
An unusual C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C⋯CO interaction in (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates†
C⋯CO interaction in (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates†
Zhenfeng
Zhang
*,
Nana
Ma
and
Xiaopeng
Xuan
School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, P. R. China. E-mail: zzf5188@163.com; Fax: +86 373-3326336; Tel: +86 373-3326335
First published on 20th August 2015
Abstract
An unusual C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C⋯CO interaction has been discovered in (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates and rationalized by the density functional theory calculations. Second order perturbation theory analysis based on the NBO method and LMOEDA energy decomposition further reveals that the CC⋯CO interaction is mainly dominated by dispersion and electrostatic energies, and two orbital interactions.
C⋯CO interaction has been discovered in (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates and rationalized by the density functional theory calculations. Second order perturbation theory analysis based on the NBO method and LMOEDA energy decomposition further reveals that the CC⋯CO interaction is mainly dominated by dispersion and electrostatic energies, and two orbital interactions.
Noncovalent interactions play a dominant role in chemical interactions between a protein and a drug,1 or an enzyme and its substrate,2 self-assembly of nanomaterials,3 and even some chemical reactions.4,5 A complete understanding of these chemical interactions will often demand a complete understanding of the noncovalent interactions too. Therefore, it is not surprising that a great deal of interest has been generated in the study of noncovalent interactions. Experimental and theoretical results have shown that unsaturated carbon atoms can act as an electron donors6–8 as well as an electron acceptors9 forming various kinds of noncovalent interactions, such as C⋯π, cation⋯π and anion⋯π, etc. Considering the complementary roles of carbon atoms, C⋯C interactions are expected to occur between electron-rich vinyl and electron-poor carbonyl fragments. Our interest in β-enamino esters, (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates, mainly stems from this expectation. In this context, we have prepared a series of such β-enamino esters and studied their X-ray crystal structures.
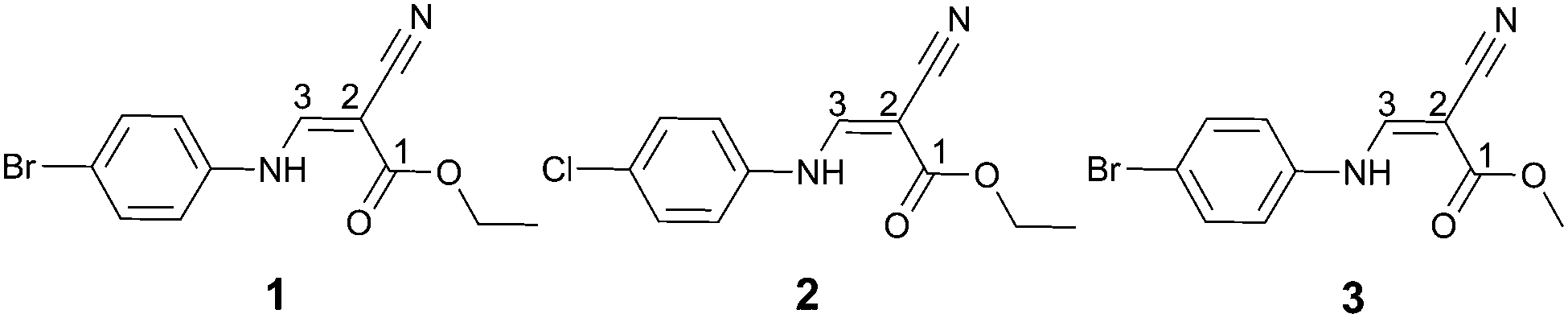
Interestingly we have discovered a hitherto unreported CC⋯CO interaction in the supramolecular structure of ethyl (Z)-3-[(4-bromophenyl)amino]-2-cyanoprop-2-enoate, 1 (Fig. 1). As shown in the figure, the vinyl atom C3 resides orthogonally above the pseudotrigonal axis of the ester carbonyl group. The distance between C3 and C1i is only 3.277(3) Å, which is significantly shorter than the sum of van der Waals radii, 3.4 Å. The characteristic geometry suggests a pair of C⋯C interactions between the vinyl and carbonyl groups.
