
An efficient ICT based fluorescent turn-on dyad for selective detection of fluoride and carbon dioxide†
Rashid
Ali
,
Syed S.
Razi
,
Ramesh C.
Gupta
,
Sushil K.
Dwivedi
and
Arvind
Misra
*
Department of Chemistry, Faculty of Science, Banaras Hindu University, Varanasi - 221 005, UP, India. E-mail: arvindmisra2003@yahoo.com; amisra@bhu.ac.in
First published on 19th October 2015
Abstract
A new intramolecular charge transfer (ICT) based fluorescent turn-on ratiometric probe 2 (D–π–A type) has been designed and synthesized by bridging imidazole (donor, D) and benzothiazole (acceptor, A) moieties through a phenyl ring. The photophysical behavior of probe 2 in solvents of different polarities and at different pH values has been investigated. Upon interaction with different types of anions probe 2 showed selective high affinity for fluoride anions (F−) in aqueous DMSO (20%) solution. The ratiometric fluorescence turn-on behavior displayed by 2 with F− is attributed to changes in the ICT process. Job's plot analysis revealed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry for 2 + F− interactions with a high binding constant and detection sensitivity (30 ppb). Moreover, a solution of 2 + F− enabled the detection of CO2 (∼100 ppb; 2.29 μM) through enhanced emission. The mode of interaction has been confirmed by 1H NMR titration studies which suggested the deprotonation of the –NH fragment of an imidazolyl unit in the presence of F−.
:1 binding stoichiometry for 2 + F− interactions with a high binding constant and detection sensitivity (30 ppb). Moreover, a solution of 2 + F− enabled the detection of CO2 (∼100 ppb; 2.29 μM) through enhanced emission. The mode of interaction has been confirmed by 1H NMR titration studies which suggested the deprotonation of the –NH fragment of an imidazolyl unit in the presence of F−.
Introduction
Construction of good organic molecular scaffolds for recognition and sensing of anions with high sensitivity and selectivity is an interesting research area in the field of supramolecular chemistry.1 Recently, great effort has been made to develop abiotic receptors for anionic species.2–5 Among the various types of anions the recognition of fluoride anions (F−) is of paramount importance due to their important roles in different biological, environmental and industrial processes.3,4 Fluoride (F−) is useful in dental care and in treatment of osteoporosis.5 However, a high intake of fluoride from drinking water is believed to cause fluorosis, nephrotoxic changes, urolithiasis in organism and even cancer.6 Moreover, fluoride ions are also associated with the refinement of uranium used in the manufacture of nuclear weapons.7 The Environmental Protection Agency (EPA, USA) has recommended the 2 ppm permissible level of fluoride in water.8 Thus the development of naked eye sensitive colorimetric and fluorogenic anion sensing systems has great implication in obtaining qualitative and quantitative information, and is challenging because of structural diversity and physical properties of different types of anions.4–9In the recent past considerable effort has been made to detect fluoride using different chromophoric and fluoroionophore units such as BODIPY, urea or thiourea, amide, phenol, cationic borane, and silicon.5 However, examples of a good sensing system which can detect F− ions via intramolecular charge transfer (ICT) and ratiometric fluorescence response are limited in number.10 Ratiometric fluorescent probes/sensors provide the practical advantage of built in corrections for environmental effects by allowing simultaneous detection at two signals.11 Fluorescent based chemosensors, due to their high sensitivity, simplicity and cost effectiveness are widely applicable in various areas such as medicine, industry and the environment.12
Furthermore, the imidazole derivatives due to their favorable properties such as stability, high tuneable photophysical properties, extinction coefficients, and ease of synthesis enable the application of these chromophores in different areas.13 Recently, a limited number of imidazole based systems have been exploited to develop OLED materials14 and chemosensors.15 A receptor system containing imidazole type ionophores is susceptible to detect anions either by H-bonding interaction or deprotonation of potential –NH fragments.3,16 Moreover, the typical structural change in donor and acceptor moieties and the change in photophysical behavior due to a push–pull mechanism17 may be easily characterized by UV-vis, fluorescence and NMR studies.18
Additionally, carbon dioxide (CO2) plays a vital role in human physiology and has important diagnostic application in medical sciences.19,20 On the other hand, CO2 is an important green house gas which is a major factor for global climate changes including critical global warming.21 Thus, it is highly desirous to monitor and detect CO2 sensitively in the environment. As well, quantification of CO2 before its release into the atmosphere, in high CO2 level containing anthropogenic gas streams (e.g., flue gas, syngas, biogas, etc.) is equally important.21 Among various analytical methods, optical chemosensors for CO2 have attracted considerable interest due to their simple, inexpensive and rapid sensing abilities.19–24 Gunnlaugsson et al. had earlier suggested that an anionic amine function generated in situ by fluoride anions is a suitable substrate to react with CO2.22 Recently, Yoon et al. utilized benzobisimidazolium and bispyrene derivatives as a new class of CO2 sensing probes, in which in situ generated carbene and anionic amide functions displayed a unique anion-activated CO2 sensing mechanism through a simple colorimetric and fluorescence changes.19,21
Keeping these facts in mind our ongoing research is currently paying attention toward the development of some efficient fluorescent organic scaffolds/motifs to detect anions sensitively in different media.25 Through this contribution we present the design and synthesis of a new type of intramolecular charge transfer (ICT) probe 2 and its potential application to recognize F−. The objective of designing a selective and sensitive F− responsive ratiometric probe has been achieved by developing a conjugated π-electron system containing specific ionophores, the –NH fragments, in which the electron rich imidazole (the donor) and electron deficient thiazole (as the acceptor) units are linked through a phenyl ring. As expected, probe 2 upon interaction with different anions has shown ratiometric fluorescence response toward F− selectively, in aqueous-dimethylsulfoxide (20% solution), while the anion activated probe, 2 + F−, showed unique chromo and fluorogenic response to detect CO2 with high sensitivity.
Results and discussion
Synthesis and photophysical behavior of probe 2
The multi-component reaction (MCR) involving three or four different substrates has emerged as a powerful one-pot transformation strategy for the synthesis of chemically and biologically important organic frameworks.26,27 The one-step synthesis of imidazole derivatives encouraged us to involve carbonyl compounds that may be further utilized to introduce other functionalities and extend the π-conjugation for better photophysical properties. Probe 2 was synthesized in two steps (Scheme 1). The synthesis of imidazole derivatives 1a and 1b was carried out by one pot multi-component reaction27 in which benzaldehyde and 4-formylbenzoic acid, and benzyl and NH4OAc were refluxed in glacial acetic acid. Subsequently, compound 1b was refluxed with 2-aminothiophenol in polyphosphoric acid (PPA) to obtain probe 2 in quantitative yield. The compounds were well characterized by 1H NMR, 13C NMR, IR and ESI-MS spectroscopy data (Fig. S5–S8, ESI†). | ||
| Scheme 1 (i) CH3COONH4/glacial acetic acid/110 °C. (ii) PPA/Δ. | ||
The photophysical behavior of compound 2 (10 μM) was investigated in solvents of different polarities and data are summarized in Table 1. Probe 2 possesses a typical D–π–A type structural motif and displayed different photophysical properties and good solvatochromism in solvents (such as hexane, 1,4-dioxane, chloroform tetrahydrofuran, dimethylformamide, acetonitrile and dimethylsulfoxide) of different polarities28 (Fig. S9, ESI† and Table 1). Moreover, in DMSO upon increasing the percentage of water (>30%) the intensity of the probe decreased gradually along with the change in color of solution (Fig. S10, ESI†). This is due to the dominance of H-bonding interaction between fluoroionophores and water molecules as a result of the decrease in the extent of intermolecular charge transfer between the antibonding orbital of the –NH fragment of probe 2 and the lone pair of electrons available in DMSO.29 Therefore, the selectivity of probe 2 toward different types of anions has been studied in aqueous-DMSO (20% solution). The pH of the medium was found to be ∼7.5.
| Entry | λ max (nm) | Molar absorptivity ε = M−1 cm−1 | λ em (nm) | Stoke's shift Δ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) (cm−1) (cm−1) |
Quantum yield Φ |
|---|---|---|---|---|---|
| Hexane | 368 | 19179 |
442 | 4550 | 0.39 |
| 298 | 14518 |
||||
| Dioxane | 367 | 18197 |
458 | 5414 | 0.49 |
| 294 | 13289 |
||||
| CHCl3 | 369 | 16851 |
461 | 5408 | 0.52 |
| 301 | 12509 |
||||
| THF | 371 | 18530 |
466 | 5495 | 0.49 |
| DMF | 370 | 20646 |
482 | 6280 | 0.44 |
| 284 | 17517 |
||||
| ACN | 357 | 19087 |
484 | 7350 | 0.48 |
| 296 | 13772 |
||||
| DMSO | 370 | 20082 |
488 | 6462 | 0.53 |
| 302 | 16086 |
||||
| MeOH | 356 | 20215 |
490 | 7681 | 0.45 |
| 296 | 15325 |
Anion selectivity of probe 2
The UV-vis spectrum of 2 (5 μM) showed an intramolecular charge transfer (ICT) band at 370 nm (ε = 1.90 × 104 M−1 cm−1) and a high energy band at 302 nm (ε = 1.55 × 104 M−1 cm−1) in H2O–DMSO (20%). Similarly, upon excitation at 370 nm probe 2 showed emission maxima at 488 nm (Φ2 = 0.53). The affinity of probe 2 toward different types of anions (140 equiv.) such as F−, Cl−, Br−, I−, NO3−, N3−, SO42−, H2PO4−, CO32−, AcO−, CN−, and SCN− has been examined through absorption and emission spectroscopy. Notably, upon interaction with different anions probe 2 displayed high selectivity for F− in which the ICT band disappeared completely and a new transition band appeared at 455 nm (ε = 1.82 × 104 M−1 cm−1) and naked-eye sensitive orange-red color appeared in the solution. Moreover, CN− and AcO− anions also showed affinity towards the probe, relatively less, in which molar absorptivity at the ICT band decreased around ∼30 to 40% (Fig. 1a). | ||
| Fig. 1 (a) Absorption (5 μM) and (b) emission spectra of 2 (1 μM) upon interaction with tested anions (140.0 equiv.) in H2O–DMSO (20%). Insets: Absorption and emission spectra of 2 + F− upon interference with competitive anions. | ||
Similarly, upon interaction with tested anions the fluorescence intensity of 2 (1 μM, λex = 370 nm) centered at 488 nm exhibited fluorescence quenching with AcO− (∼17%) and CN− (∼52%) anions. However, upon interaction with F− the emission centered at 488 nm diminished and a dual emission band appeared at 460 and 586 nm (Φ2+F− = 0.1) (Fig. 1b). The intense blue color of the solution, under UV light, switched-on immediately with F− to a naked eye sensitive light pink color (Fig. 2, images A). The interaction with other tested anions revealed an insignificant change in the photophysical behavior of 2. Further to ascertain high selectivity of 2 toward F− competitive anion interference studies have been performed by the addition of excess of tested anions (150 equiv.) to a probable solution of 2 + F− and reversibly, by the addition of F− to a solution of 2 containing another tested anions. Insignificant changes in the absorption and emission spectra of 2 + F− suggested the high selectivity of 2 for F− (see the inset of Fig. 1a, b and 2, images B).
 | ||
| Fig. 2 Chromogenic and fluorogenic response of 2 (10 μM) upon interaction (images A) and interference (images B) with different anions (140.0 equiv.) in H2O–DMSO (20%; pH ∼7.5). | ||
Furthermore, the binding affinity of 2 toward F− has been realized through the absorption and emission titration experiments (Fig. 3). Upon a gradual addition of F− (0–140 equiv.) to a solution of 2 the absorption spectra displayed ratiometric behavior in which the band centered at 370 nm decreased while the absorption of a new band at 455 nm enhanced concomitantly (Fig. 3a). Similarly, the emission titration studies displayed ratiometric behavior in which fluorescence intensity of 2 (λex 370 nm) centered at 488 nm diminished gradually and two new emission bands appeared at 455 nm and 586 nm (Fig. 3b). The appearance of isosbestic and isoemissive points at 405 and 584 nm, respectively, clearly supported the existence of more than one species in the medium. Moreover, probe 2 upon excitation at the isosbestic point, 405 nm, displayed emission at 488 nm while upon titration with F− (0–140 equiv.) the intensity of a new emission band enhanced ratiometrically at 578 nm (Fig. 3c). The observed significant red shift in the photophysical behavior of 2 upon interaction with F− is attributed to enhance the intramolecular charge transfer encounter due to deprotonation of –NH fragments.18 The reaction stoichiometry between 2 and F− has been realized by obtaining the absorption and emission spectra as a function of F− concentration. The maxima at 0.5 mole fractions suggested a 1:1 stoichiometry for a probe–fluoride interaction. For which, the binding constant has been estimated through the Benesi–Hildebrand (B–H) method30 and was found to be Kass(abs) = 1.01 × 104 M−1 and Kass(em) = 1.28 × 104 M−1, respectively (Fig. 3d and Fig. S11b, ESI†).
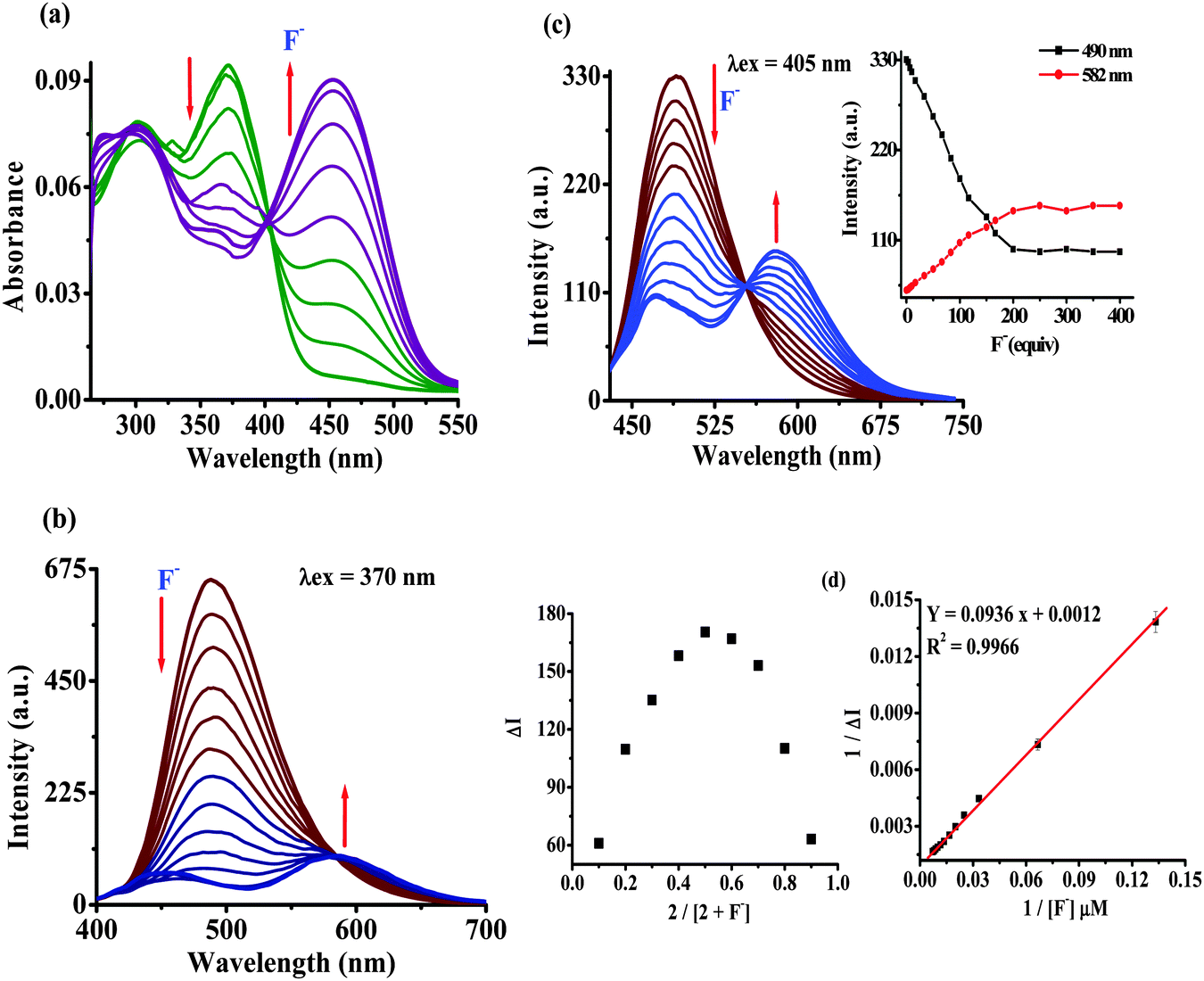 | ||
| Fig. 3 Absorption (5 μM) (a) and emission (b) at λex = 370 nm and (c) at λex = 405 nm; titration spectra of 2 (1 μM) upon sequential addition of F− (0–140.0 equiv.) in H2O–DMSO (20%). (d) Job's plot and the Benesi–Hildebrand plot based on emission spectra. | ||
The limit of detection (LOD) of probe 2 for F− has been estimated as reported previously.31 An approximately straight line calibration curve, obtained by taking the emission spectra of 2 at different concentrations (1.0 to 0.4 μM) suggested a linear correlation between intensity and concentrations of the probe with a standard deviation of 0.6987 (Fig. S12a, ESI†). Furthermore, from the slope of the fluorescence curve, obtained between ΔI (I − I0) (where I0 and I illustrate the emission intensities of 2 in the presence and the absence of F−) and concentration of the anions in the aforementioned range, the calibration sensitivity (m) was estimated and was found to be 1.3228 (Fig. S12b, ESI†). Here employing eqn (S3) (ESI†) the limit of detection (LOD) for F− was calculated and was found to be 1.58 μM (30 ppb) which is well below the recommended level and comparable to other reported methods.5
pH study of probe 2 in HEPES buffer
The photophysical behavior displayed by 2 in the presence of F− suggested the deprotonation of –NH fragments of the imidazolyl unit.18 Consequently, the observed ratiometric enhanced emission is attributed to polarization and/or charge propagation of an increase electron density on the imidazolyl unit. Moreover, the moderately weak acidic –NH function of the imidazole unit is susceptible to protonation and deprotonation at acidic and alkaline pH. Therefore, to rationalize the effect of F−, the pH dependent photophysical behavior of 2 was investigated in HEPES buffer (10 mM, pH 7.04; 20% aqueous DMSO).The absorption spectrum of 2 (5 μM) showed electronic transition bands at 368 nm (ε = 1.82 × 104 M−1 cm−1) with a shoulder at 305 nm (1.41 × 10−4 M−1 cm−1) (Fig. S13a, ESI†). Similarly, upon excitation at 368 nm probe 2 showed an intramolecular charge transfer emission band at 496 nm (Fig. S13b, ESI†). Notably, under acidic (pH 6 to 1) and alkaline (pH 10 to 14) conditions the molar absorptivity of 2 decreased wherein, the band centered at 368 nm disappeared and a new transition band appeared at 354 nm (ε = 1.23 × 104 M−1 cm−1; blue-shift, ∼14 nm) and 430 nm (ε = 1.15 × 104 M−1 cm−1 red-shift, ∼62 nm), respectively (Fig. 4a and Fig. S13a, ESI†). The formation of isosbestic points at 360 and 396 nm in acidic and alkaline medium, respectively, suggested the existence of more than one species in the medium. Similarly, at acidic pH (<6) the emission intensity of 2 centered at 496 nm (λex = 368 nm) decreased with a blue shift of ∼10 nm while under alkaline (>10) conditions dual emission bands appeared at 450 nm (blue shift ∼46 nm) and 580 nm (red shift ∼84 nm) (Fig. 4b and Fig. S13b, ESI†) and the color of the solution becomes orange-red. Additionally, probe 2 upon excitation at the isosbestic point, 396 nm, displayed ratiometric behavior at alkaline pH (8–14) in which, the intensity of 2 centered at 496 nm decreased and concomitantly the relative fluorescence intensity of a new emission band (at 586 nm) enhanced progressively (Fig. 5). However, at acidic pH the intensity of 2 quenched gradually (Fig. 5, inset). Thus, acid–alkali titration studies clearly supported the protonation–deprotonation in the medium.
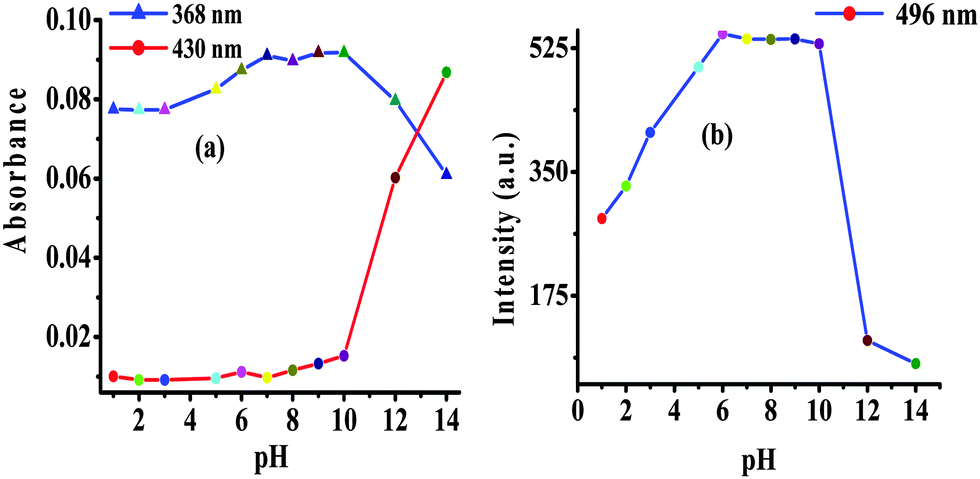 | ||
| Fig. 4 Change in (a) absorption and (b) emission (λex = 368 nm) intensity of 2 as a function of pH in HEPES buffer. | ||
 | ||
| Fig. 5 (a) Change in emission spectra of 2 (1 μM) at different pH values in HEPES buffer, λex = 396 nm and (b) change in emission intensity of 2 (1 μM) at 496 nm and 580 nm (λex = 396 nm) as a function of pH in HEPES buffer. | ||
Nature of interaction between 2 and F−
To gain deep insight into the actual mechanism of interaction between 2 and F− the 1H NMR studies have been performed in DMSO-d6. The 1H NMR spectrum of 2 (2.1 × 10−2 M) (Fig. 6 and Fig. S5, ESI†) showed aromatic proton resonance in the δ range of 8.25–7.34 ppm. The imidazolyl –NH resonance appeared at δ 12.96(s) ppm due to hydrogen bonding interaction with solvent DMSO-d6. Upon addition of F− (0–2.0 equiv.) to a solution of 2 the –NH resonances broadened (Fig. S14 and S15, ESI†). Further addition of 3.0 to 5.0 equiv. of F− the –NH resonances disappeared completely and the resonances of the phenyl ring proton shifted up-field. A triplet appeared at δ 16.43 ppm suggested the formation of HF2− species18 in the medium (Fig. S16 and S17, ESI†). Thus, the proton NMR studies clearly favored the H-bonding interaction between the –NH fragments of 2 and F− followed by deprotonation. Consequently, the ICT increased due to charge propagation from imidazole to benzothiazole units. | ||
| Fig. 6 Stacked 1H NMR spectra of 2 (2.1 × 10−2 M) upon addition of F− ions (0–5.0 equiv.) in DMSO-d6. | ||
DFT calculation
The geometry optimization and quantum chemical calculations for 2 and the corresponding deprotonated product, 3, were performed by the density functional theory (DFT) method as implemented in Gaussian 03 suits of programs32 employing basis set B3LYP/6-31G*. As shown in Fig. 7, the relevant occupied molecular orbital, HOMO (−0.30199 eV), is delocalized to the entire unit probe 2, in which the electron density is spread over the imidazole–phenyl bridge–thiazole unit while the unoccupied molecular orbital, LUMO (−0.21798 eV), is distributed more towards the thiazole–phenyl unit. Similarly, for 3 the HOMO (−0.29623 eV) is localized to the entire unit while the LUMO (−0.21833 eV) is more localized to the thiazole–phenyl bridge. Thus, the molecular orbital approximation favors the intramolecular charge transfer (ICT) from imidazole to thiazole units. The deprotonation of imidazolyl –NH led to a further polarization in electron density from the imidazole to the thiazole unit. Furthermore, the energy gap between the HOMO and the LUMO of 3 (ΔE = 0.0779 eV) was found to be less than that of probe 2 (ΔE = 0.08401 eV) and in good agreement with the red shift observed in the absorption spectra upon treatment with F−. Thus both the theoretical and experimental observations corroborated the possible mode of interaction between 2 and fluoride anions. | ||
| Fig. 7 DFT optimized minimum energy structures and HOMO–LUMO energy difference between 2 and 3. The proposed mechanism of interaction between probe 2 and F− anions. | ||
Analytical application of probe 2
 | ||
| Fig. 8 (a) Chromogenic (light blue to light yellow) and (b) fluorogenic (blue to green) responses of 2 on paper strips containing different concentrations of probe 2; (i) 5 mM, (ii) 2 mM, and (iii) 1 mM before and after addition of F−: (a) 10 × 10−6, (b) 5.0 × 10−6 and (c) 1.0 × 10−6 M. (c) Fluorogenic response of 2 on silica coated slides. | ||
 | ||
| Fig. 9 Change in absorption spectra of 2 + F− upon interaction with CO2 in H2O–DMSO (20%). Inset: Change in color of 2 with F− and CO2. | ||
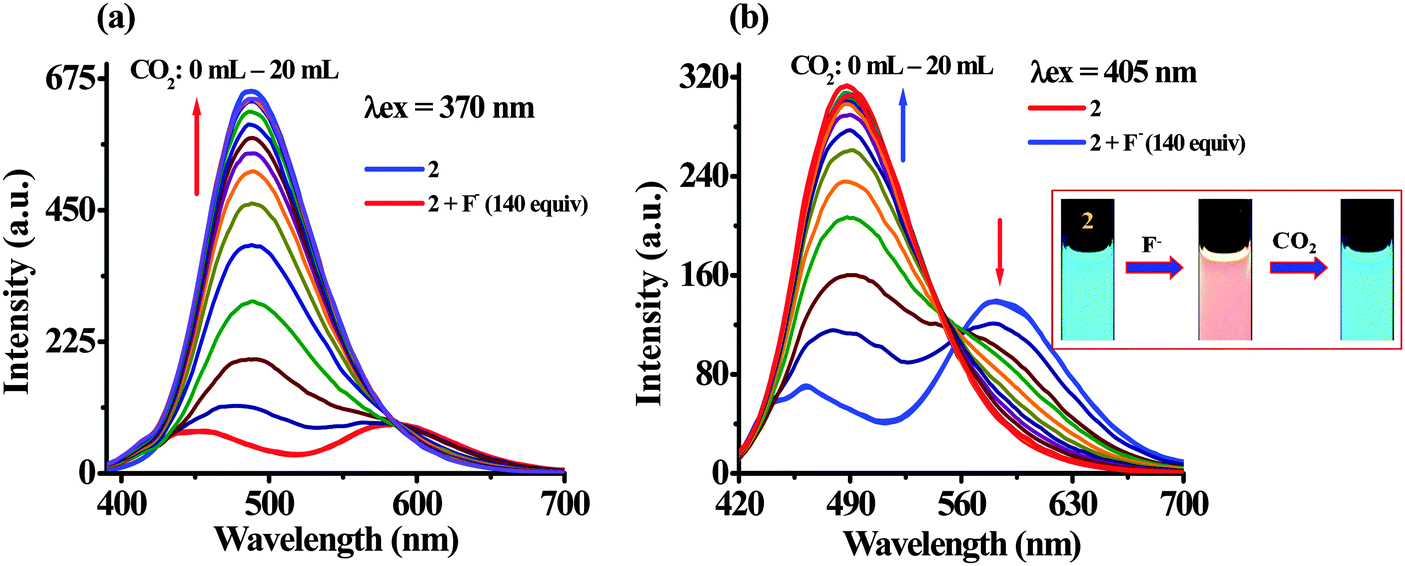 | ||
| Fig. 10 Change in emission spectra of 2 (1 μM) at (a) λex = 370 nm and (b) λex = 405 nm, upon bubbling of different volumes of CO2 to the solution of 2 + F− in H2O–DMSO (20%). Inset show the respective color change of 2 with F− and CO2. | ||
To investigate CO2 sensing first, the solution of probe 2 was treated with TBAF to generate in situ an anion induced anionic probe, 2 + F−. Upon passing the increasing volume of CO2 through a solution of 2 + F− significant naked-eye sensitive color changes were observed in which the original color of the probe revived. Similarly, the typical absorption band for 2 + F− centered at 455 nm diminished with the revival of the original absorption band of 2 (370 nm and 302 nm) along with the formation of an isosbestic point at 404 nm (Fig. 9). Moreover, the observed emission intensity of 2 + F− at 455 nm and 586 nm (at λex = 370 nm) upon bubbling the increasing volume of CO2 gas showed a gradual revival in emission intensity of 2 at 488 nm. Moreover, upon excitation at 405 nm, the emission intensity of 2 + F− centered at 578 nm diminished ratiometrically upon increasing the volume of CO2 and the original fluorescence intensity of 2 at 488 nm revived (Fig. 10) with unique naked-eye sensitive colorimetric response wherein, the color of probe 2 changed from colorless to orange-yellow by F− revived to the original color (Fig. 9, inset) while under UV light the reddish-orange color of 2 + F− upon exposure to CO2 regenerated the actual intense blue color of 2 (Fig. 10, inset). Moreover, fluorescence titration data have been acquired to estimate the detection limit of 2 + F− for CO2, which was found to be 2.29 μM (∼100 ppb) (Fig. S18, ESI†). Moreover, to understand the high selectivity of the probe for CO2 the sensitivity of 2 + F− has been investigated by passing through other gases such as H2S, SO2, and HCl (Fig. S19, ESI†). It is interesting to mention that upon interaction only in the presence of CO2 the emission intensity of the probe was revived while rest of the gases failed to exhibit any significant change in the emission behavior of 2 + F−.
Furthermore, the significant colorimetric response displayed by 2 in the presence and the absence of fluoride anions and carbon dioxide is attributed to the formation of imidazolium hydrogen carbonate or imidazolium carbamic acid or its salt, generated from the N–CO2 adduct formed by the initial attack of negatively charged imidazolium ions to CO2.19,21 Carbamic acids are usually considered as highly unstable and upon formation they may typically release CO2 to revive the corresponding free amine.33a Therefore to understand the actual mechanism of CO2 sensing involved in the present case we acquired 1H/13C NMR, FTIR and ESI-MS spectroscopic data of the probable imidazolyl N–CO2 adducts, i.e. imidazolyl carbamic acid, 3 after passing the excess of CO2 through a solution of 2 (35 mg; 0.08 mmol) and TBAF (2.0 equiv.) in acetonitrile.
The 1H NMR spectra showed a broad resonance at δ 13.01 ppm due to the partially deprotonated –NH function of the imidazole unit of the probe. However, the resonances appeared at δ 12.68 and δ 5.072 ppm may be assigned to the formation of imidazolyl carbamic acid33b (–NCOOH) (Fig. S20, ESI†). Similarly, the resonances appeared at δ 183.3 and δ 163.883 ppm in the 13C NMR spectrum are assignable to the carbonyl carbon (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of carboxylic acid and imidazolyl-N-carbamate anions (Fig. S21, ESI†). Moreover, the FTIR spectrum of a probable adduct showed resonances at 1703 and 3208–2873 cm−1 due to the CO and O–H stretching vibrations of the carboxylic function. The stretching vibration bands at 3379 and 2341 cm−1 may be assigned to –NH and CO2, respectively33c (Fig. S22, ESI†). Thus, the spectroscopy studies suggested the formation of adducts as well as the existence of H-bonding interaction in the medium in between the imidazolium N–CO2 adduct and the imidazolyl –NH unit of the second molecule as shown in Scheme 2. Nevertheless, the species formed in the medium are not very much stable33a and we attempted to acquire the mass spectrum of adduct 3 to justify our hypothesis. Interestingly, the molecular ion peak corresponding to [2 + H]+ appeared at m/z 430.1354. However we could not observe any sharp signal corresponding to the formation of imidazolium carbamate or carbonate salt probably due to poor stability of the adduct. However, a very weak molecular ion peak appeared at m/z 475.1361 [2 + CO2 + 2H]+ corresponding to the formation of imidazolyl carbamic acid in the medium (Fig. S23, ESI†). Based on the above findings we proposed a plausible mechanism of CO2 interaction, shown in Scheme 2.
O) of carboxylic acid and imidazolyl-N-carbamate anions (Fig. S21, ESI†). Moreover, the FTIR spectrum of a probable adduct showed resonances at 1703 and 3208–2873 cm−1 due to the CO and O–H stretching vibrations of the carboxylic function. The stretching vibration bands at 3379 and 2341 cm−1 may be assigned to –NH and CO2, respectively33c (Fig. S22, ESI†). Thus, the spectroscopy studies suggested the formation of adducts as well as the existence of H-bonding interaction in the medium in between the imidazolium N–CO2 adduct and the imidazolyl –NH unit of the second molecule as shown in Scheme 2. Nevertheless, the species formed in the medium are not very much stable33a and we attempted to acquire the mass spectrum of adduct 3 to justify our hypothesis. Interestingly, the molecular ion peak corresponding to [2 + H]+ appeared at m/z 430.1354. However we could not observe any sharp signal corresponding to the formation of imidazolium carbamate or carbonate salt probably due to poor stability of the adduct. However, a very weak molecular ion peak appeared at m/z 475.1361 [2 + CO2 + 2H]+ corresponding to the formation of imidazolyl carbamic acid in the medium (Fig. S23, ESI†). Based on the above findings we proposed a plausible mechanism of CO2 interaction, shown in Scheme 2.
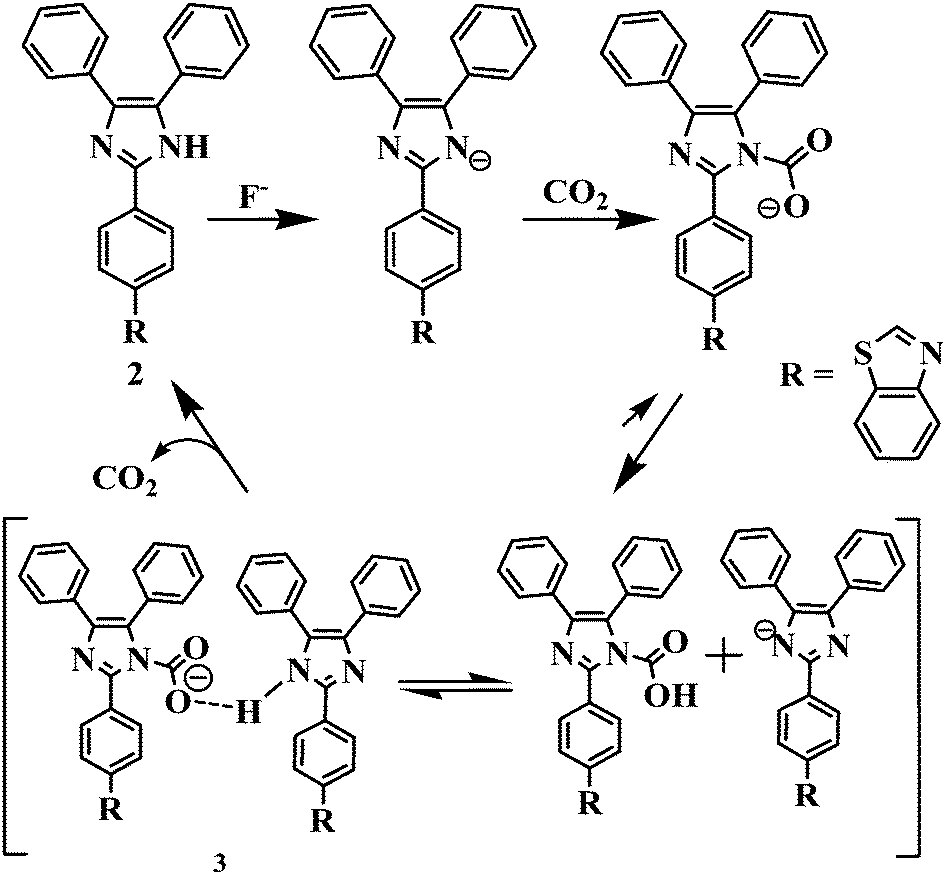 | ||
| Scheme 2 A plausible mechanism of CO2 interaction with the probe. | ||
Conclusion
In summary, an efficient donor–acceptor type intramolecular charge transfer (ICT) probe has been developed. The photophysical behavior of the probe has been examined in the solvents of different polarities. The anion interaction studies showed that the probe can be utilized as a potential chemosensor to detect F− selectively and sensitively with naked-eye visual color changes in the partial aqueous medium. Moreover, probe 2 has been utilized to detect CO2 by a chemical-based optical method. The in situ generated anionic species, 2 + F−, displayed unique colorimetric response along with revival in the typical photophysical properties of probe 2. The proton NMR studies unequivocally supported the deprotonation of a moderately acidic imidazolyl –NH fragment in the presence of F− anions and corroborated the observed changes in the photophysical behavior of the probe, consequently.Experimental
Synthesis of compounds 1a and 1b
Benzyl (1.05 g, 5 mmol), benzaldehyde or 4-formylbenzoic acid (5 mmol) and ammonium acetate (1.156 g, 15 mmol) were refluxed in glacial acetic acid (20 ml) for 4 h. After completion of the reaction (monitored on TLC) the reaction mixture was cooled down to room temperature and poured into ice-water (20 ml). The precipitate so obtained was filtered, washed with cold water, and dried in air to obtain compounds 1a and 1b.Synthesis of compound 2
Compound 1b (170 mg, 0.5 mmol) and 2-aminothiophenol (62.5 mg, 0.5 mmol) were refluxed for 16 h in the presence of PPA (0.5 g). After completion of the reaction (monitored on TLC) the reaction mixture was quenched with water and the resulting precipitate was washed with saturated solution of sodium hydrogen carbonate comprehensively to afford compound 2. Yield, 275 mg (64%). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.0 (s, 1H, –NH), 8.31–8.28 (d, 2H, J = 7.5 Hz), 8.22–8.19 (d, 2H, J = 8.4 Hz), 8.09–8.07 (d, 2H, J = 7.8 Hz), 7.57–7.47 (m, 12H). 13C NMR (75 MHz, DMSO-d6) δ (ppm): 166.80, 153.61, 144.56, 134.47, 132.16, 129.40, 128.65, 127.65, 127.21, 126.72, 125.82, 125.55, 124.49, 122.83, 122.38; FT-IR (KBr) νmax (cm−1); 3370, 3055, 1586, 1601, 1479, 1432,1388, 1251, 1227, 1181, 1125, 1071, 966, 915, 842, 758, 727, 695. ESI-MS m/z: [2 + H]+ for C28H19N3S, 430.2.Acknowledgements
The authors are thankful to the Council of Scientific and Industrial Research (CSIR) (02(0199)/14/EMR-II) and UGC New Delhi for financial assistance and fellowships (RA, SSR, RCG and SKD).References
- (a) F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609–1646 CrossRef CAS PubMed; (b) X. Lou, D. Ou, Q. Li and Z. Li, Chem. Commun., 2012, 48, 8462–8477 RSC.
- (a) M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809–2829 RSC; (b) S. Y. Kim and J.-I. Hong, Org. Lett., 2007, 9, 3109–3112 CrossRef CAS PubMed.
- M. Cametti and K. Rissanenb, Chem. Soc. Rev., 2013, 42, 2016–2038 RSC.
- (a) R. J. Carton, Fluoride, 2006, 39, 163–172 CAS; (b) R. Hu, J. Feng, D. Hu, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2010, 49, 4915–4918 CrossRef CAS PubMed.
- (a) Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed; (b) S. Goswami, A. K. Das, A. Manna, A. K. Maity, H.-K. Func, C. K. Quah and P. Saha, Tetrahedron Lett., 2014, 55, 2633–2638 CrossRef CAS.
- A. Wiseman, Handbook of Experimental Pharmacology, 20, Springer-Verlag, Berlin, 1970, pp. 48–97 Search PubMed.
- T. W. Hudnall, C.-W. Chiu and F. P. Gabbai, Acc. Chem. Res., 2009, 42, 388–397 CrossRef CAS PubMed.
- J.-A. Gu, V. Mania and S.-T. Huang, Analyst, 2015, 140, 346–352 RSC.
- R. Martinez-Manez and F. Sancenon, Chem. Rev., 2003, 103, 4419–4476 CrossRef CAS PubMed.
- (a) A. Aydogan, D. J. Coady, V. M. Lynch, A. Akar, M. Marquez, C. W. Bielawski and J. L. Sessler, Chem. Commun., 2008, 1455–1457 RSC; (b) C. Bhaumik, D. Saha, S. Das and S. Baitalik, Inorg. Chem., 2011, 50, 12586–12600 CrossRef CAS PubMed; (c) D. Maity, S. Das, S. Mardanya and S. Baitalik, Inorg. Chem., 2013, 52, 6820–6838 CrossRef CAS PubMed; (d) N. Kumari, S. Jha and S. Bhattacharya, J. Org. Chem., 2011, 76, 8215–8222 CrossRef CAS PubMed; (e) Z. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. W. Bielawski, J. L. Sessler and J. Yoon, J. Am. Chem. Soc., 2012, 134, 17846–17849 CrossRef CAS PubMed.
- (a) D. Srikun, E. W. Miller, D. W. Domaille, J. Christopher and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 4596–4597 CrossRef CAS PubMed; (b) H. Yu, Q. Zhao, Z. Jiang, J. Qin and Z. Li, Sens. Actuators, B, 2010, 148, 110–116 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- D. Burtscher, R. Saf and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6136–6145 CrossRef CAS.
- (a) K. Benelhadj, J. Massue, P. Retailleau, G. Ulrich and R. Ziessel, Org. Lett., 2013, 15, 2918–2921 CrossRef CAS PubMed; (b) X. Zhang, J. Lin, X. Ouyang, Y. Liu, X. Liu and Z. J. Ge, Photochem. Photobiol., 2013, 268, 37–43 CrossRef CAS.
- (a) K. Zheng, W. Lin and L. Tan, Org. Biomol. Chem., 2012, 10, 9683–9688 RSC; (b) K. C. Song, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Sens. Actuators, B, 2013, 176, 850–857 CrossRef CAS.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. D. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (b) J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17–55 CrossRef CAS; (c) K. Choi and A. D. Hamilton, Coord. Chem. Rev., 2003, 240, 101–110 CrossRef CAS; (d) K. M. K. Swamy, Y. J. Lee, H. N. Lee, J. Chun, Y. Kim, S.-J. Kim and J. Yoon, J. Org. Chem., 2006, 71, 8626–8628 CrossRef CAS PubMed; (e) X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., Int. Ed., 2006, 45, 5475–5478 CrossRef CAS PubMed.
- (a) P. A. Gale, Coord. Chem. Rev., 2003, 240, 191–221 CrossRef CAS; (b) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Krugerc and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC; (c) J. V. Ros-Lis, R. Martínez-Máñez, F. Sancenón, J. Soto, K. Rurack and H. Weißhoff, Eur. J. Org. Chem., 2007, 2449–2458 CrossRef CAS; (d) C. Marín-Hernandez, L. E. Santos-Figueroa, M. E. Moragues, M. M. M. Raposo, R. M. F. Batista, S. P. G. Costa, T. Pardo, R. Martínez-Mańez and F. Sancenón, J. Org. Chem., 2014, 79, 10752–10761 CrossRef PubMed.
- (a) P. Dydio, D. Lichosyt and J. Jurczak, Chem. Soc. Rev., 2011, 40, 2971–2985 RSC; (b) M. Shahid, P. Srivastava and A. Misra, New J. Chem., 2011, 35, 1690–1700 RSC; (c) A. Misra, M. Shahid and P. Dwivedi, Talanta, 2009, 80, 532–538 CrossRef CAS PubMed; (d) A. Misra, M. Shahid and P. Srivastava, Sens. Actuators, B, 2012, 169, 327–340 CrossRef CAS.
- M. Lee, S. Jo, D. Lee, Z. Xu and J. Yoon, Dyes Pigm., 2015, 120, 288–292 CrossRef CAS.
- M. Lee, J. H. Moon, K. M. K. Swamy, Y. Jeong, G. Kim, J. Choi, J. Y. Lee and J. Yoon, Sens. Actuators, B, 2014, 199, 369–376 CrossRef CAS.
- Z. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. W. Bielawski, J. L. Sessler and J. Yoon, J. Am. Chem. Soc., 2012, 134, 17846–17849 CrossRef CAS PubMed.
- T. Gunnlaugsson, P. E. Kruger, P. Jensen, F. M. Pfeffer and G. M. Hussey, Tetrahedron Lett., 2003, 44, 8909–8913 CrossRef CAS.
- (a) X. Xie, M. Pawlak, M. Tercier-Waeber and E. Bakker, Anal. Chem., 2012, 84, 3163–3169 CrossRef CAS PubMed; (b) Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Y. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951–13953 CrossRef CAS PubMed; (c) R. N. Dansby-Sparks, J. Jin, S. J. Mechery, U. Sampathkumaran, T. W. Owen, B. D. Yu, K. Goswami, K. Hong, J. Grant and Z.-L. Xue, Anal. Chem., 2010, 82, 593–600 CrossRef CAS PubMed; (d) M. Ishida, P. Kim, J. Choi, J. Yoon, D. Kim and J. L. Sessler, Chem. Commun., 2013, 49, 6950–6952 RSC.
- (a) R. Ali, T. Lang, S. M. Saler, R. J. Meier and O. S. Wolfbeis, Anal. Chem., 2011, 83, 2846–2851 CrossRef CAS PubMed; (b) D. Wencel, J. P. Moore, N. Stevernson and C. McDonagh, Anal. Bioanal. Chem., 2010, 398, 1899–1907 CrossRef CAS PubMed; (c) Q. Xu, S. Lee, Y. Cho, M. H. Kim, J. Bouffard and J. Yoon, J. Am. Chem. Soc., 2013, 135, 17751–17754 CrossRef CAS PubMed; (d) X. Zhang, S. Lee, Y. Liu, M. Lee, J. Yin, J. L. Sessler and J. Yoon, Sci. Rep., 2014, 4 DOI:10.1038/srep04593.
- (a) S. S. Razi, R. Ali, P. Srivastava and A. Misra, Tetrahedron Lett., 2014, 55, 1052–1056 CrossRef CAS; (b) S. S. Razi, R. Ali, P. Srivastava and A. Misra, Tetrahedron Lett., 2014, 55, 2936–2941 CrossRef CAS; (c) S. S. Razi, R. Ali, P. Srivastava, M. Shahid and A. Misra, RSC Adv., 2014, 4, 22308–22317 RSC; (d) S. S. Razi, P. Srivastava, R. Ali, R. C. Gupta, S. K. Dwivedi and A. Misra, Sens. Actuators, B, 2015, 209, 162–171 CrossRef CAS.
- (a) D. M. D'Souza, A. Kiel, D. P. Herten, F. Rominger and T. J. J. Muller, Chem. – Eur. J., 2008, 14, 529–547 CrossRef PubMed; (b) J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed; (c) B. B. Toure and D. G. Hall, Chem. Rev., 2009, 109, 4439–4486 CrossRef CAS PubMed.
- S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. M. Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef CAS PubMed.
- Y. Zhang, D. Li, Y. Li and J. Yu, Chem. Sci., 2014, 5, 2710–2716 RSC.
- A. Misra and M. Shahid, J. Phys. Chem. C, 2010, 114, 16726–16739 CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- A. Misra, P. Srivastava and M. Shahid, Analyst, 2012, 137, 3470–3478 RSC.
- M. J. Frisch, G. W. Truncks and H. B. Schlegel, Gaussian 03, Revision E.01, Gaussian, Wallingford, CT, 2007 Search PubMed.
- (a) E. M. Hampe and D. M. Rudkevich, Chem. Commun., 2002, 1450–1451 RSC; (b) G. A. Olah, T. Heiner, G. Rasul and G. K. Surya Prakash, J. Org. Chem., 1998, 63, 7993–7998 CrossRef CAS; (c) R. K. Khanna and M. H. Moore, Spectrochim. Acta, Part A, 1999, 55, 961–967 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, 1H, 13C NMR, FTIR, and ESI-MS. See DOI: 10.1039/c5nj01920f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |