
Absolute photoluminescence quantum yields of IR26 and IR-emissive Cd1−xHgxTe and PbS quantum dots – method- and material-inherent challenges†
Soheil
Hatami
a,
Christian
Würth
a,
Martin
Kaiser
a,
Susanne
Leubner
b,
Stefanie
Gabriel
b,
Lydia
Bahrig
b,
Vladimir
Lesnyak
bc,
Jutta
Pauli
a,
Nikolai
Gaponik
b,
Alexander
Eychmüller
b and
Ute
Resch-Genger
*a
aBAM Federal Institute for Materials Research and Testing, Richard-Willstaetter-Str. 11, 12489 Berlin, Germany. E-mail: ute.resch@bam.de
bPhysical Chemistry and Center for Advancing Electronics Dresden, TU Dresden, Bergstr. 66b, 01062 Dresden, Germany
cDepartment of Nanochemistry, Istituto Italiano di Tecnologia, via Morego, 30, 16163 Genova, Italy
First published on 30th October 2014
Abstract
Bright emitters with photoluminescence in the spectral region of 800–1600 nm are increasingly important as optical reporters for molecular imaging, sensing, and telecommunication and as active components in electrooptical and photovoltaic devices. Their rational design is directly linked to suitable methods for the characterization of their signal-relevant properties, especially their photoluminescence quantum yield (Φf). Aiming at the development of bright semiconductor nanocrystals with emission >1000 nm, we designed a new NIR/IR integrating sphere setup for the wavelength region of 600–1600 nm. We assessed the performance of this setup by acquiring the corrected emission spectra and Φf of the organic dyes Itrybe, IR140, and IR26 and several infrared (IR)-emissive Cd1−xHgxTe and PbS semiconductor nanocrystals and comparing them to data obtained with two independently calibrated fluorescence instruments absolutely or relative to previously evaluated reference dyes. Our results highlight special challenges of photoluminescence studies in the IR ranging from solvent absorption to the lack of spectral and intensity standards together with quantum dot-specific challenges like photobrightening and photodarkening and the size-dependent air stability and photostability of differently sized oleate-capped PbS colloids. These effects can be representative of lead chalcogenides. Moreover, we redetermined the Φf of IR26, the most frequently used IR reference dye, to 1.1 × 10−3 in 1,2-dichloroethane DCE with a thorough sample reabsorption and solvent absorption correction. Our results indicate the need for a critical reevaluation of Φf values of IR-emissive nanomaterials and offer guidelines for improved Φf measurements.
1 Introduction
Fundamental advances in biochemical assays, molecular sensors, optical imaging, telecommunication, and optical, electroluminescent, and photovoltaic devices require bright and stable fluorophores.1–7 A straightforward measure of fluorophore performance is the photoluminescence quantum yield (Φf),8,9 given by the number of emitted photons Nem per number of photons Nabs absorbed by the system, see eqn (1).9–11 For fluorescent nanomaterials such as semiconductor nanocrystals (NCs), i.e., quantum dots and rods,8 and for upconversion nanocrystals (NCs),12,13 where surface states and the accessibility of emissive states by quenchers largely control the accomplishable Φf values,14–18 the value of Φf provides also a straightforward tool to assess the quality of the surface shell and surface passivation and hence to evaluate new synthetic and surface functionalization strategies.19 Moreover, concentration-dependent Φf gives a direct hint of ligand desorption as recently shown for, e.g., CdTe and CdSe quantum dots,14,15,20–22 which is typically the first step to material decomposition, resulting in the release of toxic heavy metal ions.8,20,23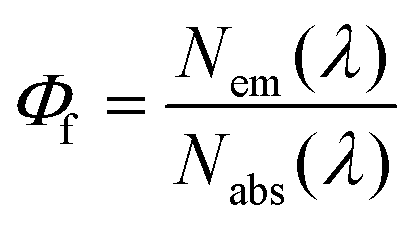 | (1) |
Φ
f can be obtained directly by optical methods either relative to a fluorescent reference with known Φf or as an absolute quantity (standard-free),9,11,24,25 or by calorimetric methods like photoacoustic spectroscopy and thermal lensing.26–28 Commonly, Φf is obtained optically, comparing the absorption-weighted integral fluorescence intensities of a sample and a reference measured under identical conditions.9,11,24 Due to the need for standards with reliably known Φf, this straightforward procedure is limited predominantly to the ultraviolet (UV), visible (vis) and NIR regions, where Φf standards have been recommended for many years9,24,25,27–32 or were recently provided by us.9,25 This included the reevaluation of Φf of IR125 in dimethylsulfoxide (DMSO), the most frequently used NIR reference dye for organic fluorophores and semiconductor NCs,33–37 yielding a Φf of 0.239 instead of the previously assumed Φf of 0.13,38 and consequently, an underestimation of all Φf values obtained relative to this dye. Nevertheless, the accurate determination of Φf> 1000 nm still presents a considerable challenge as indicated by the recently reexamined Φf of the most frequently used IR Φf reference dye IR26,39–42 demonstrating large deviations in its Φf in 1,2-dichloroethane (DCE) by a factor of up to 10.43–45 This was recently addressed in depth by the absolute measurements of its Φf by Semonin et al. using a self-made integrating sphere setup.43 These high uncertainties in the red wavelength region originate from the fact that many critical issues affecting especially photoluminescence measurements in the IR wavelength region, such as the performance and validation of instrument calibrations,46 solvent and water vapor absorption, and sample reabsorption,24,47,48 have not been really detailed so far. Moreover, semiconductor NC-specific effects like material- and surface chemistry-dependent photodecomposition and photobrightening,8,14,49 which can depend on the incident spectral radiant flux and even on the NC size, or a possible excitation wavelength dependence of Φf![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50,51 were not addressed.
50,51 were not addressed.
Aiming at the rational design of bright NIR- and IR-emissive semiconductor NCs with emission in the wavelength region of 650–1600 nm and to address the relevance of the reliable characterization of emerging luminescent reporters for the NIR II window of 1000–1700 nm for, e.g., bioimaging studies,52–54 we present the design of a new integrating sphere setup for absolute measurements of photoluminescence spectra and Φf from 600 to 1600 nm and its calibration and performance validation. Subsequently, we reexamined the Φf of IR26 and quantified measurement uncertainties. Then, we measured Φf of representative NIR- and IR-emissive semiconductor NCs, here Cd1−xHgxTe colloids with size and composition tuning of the optical and electrochemical properties,42,55,56 and differently sized PbS, presenting the most commonly used NCs in photovoltaics,3,33,57 to provide examples of achievable Φf values.