 | ||
| Fig. 1 The dimeric structure of ethyl (Z)-2-cyano-3-[(4-bromophenyl)-amino]prop-2-enoate, 1, showing both experimentally and theoretically the formation of the C⋯C interaction between C3 and C1i; the distances C3⋯C1i from experimental and DFT calculations are 3.277 and 3.186 Å, respectively. Symmetry code: (i) 1 − x, 1 − y, 2 − z. | ||
A recent study7 has shown the existence of C⋯C interactions. However, the only work on this subject still remains theoretical. A computational study shows that the C⋯C interactions exist between electron-deficient molecules (CO2 or NCCN) and electron-rich molecules, such as HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH and H2CCH2, etc., and that all the dimers formed by this interaction show C2V symmetry and correspond to energy minima. The CC⋯CO interaction in 1, we believe, is very similar to the CH2CH2⋯CO2 interaction, though the formed dimers differ in parameters and symmetry.
CH and H2CCH2, etc., and that all the dimers formed by this interaction show C2V symmetry and correspond to energy minima. The CC⋯CO interaction in 1, we believe, is very similar to the CH2CH2⋯CO2 interaction, though the formed dimers differ in parameters and symmetry.
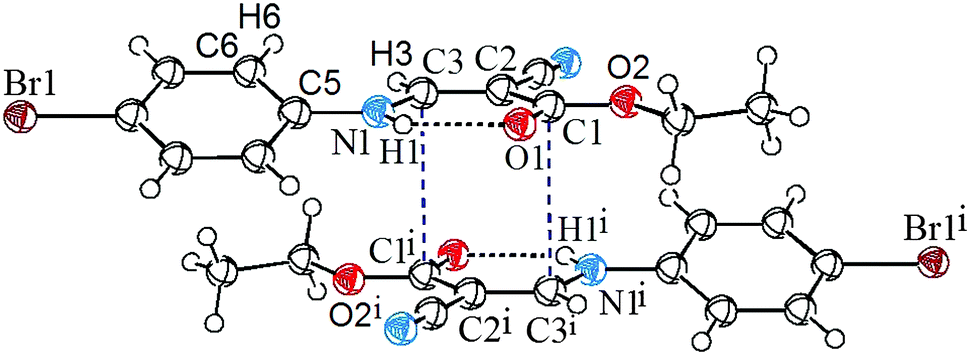
To quantitatively evaluate the strength of the CC⋯CO interactions, the density functional theory (DFT) calculations at the WB97XD10/6-311+G(d,p) level were performed on compound 1. The monomer and the dimers formed by CC⋯CO interaction were studied. All the calculations were carried out using the GAUSSIAN 0911 package. At this selected theoretical level, both the monomer and dimer of 1 correspond to energy minima, and are in good agreement with the crystallographically determined structure. The optimized dimer is also shown in Fig. 1. As shown in the figure, the atom C3 approaches the atom C1i perpendicularly to the ester carbonyl plane, and the distance between C3 and C1i is 3.186 Å. The total dimerization energy is −20.79 kcal mol−1, showing theoretically that the CC⋯CO interaction is thermodynamically most favorable.
It is widely accepted that the natural bond orbital (NBO) theory12,13 is quite useful for analyzing intermolecular interactions.14 In order to further characterize the CC⋯CO interaction via orbital interactions, we have also performed NBO analysis on the CC⋯CO dimer with NBO version 3.115 incorporated in the G09 package using the optimized ground-state geometries. Interestingly, we have found that there are four intermolecular orbital interactions corresponding to the CC⋯CO interaction, out of which only the strongest interactions (as well as strongest between the dimer), A and B, are shown in Table 1 and Fig. 2. As shown in the table and figure, the π-orbital of the carbonyl O1iC1i interacts with the π*-antibonding orbital of the vinyl C3C2, with a concomitant second-order stabilization energy of E(2) = 0.27 kcal mol−1, and on the other hand, the π-orbital of C2C3 interplays with the π*-antibonding orbital of the C1iO1i bond, with a second-order energy of 0.26 kcal mol−1. The orbital interactions involving atoms C1 and C3i are completely identical to the ones involving C1i and C3. The total stabilization energy that attributed to the CC⋯CO interactions is approximately 1.36 kcal mol−1. The results have theoretically confirmed that the orbital interaction between the vinyl and carbonyl groups, though weak, can actually occur, and may be of vital importance in controlling the directionality and geometrical details.
| Pair name | Donor NBO | Acceptor NBO | E (2) energy (kcal mol−1) |
|---|---|---|---|
| A | π(O1iCi) |
π*(C3C2) |
0.27 |
| B | π(C2C3) |
π*(C1iO1i) |
0.26 |
| A′ | π(O1C1) |
π*(C3iC2i) |
0.27 |
| B′ | π(C2iC3i) |
π*(C1O1) |
0.26 |
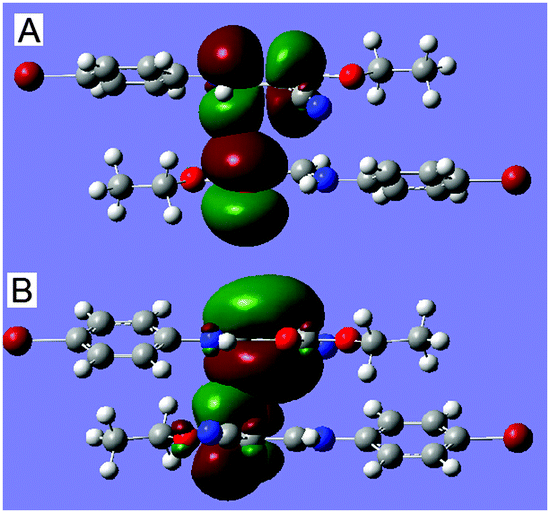 | ||
| Fig. 2 Dominant orbital interactions A, π(O1iC1i) → π*(C3C2), and B, π(C2C3) → π* (C1iO1i), showing the nature of the CC⋯CO in 1. For the sake of clarity, the same orbital interactions A′ and B′ have been omitted. | ||
Here the CC⋯CO interaction, though being bidirectional charge-transfer, leads to a redistribution of charge within the dimer. NBO calculations have revealed that during the formation of the CC⋯CO bond, the positive charge on the carbonyl atom C1i decreases from 0.821 to 0.810 e, the charge (0.182 e) on the partner atom C3 is, however, nearly unchanged, thus reducing the repulsion component between C3 and C1i. Conversely, the negative charges on atoms C2 and N2 increase from −0.384 and −0.340 e to −0.397 and −0.363 e, respectively, the former favoring to maintain the dimer in a lower electrostatic repulsion level due to the longer distance between C2 and C2i, and the latter favoring to enhance the following C–H⋯NC hydrogen bond.
In addition to the orbital interactions between the vinyl and carbonyl groups, the CC⋯CO dimer should also be stabilized by other standard energy contributions, such as dispersion, electrostatic, exchange-repulsion and polarization. To roundly explore the source of dimerization energy (−20.79 kcal mol−1), we employed the localized molecular orbital energy decomposition analysis (LMOEDA) approach of Su and Li16 to decompose the CC⋯CO interaction energy into electrostatic, exchange, repulsion, polarization, and dispersion components. The LMOEDA computations have been performed using the GAMESS package at the MP2/maug-cc-pVTZ level.17 The results shows that the CC⋯CO interaction is mainly dominated by dispersion energy and electrostatic energy, which contribute 66.6% and 25.4%, respectively, to the total dimerization energy.
Given that the CC⋯CO interactions occur in ethyl (Z)-3-[(4-bromophenyl)amino]-2-cyanoprop-2-enoate, 1, it is very probable to find this kind of interactions in ethyl (Z)-3-[(4-chlorophenyl)amino]-2-cyanoprop-2-enoate, 2 and methyl (Z)-3-[(4-bromophenyl)amino]-2-cyanoprop-2-enoate, 3.
Keeping our motivation in mind, we replace the Br1 atom (Fig. 1) with a Cl1 atom and the O2-ethyl with methyl to build the monomer and dimer of 2 and 3, respectively, for the evaluation of CC⋯CO interactions. The DFT calculations were performed for geometry optimization using the same methods as for 1. Interestingly, in the optimized dimers 2 and 3, the main geometry parameters are in perfect accordance with those found in 1 (Fig. 1); the atom C3 approaches the atom C1i perpendicularly to the ester carbonyl plane; the distances between C3 and C1i are 3.188 and 3.218 Å, respectively. The corresponding total dimerization energies are −20.76 and 19.18 kcal mol−1, respectively. The main orbital interactions involved in the CC⋯CO interaction are A, π(O1iC1i) → π*(C3C2), and B, π(C2C3) → π*(C1iO1i) (Fig. 3), which are shown by the second-order perturbation theory analysis of the Fock Matrix. The results reveals that the CC⋯CO interactions do exist universally in (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates.
 | ||
| Fig. 3 The dominant NBO orbital interactions A, π(O1iC1i) → π*(C3C2), and B, π(C2C3) → π*(C1iO1i) corresponding to the C⋯C interaction between vinyl atom C1 and carbonyl atom C3i in 2 (left) and 3 (right). For the sake of clarity, the same orbital interactions between atoms C3 and C1i have been omitted. | ||
The CC⋯CO interaction is very scarce. Searches of the Cambridge Structural Database (CSD) showed that no such interaction occurred between the vinyl and carbonyl groups despite the abundance of vinyl and carbonyl groups existing in crystal structures deposited so far in the CCDC. However, this interaction exists predominantly in (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates.
The crystal structure of 1 also exhibits strong intramolecular N–H⋯O hydrogen bonding and strong H⋯H van der Waals repulsion (Fig. 1). The distance of N1–H1⋯O1 is 2.01 Å and the associated angle is 133°. The H3⋯H6 distance and the C3–N1–C5–C6 torsion angle are 2.25 Å and 21°, respectively, suggesting strong van der Waals repulsion between C3–H and C6–H. A combination of these two interactions leads to a nearly planar conformation on the molecule's main skeleton. This conformation sets the stage for the CC⋯CO interactions observed within the crystal lattice.
In addition to the CC⋯CO interactions, there are two intermolecular noncovalent interactions in 1, one is of vinyl C–H⋯NC type (Fig. 4), and the other is of CN⋯Br type (Fig. 5). The distances of H3⋯N2ii and N2iii⋯Br1 are 2.47 and 3.36 Å, respectively, and the bond angle of C3–H3⋯N2ii is 168°. These noncovalent interactions synergistically constitute a stepped (011) sheet, the formation of which can be understood in the following way: the inversion-related molecules form a dimer via a pair of CC⋯CO interactions (Fig. 1 and 4); such dimers related by translation are further linked to a stepped molecular chain parallel to [100] via a pair of equivalent C–H⋯N hydrogen bonds (Fig. 4); chains of this type are laterally linked to a sheet by N⋯Br interactions (Fig. 5).
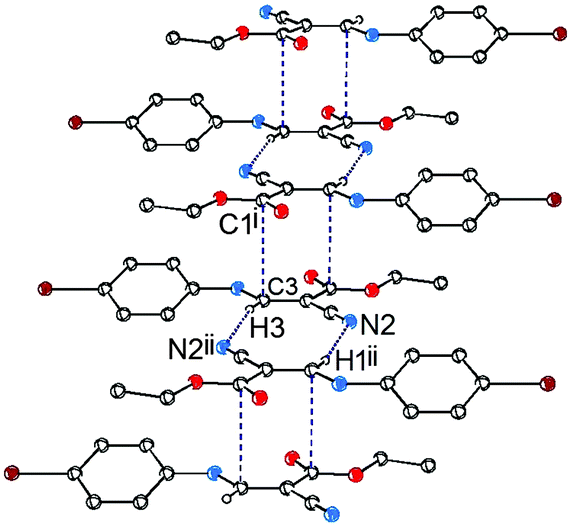 | ||
| Fig. 4 Packing diagram of 1, showing the formation of the stepped hydrogen-bonded chain along the [100] direction. For the sake of clarity, H atoms not involved in the motif shown have been omitted. Symmetry transformations: (i) 1 − x, 1 − y, 2 − z; (ii) −x, 1 − y, 2 − z. | ||
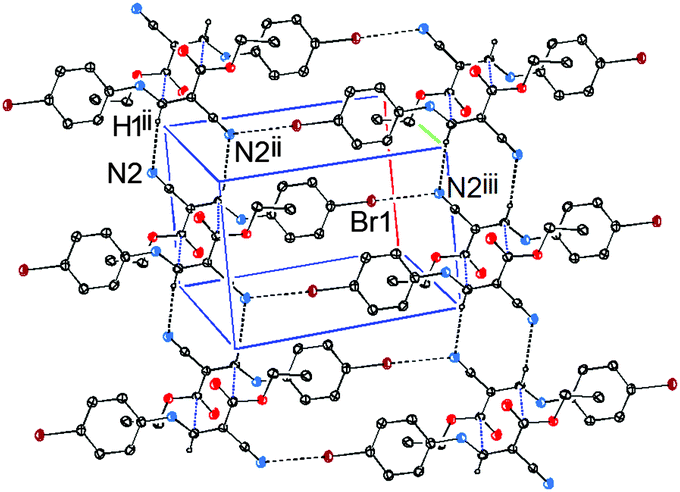 | ||
| Fig. 5 Part of the crystal structure of 1, showing the formation of a two-dimensional sheet via C⋯C, N⋯Br and C–H⋯N interactions. For the sake of clarity, H atoms not involved in the motif shown have been omitted. Symmetry codes: (ii) −x, 1 − y, 2 − z; (iii) x, y + 1, z − 1. | ||
In conclusion, the detailed structural analysis has revealed an unusual CC⋯CO interaction existing predominantly in (Z)-3-[(4-halogenphenyl)amino]-2-cyanoprop-2-enoates. The DFT computations and second order perturbation theory analysis of the Fock Matrix on the NBO basis at the WB97XD/6-311+G(d,p) level provided effective support to the interaction. The present work, we believe, may be the first systematic study on C⋯C interactions occurring between the vinyl and carbonyl fragments.
Experimental
Synthesis of ethyl (Z)-3-[(4-bromophenyl)amino]-2-cyanoprop-2-enoate, 1
A mixture of 4-bromoaniline (0.02 mol) and ethyl-2-cyano-3-ethoxyacrylate (0.02 mol) in toluene (10 ml) was refluxed for ca. 10 min. The reaction mixture was then cooled to room temperature. The precipitate was collected by filtration and washed with ethanol. Crystals suitable for X-ray analysis were obtained by slow cooling of a hot toluene solution of the crude product.X-Ray structure determination
The selected crystal was mounted with inert oil on glass fibers. Data were measured using Mo-Kα radiation on a Bruker SMART 1000 CCD diffractometer. Data collection at 296 K and reduction were performed using the SMART and SAINT software.18Absorption correction was applied using the multi-scan method (SADABS).19 The crystal structure of 1 was solved by direct methods and refined by full matrix least-squares on F2 using the SHELXTL program package.20 All non-hydrogen atoms were subjected to anisotropic refinement, and all H atoms were placed in geometrically idealized positions and constrained to ride on their parent atoms.
Crystal data
C12H11BrN2O2, M = 295.13, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 7.495 (1) Å, b = 7.525 (1) Å, c = 12.065(1) Å, α = 80.285(2)°, β = 86.767(2)°, γ = 67.729(2)°, V = 620.7(2) Å3, Z = 2, T = 293(2) K, Dc = 1.579 g cm−3; 6597 reflections collected, 2189 unique (Rint = 0.026), 1855 observed with I > 2(I); final R = 0.0309, wR2 = 0.0770, goodness-of-fit S = 1.07.
, a = 7.495 (1) Å, b = 7.525 (1) Å, c = 12.065(1) Å, α = 80.285(2)°, β = 86.767(2)°, γ = 67.729(2)°, V = 620.7(2) Å3, Z = 2, T = 293(2) K, Dc = 1.579 g cm−3; 6597 reflections collected, 2189 unique (Rint = 0.026), 1855 observed with I > 2(I); final R = 0.0309, wR2 = 0.0770, goodness-of-fit S = 1.07.
Notes and references
- (a) D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed; (b) L. Strekowski and B. Wilson, Mutat. Res., 2007, 623, 3–13 CrossRef CAS PubMed; (c) B. T. Benedetti, E. J. Peterson, P. Kabolizadeh, A. Martinez, R. Kipping and N. P. Farrell, Mol. Pharmaceutics, 2011, 8, 940–948 CrossRef CAS PubMed; (d) C. P. Selby and A. Sancar, Biochemistry, 1991, 30, 3841–3849 CrossRef CAS PubMed.
- (a) Z. Chi, R. Liu and H. Zhang, Biomacromolecules, 2010, 11, 2454–2459 CrossRef CAS PubMed; (b) M. N. Namchuk and S. G. Withers, Biochemistry, 1995, 34, 16194–16202 CrossRef CAS PubMed; (c) D. H. Williams, E. Stephens, D. P. O'Brien and M. Zhou, Angew. Chem., Int. Ed. Engl., 2004, 43, 6596–6616 CrossRef CAS PubMed.
- (a) D. A. Britz and A. N. Khlobystov, Chem. Soc. Rev., 2006, 35, 637–659 RSC; (b) B. Rybtchinski, ACS Nano, 2011, 5, 6791–6818 CrossRef CAS PubMed; (c) D. Baskaran, J. W. Mays and M. S. Bratcher, Chem. Mater., 2005, 17, 3389–3397 CrossRef CAS; (d) K. L. Vidale, D. M. Sherman, K. Hallenga, K. V. Wood and J. G. Stowell, J. Am. Chem. Soc., 2001, 123, 3854–3855 CrossRef.
- S. S. Sheiko, F. C. Sun, A. Randall, D. Shirvanyants, M. Rubinstein, H. Lee and K. Matyjaszewski, Nature, 2006, 440, 191–194 CrossRef CAS PubMed.
- G. A. DiLabio, P. G. Piva, P. Kruse and R. A. Wolkow, J. Am. Chem. Soc., 2004, 126, 16048–16050 CrossRef CAS PubMed.
- M. A. Viswamitra, R. Radhakrishnan, J. Bandekar and G. R. Desiraju, J. Am. Chem. Soc., 1993, 115, 4868–4869 CrossRef CAS.
- I. Alkorta, F. Blanco, J. Elguero, J. A. Dobado, S. M. Ferrer and I. Vidal, J. Phys. Chem. A, 2009, 113, 8387–8393 CrossRef CAS PubMed.
- (a) Z. Zhang, Y. Wu and G. Zhang, CrystEngComm, 2011, 13, 4496–4499 RSC; (b) J. L. Atwood, F. Hamada, K. D. Robinson, G. W. Orr and R. L. Vincent, Nature, 1991, 349, 683–684 CrossRef CAS.
- (a) Z. Zhang, H. Tong, Y. Wu and G. Zhang, New J. Chem., 2012, 36, 44–47 RSC; (b) G. Bhattacharjya, G. Savitha and G. Ramanathan, CrystEngComm, 2004, 6, 233–235 RSC.
- (a) S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed; (b) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz and J. Cioslowskiand D. J. Fox, GAUSSIAN 09 revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- E. A. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef.
- E. A. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef.
- L. Song, Y. Lin, W. Wu, Q. Zhang and Y. Mo, J. Phys. Chem. A, 2005, 109, 2310–2316 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, Theoretical Chemistry Institute, University of Wisconsin, Madison, USA, 1995 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- (a) H He Li Be B C N O F Ne Na Mg Al Si P S Cl Ar: E. Papajak and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 597–601 CrossRef CAS; (b) B–F: R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- SMART 5.0 and SAINT 4.0 for Windows NT, Area Detector Control and Integration Software, Bruker Analytical X-Ray Systems Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS: Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELXTL 5.1 for Program for Windows NT: Structure Determination Software Programs, Bruker Analytical X-Ray Systems, Inc., Madison, WI, 1997 Search PubMed.
Footnote |
| † CCDC 952088. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5nj01814e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |