
On-chip high power porous silicon lithium ion batteries with stable capacity over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) 000 cycles†
000 cycles†
Andrew S.
Westover
ab,
Daniel
Freudiger
a,
Zarif S.
Gani
a,
Keith
Share
ab,
Landon
Oakes
ab,
Rachel E.
Carter
a and
Cary L.
Pint
*ab
aDepartment of Mechanical Engineering, Vanderbilt University, Nashville, TN 37235, USA. E-mail: cary.l.pint@vanderbilt.edu
bInterdisciplinary Materials Science Program, Vanderbilt University, Nashville, TN 37235, USA
First published on 10th November 2014
Abstract
We demonstrate the operation of a graphene-passivated on-chip porous silicon material as a high rate lithium battery anode with over 50X power density, and 100X energy density improvement compared to identically prepared on-chip supercapacitors. We demonstrate this Faradaic storage behavior to occur at fast charging rates (1–10 mA cm−2) where lithium locally intercalates into the nanoporous silicon, preventing the degradation and poor cycling performance attributed to deep storage in the bulk silicon. This device exhibits cycling performance that exceeds 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles with capacity above 0.1 mA h cm−2 without notable capacity fade. This demonstrates a practical route toward high power, high energy, and long lifetime all-silicon on-chip storage systems relevant toward integration into electronics, photovoltaics, and other silicon-based platforms.
000 cycles with capacity above 0.1 mA h cm−2 without notable capacity fade. This demonstrates a practical route toward high power, high energy, and long lifetime all-silicon on-chip storage systems relevant toward integration into electronics, photovoltaics, and other silicon-based platforms.
From portable electronics to solar cells, power delivery systems that can be integrated into unused components in devices are critical to meet the increasing power requirements of future applications. In electronics, as the number of transistors per unit area increases, so do the corresponding power requirements for operation. For solar cells, as residential solar usage becomes more prominent, integrated energy storage allows continuous power generation amidst intermittent periods of sunshine.1–3 In both of these cases, as well as in other silicon-based on-chip applications, a key differentiating factor for integration is the notion that energy storage must be developed on a two-dimensional, planar substrate that must concurrently support operation of the component on the front side of the device. Unlike conventional energy storage systems that are packaged into three-dimensional architectures with performance assessed relative to mass or volume,4 on-chip devices must be assessed for their ability to store charge that scales with the total available chip area, or footprint. This has led to the development of on-chip devices called micro-supercapacitors or micro-batteries,5–8 which are most practical when the native chip material is used as real estate for energy storage with minimal additional manufacturing steps. Silicon is an ideal choice due to its relevance to batteries, supercapacitors, and its broad application in electronics, MEMS, solar energy conversion, and sensing.
Specifically for lithium storage applications, silicon exhibits the highest known storage capacities (up to 4200 mA h g−1), but exhibits rapid capacity fade upon cycling associated with the large volume expansion in silicon during lithiation.9–11 Whereas elegant routes have been developed to combat such degradation in nanoparticle electrodes, such as yolk–shell structures12 and pomegranate-like architectures,13 seamless integration of these techniques to on-chip platforms is not straightforward. Previous studies which have demonstrated on-chip storage using porous silicon build upon a native architecture that enables efficient volumetric expansion of the silicon during cycling, but exhibit limitations in cycle lifetime due to the porous silicon–silicon interface,14–16 even though some studies indicate some intercalation control through modulating the rates or capacities.16,17 This has motivated the development of superior macroparticle silicon particle networks.18 Therefore, despite the promise of large energy densities, the bottleneck for on-chip silicon batteries is the low power density and short cycle lifetime of these materials. On the other hand, electrochemical supercapacitors exhibit the power capability sought for on-board electronics, and are often rated for up to a million consecutive cycles, but are limited to energy densities at best 10X lower than conventional batteries due to the physical double layer charge storage mechanism.19–21 However, porous silicon materials used for supercapacitors have demonstrated unique integration capability with applications,22–24 such as the ability to form supercapacitors to store energy directly in the unused material in solar cells.25
In this work, we demonstrate hybrid performance bridging these two device platforms based on carbon-passivated porous silicon material etched into bulk silicon wafers and cycled as a lithium-ion battery electrode at high currents. Using identically structured supercapacitors as a control comparison, we demonstrate over 50X improved power density, and over 100X improved energy density in this high rate porous silicon battery which operates over 10000 cycles with no significant capacity fade and stores more total energy on chip than both an ideal deeply intercalated battery anode, or an electrochemical supercapacitor operating for 250 and a million cycles, respectively. This unique operation mode provides ideal characteristics of power, energy, and lifetime sought for on-chip integration.
In order to prepare electrodes, B-doped silicon wafers (0.01–0.02 Ω cm) were electrochemically etched using a 3:7 v/v HF (50% H2O by volume) and ethanol solution at 45 mA cm−2 for 180 seconds in either a home-built etching cell or an AMMT porous silicon etching system. This yields porous Si with 75% porosity and a ∼4 μm depth (Fig. 1A). To passivate the porous silicon from the effects of electrochemical degradation or inhibitory solid electrolyte interphase layer formation, we used chemical vapor deposition using a ramp procedure described previously22,23 that is known to generate few-layer coatings of graphene-like carbon on the surface of the porous silicon material. Briefly, this involves consecutive temperature ramps from 650–750 °C, and 750–850 °C for 10 minutes each, with a 1:20:100 ratio of C2H2–H2–Ar gas mixture carried out under atmospheric conditions. A representative TEM image of the carbon passivated silicon material used in these experiments is shown in Fig. 1B. In order to assess the storage performance of these materials, we constructed lithium-ion batteries using the passivated porous silicon as an electrode combined with Li foil, a Celgard battery separator, and electrolyte consisting of 1 M LiPF6 dissolved in ethylene carbonate/dimethyl carbonate, sealed in a coin cell assembly. One general observation is that the charging rate dictates the penetration depth of the intercalation process, and a transition exists between what we indicate as “high-rate” behavior, where the current is high enough to only allow intercalation into the nanoporous active material, and “deep-charge” behavior where lower charging currents allow the slower bulk intercalation kinetics into bulk silicon material underneath the porous silicon layer. In order to assess the non-Faradaic (surface) storage per unit footprint in a material with an identical porous structure (thickness, porosity, etc.), we utilized an electrochemical supercapacitor as an ideal control experiment. Here, a supercapacitor only stores energy by the assembly of a double-layer of ions at the surface of the porous silicon, allowing a robust comparison to Faradaic, bulk redox storage in batteries using an identical material. Supercapacitors were made in a manner reported elsewhere22,23 by sandwiching two symmetric porous silicon electrodes with a Celgard separator and 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIBF4) ionic liquid electrolyte. A visual comparison of these three different storage modes in the same material are shown in Fig. 1C representing (top) double-layer storage of ions in a supercapacitor, (middle) fast lithium intercalation into the nanoscale porous active material, and (bottom) deep intercalation at slow rates into both the porous layer and the bulk silicon wafer.
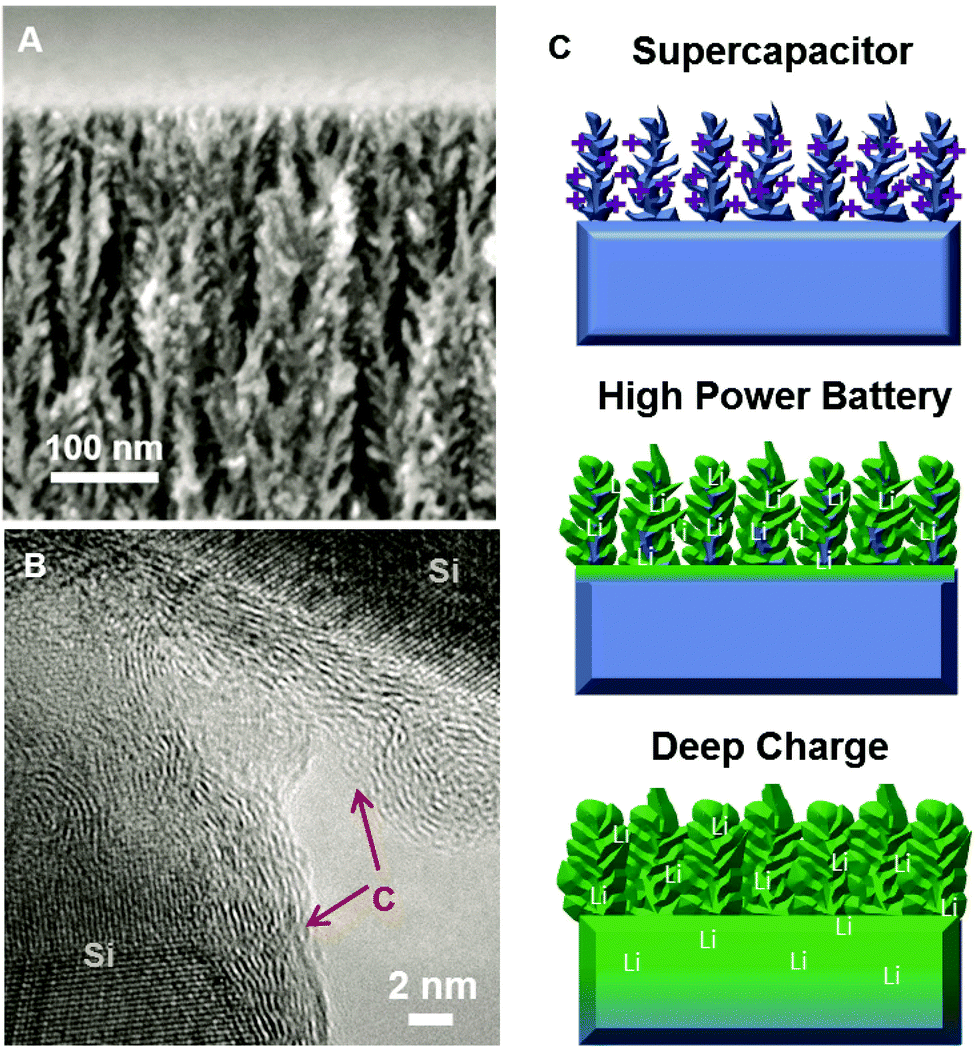 | ||
| Fig. 1 (A) Side-view SEM image of carbon-passivated porous silicon materials utilized in this work, (B) High resolution TEM image of the carbon passivated silicon material that indicates the quality of the carbon and crystalline nature of the silicon nanomaterials, and (C) Scheme visually depicting the three modes of energy storage in carbon-passivated porous silicon materials explored and discussed in this work including supercapacitors, high-power batteries, and deep intercalation batteries. | ||
In order to assess the performance of these on-chip devices, we performed both Galvanostatic charge–discharge measurements and cyclic voltammetry analysis. With this specific porous silicon material employed in a Li-ion battery, we observe a distinct transition near charging currents of 1.2 mA cm−2 separating the “high rate” and “deep-intercalation” processes. Above 1.2 mA cm−2, we observe reversible storage capacities up to ∼0.1 mA h cm−2, with energy stored at voltages vs. Li/Li+ above the energy of intercalation into bulk silicon (e.g. above 0.6 V vs. Li/Li+) extending up to ∼3 V (Fig. 2A). In comparison to a device charged at 1 mA cm−2 (inset, Fig. 2A), signatures of deep Li bulk intercalation (below 0.6 V) and Li intercalation into the passivated porous silicon layer (0.6 V–3 V) are observed, distinguishing these two intercalation processes. To better understand this, we performed CV measurements of high-rate devices at rates of 5, 10, and 20 mV s−1 (Fig. 2B). Evident from these curves is the lack of a bulk intercalation signature below 0.5 V, and a redox couple for insertion/deinsertion at voltages centered around ∼1.6 V and ∼1.2 V vs.. Li/Li+, respectively. CV curves taken at the same rates, but without the carbon passivation layer indicate similar intercalation and deintercalation kinetics, emphasizing that this behavior is not due to the graphene passivation (ESI†). Furthermore, we performed control experiments where freestanding layers of identical porous silicon were isolated on Ti substrates and assessed at high rates, with similar cyclic voltammetry profiles observed in comparison to on-chip integrated devices emphasizing the porous material as being responsible for the lithium storage. The high rate intercalation behavior is distinguished from that observed in CV scans taken at slow rates of 0.1 and 0.5 mV s−1, respectively (Fig. 2C), where sharp intercalation peaks are observed at 0.36 V and 0.56 V and a deintercalation peak is observed at 0.14 V that emphasizes the redox couples associated with bulk Li storage in silicon.26–28 Furthermore, whereas high-rate Galvanostatic measurements indicate less distinctly defined Faradaic storage energies, we can distinguish this storage from capacitive double-layer storage by direct comparison to supercapacitors formed from identical carbon passivated porous silicon. CV measurements for supercapacitors (Fig. 2D) indicate smooth, featureless curves representative of non-Faradaic storage processes, in comparison to CV curves for high-rate batteries, which implies storage through Faradaic chemical redox processes. This indicates the high-rate storage energetics and process for carbon-passivated porous silicon electrodes is distinguished both from bulk Li storage observed in conventional silicon battery electrodes and the surface double-layer storage behavior observed in electrochemical supercapacitors.
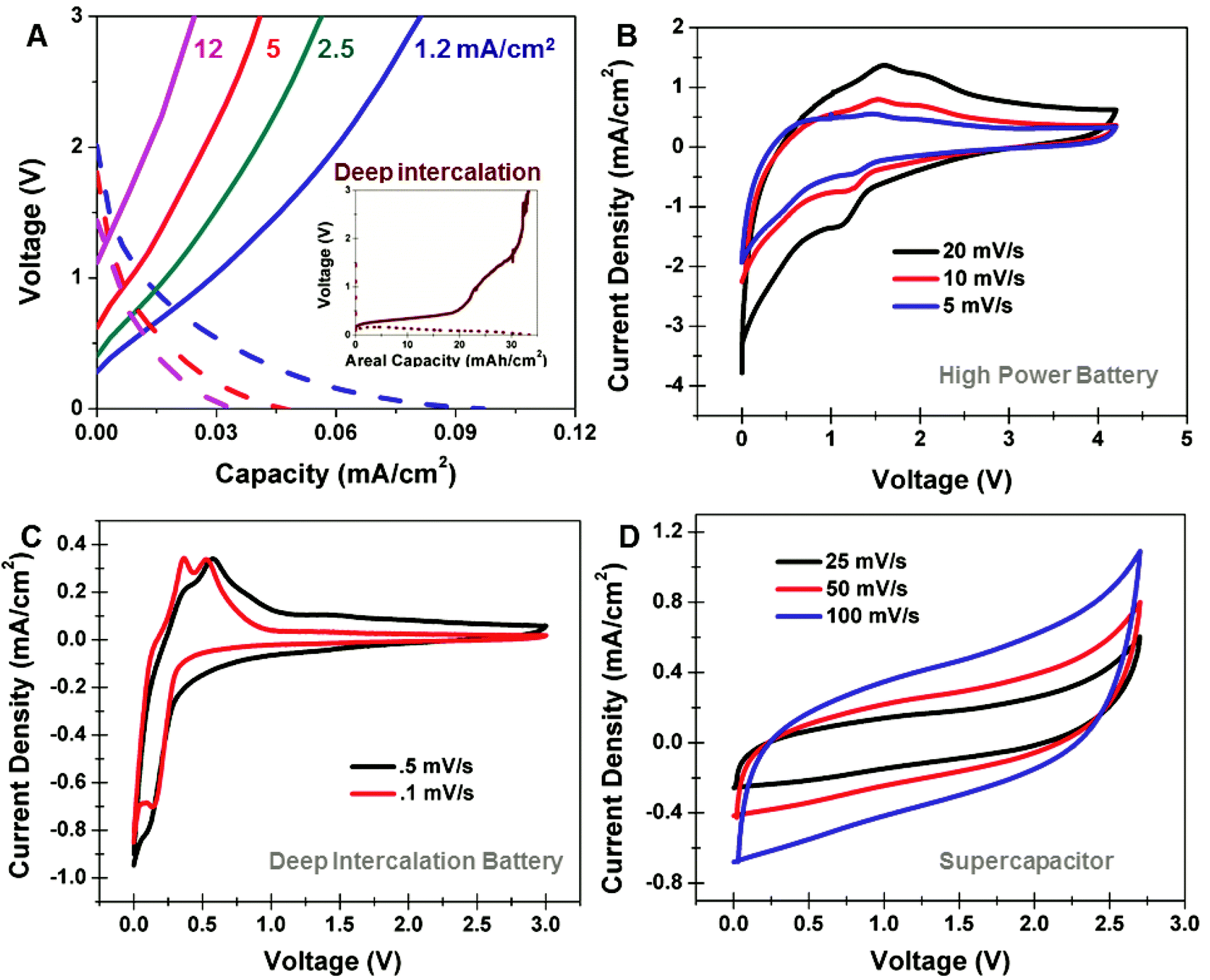 | ||
| Fig. 2 (A) Galvanostatic charge–discharge curves taken for carbon-passivated porous silicon high power batteries at rates between 1.2–12 mA cm−2. A corresponding deep intercalation battery charge–discharge curve is inset in order to distinguish bulk Si intercalation versus high power storage in the nanoscale carbon-passivated porous silicon material. (B) Cyclic voltammetry (CV) curves of carbon-passivated porous silicon batteries at fast scan rates of 5, 10, and 20 mV s−1 demonstrating the high power storage behavior of the porous silicon. (C) CV curves for carbon-passivated porous silicon batteries at slow scan rates of 0.1 and 0.5 mV s−1 indicating standard Si bulk intercalation behavior. (D) CV curves for carbon passivated porous silicon supercapacitors with EMIBF4 electrolytes at 50 mV s−1 to demonstrate charge storage through a surface double layer formation. | ||
One of the key challenges in utilizing silicon materials in batteries is the poor cycling lifetime of the active materials. Our work demonstrates that we can overcome this limitation for an on-chip carbon passivated porous silicon battery by operation in the high power mode, thus inhibiting deep bulk intercalation that leads to irreversible capacity fade. Fig. 3 shows the capacity measured as a function of the number of cycles for an on-chip porous silicon Li ion battery charged and discharged at 3 mA cm−2, following 3000 preliminary cycles for stabilization at 3 mA cm−2. We speculate that the preliminary 3000 cycles represents a continuous activation of the porous silicon electrical conductivity by formation of a Li–Si alloy, which has been observed to improve conductivity of amorphous silicon by over 3 orders of magnitude.29 As porous silicon exhibits high resistance after electrochemical etching, we anticipate that lowering the resistance of the active material in combination with the high surface area for the electrode–electrolyte interface that leads to low cell resistance, allows this device to overcome the resistance polarization that ultimately limits conventional batteries from high power cycling behavior. After capacity stabilization, the device shows excellent cycling capability, with a slightly fluctuating but constant capacity that exceeds 13000 cycles. This result is in direct contrast to the slow charged porous Si anodes which rapidly degrade over 10–30 cycles with the capacity fade showing an exponential decay upon cycling (ESI†). Whereas the best silicon nanoparticle electrode materials have demonstrated cycling lifetime up to 1000 cycles,12,13 these materials must be combined with binders and cast into coatings applied to a conductive charge collector. In our case, the electrode active material is mechanically integrated directly into a silicon wafer with no binder materials, resulting in deep intercalation processes that inhibit cyclability as observed in other studies with porous silicon materials.10,11,14 However, by using the Galvanostatic charging conditions to control the intercalation depth to only penetrate the nanoscale active layer, we observe this battery to exhibit exemplary cycling stability that significantly exceeds the capability of other Si materials. This mechanism and the device lifetime is similar to that exhibited in a conventional pseudocapacitor, except with significantly improved capacities and using conventional carbonate electrolytes. To support this concept, we performed SEM imaging of porous Si materials cycled both at high power conditions, and under deep intercalation conditions. For the deep intercalation (∼20 cycles), significant cracking that extends up to ∼40 microns into the surface is observed. However, for the high power cycling experiments, the porous structure after cycling is clearly in-tact, with no evident damage to the silicon material residing underneath the porous layer. This confirms, in agreement with cycling data performed on freestanding porous silicon films, that the high rate cycling is correlated to storage in the porous material (ESI†). To further emphasize this point, normalizing the capacity of the deep intercalation battery to only the material in the porous layer yields ∼124600 mA h g−1, which is clearly well above the maximum capacity of Si for Li storage. Similar assessment of the high rate storage normalized to the porous layer indicates a capacity of ∼570 mA h g−1, which is a reasonable capacity for silicon materials at high charge–discharge cycling rates, and still respectable in comparison to conventional anodes such as bulk carbons (maximum capacity of 372 mA h g−1).
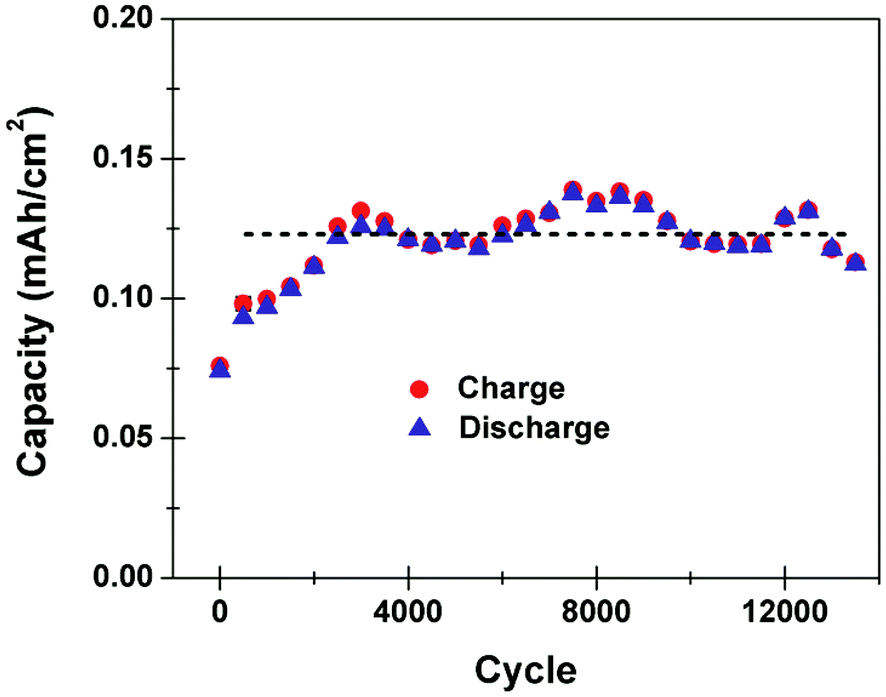 | ||
| Fig. 3 Footprint capacity as a function of cycle number over more than 13000 consecutive cycles with charge and discharge capacities plotted separately to indicate the ∼100% Coulombic efficiency achieved. A straight line is included to guide the eye. | ||
Additionally, it is important to assess the energy and power capability of these devices based on their available chip-based footprint. For devices produced using identical carbon-passivated porous silicon materials, we compared the energy and power performance of supercapacitors, high power batteries, and deep intercalation batteries (Fig. 4A). These curves were each assessed by direct integration of Galvanostatic discharge curves at different cycling rates, and analysis of the average power that is represented by the total energy divided by the discharge duration. A distinguishing factor between the high-power Li-ion battery and the electrochemical supercapacitor is that the high power battery can exhibit up to 100X the power density per footprint of the supercapacitor, and exhibit energy densities up to almost 250X greater than the supercapacitor per unit footprint. We anticipate this is due to the greater electrode–electrolyte surface area interface that minimizes electrode resistance polarization losses that inhibit bulk materials from high power storage. These relative values significantly distinguish the bulk storage in the nanoscale porous layer from the surface double-layer storage in a supercapacitor since both materials have the exact same structure. Comparing the power and energy storage capability measured in these high rate devices to the best state-of-the-art 3D graphene supercapacitors per unit active mass (40–85 Wh kg−1, 25 kW kg−1),30 the specific power and energy capability, scaled to the porous active material, still exhibits comparable power density of ∼20 kW kg−1, and much greater energy density of ∼220 Wh kg−1. It should further be noted that higher power carbon supercapacitors are usually realized from ultrathin highly porous electrodes, and the energy and power performance is known to decrease or change with electrode thickness making the comparison of such materials challenging.4 This means thick active layers of storage material, such as graphene, are likely to both inhibit good areal on-chip performance, in addition to being poorly suited for integration due to a heterogeneous van der Waals interface with a collector material – in contrast to the directly integrated high power porous silicon battery electrode. Further, comparison of the high power battery to a slow-charged, deep intercalation battery indicates a 100–150X lower energy density, but a power density that remains between 10–100X improved per unit footprint.
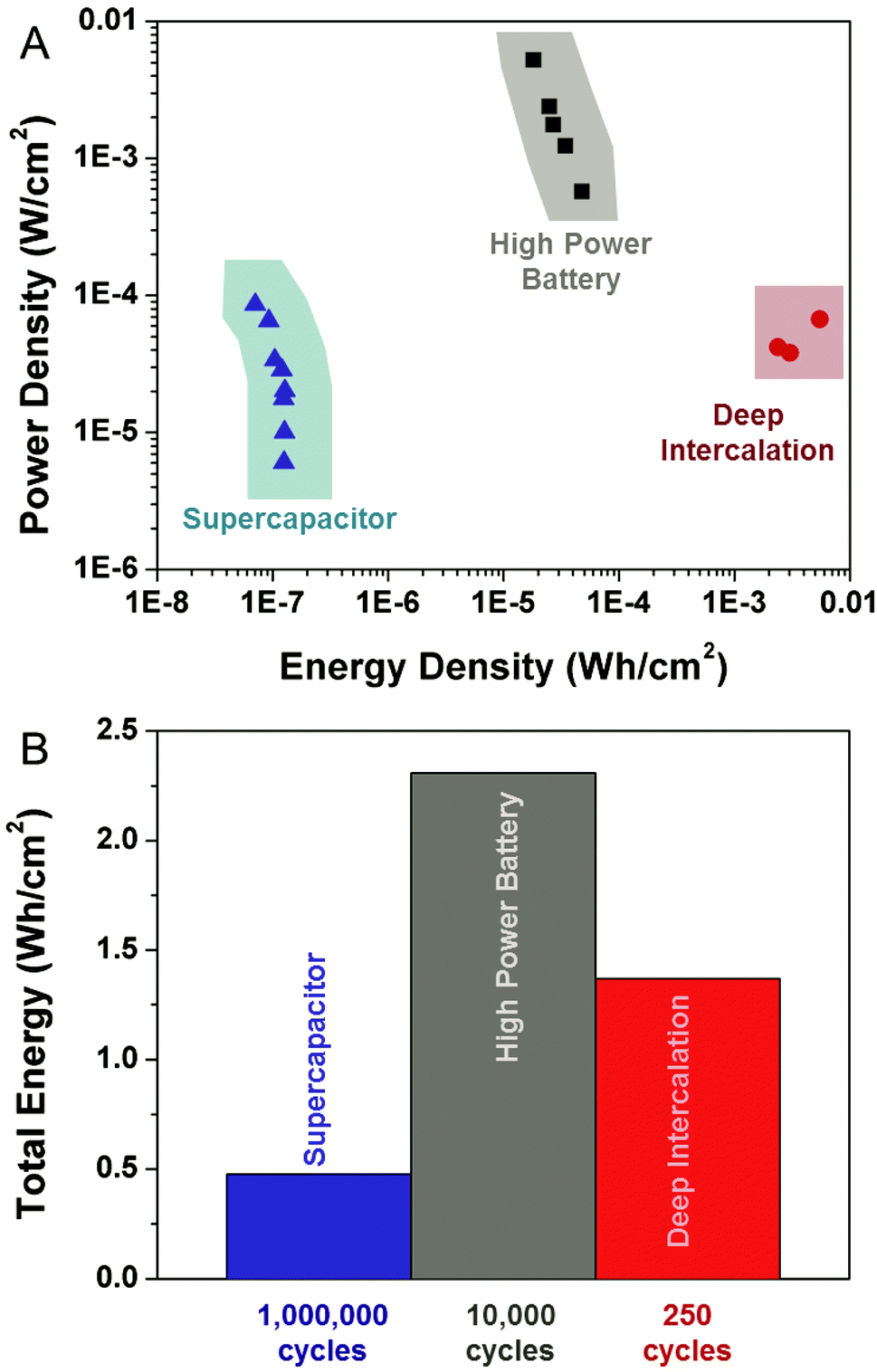 | ||
| Fig. 4 (A) Energy-power analysis based on the integration of discharge curves from devices prepared with identical carbon-passivated porous silicon materials, including supercapacitors, high-power lithium-ion batteries, and deep-intercalation lithium-ion batteries. (B) Total energy stored over the measured or extrapolated lifetime of the three devices studied in this work. For supercapacitors, the cycle lifetime is extrapolated to 1 million cycles, and for deep-intercalation batteries the cycle lifetime is extrapolated to 250 cycles. Whereas both of these are optimistic and ideal extrapolations, the total energy for the high power device is based on the measured stored energy over 10000 cycles conducted in this study. | ||
The unique nature of a secondary battery emphasizes that both the energy stored per cycle and the total stored energy over the lifetime of the device are important metrics for device performance. In order to compare the total energy stored, we provide an optimistic comparison between the electrochemical supercapacitor and deep intercalation battery performance, compared to the measured performance of the high power battery. The total energy stored by the supercapacitor, over the lifetime of the device was estimated by multiplying the maximum energy stored by the supercapacitor times the anticipated supercapacitor lifetime of 1000000 cycles. For the deep intercalation battery, we assess the maximum capacity and optimistically assume this capacity to be achievable for 250 cycles, despite our experimental measurements of ∼60% capacity fade for the first 10 cycles, emphasizing the highly conservative nature of this estimate. Finally, we calculated the total energy stored in the high power Li ion battery based on experimental analysis of the 10000 active cycles that were experimentally measured and assessed. Whereas this device is still active after 10000 cycles and our cycling experiments extend beyond 13000 cycles, we observe this high rate battery platform to exhibit significantly more total energy stored in comparison to both a conservatively estimated ideal deep intercalation battery and an ideal supercapacitor that operates based on an identical electrode structure. Furthermore, the higher power capability of the fast-rate porous silicon battery that is enabled by Li metal stored in the nanoscale silicon material enables it to exhibit better power capability in storing its energy over the course of its lifetime.
Finally, whereas our results emphasize that the on-chip porous layer is responsible for the high power, long lifetime cycling capability that we demonstrate here, we emphasize that modifications of the porous silicon porosity, pore structure, thickness, and passive layer composition are all factors that could lead to improved and controllable performance relevant to a spectrum of long-term integrated applications. As an example, for energy storage directly integrated into photovoltaics, a concept we recently demonstrated as being feasible for porous silicon supercapacitors,25 most respectable photovoltaic devices exhibit current densities greater than 5 mA cm−2 and are rated for between 20–30 years of stable operation. Such charging currents kinetically inhibit deep bulk intercalation storage in silicon (Fig. 2), requiring asymmetry in power generation and power storage footprint areas, which makes integration challenging in addition to the large mismatch between storage and generation lifetimes. For a high-power silicon battery device that can operate at the native photogenerated current density of a silicon solar cell exposed to 1 sun of illumination, and charged and discharged once per day, the ability to achieve 10000 cycles without noticeable capacity loss indicates the capability to be charged and discharged once per day through a period of almost 30 years – which is the maximum rated lifetime of a solar cell.1 Beyond this unique integration scheme for solar cells, integration of on-chip high power, long-lifetime silicon-based energy storage into other applications such as silicon-based integrated circuits and electronics, MEMS devices, and sensors, provides a unique pathway to provide on-board energy storage in excess silicon material real estate in these devices.
Acknowledgements
We thank Adam Cohn for useful discussions, Jeremy Mares for insight on electropolishing, and Sharon Weiss for early use of facilities for porous silicon etching. This work was supported by National Science Foundation (NSF) CMMI grant # 1334269, and A.S.W. was supported by NSF grant EPS 1004083.References
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- H. Lund, A. N. Andersen, P. A. Ostergaard, B. V. Mathiesen and D. Connolly, Energy, 2012, 42, 96–102 CrossRef PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Y. Gogotsi and P. Simon, Science, 2011, 334, 917–918 CrossRef CAS PubMed.
- L. Baggetto, R. A. H. Niessen, F. Roozeboom and P. H. L. Notten, Adv. Funct. Mater., 2008, 18, 1057–1066 CrossRef CAS.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480–483 CrossRef CAS PubMed.
- J. H. Pikul, H. G. Zhang, J. Cho, P. V. Braun and W. P. King, Nat. Commun., 2013, 4, 1732 CrossRef PubMed.
- H. G. Zhang and P. V. Braun, Nano Lett., 2012, 12, 2778–2783 CrossRef CAS PubMed.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 CAS.
- S. J. Lee, J. K. Lee, S. H. Chung, H. Y. Lee, S. M. Lee and H. K. Baik, J. Power Sources, 2001, 97–8, 191–193 CrossRef.
- S. Bourderau, T. Brousse and D. M. Schleich, J. Power Sources, 1999, 81, 233–236 CrossRef.
- N. Liu, H. Wu, M. T. McDowell, Y. Yao, C. M. Wang and Y. Cui, Nano Lett., 2012, 12, 3315–3321 CrossRef CAS PubMed.
- N. Liu, Z. D. Lu, J. Zhao, M. T. McDowell, H. W. Lee, W. T. Zhao and Y. Cui, Nat. Nanotechnol., 2014, 9, 187–192 CrossRef CAS PubMed.
- M. Thakur, M. Isaacson, S. L. Sinsabaugh, M. S. Wong and S. L. Biswal, J. Power Sources, 2012, 205, 426–432 CrossRef CAS PubMed.
- H. C. Shin, J. A. Corno, J. L. Gole and M. L. Liu, J. Power Sources, 2005, 139, 314–320 CrossRef CAS PubMed.
- X. D. Sun, H. Huang, K. L. Chu and Y. Zhuang, J. Electron. Mater., 2012, 41, 2369–2375 CrossRef CAS.
- E. Quiroga-Gonzalez, J. Carstensen and H. Foll, Electrochim. Acta, 2013, 101, 93–98 CrossRef CAS PubMed.
- M. Thakur, S. L. Sinsabaugh, M. J. Isaacson, M. S. Wong and S. L. Biswal, Sci. Rep., 2012, 2, 795 Search PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- A. Chu and P. Braatz, J. Power Sources, 2002, 112, 236–246 CrossRef CAS.
- I. Hadjipaschalis, A. Poullikkas and V. Efthimiou, Renewable & Sustainable Energy Reviews, 2009, 13, 1513–1522 Search PubMed.
- S. Chatterjee, R. Carter, L. Oakes, W. R. Erwin, R. Bardhan and C. L. Pint, J. Phys. Chem. C, 2014, 118, 10893–10902 CAS.
- L. Oakes, A. Westover, J. W. Mares, S. Chatterjee, W. R. Erwin, R. Bardhan, S. M. Weiss and C. L. Pint, Sci. Rep., 2013, 3, 3020 Search PubMed.
- A. S. Westover, J. W. Tian, S. Bernath, L. Oakes, R. Edwards, F. N. Shabab, S. Chatterjee, A. V. Anilkumar and C. L. Pint, Nano Lett., 2014, 14, 3197–3202 CrossRef CAS PubMed.
- A. S. Westover, K. Share, R. Carter, A. P. Cohn, L. Oakes and C. L. Pint, Appl. Phys. Lett., 2014, 104, 213905 CrossRef PubMed.
- Y. Yao, M. T. McDowell, I. Ryu, H. Wu, N. A. Liu, L. B. Hu, W. D. Nix and Y. Cui, Nano Lett., 2011, 11, 2949–2954 CrossRef CAS PubMed.
- X. S. Zhou, Y. X. Yin, L. J. Wan and Y. G. Guo, Chem. Commun., 2012, 48, 2198–2200 RSC.
- T. Jiang, S. C. Zhang, X. P. Qiu, W. T. Zhu and L. Q. Chen, Electrochem. Commun., 2007, 9, 930–934 CrossRef CAS PubMed.
- E. Pollak, G. Salitra, V. Baranchugov and D. Aurbach, J. Phys. Chem. C, 2007, 111, 11437–11444 CAS.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr04720f |
| This journal is © The Royal Society of Chemistry 2015 |