2 Methods
2.1 Reagents and materials
The Φf standards HITCI (batch number 029006) and IR125 (batch number 10970) used for relative measurements and IR140 (batch number 9310) were obtained from Lambda Physics, the spectral emission standard Itrybe from Otava (batch number OTVD_0037), and IR26 from Acros (batch number 409401000). The chemical structures are given in the ESI† (Fig. 1S). The solvents used for the spectroscopic studies, i.e., ethanol for Itrybe and HITCI, dimethylsulfoxide (DMSO) for IR125 and IR140, 1,2-dichloroethane (DCE) for IR26, and tetrachloroethylene (TCE) for PbS, were of spectroscopic grade and were purchased from Sigma Aldrich and Merck. For Cd1−xHgxTe, deuterated water (D2O) from Merck was employed.2.2 Synthesis of semiconductor NCs
Cd1−xHgxTe of different compositions and sizes were synthesized in water as previously described using mercaptopropionic acid (MPA) as a ligand42 and stored in the dark in air. Differently sized PbS colloids were prepared according to Poppe et al.58 under inert gas, purified, and stored as stock solutions in toluene either in a glove box or under an inert gas atmosphere or in air in the dark in the refrigerator.2.3 Characterization
Dye purity was determined by high performance liquid chromatography (HPLC) with the Agilent 1200 HPLC system (Agilent) using a previously reported method25 and yielded the following purities (100% method): HITCI: 97.9% (760 nm),25 IR125: 99.1% (800 nm),25 Itrybe: 99.8% (600 nm), IR140: 90.2% (700 nm), and IR26: 99.5% (750 nm). The size of the PbS NCs was determined from absorption spectra to be 2.3 nm, 4.1 nm, and 4.4 nm, respectively, as described by Cademartiri et al.59 The size and composition of the Cd1−xHgxTe NCs was determined to be 4.8 nm, 6.3 nm, and 7.1 nm for colloids with Cd:Hg ratios of 99:1, 98:2, and 97:3, respectively, using TEM and ICP-OES. Absorption spectra were recorded on a calibrated Cary 5000 spectrometer. Fluorescence emission spectra and Φf were measured with a FSP920 spectrofluorometer (Edinburgh Instruments; 300–1700 nm; relative measurements with HITCI (Φf = 0.30; ethanol) and IR125 (Φf = 0.23; DMSO) as the references),9,24,25 a custom made UV/vis/NIR integrating sphere setup (350–1050 nm),25 and our newly designed NIR/IR integrating sphere setup (600–1600 nm), all independently calibrated (see ESI† for details of instrumentation and calibration including the transfer standards employed). All absorption and fluorescence measurements were performed with freshly prepared dye or NC solutions (absorbances A of 0.02 to 0.1 at the longest wavelength absorption maximum or the first excitonic peak) at T = (25 ± 1) °C in air using either 10 mm × 10 mm quartz cuvettes from Hellma GmbH or 10 mm × 4 mm cells to minimize dye or NC reabsorption and/or solvent absorption. For IR26, absorbances of up to 0.8 were used. Stock solutions of Cd1−xHgxTe NC stored in air in the dark at room temperature were diluted with D2O by a factor of at least 1000:1 before each measurement. For potentially air-sensitive PbS colloids, aging studies were performed with the stock solutions of the three PbS colloids in toluene, stored in air in the dark in the refrigerator. To representatively address the influence of oxygen, the 4.4 nm sized PbS NC batch was split into two portions, one kept always under inert gas in the dark at room temperature and the other one under identical conditions to the stock solutions of the smaller PbS colloids. For the spectroscopic studies, the PbS stock solutions were always diluted by a factor of at least 1000:1 with TCE and measured in air, except for 4.4 nm PbS stored and aged under inert gas. In this case, TCE dilution was performed in a glove box and sealed cells were used for subsequent spectroscopic measurements. All fluorescence emission spectra shown were blank-corrected and corrected for instrument-specific effects relative to the spectral photon radiance scale.46,60,61 Relative standard deviations of fluorescence measurements were obtained from four independent measurements. Spectral corrections such as a reabsorption correction9,24,25 and solvent absorption correction are detailed in the ESI.†
3 Results and discussion
3.1 Design of an integrating sphere setup for the NIR and IR
The design of our integrating sphere setup for absolute Φf measurements from 600 to 1600 nm with versatile excitation, reasonable spectral resolution, and high sensitivity is shown in Fig. 1 (for more details of setup components, see ESI†). To optimize the sensitivity of the setup, which should enable the determination of not only the moderate to high Φf of semiconductor NCs exceeding 0.1, but also those of organic dyes with emission >1000 nm like IR26 with very small Φf < 5 × 10−3,43 we chose a small integrating sphere (diameter of ca. 11 cm) with a high reflectivity hydrophobic Spectralon coating (99% from 400 to 1500 nm) as the sample compartment, thereby minimizing water adsorption from air. This sphere, which is large enough to minimize distortions of the radiating field in the sphere by the sample, was equipped with six ports for sample illumination, positioning of the sample holder, emission detection, and purging with dry nitrogen to remove water vapor absorbing at around 950 nm, 1130 nm, and 1450 nm (see ESI, Fig. 7S†). This avoids distortions of the measured transmitted excitation light and photoluminescence, which become otherwise dependent on air humidity (and ambient temperature) and optical path length. As excitation light sources, we chose different high stability (intensity fluctuations <1%; see ESI†) intense lasers (HeNe laser: 633 nm), laser diodes (690 nm, 730 nm, 785 nm, 808 nm, and 980 nm), and a diode-pumped solid-state laser (914 nm), to enable the measurement of dye and NC samples absorbing and emitting over a broad wavelength region with strongly varying photoluminescence efficiencies. These light sources were coupled via fiber optics (Fig. 1, top) to the integrating sphere (Fig. 1, bottom) or directed either directly into the middle of the sphere (sample position) or on the sphere wall with a flat mirror. This enables direct and indirect sample illumination and their combination.62 The small spot diameters of 1–4 mm of these excitation light sources provide control of the illuminated volume to minimize solvent absorption and reabsorption effects. This is of special relevance for the measurement of solutions and dispersions >950 nm due to the absorbance of many solvents and matrices in this wavelength region (Fig. 3, right panel). For signal detection, we used an imaging spectrograph attached to a Peltier cooled, thinned back side illuminated deep depletion InGaAs CCD with high quantum efficiencies of >85% (1100–1500 nm). The N2-purged spectrograph-detector ensemble was coupled to the integrating sphere with an IR-suitable quartz fiber bundle (LLB552-IR-0,12, IR Quartz, LOT Oriel, transmission >50%) shielded from direct reflexes with several baffles. We placed a filter wheel with neutral density (ND) filters (known wavelength-dependent transmission) in front of the integrating sphere for the control and variation of the incident radiant power and a filter wheel with several long pass (LP) interference filters (cut-on wavelengths of 600 nm, 800 nm, 950 nm, and 1000 nm) in front of the spectrograph–CCD ensemble to eliminate signal distorting second order diffractions and stray light. Dye solutions and NC dispersions in quartz cells were center-mounted in the sphere with a custom designed Spectraflect-coated cuvette holder positioned with a HeNe laser.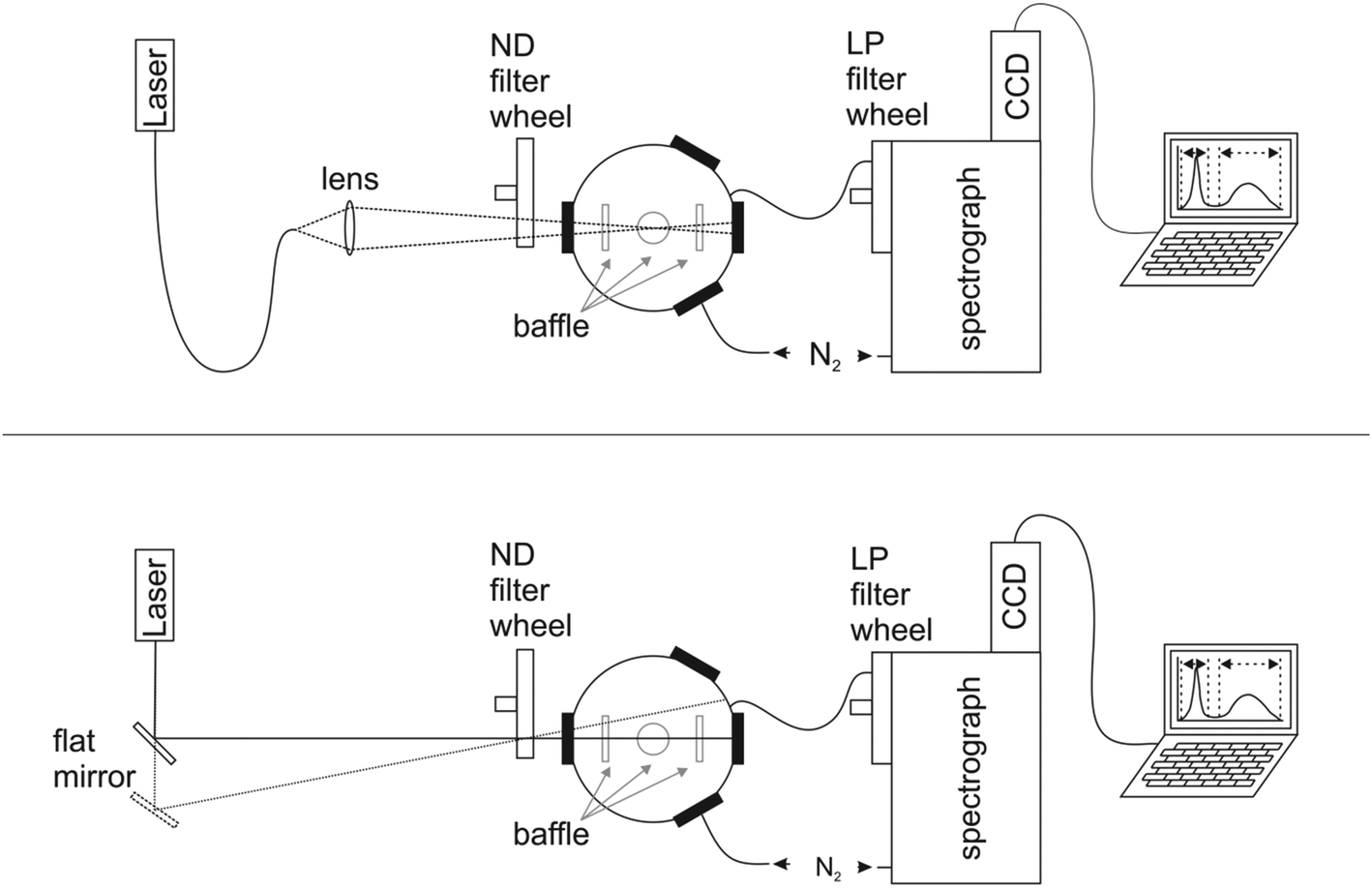 | ||
| Fig. 1 NIR/IR integrating sphere setup for absolute measurement of photoluminescence in the wavelength region of 600 nm to 1600 nm consisting of several high stability (intensity fluctuations <1%) lasers and laser diodes, coupled either via fiber optics (upper panel; 808 and 980 nm laser diodes) or directly (HeNe laser, 690 nm, 730 nm, and 785 nm laser diodes; 914 nm solid-state laser) with a flat mirror (lower panel) to a N2-purged 12 cm integrating sphere attached via an IR-quartz fiber bundle to an imaging spectrograph equipped with an InGaAs CCD. The mirrors can be used for direct or indirect sample illumination. Two filter wheels, one equipped with neutral density (ND) filters placed between the sphere and the light sources, and one equipped with different long pass (LP) interference filters (cut-on wavelengths of 600, 800, 950, and 1000 nm) in front of the detection system, enable control of the exciting radiant power and remove second order diffractions. Dye solutions and NC dispersions in cuvettes are placed in the center of the sphere using a custom designed Spectraflect-coated cell holder. | ||
3.2 Setup characterization
We subsequently determined the wavelength accuracy of the excitation and detection channel(s), the linearity of the detection channel, and the wavelength-dependent spectral responsivity of the integrating sphere-spectrograph–CCD ensemble (s(λem)).46 The wavelength accuracy of the emission monochromator revealed maximum deviations <1.1 nm (see ESI, Fig. 3S†). Relative deviations from a linear behavior of the InGaAs CCD, measured at a constant integration time for varying excitation radiant powers, amounted to 0.3% (see ESI, Fig. 4S†) and for a constant radiant power and varying integration times to 0.5%. To reduce calibration uncertainties in s(λem) caused by stray light and second order effects and to account for the lack of suitable emission standards >950 nm, we employed two calibration lamps, a conventional spectral radiance transfer standard (SDS)63 with an emission maximum at 1050 nm and a blackbody radiator (BBR) with a temperature-controlled spectral radiance, with an emission maximum at 1970 nm (for T = 1200 °C). An example of an emission correction curve (equaling 1/s(λem)) derived from the combination of these measurements (see ESI, Fig. 6S†) is shown in Fig. 2 (top panel; SDS: emission correction curve for 600–950 nm; BBR: emission correction curve measured for 950–1600 nm). The relative uncertainty for the determination of s(λem) from such combined calibration curves amounts to maximum 12%. This value includes uncertainties of the certified values of the calibrated light sources, the wavelength accuracy, linearity of the detection system, contributions from stray light, specific uncertainties arising from the different types of light sources and merging of the emission correction curves. Finally, the long term stability of the spectral radiant power of the different light sources of the integrating sphere setup was obtained as this is a critical parameter for all absolute measurements of Φf.25 Multiple measurements over time using typical measurement conditions (integration time, number of accumulations; see ESI, Fig. 5S† for exemplary light sources) revealed relative deviations of the mean values (integration over the entire excitation peak) of <1%.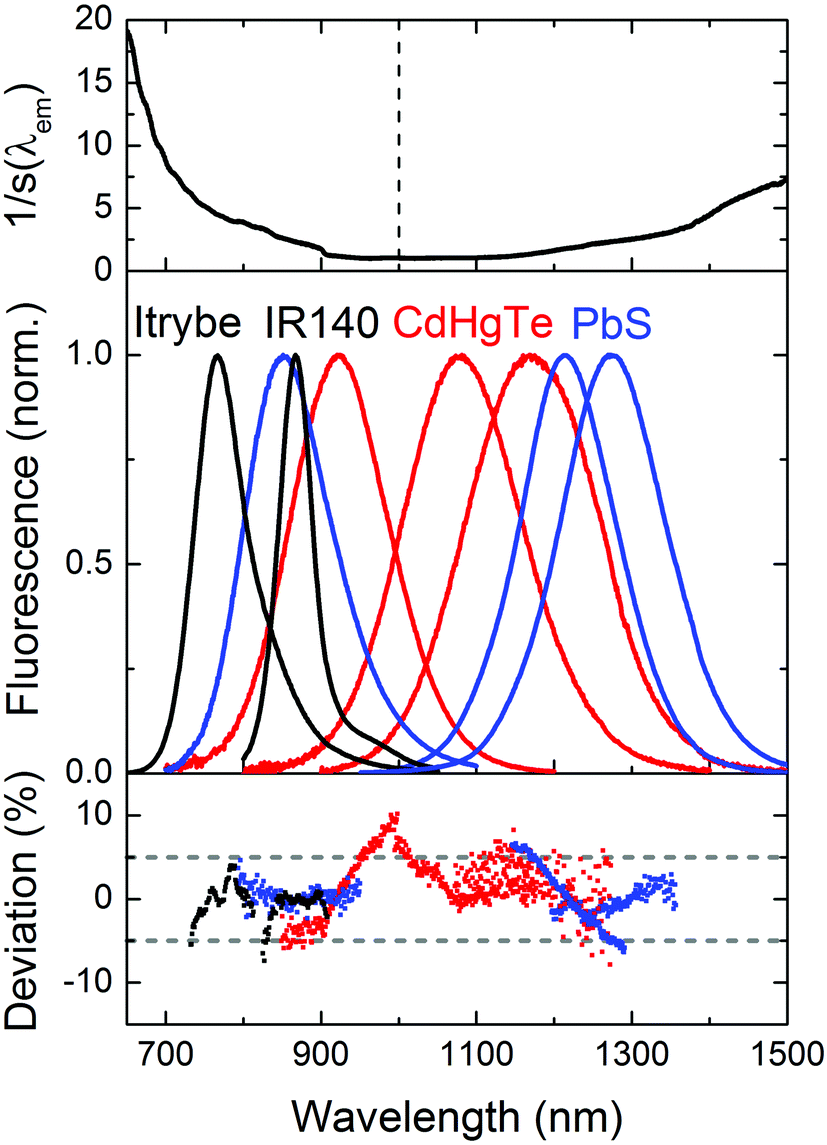 | ||
| Fig. 2 Top: inverse spectral responsivity 1/s(λem) of the NIR/IR integrating sphere setup determined with two calibration lamps. Middle: normalized corrected emission spectra of the NIR dyes Itrybe (solvent ethanol; black line) and IR140 (solvent DMSO; black line), MPA-stabilized CdHgTe NC colloids of various compositions and sizes dispersed in air-saturated D2O (red lines), and PbS colloids of various sizes in TCE (blue lines) in air obtained with our NIR/IR integrating sphere setup. Bottom: relative spectral deviations between the corrected spectra measured with the integrating sphere setup and an independently calibrated spectrofluorometer (data given for intensities up to 50% of the intensity at the respective emission maximum). The dotted grey lines indicated relative deviations of 5%. | ||
3.3 Setup validation – comparison of emission spectra
Proper validation of the calibration of the wavelength and intensity scale is the only way to obtain reliable emission spectra and Φf values. The good agreement between the emission correction curves obtained with SDS and BBR (see ESI, Fig. 6S†) provides already a first hint of the accuracy of our emission correction curve. For the control of s(λem) in the wavelength region of 600–1000 nm, we acquired the corrected emission spectra of the NIR dyes Itrybe and IR140 in ethanol and DMSO, respectively, and for 900–1600 nm, the corrected emission spectra of different IR-emissive semiconductor NCs, here mercaptopropionic acid (MPA)-stabilized Cd1−xHgxTe NCs of various compositions and sizes dispersed in D2O and PbS colloids of various sizes in TCE with the NIR/IR sphere setup and our independently calibrated fluorometer. The generally good match between these emission spectra (Fig. 2, middle panel), indicated by the small relative spectral deviations of the order of typically 5% (Fig. 2, lower panel), underlines the reliability of our emission correction curve.3.4 Fluorescence quantum yields of NIR dyes with emission <1000 nm
Subsequently, we measured Φf of the NIR dyes Itrybe (solvent ethanol; emission: 680–950 nm; λex = 633 nm)64 and IR140 (solvent DMSO; emission: 830–1050 nm; λex = 808 nm) with our NIR/IR and/or UV/vis/NIR integrating sphere setups and our calibrated fluorometer FSP920. Itrybe and IR140 were chosen because of their different Stokes shifts providing examples of small (Itrybe)64 and large (IR140) spectral overlap between absorption and emission and their commercial availability in high purity as well as their subsequently derived good photochemical stability (see ESI, Fig. 9S†) and their excitation wavelength-independent Φf. The good agreement between the different methods and setups used underlines the reliability of Φf measurements with our new integrating sphere in the wavelength region of 630–1050 nm. We previously provided an uncertainty budget for Φf measurements in the wavelength region of 400–1000 nm.9,24,253.5 Reassessment of Φf of IR26 with emission >1000 nm
For the control of the reliability of Φf measurements >1000 nm, we chose IR26 in DCE (excitation range: 800–1000 nm, emission: 1010–1450 nm; see Fig. 3, left panel). Together with IR125 in DMSO, this dye presents the most common reference for red emissive NCs. Moreover, its Φf was only recently redetermined absolutely to be 4.8 × 10−4,43 which is by a factor of ca. 10 lower than previously assumed,44,45 rendering all Φf measured relative to this dye questionable.39,40,42 Its extremely low Φf enables us to test the sensitivity of our new setup. Other challenges in the measurement of the Φf of IR26 are the strong spectral overlap between its absorption and emission bands favoring reabsorption and the absorption of the recommended solvent DCE within the dye's emission band with a sharp maximum at ca. 1170 nm (Fig. 3, left and right panels). Solvent absorption occurs for many common solvents >900 nm (Fig. 3, right); hence, uncertainties of determined quantum yields in this wavelength region exceed those of measurements at smaller wavelengths. Solvent and reabsorption effects are especially critical for integrating sphere measurements, as the size of the resulting signal distortions depends on the sphere diameter and reflectivity of the sphere coating as well as on the sample volume.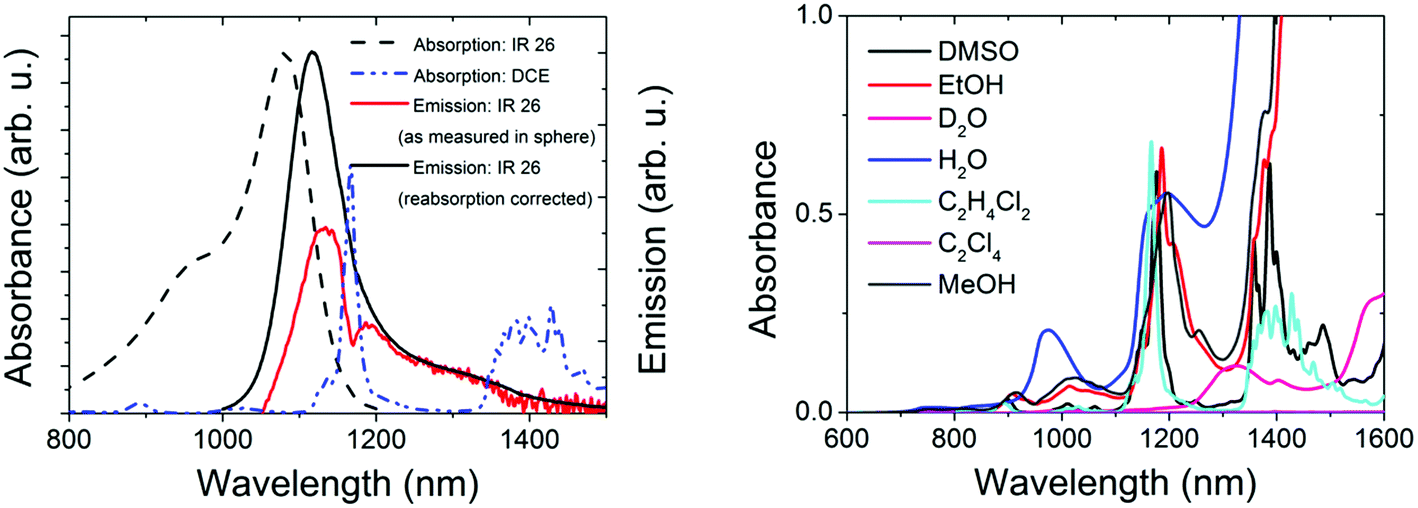 | ||
| Fig. 3 Left: normalized absorption and emission spectra of IR26 in DCE (without and with corrections for dye reabsorption and solvent absorption) and absorption of the solvent DCE within the wavelength region of dye emission. Right: absorption spectra of common organic solvents, H2O, and D2O used for the study of IR dyes and NCs, given for an optical path length of 1 cm. | ||
We determined the Φf of IR26 in DCE (see Table 1) absolutely at dye concentrations (c) of 0.7 × 10−6 to 6 × 10−6 mol L−1 (equaling absorbances of 0.09–0.80 at the absorption maximum of 1082 nm using a molar absorption coefficient ε = 1.4 × 10−5 L mol−1 cm−1 (ref. 65)) and with our spectrofluorometer relative to IR125 in DMSO9,25 (IR26: λex = 980 nm; IR125: λex = 808 nm; see ESI, Fig. 2S†).24,46 Absorption spectra of these dye solutions revealed the absence of dye aggregates at these concentrations (see ESI, Fig. 8S†) and the photochemical stability of IR26 was sufficient for excitation with a 980 nm laser diode (see ESI, Fig. 9S†). The sharp dip in the measured emission band of IR26 at ca. 1170 nm shown in Fig. 3 (left panel, red spectrum) demonstrates the considerable influence of solvent absorption. The influence of dye reabsorption on IR26 fluorescence follows from the red shift and the reduced intensity of the as-measured emission band compared to the reabsorption- and solvent-absorption-corrected fluorescence spectrum (Fig. 3, left panel, black spectrum). This is reflected by the corresponding Φf data, i.e., a Φf of 0.7 × 10−3 as measured, which amounts to 1.1 × 10−3 after consideration of solvent absorption and dye reabsorption (see ESI;† Fig. 8S).9,24,25 We determined the relative uncertainty of this value to be 21% (2.3 × 10−4). This includes contributions from setup calibration (12%) and applied corrections (15%) as well as the relative standard deviation of fluorescence measurements with our NIR/IR integrating sphere setup (9%). Relative measurements yielded a Φf of 0.7 × 10−3 (Table 1). We estimated the relative uncertainty of this value to be 30% (2.1 × 10−4) taking into account uncertainties from instrument calibration (emission correction from the instrument manufacturer, two point self-made excitation correction, and Φf uncertainty of the reference dye IR125), dye reabsorption and solvent absorption and the relative standard deviation of fluorescence measurements. Dye purity was not considered in both cases.
| Dye | Solvent | Φ f Absolute 1 | Φ f Absolute 2 | Φ f Relative | Φ f Lit. |
|---|---|---|---|---|---|
| Itrybe | EtOH | 0.22 | — | 0.23 | — |
| IR140 | DMSO | 0.20 | 0.20 | 0.20 | — |
| IR26 | DCE | 0.0011 | — | 0.0007 | 0.00048 |
We attribute our finding of a higher Φf for IR26 than the value of 0.48 × 10−3 reported by Semonin et al.43 to different calibration and correction procedures. Nevertheless, our value still equals the absolute upper limit of Φf of this dye assumed by Semonin. In addition, the fact that we could measure Φf absolutely at concentrations of 0.7 × 10−6 to 6 × 10−6 mol L−1 demonstrates the higher sensitivity of our integrating sphere setup compared to the setup reported by Semonin et al. who used dye concentrations of 2 × 10−6 to 11 × 10−6 mol L−1 (equaling the absorbances of 0.3–1.5 at the absorption maximum of 1082 nm) for the integration sphere measurement of Φf of IR26. In this respect, the measurement uncertainties given by us for the absolute determination of Φf of IR26 exceed only apparently the uncertainty provided by Semonin et al. of 0.2 × 10−4 (equaling a relative uncertainty of 4.2%) that was not really deduced from the actual integrating sphere measurements. This uncertainty was derived by Semonin et al. only from a fit considering dye reabsorption, but not solvent absorption, which was made for a 0°/90° measurement geometry and not for the integrating sphere measurements. Most likely, the small uncertainties given by Semonin et al. consider solely the standard deviation of the fluorescence measurements, but not a complete uncertainty budget as given by us.24,25
Nevertheless, our data verify the considerable overestimation of many Φf of NIR- and IR-emissive NCs like PbS, PbSe, and Cd1−xHgxTe.39,40,42 This finding, together with the Φf values absolutely measured for PbS in different solvents of the order of 0.05 to 0.6043,66–68 and relative to IR125 (using a reference Φf of 0.13)38 of the order of 0.20 to 0.40,33,35 clearly demonstrates the need for a critical reexamination of Φf values of representative IR-emissive semiconductor NCs.
3.6 Φ f of IR-emissive semiconductor Cd1−xHgxTe ternary alloys
Especially promising IR-emissive NCs are Cd1−xHgxTe ternary alloys as well as PbS and PbSe.3,33,42,55–57 As PbSe NCs can be more prone to surface oxidation as PbS NCs,69–72 we decided to evaluate only the Φf of dispersions of representative Cd1−xHgxTe ternary alloys of different compositions and sizes and exemplary differently sized PbS colloids detailed in the following section to provide examples of achievable Φf values for IR-emissive quantum dots and to identify NC-specific challenges and sources of uncertainty.We determined the Φf of three MPA-capped CdxHg1−xTe NC samples with comparatively broad emission spectra in D2O spanning a wavelength region of ca. 750–1350 nm shown in Fig. 2 and 4 (inset) at different excitation wavelengths between 633 and 785 nm relatively and absolutely. Prior to these measurements, we measured time traces with our integrating sphere setup to exclude photodecomposition and photobrightening for excitation with intense laser diodes, the radiant flux of which considerably exceeded that of a xenon lamp monochromator ensemble used for relative photoluminescence measurements. Moreover, Φf were determined relatively before and after each absolute measurement. These studies did not reveal light-induced changes of the Cd1−xHgxTe NC colloids.
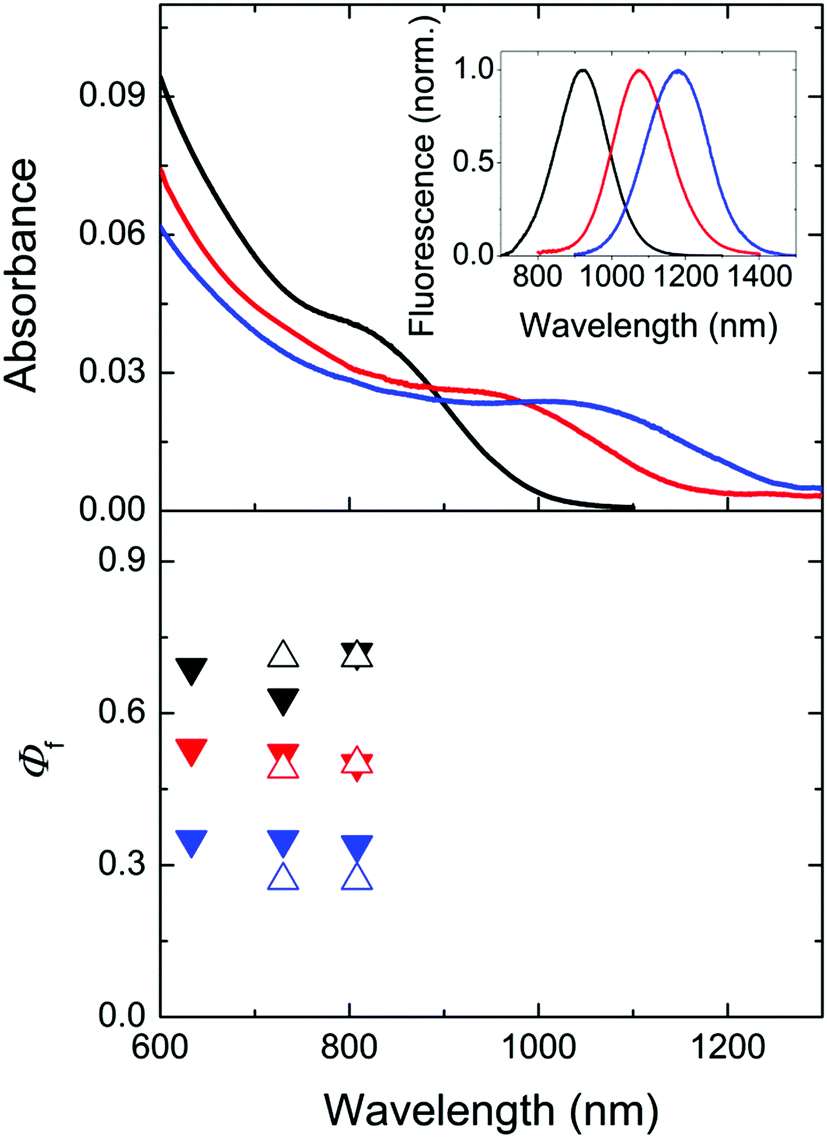 | ||
| Fig. 4 Top: normalized absorption spectra of three representative Cd1−xHgxTe NC colloids of different sizes and compositions (black: 4.8. nm, Cd:Hg = 99:1, c = 1.3 × 10−6 mol L−1; red: 6.3 nm, Cd:Hg = 98:2, c = 1.3 × 10−6 mol L−1; blue: 7.1 nm, Cd:Hg = 97:3, c = 5.2 × 10−7 mol L−1) in D2O in air. The corresponding corrected emission spectra are shown in the inset and in Fig. 2 (middle panel). The concentration of the Cd1−xHgxTe NC was determined by measuring the concentration of the respective metal ions (Cd2+ and Hg2+) via ICP-OES as previously described15,73 and considering the NC diameter derived from TEM data. Bottom: absolutely measured Φf (solid symbols) and relatively measured Φf (open symbols) at different excitation wavelengths. Considering the uncertainties of both methods, the determined values overlap. | ||
As follows from Fig. 4, the absolutely and relatively determined Φf of the three Cd1−xHgxTe colloids agree reasonably well. We determined the relative uncertainties of absolutely measured Φf to be 16%, considering uncertainty contributions of setup calibration (12%) and applied reabsorption and stray light corrections (5%; see ESI†) as well as of the relative standard deviation of fluorescence measurements with the NIR/IR integrating sphere setup (10%). The uncertainties of relatively measured Φf are estimated to be 15% (no excitation correction, only small absorption of D2O, less pronounced reabsorption as found for IR26). None of the colloids studied showed a dependence of its Φf on the excitation wavelength within the uncertainties. The comparatively high Φf between ca. 0.3 and 0.65 of our colloids underline the suitability of our simple one pot aqueous synthesis42 for the preparation of bright CdxHg1−xTe NCs especially with the ligand MPA.73 Whether the observed trend of a decrease in Φf with increasing size and/or Hg content is really representative, possibly originating from a confinement reduction with increasing particle size and/or the introduction of an increasing number of crystal and surface defects with larger numbers of Hg2+ ions (with sizes exceeding that of Cd2+) favoring luminescence quenching, remains to be shown for a larger set of samples.
3.7 Aging studies and Φf of IR-emissive PbS colloids
Subsequently, we investigated the spectroscopic properties of three oleate-capped PbS colloids of different sizes, i.e., 2.3 nm, 4.1 nm, and 4.4 nm, in TCE which does not absorb within the wavelength region of NC emission (Fig. 2 and 3, right panel; ESI, Fig. 10S†). To account for a possible influence of oxygen and hence NC oxidation, which is manifested by a blue shift in absorption and emission and can depend on particle size,72 the 4.4 nm-sized PbS colloid batch was split into two portions immediately after synthesis. One portion was kept always under inert gas and the other sample was stored in air.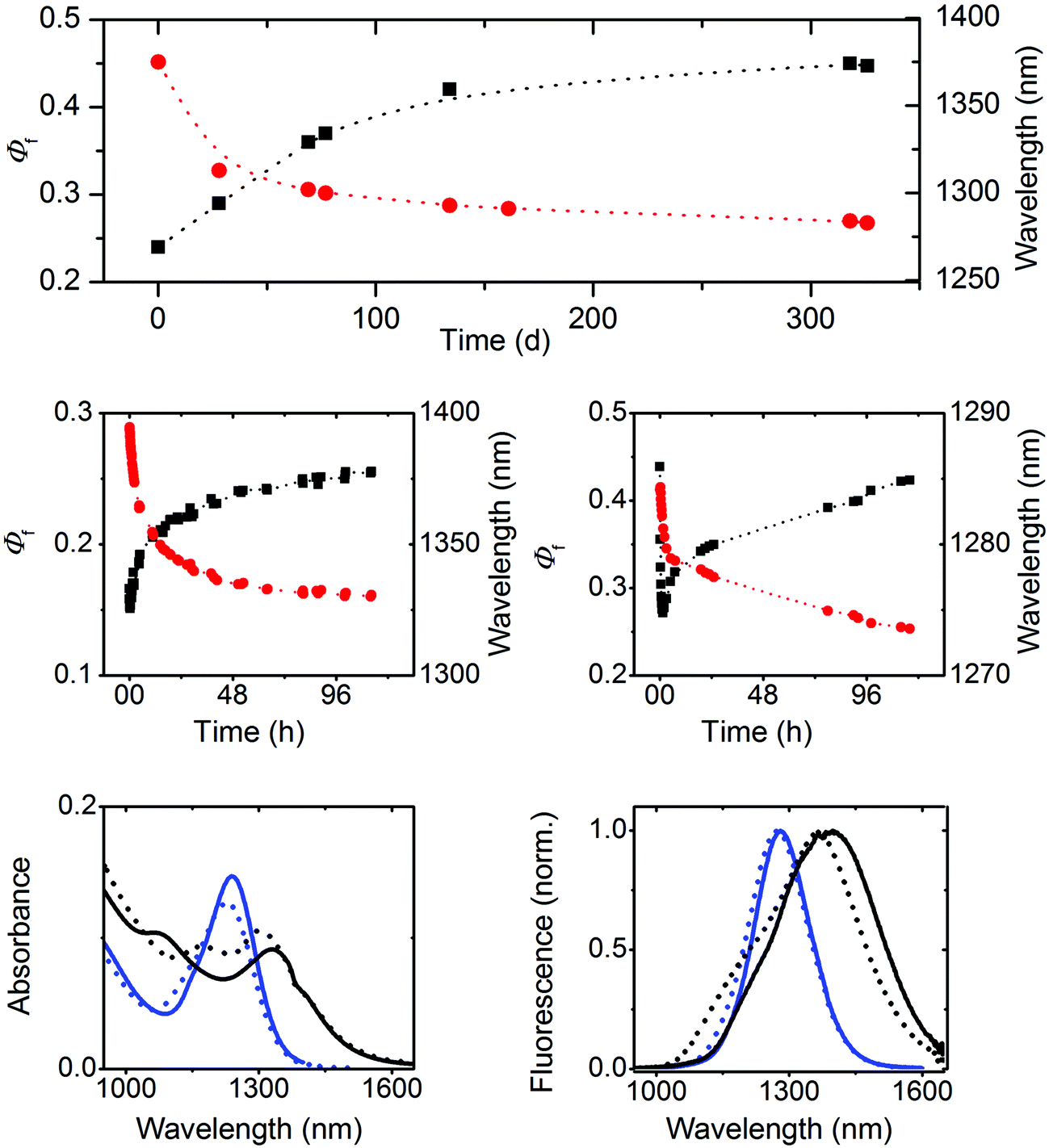 | ||
| Fig. 5 Top: aging of 4.4 nm PbS NCs in air. Blue shift of the spectral position of the emission band (red circles) and the relatively measured Φf (black squares) of 4.4 nm PbS NCs as a function of time after colloid preparation. Middle: absolutely measured Φf and photoeffects. Change in absolutely measured Φf (black squares) and in the spectral position of the emission band (red circles) of 4.4 nm PbS NCs kept under inert gas (left panel) and of 4.4 nm PbS NCs, both aged in air for ca. 300 days, as a function of illumination time. Illumination was performed with the 808 nm laser diode of the integrating sphere setup (radiant power of 62 mW). Bottom: aging and light-induced spectral changes. Change in the absorption spectra (left panel) of 4.4 nm PbS NCs aged/stored under inert gas for ca. 300 days (black) and in air (blue) and diluted with TCE, before (solid lines) and after illumination (dashed lines) with the 808 nm laser diode (radiant power of 62 mW) for 120 hours. The initial absorption spectrum of 4.4 nm PbS NCs stored under an inert gas atmosphere (black solid line) closely matches the initial absorption spectrum of this colloid prior to aging in air (top panel). | ||
Our explanation for the aging-induced luminescence enhancement is supported by the improved stability of 4.4 nm PbS NCs kept under inert gas. This is reflected by only very small time-dependent changes in the spectral position of the emission maximum located at 1390 nm, matching the initial value found for the freshly synthesized 4.4 nm PbS NC in the presence of oxygen (Fig. 5, upper panel), and its considerably smaller Φf of 0.15.
Similar experiments with 2.3 nm and 4.1 nm PbS NCs in air (see ESI, Fig. 12S†) revealed size-dependent photoeffects. The smallest PbS colloid showed solely photobrightening, leading to an increase of the initially obtained Φf of 0.18 to 0.71, accompanied by a blue shift of the emission band from 958 nm to 920 nm. At lower radiant powers, also fast photodarkening was observed, yet in all cases, subsequent photobrightening dominated, always yielding similarly high Φf. In contrast, 4.1 nm PbS NCs showed a similar behavior to 4.4 nm PbS particles and comparable Φf. These size-dependent differences in photochemical behavior seem to correlate with the size-dependent aging of these colloids, providing a further hint of a different surface chemistry of our three PbS NCs.
4 Conclusions and outlook
In summary, we presented the design of a new integrating sphere setup for spectrally resolved measurements of absolute photoluminescence quantum yields (Φf) of barely to moderately emissive organic dyes to highly emissive semiconductor NCs in the wavelength region of 600–1600 nm and addressed common instrument- and material-related challenges for fluorescence measurements in the NIR and IR and solutions. We demonstrated the reliability of our measurements by comparing the corrected emission spectra and the relatively and absolutely determined Φf of several organic dyes, MPA-capped Cd1−xHgxTe in D2O, and oleate-capped PbS NCs in TCE obtained with two integrating sphere setups and a spectrofluorometer, all independently calibrated. These validation strategies provide a good example of the validation of challenging fluorescence measurements in the NIR and IR for, e.g., semiconductor quantum dots and rods, carbon nanotubes, and upconversion materials.Moreover, we redetermined the Φf of the only IR reference dye IR26 to be 1.1 × 10−3 with an uncertainty of 2.3 × 10−4 derived from an uncertainty budget and introduced a procedure for the simultaneous correction for sample reabsorption and solvent absorption. These effects present common signal distortions in the IR. A further reduction in measurement uncertainties requires especially an improved stability of the excitation light sources used for our NIR/IR integrating sphere setup. We could verify the high Φf of 0.3 to 0.7 for high quality aqueous Cd1−xHgxTe NC colloids prepared via our previously reported simple one pot synthesis. For differently sized oleate-capped PbS colloids with Φf between 0.2 and 0.7, we demonstrated the influence of quantum dot-inherent photobrightening and photodarkening on Φf measurements, which can be material-specific, size-dependent, and affected by oxygen. These effects, which can be expected also, e.g. for other lead chalcogenides, together with the considerable overassessment of many previously published Φf of NIR- and IR-emissive quantum dots like PbS, PbSe, and Cd1−xHgxTe derived from our reexamined Φf of the reference dye IR26 underline the need for a critical reevaluation of Φf values of IR-emissive nanomaterials. A critical evaluation of Φf data and procedures used for the determination of Φf is generally of considerable importance with respect to the foreseeable increasing importance of luminescence measurements in the wavelength region of 1000–1600 nm, e.g. for bioimaging studies. This similarly triggers the need for broadly available and well characterized quantum yield standards also for this wavelength region.
Acknowledgements
We express our gratitude to Dr Markus Grabolle (BAM) for many fruitful discussions, to Dipl.-Ing A. Güttler (BAM) for technical assistance during the calibration of the spectrofluorometer FSP920, to Frau M. Spieles (BAM) for the HPLC measurements of the dyes, and Christine Damm (IFW Dresden e.V.) for the supports by the TEM measurements. We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG; grants RE-1203/12-1 and EY 16/14-1).References
- B. Valeur, Molecular Fluorescence: Principles and Application, Wiley-VCH, Weinheim, 2002 Search PubMed.
- D. M. Jameson, J. C. Croney and P. D. J. Moens, Biophotonics, Pt A, 2003, 360, 1–43 CrossRef CAS.
- S. V. Kershaw, A. S. Susha and A. L. Rogach, Chem. Soc. Rev., 2013, 42, 3033–3087 RSC.
- M. T. Harrison, S. V. Kershaw, M. G. Burt, A. L. Rogach, A. Kornowski, A. Eychmüller and H. Weller, Pure Appl. Chem., 2000, 72, 295–307 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CAS.
- E. H. Sargent, Adv. Mater., 2005, 17, 515–522 CrossRef CAS.
- M. Graetzel, R. A. J. Janssen, D. B. Mitzi and E. H. Sargent, Nature, 2012, 488, 304–312 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protocols, 2013, 8, 1535–1550 Search PubMed.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CrossRef CAS.
- U. Resch-Genger and K. Rurack, Pure Appl. Chem., 2013, 85, 2005–2026 CrossRef CAS.
- C. T. Xu, Q. Q. Zhan, H. C. Liu, G. Somesfalean, J. Qian, S. L. He and S. Andersson-Engels, Laser Photonics Rev., 2013, 7, 663–697 CrossRef CAS.
- S. L. Gai, C. X. Li, P. P. Yang and J. Lin, Chem. Rev., 2014, 114, 2343–2389 CrossRef CAS PubMed.
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285–6294 CrossRef CAS.
- S. Leubner, S. Hatami, N. Esendemir, T. Lorenz, J. O. Joswig, V. Lesnyak, S. Recknagel, N. Gaponik, U. Resch-Genger and A. Eychmüller, Dalton Trans., 2013, 42, 12733–12740 RSC.
- J. C. Boyer, M. P. Manseau, J. I. Murray and F. van Veggel, Langmuir, 2010, 26, 1157–1164 CrossRef CAS PubMed.
- F. Wang, J. A. Wang and X. G. Liu, Angew. Chem., Int. Ed., 2010, 49, 7456–7460 CrossRef CAS PubMed.
- J. Ziegler, A. Merkulov, M. Grabolle, U. Resch-Genger and T. Nann, Langmuir, 2007, 23, 7751–7759 CrossRef CAS PubMed.
- L. H. Qu and X. G. Peng, J. Am. Chem. Soc., 2002, 124, 2049–2055 CrossRef CAS PubMed.
- J. B. Delehanty, K. Susumu, R. L. Manthe, W. R. Algar and I. L. Medintz, Anal. Chim. Acta, 2012, 750, 63–81 CrossRef CAS PubMed.
- X. H. Ji, D. Copenhaver, C. Sichmeller and X. G. Peng, J. Am. Chem. Soc., 2008, 130, 5726–5735 CrossRef CAS PubMed.
- G. Kalyuzhny and R. W. Murray, J. Phys. Chem. B, 2005, 109, 7012–7021 CrossRef CAS PubMed.
- C. Wang, X. Gao and X. Su, Anal. Bioanal. Chem., 2010, 397, 1397–1415 CrossRef CAS PubMed.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Anal. Chem., 2011, 83, 3431–3439 CrossRef PubMed.
- C. Würth, J. Pauli, C. Lochmann, M. Spieles and U. Resch-Genger, Anal. Chem., 2012, 84, 1345–1352 CrossRef PubMed.
- C. Würth, M. G. Gonzalez, R. Niessner, U. Panne, C. Haisch and U. R. Genger, Talanta, 2012, 90, 30–37 CrossRef PubMed.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- K. Rurack, in Standardization and Quality Assurance in Fluorescence Measurements I: Techniques, ed. U. Resch-Genger, Springer, Berlin-Heidelberg, 2008, vol. 5 Search PubMed.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science+Business Media, LLC, New York, 3rd edn, 2006 Search PubMed.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- H. Du, C. L. Chen, R. Krishnan, T. D. Krauss, J. M. Harbold, F. W. Wise, M. G. Thomas and J. Silcox, Nano Lett., 2002, 2, 1321–1324 CrossRef CAS.
- L. Cademartiri, J. Bertolotti, R. Sapienza, D. S. Wiersma, G. von Freymann and G. A. Ozin, J. Phys. Chem. B, 2006, 110, 671–673 CrossRef CAS PubMed.
- J. J. Peterson and T. D. Krauss, Nano Lett., 2006, 6, 510–514 CrossRef CAS PubMed.
- F. Q. Ren, H. G. Zhao, F. Vetrone and D. L. Ma, Nanoscale, 2013, 5, 7800–7804 RSC.
- R. C. Benson and H. A. Kues, Phys. Med. Biol, 1978, 23, 159–163 CAS.
- J. E. Murphy, M. C. Beard, A. G. Norman, S. P. Ahrenkiel, J. C. Johnson, P. R. Yu, O. I. Micic, R. J. Ellingson and A. J. Nozik, J. Am. Chem. Soc., 2006, 128, 3241–3247 CrossRef CAS PubMed.
- B. L. Wehrenberg, C. J. Wang and P. Guyot-Sionnest, J. Phys. Chem. B, 2002, 106, 10634–10640 CrossRef CAS.
- M. V. Kovalenko, R. D. Schaller, D. Jarzab, M. A. Loi and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 2457–2460 CrossRef CAS PubMed.
- V. Lesnyak, A. Lutich, N. Gaponik, M. Grabolle, A. Plotnikov, U. Resch-Genger and A. Eychmüller, J. Mater. Chem., 2009, 19, 9147–9152 RSC.
- O. E. Semonin, J. C. Johnson, J. M. Luther, A. G. Midgett, A. J. Nozik and M. C. Beard, J. Phys. Chem. Lett., 2010, 1, 2445–2450 CrossRef CAS.
- W. Kranitzky, B. Kopainsky, W. Kaiser, K. H. Drexhage and G. A. Reynolds, Opt. Commun., 1981, 36, 149–152 CrossRef CAS.
- B. Kopainsky, P. Qiu, W. Kaiser, B. Sens and K. H. Drexhage, Appl. Phys. B, 1982, 29, 15–18 CrossRef.
- U. Resch-Genger and P. C. DeRose, Pure Appl. Chem., 2012, 84, 1815–1835 CrossRef CAS.
- C. Würth, C. Lochmann, M. Spieles, J. Pauli, K. Hoffmann, T. Schuttrigkeit, T. Franzl and U. Resch-Genger, Appl. Spectrosc., 2010, 64, 733–741 CrossRef PubMed.
- S. H. Xu, C. L. Wang, Q. Y. Xu, R. Q. Li, H. B. Shao, H. S. Zhang, M. Fang, W. Lei and Y. P. Cui, J. Phys. Chem. C, 2010, 114, 14319–14326 CAS.
- T. V. Duncan, M. A. M. Polanco, Y. Kim and S. J. Park, J. Phys. Chem. C, 2009, 113, 7561–7566 CAS.
- D. Tonti, F. van Mourik and M. Chergui, Nano Lett., 2004, 4, 2483–2487 CrossRef CAS.
- R. A. Cruz, V. Pilla and T. Catunda, J. Appl. Phys., 2010, 107 Search PubMed.
- Z. M. Tao, G. S. Hong, C. Shinji, C. X. Chen, S. Diao, A. L. Antaris, B. Zhang, Y. P. Zou and H. J. Dai, Angew. Chem., Int. Ed., 2013, 52, 13002–13006 CrossRef CAS PubMed.
- G. Hong, S. Diao, J. Chang, A. L. Antaris, C. Chen, B. Zhang, S. Zhao, D. N. Atochin, P. L. Huang, K. I. Andreasson, C. J. Kuo and H. Dai, Nat. Photonics, 2014, 8, 723–730 CrossRef CAS.
- D. Salo, H. R. Zhang, D. M. Kim and M. Y. Berezin, J. Biomed. Opt., 2014, 19 Search PubMed.
- V. Lesnyak, N. Gaponik and A. Eychmüller, Chem. Soc. Rev., 2013, 42, 2905–2929 RSC.
- S. Gupta, O. Zhovtiuk, A. Vaneski, Y.-C. Lin, W.-C. Chou, S. V. Kershaw and A. L. Rogach, Part. Part. Syst. Charact., 2013, 30, 346–354 CrossRef CAS.
- E. H. Sargent, Nat. Photonics, 2012, 6, 133–135 CrossRef CAS.
- J. Poppe, S. Gabriel, L. Liebscher, S. G. Hickey and A. Eychmüller, J. Mater. Chem. C, 2013, 1, 1515–1524 RSC.
- L. Cademartiri, E. Montanari, G. Calestani, A. Migliori, A. Guagliardi and G. A. Ozin, J. Am. Chem. Soc., 2006, 128, 10337–10346 CrossRef CAS PubMed.
- P. C. DeRose and U. Resch-Genger, Anal. Chem., 2010, 82, 2129–2133 CrossRef CAS PubMed.
- U. Resch-Genger, D. Pfeifer, C. Monte, W. Pilz, A. Hoffmann, M. Spieles, K. Rurack, J. Hollandt, D. Taubert, B. Schonenberger and P. Nording, J. Fluoresc., 2005, 15, 315–336 CrossRef CAS PubMed.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- U. Resch-Genger, W. Bremser, D. Pfeifer, M. Spieles, A. Hoffmann, P. C. DeRose, J. C. Zwinkels, F. o. Gauthier, B. Ebert, R. D. Taubert, C. Monte, J. Voigt, J. Hollandt and R. Macdonald, Anal. Chem., 2012, 84, 3889–3898 CrossRef CAS PubMed.
- T. Behnke, J. E. Mathejczyk, R. Brehm, C. Würth, F. R. Gomes, C. Dullin, J. Napp, F. Alves and U. Resch-Genger, Biomaterials, 2013, 34, 160–170 CrossRef CAS PubMed.
- D. Brown and D. Benfey, Comprehensive Study of Diode-Pumped Dye Lasers. Phase 1, 1991 Search PubMed.
- S. Hinds, S. Myrskog, L. Levina, G. Koleilat, J. Yang, S. O. Kelley and E. H. Sargent, J. Am. Chem. Soc., 2007, 129, 7218–7219 CrossRef CAS PubMed.
- L. Bakueva, I. Gorelikov, S. Musikhin, X. S. Zhao, E. H. Sargent and E. Kumacheva, Adv. Mater., 2004, 16, 926–929 CrossRef CAS.
- X. S. Zhao, I. Gorelikov, S. Musikhin, S. Cauchi, V. Sukhovatkin, E. H. Sargent and E. Kumacheva, Langmuir, 2005, 21, 1086–1090 CrossRef CAS PubMed.
- M. Sykora, A. Y. Koposov, J. A. McGuire, R. K. Schulze, O. Tretiak, J. M. Pietryga and V. I. Klimov, ACS Nano, 2010, 4, 2021–2034 CrossRef CAS PubMed.
- J. W. Stouwdam, J. Shan, F. van Veggel, A. G. Pattantyus-Abraham, J. F. Young and M. Raudsepp, J. Phys. Chem. C, 2007, 111, 1086–1092 CAS.
- I. Moreels, Y. Justo, B. De Geyter, K. Haustraete, J. C. Martins and Z. Hens, ACS Nano, 2011, 5, 2004–2012 CrossRef CAS PubMed.
- H. Choi, J.-H. Ko, Y.-H. Kim and S. Jeong, J. Am. Chem. Soc., 2013, 135, 5278–5281 CrossRef CAS PubMed.
- S. Leubner, R. Schneider, A. Dubavik, S. Hatami, N. Gaponik, U. Resch-Genger and A. Eychmüller, J. Mater. Chem. C, 2014, 2, 5011–5018 RSC.
- J. Tang, L. Brzozowski, D. A. R. Barkhouse, X. Wang, R. Debnath, R. Wolowiec, E. Palmiano, L. Levina, A. G. Pattantyus-Abraham, D. Jamakosmanovic and E. H. Sargent, ACS Nano, 2010, 4, 869–878 CrossRef CAS PubMed.
- S. J. O. Hardman, D. M. Graham, S. K. Stubbs, B. F. Spencer, E. A. Seddon, H.-T. Fung, S. Gardonio, F. Sirotti, M. G. Silly, J. Akhtar, P. O'Brien, D. J. Binks and W. R. Flavell, Phys. Chem. Chem. Phys., 2011, 13, 20275–20283 RSC.
- J. J. Peterson and T. D. Krauss, Phys. Chem. Chem. Phys., 2006, 8, 3851–3856 RSC.
- T. W. F. Chang, A. Maria, P. W. Cyr, V. Sukhovatkin, L. Levina and E. H. Sargent, Synth. Met., 2005, 148, 257–261 CrossRef CAS PubMed.
- J. S. Steckel, S. Coe-Sullivan, V. Bulovic and M. G. Bawendi, Adv. Mater., 2003, 15, 1862–1866 CrossRef CAS.
- E. Lifshitz, M. Brumer, A. Kigel, A. Sashchiuk, M. Bashouti, M. Sirota, E. Galun, Z. Burshtein, A. Q. Le Quang, I. Ledoux-Rak and J. Zyss, J. Phys. Chem. B, 2006, 110, 25356–25365 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr04608k |
| This journal is © The Royal Society of Chemistry 2015 |