 0.2. Pore volume was calculated from the adsorption isotherm at the relative pressure of 0.98 and mean pore diameter were determined by the Barrett–Joyner–Helenda (BJH) method using either adsorption branch or desorption branch depending on the isotherm hysteresis type.40 Pore size distribution and mesopore volume were calculated from the adsorption and desorption data using a newly developed method involving slit geometry for the Kelvin equation and structural corrections for area and volume, while fitting the data to a log normal distribution function.41
C. Results
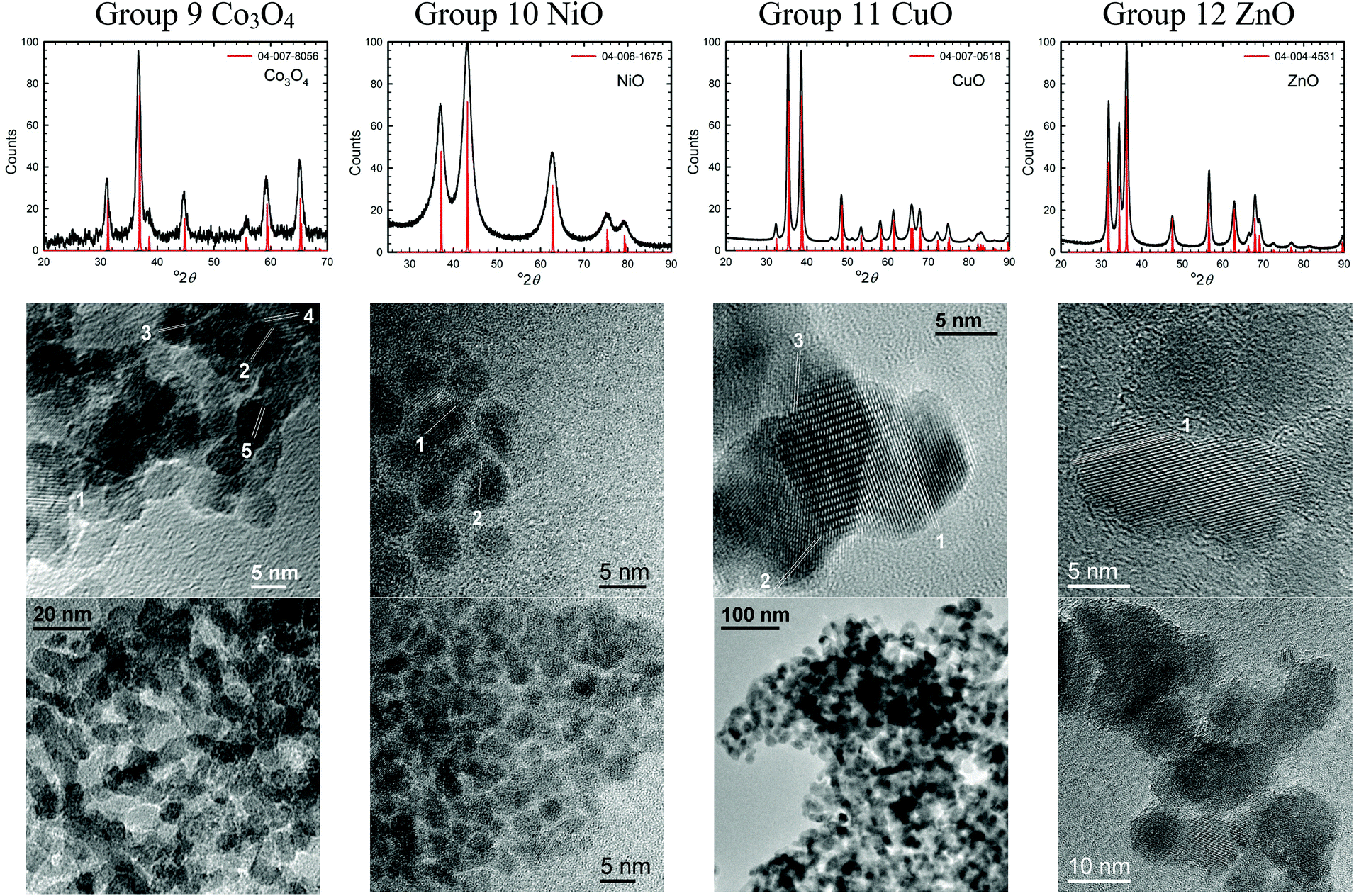
TEM images of several of the products and the XRD patterns confirming their identity/phase are shown in Fig. 2–4. (The remainders are given in Fig. S1 of the ESI.†) Summary of the synthetic parameters for each nanomaterials are listed in Table 1 and their crystallite size, agglomerate size, porosity are provided in Table 2.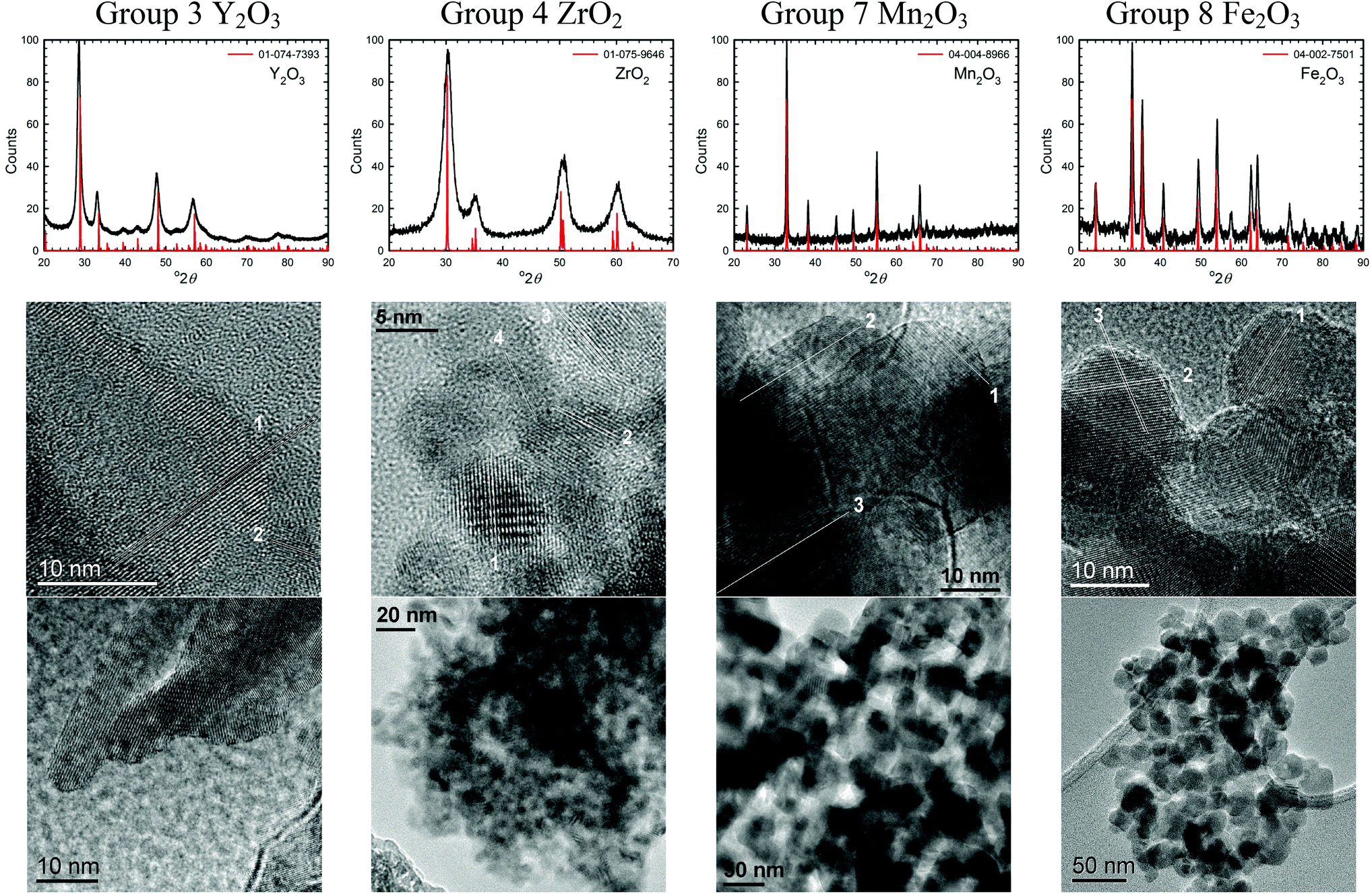 | ||
| Fig. 2 XRD patterns and TEM images of some transition metal oxide nanomaterials (group 3, 4, 7, and 8) synthesized using the solvent-deficient method (the rest are shown in Fig. S1 in the ESI†). The XRD plots show the ICDD standard pattern (red line) matched to the product (black line), with the standard's PDF number given in the legend. The numbered lines in the high-resolution TEM images highlight the lattice fringes whose d-spacings and crystal plane indices are given in the ESI.† | ||
 | ||
| Fig. 3 XRD patterns and TEM images of some transition metal oxide nanomaterials (group 9–12) synthesized using the solvent-deficient method. | ||
 | ||
| Fig. 4 XRD patterns and TEM images of some semi-metal oxide nanomaterials (group 13 and 14) and lanthanide metal oxides (CeO2) synthesized using the solvent-deficient method. | ||
| Group | Product | Crystallite size (nm) | Agglomerate size (nm) (d50, d90) | Surface area (m2 g−1) | Pore diam. (nm) | Impurity contents | |||
|---|---|---|---|---|---|---|---|---|---|
| %C | %H | %N | Cl– | ||||||
| a Agglomerate refers to aggregate of nanoparticles after dispersion in water without agitation and measured by Nanosight. Purity characterizations were not performed for all samples due to the high costs of these analyses, but the given analyses demonstrate the consistently low impurity content. Surface areas and pore sizes are not given for PdO and Ag2O because theses expensive materials were not synthesized in sufficient quantities to perform the necessary BET experiments. | |||||||||
| Transition metal oxides | |||||||||
| 3 | Y2O3 (rods) | 8(±1) × 40(±10) | 81, 302 | 3 | 97 | 0.70 | 1.10 | 0.68 | — |
| 4 | TiO2anatase | 8 (±2) | 71, 104 | 80 | 7 | <0.5 | <0.5 | <0.5 | 80ppm |
| 4 | ZrO2 | 7 (±3) | 73, 206 | 43 | 4 | ||||
| 6 | MoO3 | 25 (±15) | 70, 98 | 2 | 26 | <0.5 | <0.5 | <0.5 | 41ppm |
| 7 | α-Mn2O3 | 25 (±5) | 55, 85 | 29 | 42 | <0.5 | <0.5 | <0.5 | |
| 8 | Fe2O3 | 8 (±3) | 72, 169 | 178 | 4 | <0.5 | <0.5 | <0.5 | — |
| 8 | Fe3O4 | 50 (±20) | 52, 67 | 5 | 10 | ||||
| 9 | CoO | 12 (±4) | 86, 325 | 21 | 4 | <0.5 | <0.5 | <0.5 | — |
| 9 | Co3O4 | 10 (±4) | 44, 76 | 22 | 10 | ||||
| 10 | NiO | 4 (±1) | 67, 94 | 129 | 4 | 1.73 | 1.02 | <0.5 | — |
| 10 | PdO | 3 (±1) | |||||||
| 11 | CuO | 15 (±5) | 62, 89 | 40 | 41 | <0.5 | <0.5 | <0.5 | — |
| 11 | Ag2O | 42 (±20) | 37, 60 | ||||||
| 12 | ZnOwhite | 8 (±5) | 65, 79 | 52 | 34 | <0.5 | <0.5 | <0.5 | — |
| 12 | ZnOpink | 60(±25) | 52, 110 | 1 | 0 | ||||
| Semi-metal oxides | |||||||||
| 13 | γ-Al2O3 | 4 (±1) | 73, 102 | 267 | 4 | <0.5 | 1.57 | <0.5 | — |
| 13 | α-Al2O3 | 38 (±8) | 61, 121 | 7 | 42 | ||||
| 13 | In2O3 | 12 (±5) | 71, 102 | 57 | 7 | ||||
| 14 | SnO2 | 4 (±1) | 69, 187 | 145 | 3 | <0.5 | 1.51 | <0.5 | 0.2% |
| 15 | Bi2O3 | 30 (±20) | 65, 91 | 11 | 90 | <0.5 | <0.5 | <0.5 | — |
| Lanthanides | |||||||||
| CeO2 | 13 (±3) | 57, 80 | 31 | 71 | <0.5 | <0.5 | 1.22 | — | |
| PrO2 | 18 (±7) | 45, 121 | 4 | 7 | |||||
| Nd2O3 | 18 (±7) | 47, 92 | 10 | 10 | |||||
D. Discussion
I. Synthesis method
Briefly described, the “solvent-deficient” method for synthesizing metal oxide nanomaterials consists of 2–3 simple steps. First, a hydrated nitrate or chloride metal salt is ground with ammonium bicarbonate, causing the typically solid reagents to liquefy and bubble vigorously (see Fig. 1a–d). The reagents are ground until this bubbling ceases (∼10–30 min) and a slurry/paste containing a solid precipitate, or the precursor, has formed (Fig. 1e). For some samples, the precursor is then rinsed. Finally, the precursor is calcined (∼220–550 °C) in air (unless otherwise specified) for 1–3 hours.The method thus has several variable parameters: (1) hydration level of the metal salt reagent, (2) ratios of the metal salt and bicarbonate reagents, (3) grinding time, (4) inclusion of a rinsing step, (5) calcination temperature, (6) calcination time, and (7) atmosphere during calcination. In the following sections, we discuss each variable in turn, using our understanding of the fundamental reactions taking place to explain/rationalize the values chosen (see Table 1) and thereby provide a general methodology for applying the process to new materials.
i. Metal salt hydration level. The hydration level of the metal salt plays a key role in the first step of the process. As Fig. 1a–c illustrates, a bubbling liquid is formed as the solid reagents are ground together even though no solvent is added. This reaction occurs more quickly and is more vigorous for more hydrated metal salts, and in fact, if the metal salt is anhydrous or has ≤5 moles of hydration per mole of salt, a few drops of water must be added to initiate the reaction.
To understand the role and proper amount of metal salt hydration, we explored the reactions occurring during grinding. From the MS data shown in Fig. 5a, we know that the bubbles evolved from these reactions consist of CO2 gas. For CO2 gas to be released, the bicarbonate ion (HCO3−) must be decomposing into CO2 (g) and OH− (aq.), which is a well-known reaction that occurs rapidly when bicarbonate is dissolved in water. We therefore infer that the waters of hydration are acting as a solvent and the grinding action must free the loosely bound waters of hydration (eqn (2)), enabling the soluble NH4HCO3 and metal salt to begin dissociating into partially solvated ions (eqn (3)). This allows the bicarbonate ion to further decompose into CO2 gas (eqn (4)), producing the bubbles seen in the rapidly forming liquid.
| [M(NO3)y·xH2O](s) + yNH4HCO3(s) → M(NO3)y(s) + xH2O(l) + yNH4HCO3(s) | (2) |
| xH2O(l) + [My+ + yNO3− + yNH4+ + yHCO3−](aq.) | (3) |
| yCO2(g) + yOH− (aq.) | (4) |
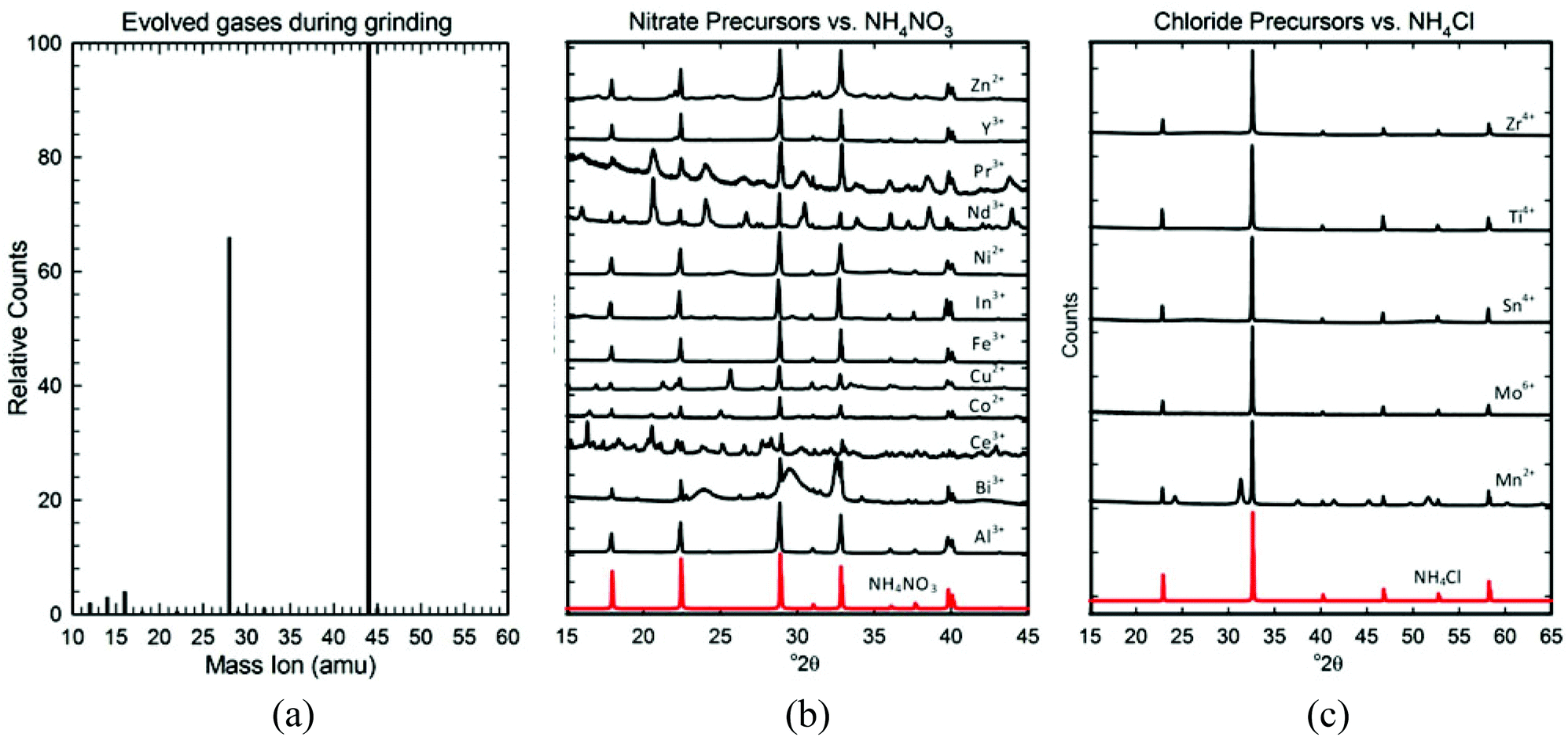 | ||
| Fig. 5 Characterization of the first step of the reaction. (a) MS data showing the CO2 (44amu) evolved during grinding, accompanied by fragment ions 12 and 16 amu in the expected 1:2 ratio. The 28 and 14 amu peaks correspond to the N2 carrier gas and its fragment. (b) XRD data for multiple precursors (black) showing that NH4NO3 (red) is formed when nitrate metal salt reagents are used. (c) XRD data for multiple precursors (black) showing that NH4Cl (red) is formed when chloride metal salt reagents are used. | ||
We have found that 6 moles of hydration is the minimum hydration level necessary to provide sufficient solvent and enable the reagents to dissociate. However, the reaction proceeds much faster and easier (5–15 vs. 20–30 minutes of grinding) if 9 moles of hydration are present. Thus, for metals whose salts are anhydrous or have less than 9 moles of hydration, we typically add enough water to make the hydration level of the salt equal to that of a nona-hydrated salt. (Adding water can influence the morphologies and corresponding surface areas of the agglomerates, however, so depending on the application it may or may not be desirable to add water to salts containing at least 6 moles of hydration.) Because this amount of “solvent” is still considerably less than that required to fully solvate all of the ions in the solution and is less than that used in other aqueous solution methods, we have named the process “solvent-deficient”.
ii. Reagent ratios. As the reagents dissociate, the mixture dissolves into a bubbling slurry with the following components:
| [My+ + yNO3−/Cl− + yNH4+ + y(HCO3− + OH− + CO2) ](aq.) + H2O(l) |
These partially solvated ions begin reacting almost immediately, as illustrated in Fig. 1 by the rapid formation of the brown-colored precipitate. To understand the appropriate reagent ratios, we used XRD to identify the precipitate formed, which we call the “precursor”.
As Fig. 5b and c show, all the precursors (un-rinsed and dried) contain an ammonium salt; NH4NO3 is formed if a nitrate metal salt was used, and NH4Cl is formed if a chloride metal salt was used. The strong diffraction signals of these ammonium salts mask the broad and somewhat diffuse XRD patterns of the nano-sized metal compounds in each precursor, but by rinsing out the soluble NH4NO3 and NH4Cl, we could identify them. Table 3 lists the metal compound(s) identified in each precursor (for the actual XRD patterns/analyses see Fig. S3 of the ESI†). As expected, most precursors contained a metal hydroxide of some sort: a simple hydroxide (Co2+, Ni2+), an oxyhydroxide (Al3+, Fe3+), a hydroxide nitrate (Co2+, Ni2+, Cu2+, Ce4+), or a hydroxide carbonate (Y3+, Zn2+, In3+, Ce4+, Nd3+, Pr3+). However, a few precursors formed carbonates (Mn2+, Ag+, Bi3+, Nd3+), and several others formed nearly amorphous hydrated oxides (Y3+, Ti4+, Zr4+, Mo5+, Sn4+). A number of precursors contained mixtures of several of these compounds (Y3+, Co2+, Ni2+, Cu2+, Zn2+, Bi3+, Ce4+, Nd3+).
| Group | Metal reagent | Dried precursor | Rinsed/dried precursor |
|---|---|---|---|
| a MS data suggest the In3+ precursors is a carbonate or hydroxide-carbonate, but no standard patterns of these were available. b These compounds are nearly amorphous. | |||
| Transition metal precursors | |||
| 3 | Y3+ (nitrate) | NH4NO3 + Y2O3·xH2O + Y(OH)(CO3)b | |
| 4 | Ti4+ (chloride) | NH4Cl + TiO2·xH2O(anatase)b | |
| 4 | Zr4+ (chloride) | NH4Cl + ZrO2·xH2Ob | |
| 6 | Mo5+ (chloride) | NH4Cl + HMoO3·H2O b | |
| 7 | Mn2+ (chloride) | NH4Cl + Mn(CO3) | |
| 8 | Fe3+ (nitrate) | NH4NO3 + FeOOH·0.4H2O | |
| 9 | Co2+ (nitrate) | NH4NO3 + Co(OH)2 + Co2(OH)3(NO3) + Co(OH)(NO3)·H2O | Co(OH)2b |
| 10 | Ni2+ (nitrate) | NH4NO3 + Ni(OH)2 + Ni3(OH)4(NO3)2 | Ni(OH)2b |
| 11 | Cu2+ (nitrate) | NH4NO3 + Cu2(OH)3(NO3) + Cu(NO3)2(NH3)4 | Cu2(OH)3(NO3) |
| 11 | Ag+ (nitrate) | NaNO3 + Ag2CO3 | |
| 12 | Zn2+ (nitrate) | NH4NO3 + Zn5(OH)6(CO3)2 + Zn(OH)(NO3)·NH3 +Zn(OH)(NO3)·H2O + (NH4)Zn(CO3)(NO3)·NH3 | Zn5(OH)6(CO3)2 + Zn4(OH)6CO3·H2O |
| Semi-metal precursors | |||
| 13 | Al3+ (nitrate) | NH4NO3 + AlOOH | |
| 13 | In3+ (nitrate) | NH4NO3 + In hydroxide-carbonatea | |
| 14 | Sn4+ (chloride) | NH4Cl + SnO2·xH2Ob |
|
| 15 | Bi3+ (nitrate) | NH4NO3 + Bi2O2CO3 + Bi2O3·CO2 and/or Bi(OH)3 | |
| Lanathanide metal precursors | |||
| Ce3+ (nitrate) | NH4NO3 + Ce(OH)(CO3) + Ce2(CO3)6·6H2O + (NH4)3Ce2(NO3)9 + (NH4)2Ce(NO3)5(H2O)2·2H2O | Ce(CO3)(OH)+CeO2 | |
| Pr3+ (nitrate) | NH4NO3 + Pr(OH)(CO3) | ||
| Nd3+ (nitrate) | NH4NO3 + Nd(OH)(CO3) + Nd2(CO3)3·2.5H2O | ||
Based on the precipitates formed during the reaction, we can infer that one or more of the following acid/base reactions occur once the reagents dissociate:
| My+ + yOH− → M(OH)y(s) | (5) |
| M3+ + 2OH− + H2O → MOOH(s) + H3O+ | (6) |
| M2+ + OH− + NO3− → M(OH)(NO3)(s) | (7) |
| M3+ + OH− + HCO3− + H2O → M(OH)(CO3)(s) + H3O+ | (8) |
| M2+ + CO32− + H2O → M(CO3)(s) | (9) |
| M4+ + 4OH− + 4H2O → MO2(s) + 4H3O+ | (10) |
Then, as the excess water evaporates (during drying or calcination) the ammonium salt crystallizes:
| NH4+(aq.) + NO3−(aq.) → NH4NO3(s) | (11) |
| NH4+(aq.) + Cl−(aq.) → NH4Cl(s) | (12) |
The appropriate reagent ratios are thus those that allow both the metal hydroxide/carbonate and the ammonium salts to form. We therefore keep a 1:1 mole ratio between the ammonium cation from the NH4HCO3 and the metal salt anion (as shown in eqn (1)). This enables the NH4NO3 or NH4Cl salts to form at the conclusion of the grinding reaction while simultaneously ensuring that the appropriate ratios of metal cation to hydroxide (and/or carbonate) anion are present for a metal hydroxide (and/or carbonate) to form.
Because the reactions merely involve simple acid–base chemistry, other bases can be used, such as sodium bicarbonate or even hydroxide bases (in cases where hydroxides and not carbonates form). In fact, sodium bicarbonate must be used in reactions involving noble metals, such as Ag and Pd, to avoid ammine complexes. However, an advantage of using ammonium bicarbonate with nitrate metal salts is that the NH4NO3 formed in the precursor decomposes easily in the calcination step and leaves no Na+ impurities.
iii. Grinding time. Grinding time is determined by the time it takes for the metal hydroxide and/or carbonate precursor compounds to form. These precursor compounds form rapidly and appear to form completely by the time bubbling has ceased (as illustrated in Fig. 1). Based on the well-studied mechanisms of nanoparticle nucleation, growth, and aging from other aqueous precipitation methods,1,14 we attribute the rapidity of the reaction to the solvent-deficient environment; the scarcity of solvent ensures that as soon as the reagents dissociate and the hydroxide/carbonate molecules begin to form they are in a supersaturated environment, allowing nucleation to begin essentially immediately. The solvent deficiency then inhibits migration/mixing of the reagent materials, making growth of the hydroxide/carbonate nanoparticles so extremely diffusion-limited that growth is choked almost as soon as nucleation begins. Because nanoparticle growth thus only requires as much time as it takes for the reagents to dissociate, we base the grinding time solely on the time it takes for bubbling (the evidence of bicarbonate dissociation) to cease. Grinding time thus decreases with increasing hydration levels of the metal salt because dissociation occurs faster when more “solvent” is present.
After initial nucleation and growth, the hydroxide and/or carbonate precursor crystallites can undergo two well-known aging processes, Ostwald ripening and aggregation,1 to assuage their high surface energies.42 Because surfactants are not employed, aggregation is inevitable, but we have found that Ostwald ripening (coarsening) is greatly minimized if the precursors are not dried prior to calcination. Thus, to obtain smaller crystallite sizes, we generally calcine the wet precursors directly.
During aggregation, the solvent-deficient environment again produces some interesting and fortuitous results. Because the agglomerates begin to form before the excess water evaporates and the ammonium salts crystallize, the spectator ions/molecules (H2O, NH4+, NO3−/Cl−) seem to provide a spatial buffer or possibly even a sort of template around which the agglomerates form, causing the agglomerates to be mesoporous after these ions are removed via calcination and/or rinsing. (Preliminary results showing that the pores have an unusually narrow size distribution and that the average size varies with the size of the spectator ions encourage this hypothesis. This work will be published elsewhere.) This mesoporous character combined with the high surface areas of the agglomerates make the products promising candidates for several catalyst or gas-sensing applications (also to be published elsewhere).
i. Calcination temperature and time. To produce the desired nano metal oxide, the nano-crystallite precursors produced by the grinding reaction (Table 3) must be calcined to decompose the ammonium salt and dehydrate/decompose the metal compound. The decomposition reactions of both NH4NO3 and NH4Cl are well-known; above ∼160 °C and 250 °C, NH4NO3 liquefies and can decompose (non-explosively) via two, well-studied routes,43 respectively:
| NH4NO3(l) → NH3(g) + HNO3(g) | (13) |
| NH4NO3(l) → N2O(g) + 2H2O(g) | (14) |
Above ∼338 °C, NH4Cl sublimes:
| NH4Cl(s) → NH3(g) + HCl(g) | (15) |
The decompositions of the metal compounds in the precursors (hydroxides, oxyhydroxides, hydroxide-nitrates, hydroxide-carbonates, and carbonates) are also predictable; precursors containing hydroxide ligands should dehydrate (evolve H2O), precursors containing carbonate ligands should decarbonate (evolve CO2), and the few precursors containing nitrates could evolve various NOx gases.
To confirm the reactions occurring and determine the temperatures/times required, the gaseous byproducts evolved during the calcination of each precursor were identified using mass spectrometry (MS) data collected in tandem with TG/DTA data. provides representative TG/DTA-MS data sets. (TG/DTA-MS data for each sample are given in Fig. S4 and S5 of the ESI,† along with a more detailed discussion of the results).
As Fig. 6a illustrates, precursors containing an ammonium nitrate salt evolve large amounts of N2O (44 amu) and H2O (18 amu) at temperatures between 250–300 °C, confirming the NH4NO3 decomposition route given in eqn (14). The evolution of a small amount of NH3 (17 amu) is also observed, but the HNO3 predicted by eqn (13) is never detected. Instead, substantial amounts of NO (30 amu) and even small amounts of NO2 (46 amu) are observed, suggesting that any HNO3 formed reacts with NH3 (or other species) in several side reactions to form NOx gases and/or additional N2O and H2O gases. The following is thus a potentially more accurate depiction of the ammonium nitrate decomposition reaction in the calcination of (un-rinsed) nitrate precursors:
| NH4NO3(l) → N2O(g) + H2O(g) + NH3(g) + NO(g) + NO2(g) | (16) |
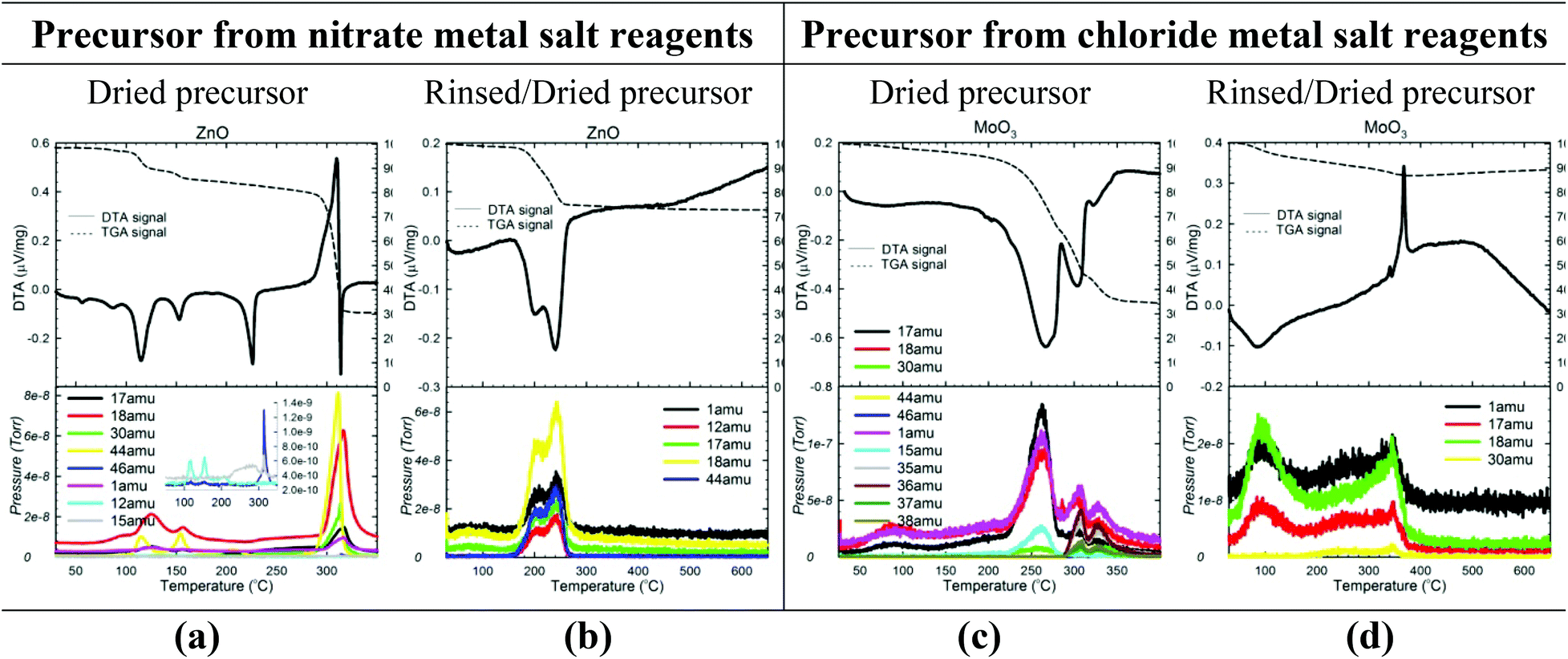 | ||
| Fig. 6 Characterization of the calcination step of the reaction. TG/DTA-MS data for both un-rinsed (a, c) and rinsed (b, d) precursors of 2 representative samples (one made from nitrate (ZnO) and one made from chloride (MoO3) metal salt reagents). See Fig. S4 and S5 in the ESI† for TG/DTA-MS data for all samples. | ||
Similarly, Fig. 6c illustrates the NH3 (17 amu) and HCl (35, 36, 37, 38 amu) mass ions observed at temperatures between 300–350 °C in the calcinations of most chloride precursors, confirming that the decomposition reaction in eqn (15) occurs to a certain extent. However, the HCl mass ions often appear in much smaller quantities than expected based on the NH3 quantity, and in fact, no HCl is observed for the Ti4+ and Mn2+ precursors (Fig. S4, ESI†). Thus, for most chloride precursors, the majority of the Cl− is not evolved as gaseous HCl but rather seems to remain as an impurity in the oxide. The NH3 (17 amu) also seems to participate in side reactions because NO (30 amu) is often observed.
Thus, a more accurate depiction of the ammonium chloride decomposition may be as follows:
| NH4Cl(s) → NH3(g) + NO(g) + HCl(g) + Cl− (impurity) | (17) |
Because both NH4NO3 and NH4Cl thus produce small quantities of noxious gases (NOx, NH3, HCl) during calcination, the gaseous byproducts from all un-rinsed precursors should be directed either through a ventilated hood or an aqueous filtration system.
The MS data for the rinsed precursors (Fig. 6b and d) display the anticipated H2O and CO2 byproducts, confirming that the following dehydration/decarbonation reactions occur:
| aM(OH)x(s) → MaO0.5ax(s) + 0.5axH2O(g) | (18) |
| MO(OH)(s) → M2O3(s) + H2O(g) | (19) |
| (M2+)x(OH)y(NO3)z(s) → xMO(s) + 0.5yH2O(g) + z(NO2 + NO)(g) | (20) |
| (M2+)x(OH)y(CO3)z(s) → xMO(s) + 0.5yH2O(g) + zCO2(g) | (21) |
| 2(M2+)(OH)(CO3)(s) → M2O3(s) + H2O(g) + 2CO2(g) | (22) |
| Mx(CO3)y(s) → MxOy(s) + yCO2(g) | (23) |
Because these reactions often occur at temperatures ≤300 °C, calcination temperatures of 300 °C and 350 °C are usually sufficient for precursors containing NH4NO3 and NH4Cl, respectively. For rinsed materials and those materials whose metal precursor compound(s) (see Table 3) require higher temperatures to form or crystallize into the oxide (see Table 1), the calcination temperature is determined by the dehydration/decarbonation temperature of the metal compound. The time generally required to decompose the NH4NO3 or NH4Cl components is 1–2 hours, as revealed by the TGA data.
However, the oxide may require 1–3 hours to form/crystallize.
These temperatures and times are sufficient initial estimates, but the minimum calcination time and temperature should be determined and employed to prevent unnecessary grain growth via Ostwald ripening. As Table 1 and 2 indicate, there is a significant amount of variation in the optimal values for each material. For example, the NH4NO3 and NH4Cl decomposition temperatures vary between 240–310 °C and 290–330 °C, respectively, depending on the precursor involved. Similarly, the dehydration of FeOOH·0.4H2O into Fe2O3 occurs between 250–300 °C in the presence of NH4NO3 (Fig. S4†) but occurs at T ≥ 350 °C when NH4NO3 is absent (as in the rinsed precursor in Fig. S5†). We suspect that these synergistic effects stem from some level of complexation between the metal precursor compound(s). Based on previous studies,44 the differing acidities of the metal cations/compounds in these suspected complexes may cause the variation in the salt decomposition temperature. Because of the variable synergistic interactions for each precursor, the calcination times and temperatures should be optimized for each material individually.
ii. Rinsing. Studying the calcination reactions and products revealed the need for a rinsing step. As previously mentioned, significant Cl− impurities remain in the oxide products if the NH4Cl is not removed prior to calcination. Thus, all precursors made from chloride metal salts (Mn2+, Mo5+, Ti4+, Sn4+, Zr4+) should be rinsed. Also, the noble metal precursors such as Ag+ and Pd2+ (for which NaHCO3 must be used instead of NH4HCO3) require rinsing to remove the NaNO3 salt formed. Unlike NH4Cl and NaNO3, the NH4NO3 in nitrate precursors only leaves noticeable impurities in one case; ZnO nanoparticles are a salmon/pink color indicative of nitrogen incorporation unless the Zn precursor is rinsed prior to calcination (which may actually be useful for certain applications).
Besides purity, a second reason for inserting a rinsing step is to obtain smaller crystallite sizes. For some samples, the synergism previously mentioned between the decompositions of the metal precursor compound(s) and NH4NO3 results in a large exothermic release of energy (> 0.4 μV mg−1) that enables grain growth. These exothermic events are uncharacteristic of either the endothermic salt decompositions or the endothermic/weakly exothermic metal compound decompositions (eqn (16–23)), and as Fig. 7 shows, rinsing the NH4NO3 out of these precursors (Fe3+, Co2+, Ni2+, Cu2+, Zn2+) prior to their calcination eliminates these large exothermic reactions, allowing crystallite sizes <20 nm to be obtained.
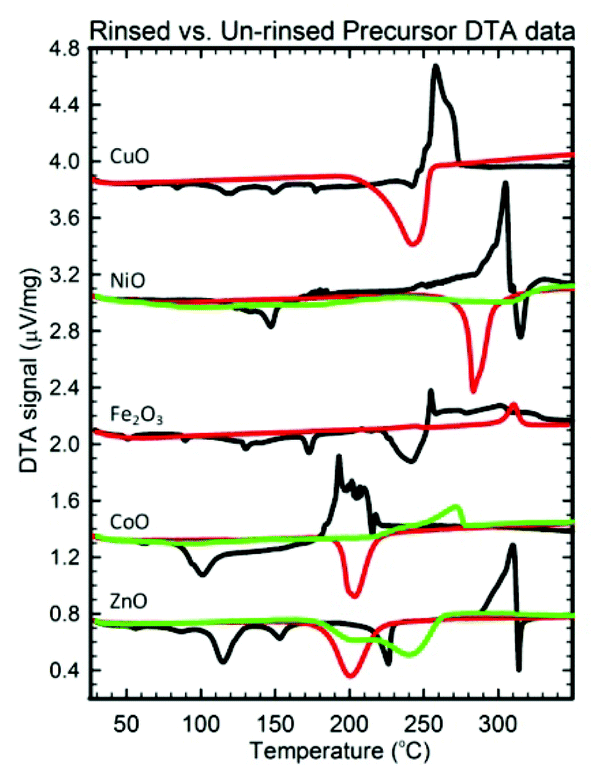 | ||
| Fig. 7 DTA data for the dried (black), rinsed/dried (green), and dried/rinsed/dried (red) precursors of oxides whose crystallite size is minimized if the precursor is rinsed prior to drying and/or calcination. | ||
Rinsing the Co2+, Ni2+, and Zn2+ precursors has another size-decreasing effect. As Table 3 indicates, these precursors (un-rinsed) typically contain a metal hydroxide-nitrate compound. These hydroxide-nitrate compounds limit the crystallite size of CoO, NiO, and ZnO to be >10 nm. However, rinsing prior to drying/calcining prevents these nitrate compounds from forming (see Table 3), meaning the nitrate ligand substitutes for one of the hydroxide ligands as the H2O solvent evaporates. By rinsing and thus preventing these compounds from forming, the crystallite size of these materials tends to be <10 nm.
The third reason for inserting a rinsing step is that it prevents uncontrolled oxidation/reduction of the metal cation during calcination. As the MS results indicate, numerous oxidizing and/or reducing agents (HNO3, N2O, NO, NO2, NH3) are released during the decompositions of un-rinsed precursors. As a result, mixed-oxidation state oxides can form for cations with multiple oxidation states (such as Bi3+, Co2+, Fe2+, Mn2+, Mo5+, Sn4+). Rinsing allows the formation of phase pure oxides for these multi-valence metal cations.
iii. Calcination atmosphere. In addition to rinsing, we have found it important to control the atmosphere during calcination for certain materials to maintain the metal oxidation state. Specifically, the CoO and Fe3O4 precursors need to be calcined in an inert atmosphere to prevent these multi-valence cations from being oxidized by O2 in the air to form Co3O4 and Fe2O3.
Though we have not explored it to the fullest extent, controlling the atmosphere during calcination opens a pathway to producing more nanomaterials than those listed in Table 1. For example, some preliminary work has shown that nano-metals can be formed from the Pd2+, Pt2+, Cu2+, Ni2+, and Co2+ precursors if they are calcined in a highly reducing atmosphere such as H2. We also hypothesize that calcining the Pr(III) and Mn(II) precursors in an inert environment might produce Pr2O3 and MnO instead of the oxidized PrO2 and Mn2O3 produced by calcination in air. Similarly, Co2O3, AgO, and either MnO2 or Mn5O8 might be formed if the Co(II), Ag(I), and Mn(II) precursors were calcined in an oxidizing environment, and conversely, SnO or Ce2O3 might be formed if the Sn(IV) and Ce(IV) precursors were calcined in a slightly reducing atmosphere. In essence, we predict that controlling the calcination atmosphere will add another valuable dimension of versatility to the solvent-deficient method.
II. Product characterization
The solvent-deficient method developed here has produced over 20 different metal oxide nanomaterials including, as Table 1 shows, at least one oxide from nearly every transition metal and semi-metal group in the periodic table (groups 3–4 and 6–15) as well as several oxides from the lanthanide group. With this wide range of products, the solvent-deficient method is one of the most versatile synthetic methods for making metal oxide nanoparticles.The XRD analyses shown in Fig. 2–4 (and Fig. S1 of the ESI†) indicate that the products are generally phase-pure and crystallize in the most thermodynamically favorable nano-scale phase. The high-magnification TEM images in Fig. 2–4 (and Fig. S1†) highlight the crystallinity of the nanoparticles produced using the solvent-deficient method; regular atomic planes are clearly visible and extend to the edges of the crystallites. Thus, low-crystallinity is not a problem exhibited by the solvent-deficient method, in contrast to many other aqueous solution methods. Additionally, the good agreement between the measured and expected d-spacings of the crystal planes (given in Table S1 in the ESI†) indicates that the particles exhibit little or no stress/strain in the crystal structure.
The TEM images also reveal the morphologies of the crystallites produced by the solvent-deficient method and enable direct measurement of their size and size distribution. As Fig. 2–4 illustrates, the crystallites have roughly spherical morphologies with the exception of the Y2O3 particles which are rod-like. The visible diameters of the roughly spherical crystallites verify the XRD size calculations given in Table 2, confirming that all of the products have average crystallite diameters well below 100 nm with about 75% having average diameters below 20 nm thereby satisfying the stringent size requirements for most applications of metal oxide nanomaterials.
As Table 2 shows, the absolute size distribution of the crystallites increases as the average crystallite size increases, but the relative percent of the size distribution is roughly between 20–45% for nearly all samples. These size distributions are small compared to those obtained by mechanical processing methods and are similar to those produced by other aqueous methods, but they are larger than those attainable using solution or vapor phase methods that employ capping agents. The lack of capping agents combined with the rapid crystallite formation and inequitable distribution of reagent materials are the most likely causes of the somewhat irregular particle morphologies and crystallite size distributions.
In turn, the absence of capping agents in the solvent-deficient method results in excellent chemical purity. The C, H, and N impurity contents (Table 2) were immeasurable (less than 0.5%) for most samples. For the few samples that displayed measurable quantities, the amounts were small (1–2%) and thus could also be attributed to the adsorption of species from the atmosphere. Small chloride impurities (<100 ppm) were present in a few of the samples made from chloride metal salt reagents, but longer/additional rinsing steps should remove these.
Another result of the absence of capping agents can be seen in Fig. 2–4; the individual crystallites visible in the high-magnification TEM images stick together to form the agglomerates seen in the lower-magnification images. The sizes and morphologies of these agglomerates vary considerably; as Table 2 indicates, the agglomerate sizes can range from roughly 50 nm to over 300 nm in size. Fortuitously, these agglomerates are mesoporous with very high surface areas and so are promising candidates for applications requiring high surface area metal oxides such as catalysis and gas sensing.
For applications in which aggregation is not advantageous, the agglomerates can be broken up relatively easily using a mill, as indicated by the preliminary Netzsch milling results given in Fig. S2 of the ESI.† This milling procedure probably introduces the same types of impurities as milling in solid-state methods, but the resulting average particle sizes and size distributions are smaller than those attained by mechanical processing alone.
A few other notable characteristics of the method can be seen in Tables 1 and 2. First, the reagents and equipment involved are common and relatively inexpensive materials, making the process itself fairly inexpensive. Next, the total time for the reaction ranges between 2–5 hours, thus the method is rapid. The average yield for most products is ≥95%, and all yields are above 60%. (The main limitation in the yield is inefficient transferring of the slurry/paste of the wet precursor from the grinding reaction vessel to a filter and/or a calcination container.) Encouragingly, the yield remains high when the reaction is scaled-up to produce large quantities; in single, 3-hour batches, over 100 g of Al2O3, Fe2O3, and ZnO nanomaterials have been produced with the same average 95% yield. The high yields combined with the low costs, scalability, versatility, speed, and ease of the method make it a prime candidate for application in the industrial production of high surface area metal oxide nanomaterials.
D. Conclusion
In summary, we have developed a general synthetic method for producing a vast range of metal oxide nanomaterials. Essentially, the method is a 2–3 step, solvent-deficient process in which a hydrated metal salt (usually a nitrate or chloride salt) is ground with bicarbonate (usually NH4HCO3) for typically 10–30 minutes to form a precursor which can be either rinsed or untreated before being calcined at relatively low temperatures (220–550 °C) in air (unless otherwise specified) for roughly 1–3 hours. The waters of hydration present on the metal salts act as a solvent in deficient quantities during grinding, allowing the metal salt and bicarbonate to dissociate and participate in a rapid replacement reaction resulting in the formation of an ammonium salt and a metal hydroxide, oxyhydroxide, hydroxide-nitrate, hydroxide-carbonate, carbonate, or amorphous oxide/hydroxide (depending on the metal cation used). In the second step, this “precursor” is calcined to dehydrate/decompose both the metal compound and the ammonium salt (if it has not already been rinsed away) to form the metal oxide nanomaterial.This solvent-deficient method has produced at least one metal oxide nanomaterial from nearly all the transition metal and semi-metal groups (groups 3–4, 6–15) as well as from the lanthanides, and it may yet be optimized to produce additional oxides. The nanomaterials produced are generally phase pure, chemically pure, crystalline, and well below 100 nm if not 20 nm in crystallite size. Capping agents are not employed, so these small crystallites aggregate into 50–300 nm agglomerates. Fortuitously, the solvent-deficient environment seems to endow these high surface area agglomerates with a mesoporous nature, making the products promising candidates for use in catalyst and gas sensing applications. The potential applicability of the products combined with the versatility, high yields, low costs, scalability, speed, and ease of the solvent-deficient method make it an attractive candidate for use in the industrial production of metal oxide nanomaterials.
Acknowledgements
We thank Dr Jeff Farrer for his assistance with TEM imaging/indexing and Dr Roger Harrison for helpful discussions and critiques of the paper. Funding for this work was provided by the U.S. Department of Energy under grant DE-FG02–05ER15666 and by the BYU ORCA program.References
- G. Oskam, J. Sol-Gel Sci. Technol., 2006, 37, 161–164 CrossRef CAS PubMed.
- C. B. Almquist and P. Biswas, J. Catal., 2002, 212, 145–156 CrossRef CAS.
- M. Anpo, T. Shima, S. Kodama and Y. Kubokawa, J. Phys. Chem., 1987, 91, 4305–4310 CrossRef CAS.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- N. Pinna, G. Neri, M. Antonietti and M. Niederberger, Angew. Chem., Int. Ed., 2004, 43, 4345–4349 CrossRef CAS PubMed.
- D. Jariwala, V. K. Sangwan, C.-C. Wu, P. L. Prabhumirashi, M. L. Geier, T. J. Marks, L. J. Lauhon and M. C. Hersam, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18076–18080 CrossRef CAS PubMed.
- F. Verbakel, S. C. J. Meskers, D. M. de Leeuw and R. A. J. Janssen, J. Phys. Chem. C, 2008, 112, 5254–5257 CAS.
- M. Wang, C. Xing, K. Cao, L. Zhang, J. Liu and L. Meng, J. Mater. Chem. A, 2014, 2, 9496–9505 CAS.
- J. M. Yuk, H. K. Seo, J. W. Choi and J. Y. Lee, ACS Nano, 2014, 8(7), 7478–7485 CrossRef CAS PubMed.
- M. S. Mauter, Y. Wang, K. C. Okemgbo, C. O. Osuji, E. P. Giannelis and M. Elimelech, ACS Appl. Mater. Interfaces, 2011, 3, 2861–2868 CAS.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- G. Skandan and A. Singhal, Perspectives on the science and technology of nanoparticle synthesis, CRC Press LLC, 2006, pp. 13–27 Search PubMed.
- M. Drofenik, D. Lisjak and D. Makovec, Mater. Sci. Forum, 2005, 494, 129–136 CrossRef CAS.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS PubMed.
- I. Capek, Colloids Interface Sci. Ser., 2007, 3, 1–60 CAS.
- J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramirez and M. J. Yacaman, Nanotechnology, 2005, 16, 2346–2353 CrossRef CAS PubMed.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS.
- C. C. Koch, Rev. Adv. Mater. Sci., 2003, 5, 91–99 CAS.
- M. Niederberger and G. Garnweitner, Chem. – Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed.
- S. C. Kuiry and S. Seal, in Synthesis of nanomaterials using microemulsion process, American Scientific Publishers, 2004, pp. 369–379 Search PubMed.
- N. Pinna and M. Niederberger, Angew. Chem., Int. Ed., 2008, 47, 5292–5304 CrossRef CAS PubMed.
- I. Bilecka and M. Niederberger, Nanoscale, 2010, 2, 1358–1374 RSC.
- B. Buesser and S. E. Pratsinis, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 103–127 CrossRef CAS PubMed.
- G. Garnweitner and M. Niederberger, J. Mater. Chem., 2008, 18, 1171–1182 RSC.
- K. Holmberg, J. Colloid Interface Sci., 2004, 274, 355–364 CrossRef CAS PubMed.
- D. G. Lamas and N. E. Walsoe de Reca, Ceram. Crist., 2006, 46, 46–49 CAS.
- S. Li, S. Shan, Q. Jia, Y. Hu, L. Jiang and Y. Wang, Adv. Mater. Res., 2012, 534, 89–92 CrossRef CAS.
- C. R. Martin, Science, 1994, 266, 1961–1966 CAS.
- V. Valtchev and L. Tosheva, Chem. Rev., 2013, 113, 6734–6760 CrossRef CAS PubMed.
- Y. Xia, X. Xia, Y. Wang and S. Xie, MRS Bull., 2013, 38, 335–344 CrossRef CAS.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS PubMed.
- B. F. Woodfield, S. Liu, J. Boerio-Goates, Q. Liu and S. J. Smith, US Patent, 2007-US4279, 2007098111, 20070216, 2007 Search PubMed.
- R. E. Olsen, C. H. Bartholomew, B. Huang, C. Simmons and B. F. Woodfield, Microporous Mesoporous Mater., 2014, 184, 7–14 CrossRef CAS PubMed.
- B. Huang, C. H. Bartholomew, S. J. Smith and B. F. Woodfield, Microporous Mesoporous Mater., 2013, 165, 70–78 CrossRef CAS PubMed.
- B. Huang, C. H. Bartholomew and B. F. Woodfield, Microporous Mesoporous Mater., 2013, 177, 37–46 CrossRef CAS PubMed.
- B. Huang, C. H. Bartholomew and B. F. Woodfield, Microporous Mesoporous Mater., 2014, 183, 37–47 CrossRef CAS PubMed.
- A. H. Lu, E. L. Salabas and F. Schueth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- M. G. Bawendi, A. R. Kortan, M. L. Steigerwald and L. E. Brus, J. Chem. Phys., 1989, 91, 7282–7290 CrossRef CAS PubMed.
- D. A. Selck, B. F. Woodfield, J. Boerio-Goates and D. E. Austin, Rapid Commun. Mass Spectrom., 2012, 26, 78–82 CrossRef CAS PubMed.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by powders and porous solids, Academic Press, London, 1999 Search PubMed.
- B. Huang, C. H. Bartholomew and B. F. Woodfield, Microporous Mesoporous Mater., 2014, 184, 112–121 CrossRef CAS PubMed.
- A. Navrotsky, Int. J. Quantum Chem., 2009, 109, 2647–2657 CrossRef CAS.
- G. Feick and R. M. Hainer, Nature, 1954, 173, 1188–1189 CrossRef CAS.
- A. O. Vuori, in 2002 IFA Technical Conference, Chennai, India, 2002.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) Preliminary Netzsch milling results for Al2O3 and CeO2, (2) XRD patterns/analyses of the dried and rinsed precursors plotted with the ICDD standard patterns of the materials they contain, (3) all TG/DTA-MS data. See DOI: 10.1039/c4nr04964k |
| ‡ Current address: MegaDiamond Inc., A Schlumberger Company, Provo, UT 84604. qliu@smith.com |
| This journal is © The Royal Society of Chemistry 2015 |