
On the nature of the phase “η-TiO2”
Inga
Vasilyeva
a,
Galina
Kuz'micheva
b,
Alena
Pochtar
c,
Asiya
Gainanova
*b,
Olesya
Timaeva
b,
Andrey
Dorokhov
b and
Vadim
Podbel'skiy
d
aInstitute of Inorganic Chemistry, Siberian Branch of the Russian Academy of Sciences, Novisibirsk, Russia
bLomonosov State University of Fine Chemical Technologies, Moscow, Russia. E-mail: ms.asenka1984@mail.ru
cBoreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences, Novisibirsk, Russia
dNational Research University “Higher School of Economics”, Moscow, Russia
First published on 19th October 2015
Abstract
Conditions for a sulfate method for the synthesis of a metastable modification, which has been previously described as “η-TiO2” (Dadachov, 2006), were found and the stability region of this phase in the hydrolysis temperature–hydrolysis duration coordinates was determined. Investigation by a number of methods (X-ray powder diffraction, a differential dissolution method, thermogravimetry, IR spectroscopy, Raman spectroscopy) showed that the η-phase is not a polymorph of TiO2 but is a pseudo-polymorph of titanium dioxide hydrate. It was demonstrated that nanoparticles of the low-temperature η-phase consist of the [TiO2−x·mH2O] core, the structure of which can be described as a superstructure in relation to anatase, and an amorphous shell containing TiO2−x (trace amount), OH, HSO4 and water. The average crystallite size depends on the ratio of the constituents.
Introduction
Polymorphism is the ability of compounds to exist in more than one form while having different crystal structures and properties, the chemical composition being the same (in some cases, in the homogeneity region), depending on the external conditions. The latter include temperature, pressure, type of field (electric, magnetic, etc.), mode of treatment of the material (heat, microwave, ultrasound, etc.), duration of treatment and so on, i.e., the thermodynamic and kinetic factors. Therefore, polymorphism is one of the key properties of compounds that enables their structures to adapt to varying external conditions.Theoretical and experimental studies of the thermodynamics of small particles showed that particle size is an active variable responsible, together with other thermodynamic variables, for the state of the system. Changes in the particle size are associated with some thermodynamic properties of nanoparticles (the concentration of vacancies in nanoparticles increases with a decrease in their size, which is accompanied by a reduction of the polymorphic transition temperature and the unit cell parameters, and an increase in compressibility and solubility) and physicochemical properties (stability, heat capacity, melting point, electric and magnetic characteristics, reactivity, etc.). The particle size can be considered as a specific equivalent of the temperature. In this case, the Gibbs potential will differ from the standard parameters of the bulk phase. The nature of the dependence, G = f(L) (G is the Gibbs free energy and L is the crystallite size) with a minimum at Lcrys implies that the formation of crystallites in the range L < Lcrys becomes thermodynamically unfavorable.1 Therefore, it can be suggested that energy minima (both global and local) can appear at different values of L for different nanosized polymorphic modifications as stable and metastable, respectively. In other words, rather large sets of polymorphs or modifications can be obtained by controlling the nanostructuring processes. Consequently, physicochemical concepts are supplemented with the terms “size” and “self-organization”.
Currently, titanium(IV) oxides seem to be the most interesting from the theoretical and practical points of view. Twelve polymorphs of titanium dioxide, which were prepared either under ambient conditions or at high temperatures and elevated pressures, are described in the literature. These polymorphs include metastable modifications (for example, anatase β-TiO2) and stable modifications (for example, rutile), which either have or do not have homogeneity regions and which were synthesized as nanosized (primarily anatase, brookite, rutile, β-TiO2) and bulk (all 12 modifications) samples.
Nano-TiO2 with an anatase structure is more widely used in practice compared to polymorphs with other modifications. Thus, it possesses a photocatalytic activity (PCA) in the UV region of the solar spectrum,2–5 has a high chemical stability, is inexpensive and non-toxic and consequently is a promising compound for the design of solar photoelements6 and photocatalysts active in the visible region of the spectrum.3,4,7,8
A new modification of titanium dioxide, called “η-TiO2”, which has been synthesized relatively recently only as a nanosized form,9 largely outperforms anatase in terms of adsorption properties.10 In ref. 11 this phase was shown to have a high PCA at pH > 7, where the reaction rate of the decomposition of organic dyes under UV irradiation is an order of magnitude higher than that in the presence of photocatalysts based on other modifications of titanium dioxide (anatase and/or rutile) studied earlier. It was hypothesized9,10 that the high reactivity of the surfaces of samples with the η-phase is due to a larger amount of OH groups on the surfaces of nanoparticles compared to anatase.
It should be noted that “η-TiO2” is merely the molecular formula, whereas the structure and the real composition of this phase are unknown. Therefore, it is impossible to assign this phase to the polymorphs of titanium dioxide and explain its specific properties. The goal of the present study is to answer these questions.
Experimental
Synthetic procedures
Samples of the η-phase were prepared by the hydrolysis of titanyl sulfate, TiOSO4·xH2SO4·yH2O, at a constant pH value of the solution (pH ∼ 2) and at a constant concentration of the starting reagent (c0 = 0.65 mol L−1). After completion of the hydrolysis (τ = 5–80 min, t = 50–95 °C), coagulation was performed using a 1.3 mol L−1 KCl aqueous solution followed by filtration of the precipitate, washing it with water (until titanium and sulfate ions were not found in the washing waters) and acetone, and drying in an oven for 1 h at 50 °C.12,13Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, where λ = 1.54051 Å is the wavelength, 2θ ∼ 25° (d ∼ 3.5 Å; anatase, 101 diffraction reflection) or ∼ 27.5° (d ∼ 3.25 Å; rutile, 110 diffraction reflection) and β-TiO2 is the broadening of the reflection compared with the instrumental width (β = [(FWHM)exp2 − (FWHM)R2]1/2, where (FWHM)exp and (FWHM)R are the peak widths at half maximum for titanium dioxide and the reference α-Al2O3, respectively); the coefficient K (Scherrer coefficient, the shape factor of crystallites) was taken to be equal to 0.9. The standard deviation was ±5%. The percentage of anatase in samples with rutile was calculated by the equation xR, % = [1 + 0.65(IA/IR)]−1, xA, % = 100 − xR, where IA and IR are the integrated intensities of anatase for the 101 diffraction reflection, and of rutile for the 110 reflection, respectively.
cosθ, where λ = 1.54051 Å is the wavelength, 2θ ∼ 25° (d ∼ 3.5 Å; anatase, 101 diffraction reflection) or ∼ 27.5° (d ∼ 3.25 Å; rutile, 110 diffraction reflection) and β-TiO2 is the broadening of the reflection compared with the instrumental width (β = [(FWHM)exp2 − (FWHM)R2]1/2, where (FWHM)exp and (FWHM)R are the peak widths at half maximum for titanium dioxide and the reference α-Al2O3, respectively); the coefficient K (Scherrer coefficient, the shape factor of crystallites) was taken to be equal to 0.9. The standard deviation was ±5%. The percentage of anatase in samples with rutile was calculated by the equation xR, % = [1 + 0.65(IA/IR)]−1, xA, % = 100 − xR, where IA and IR are the integrated intensities of anatase for the 101 diffraction reflection, and of rutile for the 110 reflection, respectively.
In order to perform a theoretical analysis of powder diffraction patterns of two-phase mixtures and determine the percentage of anatase and “η-TiO2” in the samples, we wrote the supplemental program Technol-1,15 which can be used to obtain a linear combination of the initial diffraction patterns of single-phase samples. The program was written in C# oriented to the Net Framework platform (versions 3.5 and higher). Text files containing numerical data from powder diffraction patterns serve as the starting data. When analyzing the diffraction patterns and their linear combinations, the maximum intensity at a particular value of the variable xi (2θ°) was estimated for each diffraction reflection taking into account the background level.
Solvents, in which phases of different natures can be successively dissolved, were used for the DD analysis of samples. Since hydrated forms of Ti4+ are easily dissolved in an aqueous or a weak nitric acid medium, whereas titanium dioxide can be dissolved only in strong hydrofluoric acid, the composition of the solvent was gradually changed from water at 25 °C to HNO3 (pH = 2) at 25–40 °C, 3 N HNO3 at 40–70 °C and finally 4 N HF at 80 °C. In the case of the dissolution of titanium compounds with different compositions, an ICP AES detector was adjusted to enable the determination of titanium, sulfur and potassium content because the samples were synthesized in the presence of sulfuric acid, which is known17–19 to act as a good coordinating ligand, and KCl was added as a coagulant. Preliminary experiments showed that the potassium content was low (<2%) and this element was randomly captured by the bulk of the product that precipitated. This is evidence that potassium chloride is not involved in the formation of coagulation phases.
Results and discussion
In the present paper, the studied samples were prepared by the hydrolysis of titanyl sulfate TiOSO4·xH2SO4·yH2O at different durations (τ = 5–80 min) and temperatures (t = 50–95 °C). As was found earlier,9,20 the η-phase is characterized by a strong diffraction reflection with an interplanar spacing in the range d ∼ 17–21 Å and a less intense reflection with d ∼ 2.7–2.9 Å, the other reflections (d ∼ 3.5, ∼1.9, ∼1.7, ∼1.5 Å) being the same as those of TiO2 with an anatase structure (space group I41/amd, a ∼ 3.785 Å, c ∼ 9.50 Å; JCPDS 89-4921). It was impossible to determine the crystal structure of this compound because the η-phase is characterized by very small sizes of coherent scattering regions (D ∼ 30–50 Å) related to crystallite sizes,9 as well as by a limited number of reflections. It cannot be ruled out12,21–23 that the structure of the η-phase can be considered as a superstructure with respect to the anatase structure with the unit cell parameters a ∼ a0, c ∼ 2c0 (a0 and c0 are the unit cell parameters of anatase). It should be noted that the indexing of a small number of diffraction reflections of “η-TiO2” indicates the Bravais lattice P and limits the possible tetragonal space groups to P4/mmm (P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2m, Pm2; P4mm, P422), P21m; P4212.12 It was noted9 that the absence of a diffraction reflection with d ∼ 2.4 Å and the asymmetry of the peak with d ∼ 3.5 Å toward smaller angles attest to the single-phase state of the sample (η-phase), whereas the asymmetry of the peak with d ∼ 3.5 Å toward higher angles, along with the presence of the reflection with d ∼ 2.4 Å, are indicative of a two-phase state of the sample (anatase + η-phase). The average crystallite sizes of the η-phase (L ∼ 55–90 Å) determined by transmission electron microscopy20,21 are inconsistent with the average sizes of the coherent scattering regions (D ∼ 30–50 Å).12,20–23 Therefore, a defect structure of the sample cannot be ruled out.
2m, Pm2; P4mm, P422), P21m; P4212.12 It was noted9 that the absence of a diffraction reflection with d ∼ 2.4 Å and the asymmetry of the peak with d ∼ 3.5 Å toward smaller angles attest to the single-phase state of the sample (η-phase), whereas the asymmetry of the peak with d ∼ 3.5 Å toward higher angles, along with the presence of the reflection with d ∼ 2.4 Å, are indicative of a two-phase state of the sample (anatase + η-phase). The average crystallite sizes of the η-phase (L ∼ 55–90 Å) determined by transmission electron microscopy20,21 are inconsistent with the average sizes of the coherent scattering regions (D ∼ 30–50 Å).12,20–23 Therefore, a defect structure of the sample cannot be ruled out.
A detailed study of the effect of the conditions for the preparation of the samples (the hydrolysis temperature and duration, and the presence or the absence of the coagulant KCl at a constant concentration of the precursor c0 = 0.65 mol L−1) on the formation of nano-anatase and the η-phase revealed two parameters – the hydrolysis temperature (t) and the hydrolysis duration (τ) – that play a key role in the formation of these phases. Based on the results of the synthesis in the t range from 50 to 95 °C and the τ range from 5 to 80 min, and the characterization of 40 samples of titanium dioxide by X-ray powder diffraction, the regions where nano-anatase, η-TiO2, and their mixture exist were found, as shown in Fig. 1.
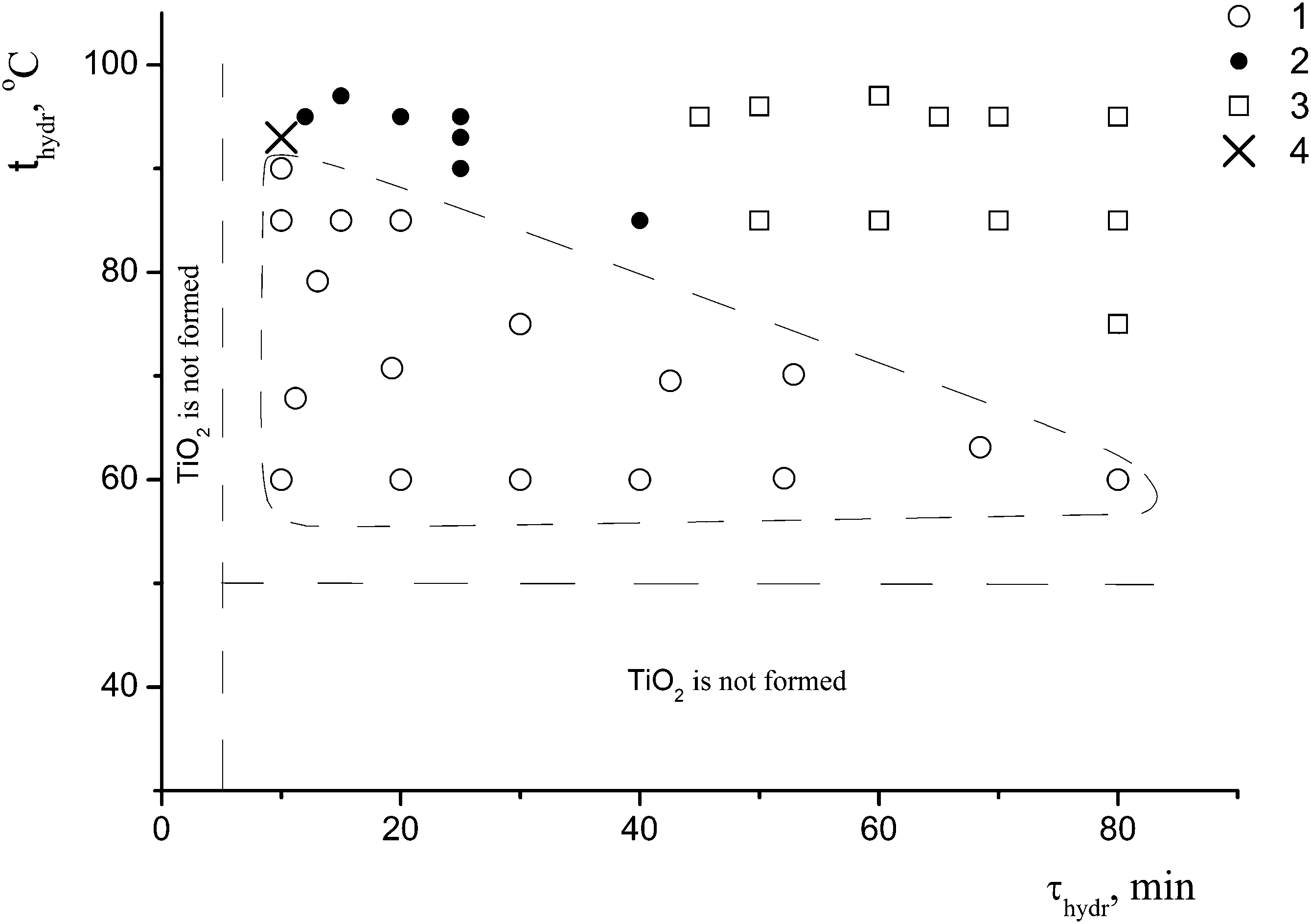 | ||
| Fig. 1 Regions of the existence of anatase and the η-phase in the hydrolysis temperature (thydr)–hydrolysis duration (τhydr) coordinates: 1 – η-phase; 2 – anatase + η-phase; 3 – anatase; 4 – sample I. | ||
For the subsequent detailed study, we chose sample I (it is marked with a cross in Fig. 1) from the two-phase region. Fig. 2a shows the powder diffraction pattern of this sample with diffraction reflections at 2θ ∼ 5° (d ∼ 18 Å), ∼25° (d ∼ 3.55 Å) and ∼33° (d ∼ 2.7 Å), a very diffuse reflection at 2θ ∼ 38° (d ∼ 2.4 Å) and a reflection at ∼48° (d ∼ 1.9 Å).
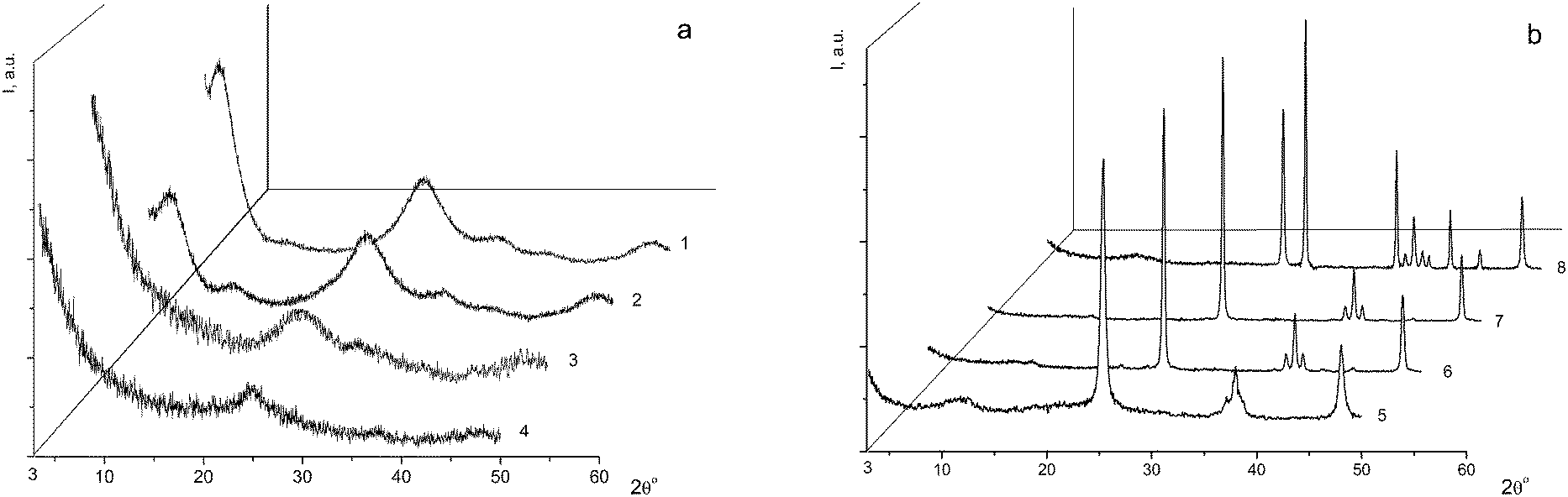 | ||
| Fig. 2 X-ray powder diffraction patterns: (a) of the initial sample I (1), the theoretical powder diffraction pattern of a mixture of anatase (20%) and η-TiO2 (80%) (2), and of sample I annealed for 1 h at 150 °C (3) and 300 °C (4); (b) of sample I annealed at 650 °C (5), 700 °C (6), 800 °C (7) and 950 °C (8). | ||
According to the X-ray diffraction data for sample I (Fig. 2a) and based on a comparison of the diffraction pattern of this sample with the theoretical diffraction pattern (Fig. 2a, 2), this sample is composed of a mixture of anatase and the η-phase in amounts of 20% and 80%, respectively. The additional diffraction peak at 2θ ∼ 12° can be assigned either to an amorphous phase of the composition H2TixO2x+1 with x = 6 or 7, or to amorphous hydrated titanium dioxide Ti4+O2−y/2(OH)1−y·nH2O (n ∼ 1).9 The average sizes of the coherent scattering regions (D = 48(2) Å) are indicative of a large fraction of the amorphous component, which is characteristic of all samples with the η-phase.12,20–23
Sample I was annealed in air for 1 h at different temperatures: 150 °C (sample I-150, Fig. 2a, 3), 300 °C (sample I-300, Fig. 2a, 4), 650 °C (sample I-650, Fig. 2b, 5), 700 °C (sample I-700, Fig. 2b, 6), 800 °C (sample I-800, Fig. 2b, 7) and 950 °C (sample I-950, Fig. 2b, 8). As can be seen in Fig. 2, the fraction of the amorphous component in samples I-150 and I-300 is larger, being most pronounced in I-150. It should be noted that the peak at 2θ ∼ 5° (d ∼ 18 Å) is absent in the X-ray diffraction pattern of sample I-150, although the diffuse peak at 2θ ∼ 33° (d ∼ 2.7 Å) characteristic of the η-phase is still observed. The latter peak is absent in the diffraction pattern of sample I-300 (Fig. 2a, 4). Samples I-650, I-700, and I-800 contain nano-anatase with crystallite sizes D = 195(7) Å, D = 270(10) Å and D = 330(15) Å. Sample I-950 consists of two phases and contains 63% rutile (D = 460(20) Å) and 37% anatase (D = 445(20) Å). Therefore, the η-phase can be assigned to the low-temperature metastable form of titanium(IV) oxide.
It is known that the formation of single-phase anhydrous TiO2 with an anatase structure in sulfuric acid solutions of titanyl sulfate occurs only in the case of the complete hydrolysis. If the hydrolysis is not complete (due to a limited duration of hydrolysis), solid products of varying and unpredictable compositions are obtained depending on the degree of hydrolysis and polycondensation of the chemical forms of titanium existing in solution.17,24–26 We chose the composition of the ultimate polymer {(TiO)10(HSO4)(OH)19}x, which was deduced taking into account coordinative and chemical saturation, and which was further confirmed by direct chemical analysis17 as the model of the expected partial hydrolysis product obtained under the conditions of the synthesis of sample I. This formula bears anatase structural information at the molecular level, where the (TiO2+)n fragment reflects the planar structure of the –O–Ti–O–Ti– chains in anatase and the HSO4 and OH groups coordinated to titanium.17
The overall composition of sample I was determined by ICP AES after the dissolution of five samples of different weight in hot 4 N HF and the adjustment of the detector to enable the determination of Ti and S. The element content expressed in the content of the Ti → TiO and S → HSO4 fragments were summarized and subtracted from the weight of the sample. The difference was related to the hydroxyl group content. According to the results of the calculations, the overall composition of the sample determined by the DD method is (Ti4+O)(HSO4)0.38(OH)1.03, and the chemical unsaturation attests to the heterogeneity of sample I.
Then we employed the DD method, which makes it possible, by selectively dissolving phases, to determine the spatial variation in the composition as a function of the volume during the dissolution of each phase comprising sample I.
Fig. 3a shows kinetic curves for the elements Ti and S that are transferred to a solution at every point in time during the complete dissolution of 0.80 mg of the starting sample I. The kinetic curves were constructed from the results of the analysis of 650 aliquots of the solution.
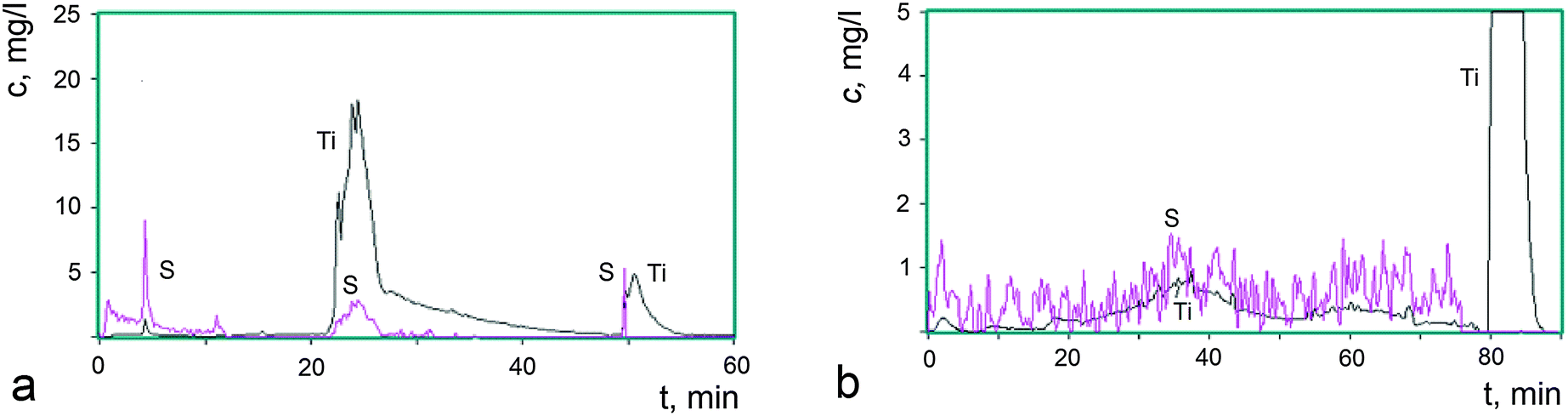 | ||
| Fig. 3 Kinetic curves for the elements Ti and S during the dissolution of sample I (a) and sample I-150 (b): (a) sample I. The solvent: H2O up to 10 min; HNO3, pH = 2, at 75 °C up to 25 min; 3 N HNO3 at 80 °C up to 48 min; 3.8 N HF at 80 °C up to 60 min. (b) Sample I-150. The solvent: H2O up to 5 min; HNO3, pH = 2, at 75 °C up to 20 min; 3 N HNO3 at 80 °C up to 80 min; 3.8 N HF at 80 °C up to 90 min. | ||
As can be seen in Fig. 3a, the phases are dissolved successively without overlapping, the dissolution of the next phase each time starting after the dissolution of the previous one. This is evidence that sample I is composed of three phases, which differ from each other in their chemical potentials of dissolution. The first phase (A) is dissolved in aqueous and weak acid media, during which titanium and sulfur are transferred to the solution. The dissolution of the second phase (B) is a more complicated process: one part (B1) is dissolved in the medium-strength acid HNO3, during which titanium and sulfur ions are transferred to the solution, whereas another part (B2) is dissolved in 3 N HNO3 at 80 °C and provides only titanium ions to the solution. This characteristic of the dissolution of phases is typical of core–shell nanosized assemblies.27 In the case under consideration, the phase B2, being a core, has a pronounced oxide nature, whereas the shell B1 is composed of titanium(IV) oxide modified by sulfate and hydroxyl groups. The latter are not directly determined by the DD method, but the presence of fluctuations in the dissolution curves is indicative of the partial occupancy of the surface by hydroxyl groups as well. The third phase (C) is dissolved in HF, resulting in the transfer of titanium and sulfur ions to the solution, although in limited amounts. The largest amount of sulfate groups was found in phase A. The amount of these groups is substantially smaller in phase B1 and particularly smaller in phase C. All phases are characterized by an essential difference between the bulk composition of dissolved particles and the composition of the surface modified by sulfate and hydroxyl groups.
The compositions of the phases were determined from the ratio of the averaged molar TiO/HSO4 fractions, and the content of the phases was expressed as the total weight of titanium and sulfur ions that were transferred to a solution during the single-phase dissolution. These quantitative data are summarized in Table 1 as a balance matrix, which reflects the distribution of the total number of moles of each fragment in the formula (TiO)(HSO4)0.38(OH)1.03 between the three phases.
In order to obtain additional data on the nature of the phases found in sample I, the thermal stability of the sample was studied after storage under different isothermal conditions. The loss of volatile anionic groups was detected and the solid residues were analyzed to determine their overall composition by ICP AES and their phase composition by the DD method. The kinetic dissolution curves for sample I-150 are shown in Fig. 3b, where 9% of the weight loss leads to a significant change in the phase state of the sample compared to the starting sample I (Fig. 3a). The phase A was depleted in sulfur, the phase B2 disappeared, the composition of the phase B1 changed with a simultaneous loss of HSO4 and OH groups, and the reactivity of the latter was reduced, which indicates that the composition acquired an oxide nature. This phase is slowly dissolved even in hot 3 N HNO3. After storage at 300 °C (sample I-300, Fig. 2a, 4) accompanied by 18% weight loss, the phase A disappeared, the phase B1 also almost completely disappeared, the fraction of the phase C substantially increased and the amorphization of the product was enhanced. The phase changes in these samples were studied by X-ray powder diffraction and the patterns are displayed in Fig. 2. The Raman spectra of the samples presented below confirm this fact.
The nature of the phase B is of the most interest. This phase displays a co-dissolution of phases of different composition which form the basis of sample I. The content of this phase was estimated by the DD method to be about 86(2)% (80(2)% according to the X-ray powder diffraction data). The quantitative changes in the phase composition of the starting sample versus the temperature of the synthesis are shown in Fig. 4.
 | ||
| Fig. 4 Changes in the phase composition of sample I after a one-hour annealing: 1 – phase A; 2 – phase B1; 3 – phase B2; 4 – phase C. | ||
Therefore, the phase characterization of sample I by the DD method based on the chemical potentials of dissolution of the phases as signs of their individuality involves the detection of three phases: phase A containing a small amount of titanium(IV) oxide (1–2%) and a large amount of sulfate and hydroxyl groups, phase B (86%), which is the η-phase composed of a core (B2) containing titanium(IV) oxide and a shell (B1) in the form of an intermediate phase of variable structure (TiO)(HSO4)x(OH)y (Fig. 5a) and phase C with an anatase structure (12%). At a temperature below 600 °C, the latter phase is also most likely composed of a core containing titanium(IV) oxide and a sulfate shell; the size of the latter is much smaller than that of the η-phase (Fig. 5b). We attribute this fact to the different dimensionality of the particles of both phases, which is confirmed by the average sizes of D: these sizes for the samples with nano-anatase prepared by the sulfate method are substantially larger (D ∼ 80–90 Å)21 compared to those of the samples containing the η-phase (D ∼ 30–50 Å). It is not improbable that the η-phase is partially transformed into nano-anatase accompanied by the formation of a defect structure (Fig. 5c) with D ≠ L, which is confirmed by the results of earlier studies.20,21
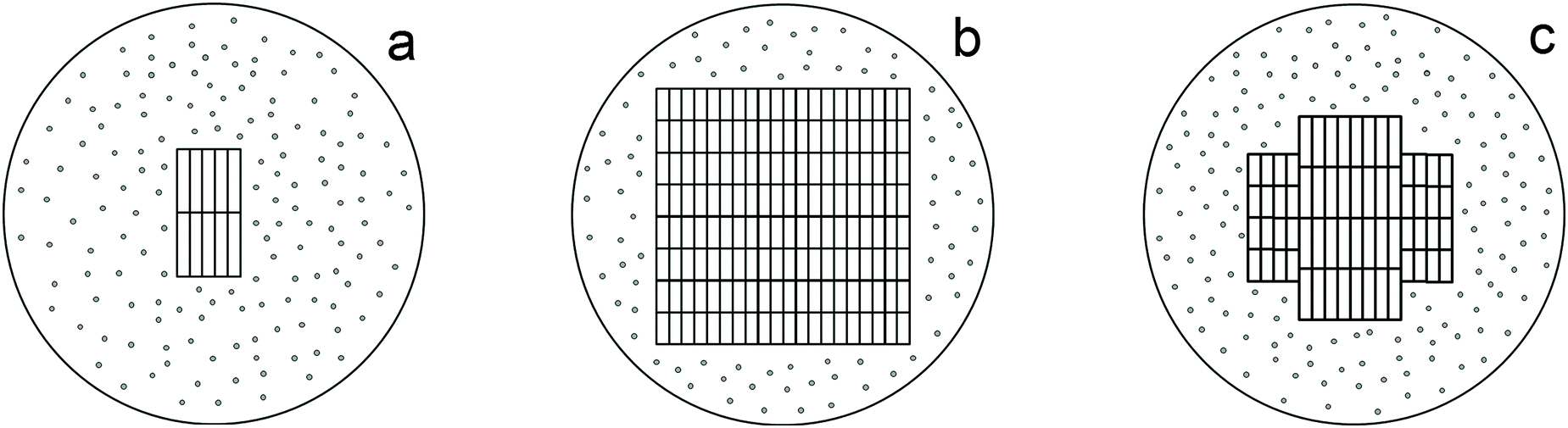 | ||
| Fig. 5 Models of the structures of nanoparticles of the η-phase (a), anatase (b), and the defect structure of the η-phase (c). | ||
Therefore, based on the X-ray powder diffraction data and the results of the DD method, we propose a model of the three-dimensional structures of the main phases comprising sample I (Fig. 5a).
The limited possibilities of X-ray powder diffraction and the DD method for the determination of the nature and amount of water in sample I, which is also of importance for an understanding of the nature of the phases, were supplemented by the results of conventional thermal analysis using polythermal heating (Fig. 6) and storage under isothermal conditions for one hour in air at different temperatures.
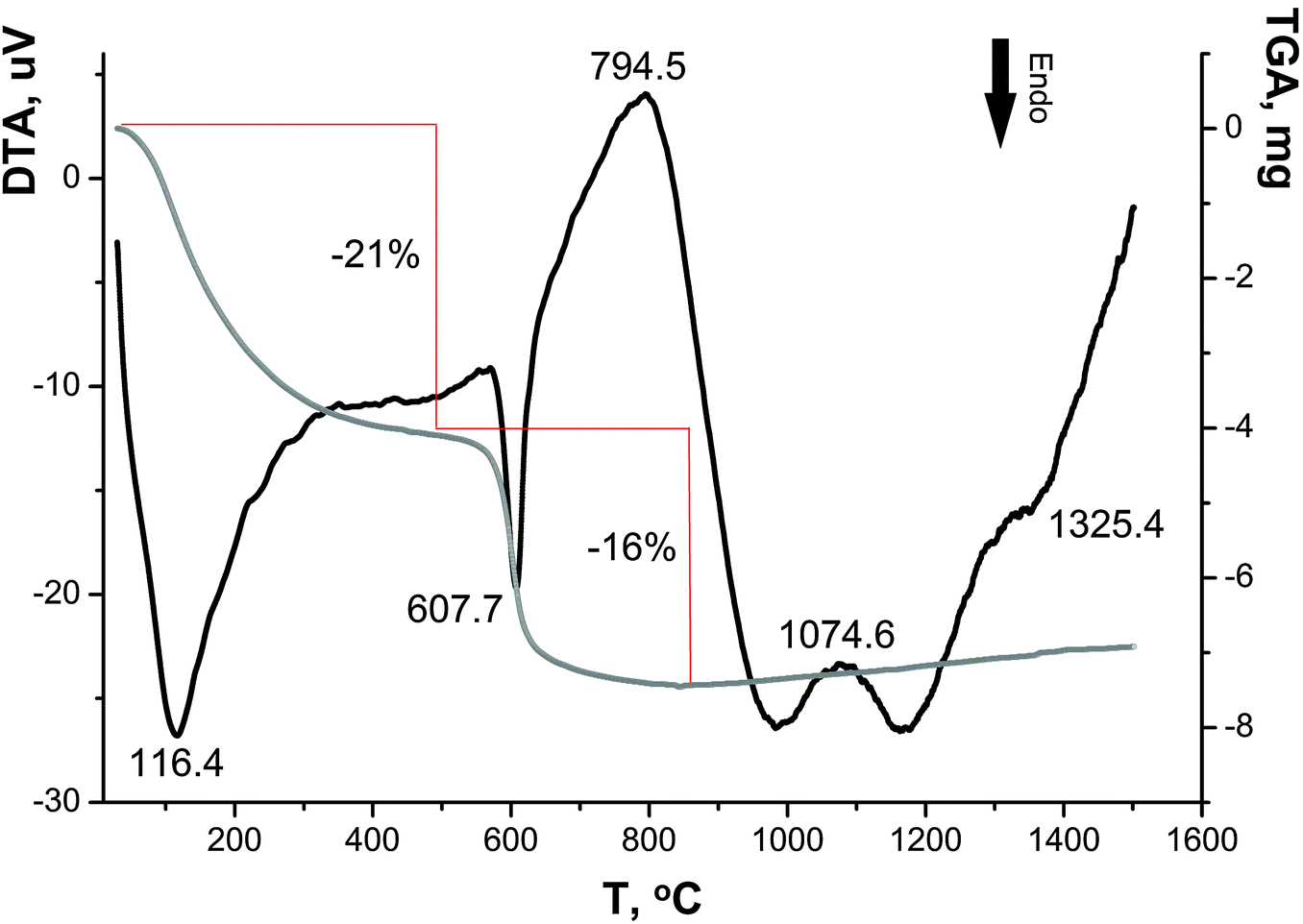 | ||
| Fig. 6 DTA–TGA curves for sample I in air. | ||
As can be seen in the DTA–TGA curves (Fig. 6), the transformations of sample I are associated with continuous changes in its weight over two temperature ranges (50–350 °C and 500–650 °C), and the decomposition processes appear as endothermic peaks; the maximum rates of these processes are characterized by the peaks at ∼90 °C and 615 °C.
Based on the literature data,28–31 the weight loss in the range of 50–350 °C is associated with the dehydration of the sample: adsorbed water molecules are removed at ∼120 °C32 and ∼200 °C,33,34 and water molecules of crystallization and constitutional water are eliminated at ∼250 °C and ∼330 °C, respectively.32 In the range of 500–650 °C, the chemisorbed OH groups and the sulfate component are lost, which is accompanied by a weight loss of ∼21 and ∼16 wt%, respectively (in ref. 35 the temperatures ∼600–700 °C were reported). Similar data on the total weight loss (∼40 wt%) and the presence of sulfate ions in samples at <500 °C were reported in ref. 34. Three small exothermic peaks at 794 °C, 1070 °C and 1330 °C, which are almost not accompanied by weight loss, are apparently attributed to the transition of nanocrystalline anatase to polycrystalline anatase and then to rutile.36,37
Therefore, the DTA–TGA curves confirm the presence of sulfate ions and OH groups in sample I determined by the DD method and are also indicative of the presence of water in this sample.
The absorption bands at 3320–3400 cm−1 are assigned to OH stretching vibrations of water molecules hydrogen-bonded to TiO2, and the bands at 3200–3300 cm−1 belong to the stretching vibrations of adsorbed water molecules (Table 2 and Fig. 7).
| Assignment | Sample/wavenumber (cm−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I | I-150 | I-200 | I-300 | I-600 | I-650 | |||||
| KBr | KBr | Nm | fo | Nm | fo | Nm | fo | KBr | Nm | |
| Abbreviations: br, broad; ben, bending; w, weak; sh, shoulder; s, strong; m, medium; Nm, Nujol mulls; fo, fluorinated oil. | ||||||||||
| ν(OH), (H2O⋯Ti) | 3348 | 3338 | 3326 | 3379 | 3433 | 3400–3200 | 3430 | |||
| ν(OH) (adsorbed H2O) | 3247 | 3271 | 3187 | 3296 | ||||||
| δ(HOH) | 1627 | 1625 | 1625 | 1623 | 1624 | 1619 | 1627 | 1622 | 1600 | 1630 |
| ν(S–O) | 1200 | 1193 | 1188 | 1197 | ||||||
| δ(SO42−), δ(TiOH) | 1144ben | |||||||||
| 1131 | 1128 | 1123 | 1121sh | 1123s | 1127w | |||||
| 1075 | 1075 | 1089sh | ||||||||
| 1059 | ||||||||||
| δ(TiOH) | 758 | 758 | 759 | |||||||
| 745 | 745 | |||||||||
| δ(TiOH), ν(TiO) | 660 | |||||||||
| 602 | 597m | 600w | ||||||||
| 512br | 498 | 500br | ||||||||
| 485br | 484 | 480br | ||||||||
| 461 | ||||||||||
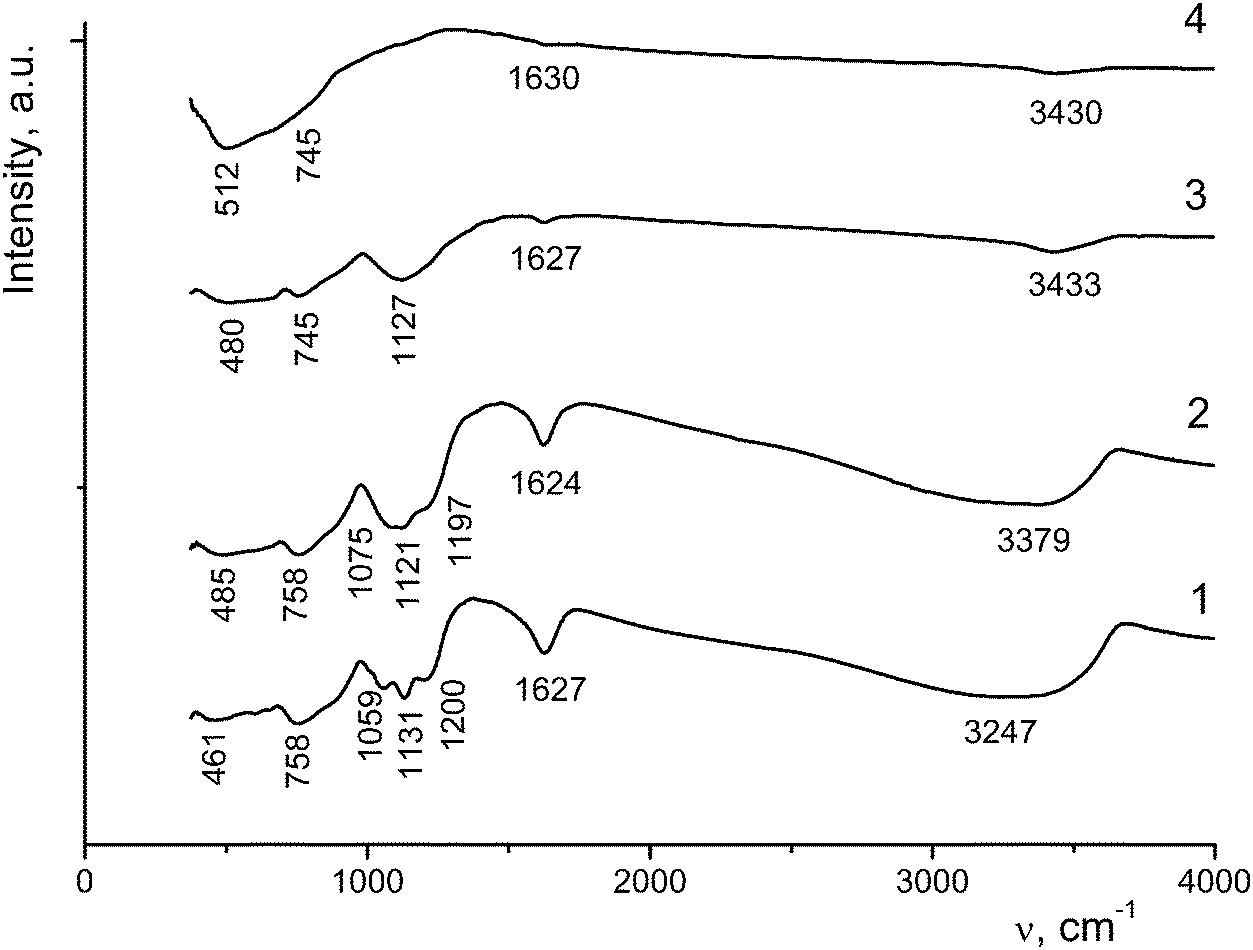 | ||
| Fig. 7 IR spectra of the initial sample I (1), I-200 (2), I-300 (3) and I-650 (4). | ||
As the annealing temperature of sample I is increased, the amount of water molecules decreases, resulting in a decrease in the intensity and/or the disappearance of the corresponding vibrations in the IR spectra. Bending vibrations of water molecules appear as bands at 1619–1630 cm−1 (Fig. 7). In the region of 1200–1055 cm−1 there are stretching and bending vibrations involving sulfate ions chemically bonded to titanium ions. An increase in the annealing temperature to 600–650 °C leads to a decrease in the intensity of these bands down to their complete disappearance, which is indicative of the removal of sulfate ions from the sample. In the region of 460–760 cm−1 bending vibrations of Ti⋯OH groups and stretching vibrations of TiO6 octahedra (at frequencies lower than 600 cm−1) are observed.
In order to get a better understanding of the structures of the phases, we used Raman spectroscopy, which identifies these phases based on the local symmetry of chemical bonds. The Raman spectrum of sample I shows peaks at ∼156 cm−1 (Eg), 415 cm−1 (B1g), ∼506 cm−1 (A1g B1g), and 625 cm−1 (Eg) providing evidence that the local bond order is similar to that in the anatase structure (Fig. 8).
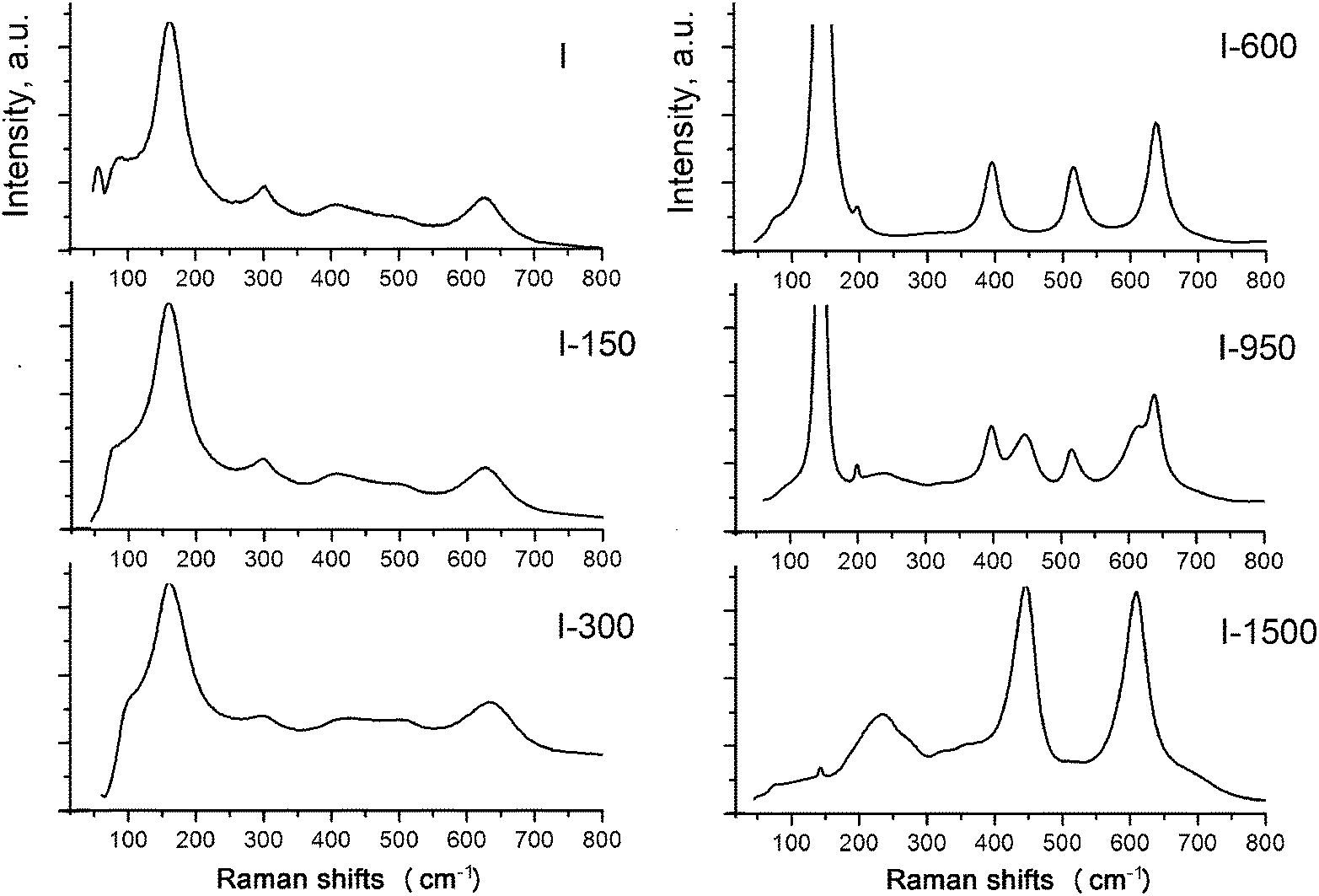 | ||
| Fig. 8 Raman spectra of the starting sample I and the samples annealed at different temperatures. | ||
However, judging from the diffuse character and low intensities of the bands (particularly, the peak at 506 cm−1), particles of the phases are amorphous and nanocrystalline. The broadened and asymmetric shape of the peak at 156 cm−1 results from the amorphous state of the phase B1, which is usually attributed to disorder in Ti–O distances of TiO6 octahedra due to low dimensionality of the particles and the reconstruction of their surfaces via sulfurization and hydration.30,34,41,42 The diffuse character of the lines in the regions of 400–425 and 624–640 cm−1 is attributed to the presence of nanocrystallites of B2. In the absence of the peak at 226 cm−1 characteristic of the hydrated form of titanium dioxide, the B2 phase can be assigned with confidence to a dioxide nature, in particular, to the η-phase.22,23 In ref. 30 the bands at 430 and 605 cm−1 are considered as a sign of the amorphous state of the product.
The character of the Raman spectrum of sample I-150 is almost identical to that of the initial sample. A weak tendency of the band at 156 cm−1 to be shifted to lower frequencies, and the shift of the bands at 430 and 605 cm−1 to higher frequencies, are due to a decrease in disorder in the Ti–O distances. The band at 300 cm−1 that disappears after the annealing is assigned to Ti–OH vibrations.
The set of peaks in the Raman spectrum of sample I-600 corresponds to anatase. A further increase in the annealing temperature leads to the polymorphic transformation of anatase to rutile accompanied by the appearance of vibrational modes at 440 and 610 cm−1 characteristic of rutile. The bands at 85 cm−1 and at lower frequencies apparently correspond to boson vibrations associated with elastic vibrations in inhomogeneities of the structure, or with certain weak, probably interionic, interactions between oxygen-containing groups in the titanium oxygen network.
The results of the investigation of sample I by a number of methods prove that this sample is not single-phase, and the surfaces of low-dimensional amorphous nanoparticles of two of the three identified phases were non-uniformly coated with hydroxyl and sulfate groups. The fact that the sample is not single-phase is attributed to the incomplete hydrolysis and fast coagulation, resulting in the precipitation of all chemical forms of titanium of different degrees of polymerization that are present at the moment of precipitation.
The overall composition of the η-phase based on the results of investigations by all methods can be written as [TiO2−x·mH2O](OH)y(HSO4)z·nH2O. In ref. 9 it was noted that the η-phase contains sulfur, which exists apparently in the form of SO4. After washing with water followed by neutralization, sulfate groups are replaced by hydroxyl groups without the degradation of the phase [TiO2−x·mH2O](OH)y·nH2O, i.e., the η-phase has a variable composition. In fact, a similar composition is assigned to hydrated titanium(IV) oxide Ti4+O2−y/2(OH)y·nH2O,9 as well as to (TiO1.9)(OH)0.2·nH2O with a distorted anatase structure,43 in which OH groups can be partially replaced by sulfate groups (phase A)9 when employing the sulfate method for the preparation of samples (Table 1 and Fig. 4).
A sample with the η-phase can be prepared not only by the hydrolysis of titanyl sulfate TiOSO4·xH2SO4·yH2O but also by the hydrolysis of an aqueous solution of TiOSO4·2H2O in the presence of strong acids such as HClO4, HNO3,44 and H2SO4. It should be noted that in the absence of acid or in the presence of the weak acid CH3COOH, the reaction affords anatase, whereas in the presence of the weak acid H3PO4 titanium dioxide is not formed.44 These experimental data indicate that the regions of the existence of the η-phase and nano-anatase are controlled by the pH value of the reaction system.
The η-phase–nano-anatase phase transitions can be visualized based on the disappearance of the diffraction reflection at 2θ = ∼4.3–5.3° and the splitting of the diffraction reflection at 2θ ∼ 38°: amorphous TiO2 (D < ∼30 Å), nano-η-TiO2 (D ∼ 30–50 Å; 2θ ∼ 5°, ∼25°, ∼33°, ∼38°), nano-anatase (I) (D ∼ 50–100 Å; 2θ ∼ 38°) and nano-anatase (II) (D ∼ 100–500 Å; 2θ ∼ 37°, ∼38°, ∼39°) (Fig. 9).
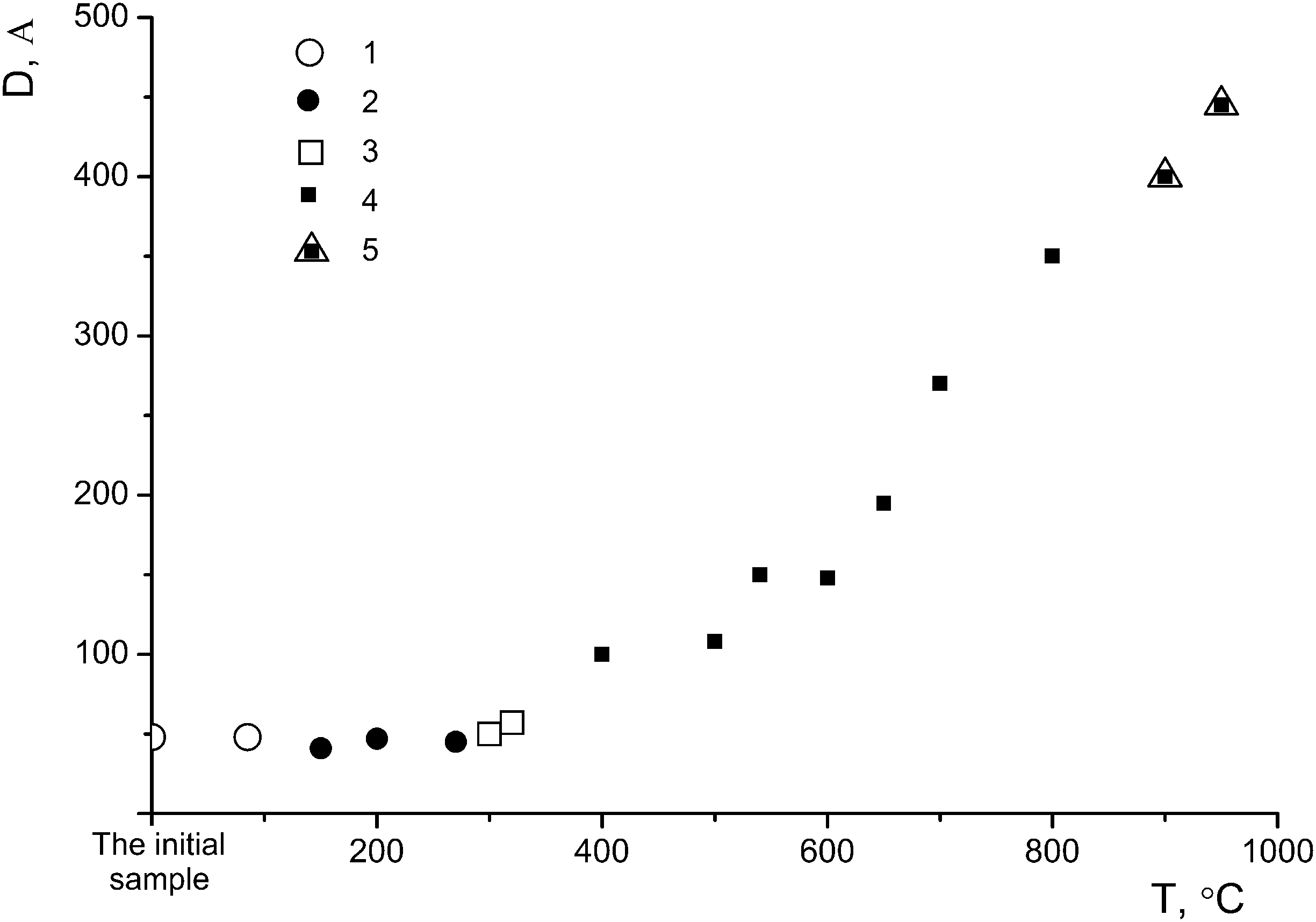 | ||
| Fig. 9 Dependence of the average sizes of coherent scattering regions (D, A) on the annealing temperature of the sample with the η-phase: 1 – η-phase; 2 – η-phase + nano-anatase (I); 3 – nano-anatase (I); 4 – nano-anatase (II); 5 – nano-anatase (II) + nano-rutile. | ||
Therefore, as can be seen in Fig. 2a (3) and 5, and according to the results of a previous study,32 adsorbed water molecules are responsible for a small-angle peak characteristic of the η-phase, which is absent in the diffraction pattern of sample I-150. It is the reflection with d ∼ 17–21 Å in the diffraction patterns of the samples with the η-phase that changes in the angle range 2θ= ∼4.3–5.3° with a change in the intensity,9,20 whereas the other diffraction peaks characteristic of the η-phase are stable to a certain extent. Hence, it can be concluded that the structure of the η-phase is stabilized by water molecules located between the layers, thus facilitating the formation of a quasi-layered structure with the large unit-cell parameter c, the crystallites being platelet-like20 (Fig. 5a). In other words, the structure of the η-phase can be considered as an intercalate. Thus, the structure consists of layers of TiO6 octahedra (probably TiO5 as well) with an anatase-like structure, and water molecules, which are located between the layers and are hydrogen-bonded to oxygen atoms. These data suggest that the η-phase is a pseudopolymorph, viz., titanium dioxide solvate (hydrate), rather than a polymorph of TiO2.
Nevertheless, the formation of low-dimensional titanium(IV) oxides, the overall compositions of which can be described as [TiO2−x(OH)x](OH)y(HSO4)z·nH2O and [TiO2−x·mH2O](OH)y(HSO4)z·nH2O with z ≥ 0, cannot be ruled out as well. The structures of these phases are determined by the regular structure of [TiO2−x(OH)x] (nano-anatase with x = 0 and distorted nano-anatase, apparently, with x > 0) and [TiO2−x·mH2O] (η-modification) containing different amounts of adsorbed interlayer water (characterized by the peak at d ∼ 17–21 Å and a variation in the unit cell parameter c), as well as by the specificity of the structure that differs from anatase (which is probably characterized by the reflection at 2θ ∼ 33° with d ∼ 2.7 Å). These phases are composed of a disordered amorphous layer with titanium(IV) oxide modified by sulfate and/or hydroxyl groups and adsorbed water. The ratio of these constituents (the core and the shell, respectively) determines primarily the average size of coherent scattering regions (crystallites) (Fig. 5), whereas the composition of the shell is responsible mainly for the specific surface area and consequently, photocatalytic, sorption, bactericidal, etc. properties.
Conclusion
Samples with the η-phase were prepared by the hydrolysis of titanyl sulfate TiOSO4·xH2SO4·yH2O. The stability region of this phase in the hydrolysis temperature–hydrolysis duration coordinates was determined. It is shown that nanoparticles of the η-phase consist of a core and a shell. The core has a [TiO2−x·mH2O] composition, the structure of which can be described as a superstructure in relation to the anatase structure. The structure of the η-phase can be considered as an intercalate with water molecules located between the layers of TiO6 octahedra. The cell parameter c depends on the content of water: it increases (decreases) with an increase (reduction) of water molecules in the interlayer space. The amorphous shell of the η-phase contains TiO2−x (trace amount), OH, HSO4 and water. According to this composition, this phase is not a polymorph of TiO2 and is instead a pseudo-polymorph of titanium dioxide hydrate. The average crystallite size depends on the ratio of the core thickness to the shell thickness of the nanoparticles. As temperature increases (to ∼250 °C), the η-phase passes into a phase with an anatase structure accompanied by water loss. It is followed by an increase in the sizes of the crystallites.Acknowledgements
The present study was financially supported by RFBR (research project no. 13-03-00367).Notes and references
- R. A. Andrievskiy and A. M. Glezer, Phys. Met. Metallogr., 1999, 88(1), 50–73 Search PubMed.
- H. Gnaser, B. Huber and C. Ziegler, in Encyclopedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific Publishers, Stevenson Ranch, 2004, vol. 6, pp. 505–535 Search PubMed.
- H. J. Chen, P. C. Jian, J. H. Chen, W. Leeyih and W. Y. Chiu, Ceram. Int., 2007, 33, 643 CrossRef CAS.
- V. Kokila, K. Senthilkumar and P. Nazeer, Arch. Phys. Res., 2011, 2(1), 246–253 Search PubMed.
- B. Ohtani, Catalysts, 2013, 3, 942–953 CrossRef CAS.
- A. Fujishima, K. Hashimoto and T. Watanabe, TiO2 Photocatalysis: fundamentals and applications, BKC, Tokio, 1999, p. 242 Search PubMed.
- P. A. K. Reddy, P. V. L. Reddy, V. M. Sharma, B. Srinivas, V. D. Kumari and M. Subrahmanyam, J. Water Resour. Prot., 2010, 2, 235–244 CrossRef CAS.
- S. Buzby, M. A. Barakat, H. Lin, C. Ni, S. A. Rykov, J. G. Chen and S. Ismat Shah, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2006, 24(3), 1210–1214 CrossRef CAS.
- M. Dadachov, US Pat., 0171877, 2006 Search PubMed.
- M. Dadachov, US Pat., 0144793, 2006 Search PubMed.
- E. V. Savinkina, L. N. Obolenskaya, G. M. Kuz'micheva, A. V. Dorokhov and A. U. Tsivadze, Dokl. Phys. Chem., 2011, 441(1), 224–226, DOI:10.1134/S0012501611110042 , http://link.springer.com/article/10.1134%2FS0012501611110042.
- G. M. Kuz'micheva, E. V. Savinkina, D. N. Titov, P. A. Demina, L. N. Obolenskaya, L. G. Bruk, A. G. Yakovenko and M. G. Chernobrovkin, Inorg. Mater., 2011, 47(7), 753–758, DOI:10.1134/S002016851106015X , http://link.springer.com/article/10.1134%2FS002016851106015X.
- E. V. Savinkina, G. M. Kuz'micheva, L. N. Obolenskaya and E. N. Domoroshchina, Rus. Pat., 2469954, 2012 Search PubMed.
- B. E. Warren, X-Ray Diffraction, Dower Publications, New York, 1990, p. 381 Search PubMed.
- G. M. Kuz'micheva, A. A. Gainanova and V. V. Podbel'skiy, Rus. Pat., 2014660201, 2014 Search PubMed.
- V. V. Malakhov and I. G. Vasil'eva, Russ. Chem. Rev., 2008, 77(4), 351–372, DOI:10.1070/RC2008v077n04ABEH003737.
- Z. Jerman, Collect. Czech. Chem. Commun., 1966, 31, 3280–3286 CrossRef.
- L. I. Bekkerman, I. P. Dobrovol'skiy and A. A. Ivkin, Russ. J. Inorg. Chem., 1976, 31, 418 Search PubMed.
- V. U. Pervushin, A. V. Tolchev, T. A. Denisova and V. A. German, Russ. J. Inorg. Chem., 1989, 34, 1096 CAS.
- G. M. Kuz'micheva, A. A. Gainanova, A. S. Orekhov, V. V. Klechkovskaya, N. V. Sadovskaya and V. V. Chernyshov, Crystallogr. Rep., 2014, 59(6), 1008–1014, DOI:10.1134/S1063774514050101 , http://link.springer.com/article/10.1134/S1063774514050101.
- L. N. Obolenskaya, M. A. Zaporozhets, G. M. Kuz'micheva and E. V. Savinkina, Crystallogr. Rep., 2015, 60(3), 455 CrossRef.
- G. M. Kuz'micheva, E. V. Savinkina and L. N. Obolenskaya, Crystallogr. Rep., 2010, 55(5), 866–871, DOI:10.1134/S1063774510050287 , http://link.springer.com/article/10.1134%2FS1063774510050287.
- G. M. Kuz'micheva, E. V. Savinkina, L. I. Belogorokhova, B. N. Mavrin and V. R. Flid, Russ. J. Phys. Chem. A, 2011, 85(6), 1037–1040, DOI:10.1134/S0036024411060203 , http://link.springer.com/article/10.1134%2FS0036024411060203.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1988, 18, 1259–1260 RSC.
- G. Lungren, Sven. Kem. Tidskr., 1959, 71, 200–220 Search PubMed.
- L. G. Ganchenko, V. F. Kiselev and V. V. Murina, Russian Journal of Kinetics and Catalysis, 1961, 11, 877 Search PubMed.
- H. Cheng, J. Ma, Z. Zhao and L. Qi, Chem. Mater., 1995, 7, 663–671 CrossRef CAS.
- G. Li, L. Li, J. Boerio-Goates and B. Woodfield, J. Am. Chem. Soc., 2005, 127, 8659 CrossRef CAS PubMed.
- Q. Yang, C. Xie, Z. Xu, Z. Gao and Y. Du, J. Phys. Chem. B, 2005, 109, 5554–5560 CrossRef CAS PubMed.
- J. Zhang, M. Li, Z. Feng, J. Chen and C. Li, J. Phys. Chem. B, 2006, 110, 927–935 CrossRef CAS PubMed.
- S. N. Ivicheva, U. F. Kargin, S. V. Kutsev, L. I. Shvornena and G. U. Urkov, Phys. Solid States, 2013, 55(5), 1111–1119, DOI:10.1134/S1063783413050132 , http://link.springer.com/article/10.1134%2FS1063783413050132.
- A. V. Kostrikin, R. V. Kuznetsova, O. V. Kosenkova, A. N. Merkulova and I. V. Lin'ko, Problems of Contemporary Science and Practice (Russian journal), 2007, 2(8), 181–186 Search PubMed.
- M. Kanna, S. Wongnawa, P. Sherdshoopongse and P. Boonsin, J. Sci. Technol., 2005, 27(5), 1017–1026 Search PubMed.
- H. E. Benito, T. D. A. Sanchez, R. G. Alamilla, J. M. H. Enriquez, G. S. Robles and F. P. Delgado, Braz. J. Chem. Eng., 2014, 31(3), 737–745 CrossRef.
- A. S. Serikov, V. E. Gladkov, D. A. Zherebtsov, A. M. Kolmogortsev and V. V. Viktorov, Bulletin of the South Ural state University (Chemistry), 2010, 4(31), 97–101 Search PubMed.
- S. Riyas, G. A. Krishnan and P. N. Mohan Das, J. Ceram. Process. Res., 2006, 7(4), 301–306 Search PubMed.
- P. S. Ha, H.-J. Youn, H. S. Jung, K. S. Hong, Y. H. Park and K. H. Ko, J. Colloid Interface Sci., 2000, 223, 16–20 CrossRef CAS PubMed.
- L. Qian, Z. L. Du, S. Y. Yang and Z. S. Jin, J. Mol. Struct., 2005, 749, 103–107 CrossRef CAS.
- M. Yan, F. Chen, J. Zhang and M. Anpo, J. Phys. Chem. B, 2005, 119, 8673–8678 CrossRef PubMed.
- T. Bezrodna, T. Puchkovska, V. Shymanovska, J. Baran and H. Patajczak, J. Mol. Struct., 2004, 700, 175–181 CrossRef CAS.
- S. Kelly, F. H. Pollak and M. Tomkiewicz, J. Phys. Chem. B, 1997, 101, 2730 CrossRef CAS.
- N. M. Dimitrijevic, Z. V. Saponjic and B. M. Rabatic, J. Phys. Chem. Lett., 2007, 111, 7235–7241 CrossRef.
- T. A. Denisova, PhD thesis, Institute of Chemistry, Ural Branch of the Russian Academy of Sciences, Ekaterinburg, Russia, 1985.
- G. M. Kuz'micheva and A. A. Gainanova, Rus. Pat., 2014137971, 2015 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |