
The synthesis of symmetrical disulfides by reacting organic halides with Na2S2O3·5H2O in DMSO†
Mohammad
Abbasi
*,
Mohammad Reza
Mohammadizadeh
and
Narges
Saeedi
Department of Chemistry, Faculty of Sciences, Persian Gulf University, Bushehr, 75169, Iran. E-mail: abbassi@pgu.ac.ir; pgu.chem@gmail.com
First published on 20th November 2015
Abstract
A one-pot, and scalable method to prepare symmetric disulfides from their corresponding primary, secondary, allylic, and benzylic halides has been developed. In this method, a disulfide is synthesized by reacting an alkyl halide with Na2S2O3·5H2O at 60–70 °C in DMSO.
The synthesis of organic disulfides (disulfanes) is a matter of interest for the industrial production of some pharmaceuticals and agrochemicals.1 This goal can be achieved in the laboratory by means of numerous methods using diverse reagents and substrates.2 Symmetrical disulfides can be obtained by reductive coupling of sulfonyl chlorides3 by the reaction of sulfur monochloride (S2Cl2) with aromatic compounds,4 alkynes,5 and alkenes,5 treatment of organic thiocyanates with samarium,6 samarium diiodide,7 tetrabutylammonium fluoride8 or tetrathiomolybdate,9 and nucleophilic ring opening of thiiranes followed by oxidation.10 However, the most important method for the preparation of symmetrical disulfides is the oxidation of the appropriate thiols.2 Thiols are foul-smelling compounds that are usually prepared from their corresponding alkyl halides in organic synthesis. In other words, thiols are intermediates in the synthesis of disulfides from alkyl halides. Even though a wide range of methods is available for the synthesis of disulfides from thiols in the literature, investigation of a simple, rapid and inexpensive conversion of alkyl halides into disulfides is still a challenge and an interesting area of study.2 In this regard, the treatment of alkyl halides with disulfide anions,11 tetrathiomolybdate and tetrathiotungstate complexes,12 a combination of borohydride reducing agents with sulfur,13 and the sulfur element in strong alkaline media14 has been used to convert them into the related disulfides.
Also, symmetric disulfides can be achieved when Bunte salts are treated with samarium in the presence of indium(III) chloride, or molecular iodine15 or when they are heated with thiourea, substituted thioureas, iodide ion, or thiocyanate ion in strong acidic solutions.16
The synthesis of organosulfur compounds via the in situ generation of thiols has been of interest especially in recent years.17 In this regard, thia-Michael adducts have been synthesized by reacting Bunte salts with electron-deficient alkenes under acidic conditions18a and by the reaction of elemental sulfur, aryl halides and electron-deficient alkenes in the presence of copper ferrite nanoparticles very recently.18b Also, the treatment of alkyl halides with thiourea as a thiol producing reagent in the presence of a suitable oxidant has been shown to be practical for the one-pot synthesis of disulfides.19
To establish a new strategy for the in situ generation of thiols, we focused our attention on the reaction of alkyl halides with Na2S2O3. Sodium S-alkyl thiosulfates (Bunte salts) are generally prepared from the reaction of alkyl halides with thiosulfate anions.20 Their hydrolysis under acidic conditions is known as a suitable method for achieving thiols.20 It can be said that Bunte salts are intermediates in the synthesis of thiols from the reaction of alkyl halides with sodium thiosulfate.
| RX + Na2S2O3 → RS–SO3Na | (1) |
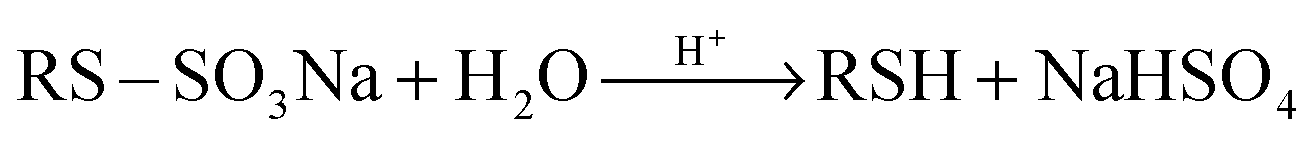 | (2) |
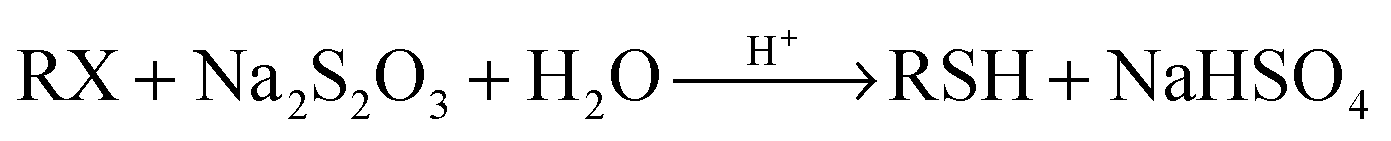 | (3) |
| Na2S2O3 + 2H+ → 2Na+ + S + SO2 + H2O | (4) |
| Entry | Solvent | T | pH | Organic product |
|---|---|---|---|---|
| a Reaction conditions: 2-phenylethyl bromide (1 mmol), Na2S2O3·5H2O (1 mmol), solvent (1–3 mL), H2O (0.1 mL), 24 h. | ||||
| 1 | THF | Reflux | Neutral | No |
| 2 | CH3CN | Reflux | Neutral | No |
| 3 | CH2Cl2 | Reflux | Neutral | No |
| 4 | H2O | 60–70 °C | Neutral | No |
| 5 | DMF | 60–70 °C | Neutral | No |
| 6 | EtOH | Reflux | Neutral | No |
| 7 | DMSO | 60–70 °C | (Acidic) | Yes (symmetric disulfide) |
After 24 h, the reaction of 2-phenylethyl bromide with Na2S2O3·5H2O to produce the corresponding Bunte salt in CH3CN, THF, and CH2Cl2 was not completed and a considerable amount of starting alkyl halide was detected in the reaction mixture (Table 1, entries 1–3). The starting halide had been completely converted to the corresponding Bunte salt during this period in H2O, DMF and EtOH (Table 1, entries 4–6). All reaction media were neutral to the litmus paper test. No thiol or any organic products were found in the reaction mixtures. However, a similar reaction in DMSO yielded a different result (Table 1, entry 7). During this period the starting halide had been completely consumed to give the corresponding symmetric disulfide in 90% yield. Also, the reaction medium had been strongly acidified. The pH was below 3 as determined by using pH cooperative paper (Universalindikator pH 0–14 Merck, Merck KgaA, 64271 Darmstadt, Germany).
Then we decided to investigate this reaction in more detail. Along this line, a mixture of 2-phenylethyl bromide (2 mmol) and well-powdered Na2S2O3·5H2O (2 mmol) in wet DMSO (2 mL DMSO + 0.2 mL H2O) was stirred magnetically at 60–70 °C. The reaction mixture was neutral to litmus paper at first. After stirring for 1 h, the starting halide was completely consumed and, the reaction medium was still neutral to wet litmus paper. After 2 h, the reaction medium had been slightly acidified and a spot of disulfide was monitored by TLC. Over time, the pink color of the litmus paper gradually turned red which can be attributed to increasing NaHSO4 in the reaction medium due to hydrolysis progression of the Bunte salt. The litmus paper became completely red after 3 h (pH < 3) nevertheless, the reaction mixture was continuously stirred for another 2 h under such conditions to ensure the reaction completion. After that the reaction was worked up by dilution with water and extracted with EtOAc. The corresponding disulfide was obtained in 91% yield after chromatography on silica gel.
Further studies indicated that the molar ratio between the substrate and sodium thiosulfate was crucial. When an equivalent of 2-phenylethyl bromide was treated with 1.2 equivalents of Na2S2O3·5H2O under the reaction conditions, the starting halide was completely consumed within 30 min but the corresponding disulfide or any organic product was not found on the TLC even after 12 h. Meanwhile, the reaction medium was neutral to wet litmus paper during this period. However, this reaction gave the corresponding disulfide after 24 h in 90% yield. Conversely, when an equivalent of Na2S2O3·5H2O was treated with 1.2 equivalents of 2-phenylethyl bromide, the reaction medium was acidified after 30 min (pH < 3) and a disulfide spot was detected on the TLC and gave the desired disulfide within 3 h in 91% yield. Considering these results, one can conclude that the hydrolysis of the in situ generated Bunte salt is retarded or at least proceeds very slowly in the presence of Na2S2O3. The hydrolysis of Bunte salts in DMSO in the absence of protons (acids) occurs but proceeds very slowly. This reaction is a non-catalyzed process at first but it produces acidic NaHSO4 as the by-product (eqn (2)), which can accelerate the process. Hence, this reaction is a self-catalyzed reaction. However, in the presence of Na2S2O3, the proton undergoes reaction with this reagent very rapidly (eqn (4)) and the hydrolysis process proceeds only via the non-catalyzed process, which is very slow and unappreciable. When thiosulfate is stoichiometric with respect to halide or is a limiting reactant, sodium thiosulfate is consumed completely by reacting with alkyl halide and subsequently the self-catalyzed hydrolysis process proceeds exponentially by increasing the protons in reaction media. When alkyl halide is the limiting reactant, the proton-catalyzed hydrolysis process is retarded by unreacted thiosulfates and thus the hydrolysis proceeds via non-catalyzed hydrolysis very slowly until the remaining thiosulfates are completely decomposed by reaction with the in situ generated protons. Because the generation of protons by non-catalyzed hydrolysis is a very slow reaction, the decomposition of remaining thiosulfates proceeds more slowly. On the other hand, the hydrolysis of S-alkylthiosulfate intermediates in the presence of Na2S2O3 destroys this key reagent due to production of acidic NaHSO4 which can cause yield loss or reaction failure. However, considering the reaction yield, it can be concluded that the non-catalyzed hydrolysis reaction of Bunte salts proceeds more slowly than the reaction between alkyl halide and sodium thiosulfate.
With these results in hand, the scope of the reaction was investigated using structurally diverse alkyl halides.22 The results are presented in Table 2. As shown in Table 2, various halides were efficiently treated with Na2S2O3·5H2O generating the corresponding disulfides. Primary aliphatic bromides and iodides reacted efficiently providing the corresponding disulfides in excellent yields (entries 1–4). Also, secondary bromides, including sec-butyl, iso-propyl, cyclohexyl and cyclopentyl bromides (entries 15–18) offered similar results. However, longer reaction times were required for the desired transformations. Using this protocol, a variety of benzyl halides including electron-rich and electron-poor benzyl bromides and chlorides were also converted into the corresponding disulfides exclusively in excellent yields (entries 5–12). Exceptionally, p-nitrobenzyl bromide (entry 7) gave the corresponding disulfide in moderate yield. With this substrate, some unidentified side products were formed. Also, allyl disulfides were produced solely when the corresponding halides were reacted with Na2S2O3 at 50–60 °C in excellent yields (entries 13 and 14).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Alkyl halide | t (h) | Isolated yield (%) | Entry | Alkyl halide | t (h) | Isolated yield (%) |
| a Reaction conditions: alkyl halide (2–2.1 mmol), Na2S2O3·5H2O (2 mmol), DMSO (2 mL), H2O (0.2 mL), 60–70 °C. b t refers to the time required to change the color of litmus paper from yellow to red (pH < 3). c The reaction was conducted at 50–60 °C. | |||||||
| 1 | 1-Bromobutane | 4 | 89 | 10 | m-CH3C6H4CH2Cl | 2.5 | 89 |
| 2 | PhCH2CH2Br | 3 | 91 | 11 | p-CH3C6H4CH2Cl | 2 | 88 |
| 3 | 1-Bromo-3-methylbutane | 3 | 86 | 12 | o-ClC6H4CH2Cl | 4 | 92 |
| 4 | 1-Iododecane | 5 | 91 | 13c | CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2Br CHCH2Br |
1.5 | 91 |
| 5 | PhCH2Br | 0.5 | 95 | 14c | CH2C(CH3)CH2Cl |
3 | 88 |
| 6 | p-BrC6H4CH2Br | 1 | 94 | 15 | 2-Bromobutane | 8 | 88 |
| 7 | p-NO2C6H4CH2Br | 1 | 60 | 16c | 2-Bromopropane | 10 | 86 |
| 8 | PhCH2Cl | 3 | 90 | 17 | Bromocyclohexane | 10 | 89 |
| 9 | o-CH3C6H4CH2Cl | 2.5 | 89 | 18 | Bromocyclopentane | 10 | 87 |
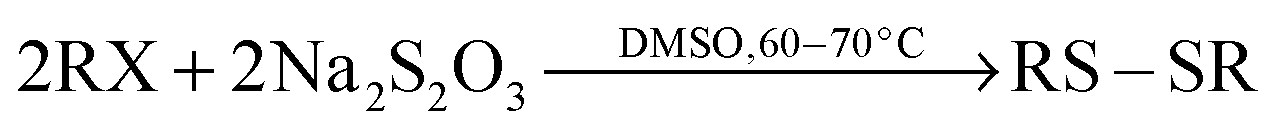
A proposed pathway for whole process is presented in Scheme 1.
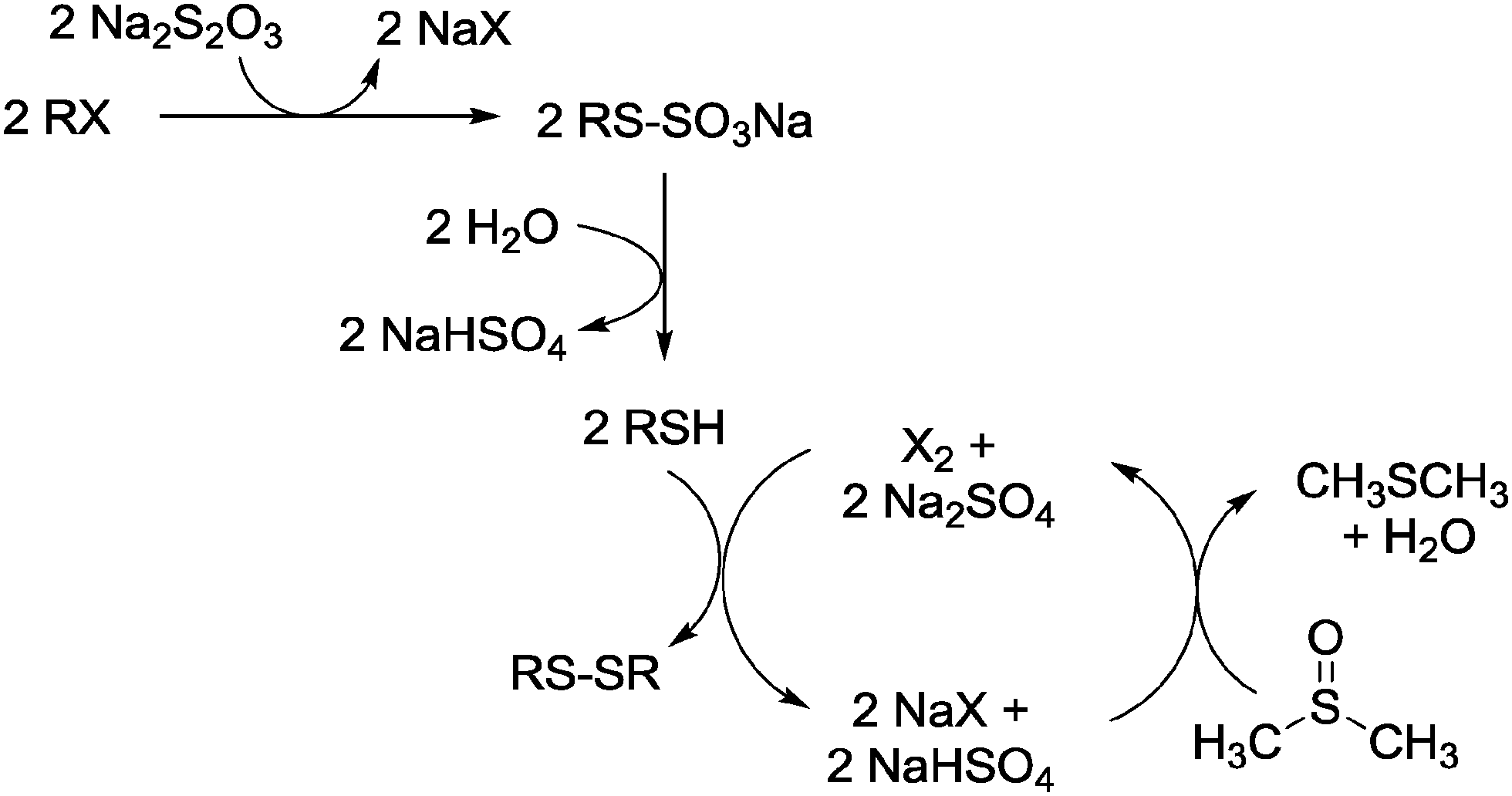 | ||
| Scheme 1 A proposed reaction pathway for the preparation of disulfides from organic halides and Na2S2O3 in DMSO. | ||
DMSO plays a dual role: it acts as a suitable solvent for the reaction and as a suitable thiol oxidizing reagent. Dimethylsulfoxide is among the employed reagents for the oxidation of thiols to disulfides.22 Because of its low oxidizing power, the conversion of thiols to disulfides with DMSO is not synthetically useful. This drawback has been circumvented by using dimethylsulfoxide in conjunction with a variety of co-reagents such as halogens and hydrogen halides,23 oxophile reagents,24 rhenium,25 or dichlorodioxomolybdenum catalysts.26
As proposed in Scheme 1, the conversion of an alkyl halide to disulfide consists of three steps. At first, the alkyl halide undergoes a nucleophilic substitution reaction with thiosulfate to yield the S-alkylthiosulfate. Next, the thiol and sodium bisulfate as its co-products are formed from the hydrolysis of salts. This stage is a self-catalyzed reaction since the hydrolysis of Bunte salts is catalyzed by acidic reagents.20 Finally, the in situ generated mercaptan undergoes oxidative coupling by DMSO producing the corresponding disulfide. This process can be catalyzed by molecular halogen, which is generated via deoxygenation of DMSO by hydrogen halide.
In conclusion, a new one-pot process for the formation of disulfides from alkyl halides has been described. This method is suitable for scale-up. As structurally diverse organic halides, sodium thiosulfate and DMSO are readily available, the preparation of structurally diverse disulfides becomes more practical than alternative protocols.
Experimental section
A typical scale-up procedure
A mixture of well-powdered Na2S2O3·5H2O (30 mmol, 7.445 g) and o-chlorobenzyl chloride (30.75 mmol, 4.952 g) in wet DMSO (30 mL DMSO + 3 mL H2O) was stirred magnetically at 60–70 °C. The progress of the reaction was examined by litmus paper. After stirring for 4 h, the color of the litmus paper changed from yellow to red. The stirring was continued for a further 2 h under such conditions. Then the reaction was worked up by adding H2O (10 mL) and extracted with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 n-hexane/EtOAc (3 × 15 mL). The product was further purified by recrystallization from 20:1 n-hexane/EtOAc to afford pure bis(2-chlorobenzyl) disulfide in 4.162 g, 88% yield.
:1 n-hexane/EtOAc (3 × 15 mL). The product was further purified by recrystallization from 20:1 n-hexane/EtOAc to afford pure bis(2-chlorobenzyl) disulfide in 4.162 g, 88% yield.
Bis(2-chlorobenzyl) disulfide (Table 2, entry 12)
Yellow crystals; m.p. 70–72 °C (lit.27 74 °C); 1H NMR (250 MHz, CDCl3): δ 7.40–7.18 (m, 8H), 3.79 (s, 4H); 13C NMR (62.5 MHz, CDCl3): δ 135.0, 134.1, 131.6, 129.7, 128.9, 126.7, 41.1. Anal. calcd for C14H12Cl2S2: C, 53.34; H, 3.84; S, 20.34%. Found: C, 53.44; H, 3.96; S, 20.19%.Acknowledgements
We are thankful to the Persian Gulf University Research Council for financial support of this work.Notes and references
- (a) P. C. Jocelyn, Biochemistry of the Thiol Group, Academic Press, New York, 1977 Search PubMed; (b) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2010, 46, 6476–6478 RSC; (c) K. Smith and M. Tzimas, J. Chem. Soc., Perkin Trans. 1, 1995, 2381–2382 RSC; (d) A. Corma, T. Ródenas and M. Sabater, J. Chem. Sci., 2012, 3, 398–404 RSC.
- (a) D. Witt, Synthesis, 2008, 2491–2509 CrossRef CAS; (b) B. Mandal and B. Basu, RSC Adv., 2014, 4, 13854–13881 RSC.
- (a) P. Dhar, R. Ranjan and S. Chandrasekaran, J. Org. Chem., 1990, 55, 3728–3729 CrossRef CAS; (b) H. Firouzabadi, N. Iranpoor and M. Jafarpour, J. Sulfur Chem., 2005, 26, 313–324 CrossRef CAS; (c) H. Firouzabadi and B. Karimi, Synthesis, 1999, 500–502 CrossRef CAS; (d) L. Kumar Pandey, U. Pathak and A. N. Rao, Synth. Commun., 2007, 37, 4105–4109 CrossRef; (e) C. Narayana, S. Padmanabhan and G. W. Kabalka, Synlett, 1991, 125–126 CrossRef CAS; (f) H. Guo, J. Wang and Y. Zhang, Synth. Commun., 1997, 27, 85–88 CrossRef CAS; (g) N. Iranpoor, H. Firouzabadi and A. Jamalian, Synlett, 2005, 1447–1449 CrossRef CAS.
- H. L. Siddiqui, M. Zia-Ur-Rehman, N. Ahmad, G. W. Weaver and P. D. Lucas, Chem. Pharm. Bull., 2007, 55, 1014–1017 CrossRef CAS PubMed.
- (a) F. K. Lautenschlaeber and N. V. Schwartz, J. Org. Chem., 1969, 34, 3991–3998 CrossRef; (b) T. Fujisawa and T. Kobori, Chem. Lett., 1972, 935–938 CrossRef CAS; (c) J. C. Hinshaw, Tetrahedron Lett., 1972, 13, 3567–3569 CrossRef.
- X.-S. Jia and Y.-M. Zhang, Chin. J. Chem., 2005, 23, 303–304 CrossRef CAS.
- X. Jia, Y. Zhang and X. Zhou, Tetrahedron Lett., 1994, 35, 8833–8834 CrossRef CAS.
- C. h. J. Burns, L. D. Field, J. Morgan, D. D. Ridley and V. Vignevich, Tetrahedron Lett., 1999, 40, 6489–6492 CrossRef CAS.
- K. R. Prabhu, A. R. Ramesha and S. Chandrasekaran, J. Org. Chem., 1995, 60, 7142–7143 CrossRef CAS.
- (a) B. Tamami, N. Iranpoor and H. Mahdavi, Synth. Commun., 2002, 32, 1251–1258 CrossRef CAS; (b) N. Iranpoor, H. Firouzabadi, M. Chitsazi and A. A Jafari, Tetrahedron, 2002, 58, 7037–7042 CrossRef CAS; (c) A. J. Pearson and J.-J. Hwang, J. Org. Chem., 2000, 65, 3466–3472 CrossRef CAS PubMed; (d) M. G. Silvestri and C.-H. Wong, J. Org. Chem., 2001, 66, 910–914 CrossRef CAS PubMed.
- (a) A. Kamyshny, I. Ekeltchlk, J. Gun and O. Lev, Anal. Chem., 2006, 78, 2631–2639 CrossRef CAS PubMed; (b) E. B. Krein and Z. Aizenshtat, J. Org. Chem., 1993, 58, 6103–6108 CrossRef CAS; (c) T. A. Hase and H. Perakyla, Synth. Commun., 1982, 12, 947–950 CrossRef CAS; (d) P. Weyerstahl and T. Oldenburg, Flavour Fragrance J., 1998, 13, 177–184 CrossRef CAS; (e) A. S. Gopalan and H. K. Jacobs, J. Chem. Soc., Perkin Trans. 1, 1990, 1897–1900 RSC; (f) S. U. Sonavane, M. Chidambaram, J. Almog and Y. Sasson, Tetrahedron Lett., 2007, 48, 6048–6050 CrossRef CAS.
- (a) P. Dhar and S. Chandrasekaran, J. Org. Chem., 1989, 54, 2998–3000 CrossRef CAS; (b) V. Polshettiwar, M. Nivsarkar, J. Acharya and M. P. Kaushik, Tetrahedron Lett., 2003, 44, 887–889 CrossRef CAS.
- (a) B. P. Bandgar, L. S. Uppalla and V. S. Sadavarte, Tetrahedron Lett., 2001, 42, 6741–6743 CrossRef CAS; (b) A. R. Kiasat, B. Mokhtari, F. Kazemi, S. Yousefi and M. Javaherian, J. Sulfur Chem., 2007, 28, 171–176 CrossRef CAS.
- (a) J.-X. Wang, C.-H. Wang, W. Cui and Y. Hu, Synth. Commun., 1995, 25, 3573–3581 CrossRef CAS; (b) A. R. Kiasat, B. Mokhtari, A. Savari and F. Kazemi, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 178–182 CrossRef CAS.
- (a) L. Wang and Y. Zhang, Tetrahedron, 1999, 55, 10695–10712 CrossRef CAS; (b) P. Li, L. Wang, Y. Yang, X. Sun, M. Wang and J. Yan, Heteroat. Chem., 2004, 15, 376–379 CrossRef CAS; (c) Y. Liu, H. Zheng, D. Xu, Z. Xu and Y. Zhang, Synlett, 2006, 2492–2494 CAS; (d) L. Wang, P. Li and L. Zhou, Tetrahedron Lett., 2002, 43, 8141–8143 CrossRef CAS.
- B. Milligan and J. M. Swan, J. Chem. Soc., 1962, 2172–2177 RSC.
- (a) H. Firouzabadi, N. Iranpoor and M. Gholinejad, Adv. Synth. Catal., 2010, 352, 119–124 CrossRef CAS; (b) L. Wang, W.-Z. Zhou, S.-C. Chen, M.-Y. He and Q. Chen, Adv. Synth. Catal., 2012, 354, 839–845 CrossRef CAS; (c) H. Firouzabadi, N. Iranpoor and M. Abbasi, Adv. Synth. Catal., 2009, 351, 755–766 CrossRef CAS; (d) H. Firouzabadi, N. Iranpoor and M. Abbasi, Tetrahedron, 2009, 65, 5293–5301 CrossRef CAS; (e) G.-P. Lu and C. Cai, Adv. Synth. Catal., 2013, 355, 1271–1276 CrossRef CAS; (f) H. Firouzabadi, N. Iranpoor and A. Samadi, Tetrahedron Lett., 2014, 55, 1212–1217 CrossRef CAS; (g) H. Firouzabadi, N. Iranpoor, M. Gholinejad and A. Samadi, J. Mol. Catal. A: Chem., 2013, 377, 190–196 CrossRef CAS; (h) M. Abbasi, Tetrahedron Lett., 2012, 53, 2608–2610 CrossRef CAS; (i) M. Abbasi, Tetrahedron Lett., 2012, 53, 3683–3685 CrossRef CAS.
- (a) Y.-M. Lin, G.-L. Lu, C. Cai and W.-B. Yi, RSC Adv., 2015, 5, 27107–27111 RSC; (b) M. Gholinejad and H. Firouzabadi, New J. Chem., 2015, 39, 5953–5959 RSC.
- (a) H. Firouzabadi, N. Iranpoor and M. Abbasi, Tetrahedron Lett., 2010, 51, 508–509 CrossRef CAS; (b) H. Firouzabadi, N. Iranpoor and M. Abbasi, Bull. Chem. Soc. Jpn., 2010, 83, 698–702 CrossRef CAS; (c) M. Abbasi, M. R. Mohammadizadeh and Z. K. Taghavi, J. Iran. Chem. Soc., 2013, 10, 201–205 CrossRef CAS.
- H. Distler, Angew. Chem., Int. Ed., 1967, 6, 544–553 CrossRef CAS.
- U. Agxrtvalca, E. Rees and G. Thode, Can. J. Chem., 1965, 43, 2802–2811 CrossRef , and references cited therein.
- C. N. Yiannios and J. V. Karabinos, J. Org. Chem., 1963, 28, 3246–3248 CrossRef CAS.
- O. G. Lowe, J. Org. Chem., 1975, 40, 2096–2098 CrossRef CAS.
- (a) B. Karimi, H. Hazarkhani and D. Zareyee, Synthesis, 2002, 2513–2516 CrossRef CAS; (b) B. Karimi and D. Zareyee, Synlett, 2002, 346–348 CrossRef CAS.
- (a) M. M. Abu-Omar and S. I. Khan, Inorg. Chem., 1998, 37, 4979–4985 CrossRef CAS PubMed; (b) J. B. Arterburn, M. C. Perry, S. L. Nelson, B. R. Dible and M. S. Holguin, J. Am. Chem. Soc., 1997, 119, 9309–9310 CrossRef CAS.
- R. Sanz, R. Aguado, M. R. Pedrosa and F. J. Arnaiz, Synthesis, 2002, 856–858 CrossRef CAS.
- S. K. Srivastava, R. Rastogi, P. Rajaram, R. J. Butcher and J. P. Jasinski, Phosphorus, Sulfur Silicon Relat. Elem., 2010, 185, 455–462 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nj01885d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |