
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strengthening the acceptor properties of thiadiazoloquinoxalines via planarization†
Shoufa
Zhou
a,
Cunbin
An
a,
Timea
Stelzig
a,
Sreenivasa Reddy
Puniredd
a,
Xin
Guo
 a,
Wojciech
Pisula
*ab and
Martin
Baumgarten
*a
a,
Wojciech
Pisula
*ab and
Martin
Baumgarten
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: martin.baumgarten@mpip-mainz.mpg.de; pisula@mpip-mainz.mpg.de
bDepartment of Molecular Physics, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
First published on 4th June 2015
Abstract
The synthesis and characterization of fused-ring thiadiazoloquinoxaline derivatives are described. The target molecules were studied by UV-Vis absorption, emission spectroscopy and cyclic voltammetry. The optical absorption maximum λmax of the new molecules in solution were shown at 714–774 nm, with the corresponding optical gaps (Eoptg) of 1.44–1.50 eV. Density functional theory calculations were applied for the design and prediction of HOMO and LUMO variations and the corresponding optical absorptions. The thiadiazoloquinoxaline with a phenanthroline moiety showed a liquid crystalline phase as found from 2D-WAXS studies and an electron transporting behavior indicating its potential as an acceptor building block.
1. Introduction
Since the first report on the synthesis of [1,2,5]thiadiazolo[3,4-g]quinoxaline (TQ),1 as an o-quinoid, strong acceptor unit, this building block has been explored in conjugated donor (D)–acceptor (A) type systems for a variety of applications, ranging from organic photovoltaics (OPVs),2 organic field-effect transistors (OFETs)3 to NIR emitters.4 The incorporation of the electron deficient TQ segment in D–A type copolymers served the purpose of lowering the LUMO level downwards making them good acceptors comparable to C60 derivatives or in combination with appropriate donors of lowering the band gap (Eg) of the materials.5 One of the consequences of a reduced Eg is the extension of the absorption and emission of the copolymers into the NIR region.6Typically, the HOMO–LUMO energy levels of conventional TQ derivatives are tuned via alkyl or aryl substituents attached to the TQ core, at positions 6 and 7 (Scheme 1, TQ, TQ1–TQ2)7 or by attaching electron donating moieties at positions 4 and 9. The substituents have profound effects on their electronic structures, charge transfer abilities, solubility, and film forming properties.7 However, the extension of π-conjugation in TQ-based derivatives and therefore the fine tuning of the HOMO and LUMO is also possible through an increase of the polycyclic molecule via an enforced planarization of the aryl units at positions 6 and 7. Despite their potentially improved photophysical and electrochemical properties, these extended fused-ring TQ analogues have been scarcely explored, apart from dibenzo[a,c][1,2,5]thiadiazolo[3,4-i]phenazine8 (analogue of TQ3, Scheme 2). The photophysical and electrochemical studies of different monomeric systems proved to be of great aid in the design of technologically useful materials.9 Therefore, it is important to assess the effect of the chemical structure on the optical and electronic properties. Within this context, a series of fused-ring TQ acceptor analogues have been developed (Scheme 2, TQ3–TQ5) and their synthesis, photophysical, and electrochemical properties are presented herein.
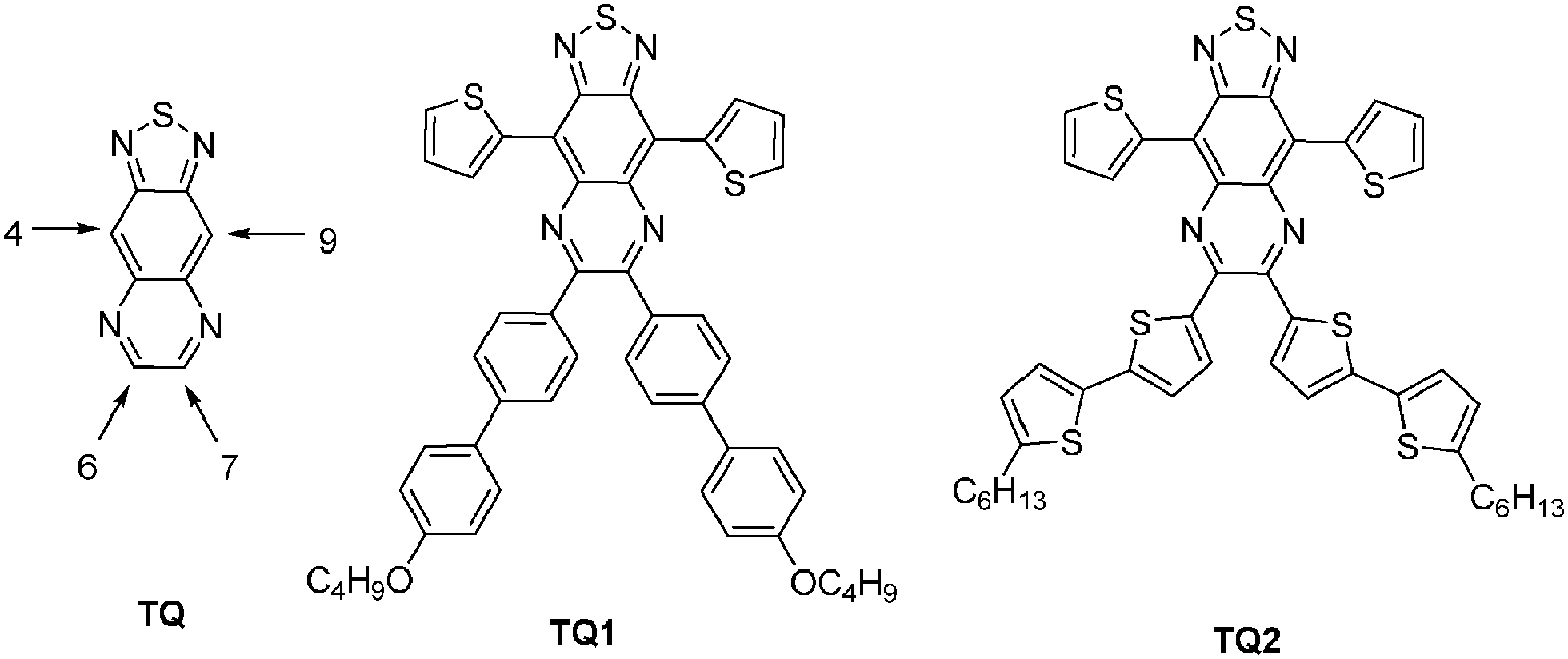 | ||
| Scheme 1 Conventional thiadiazolo[3,4-g]quinoxaline derivatives. | ||
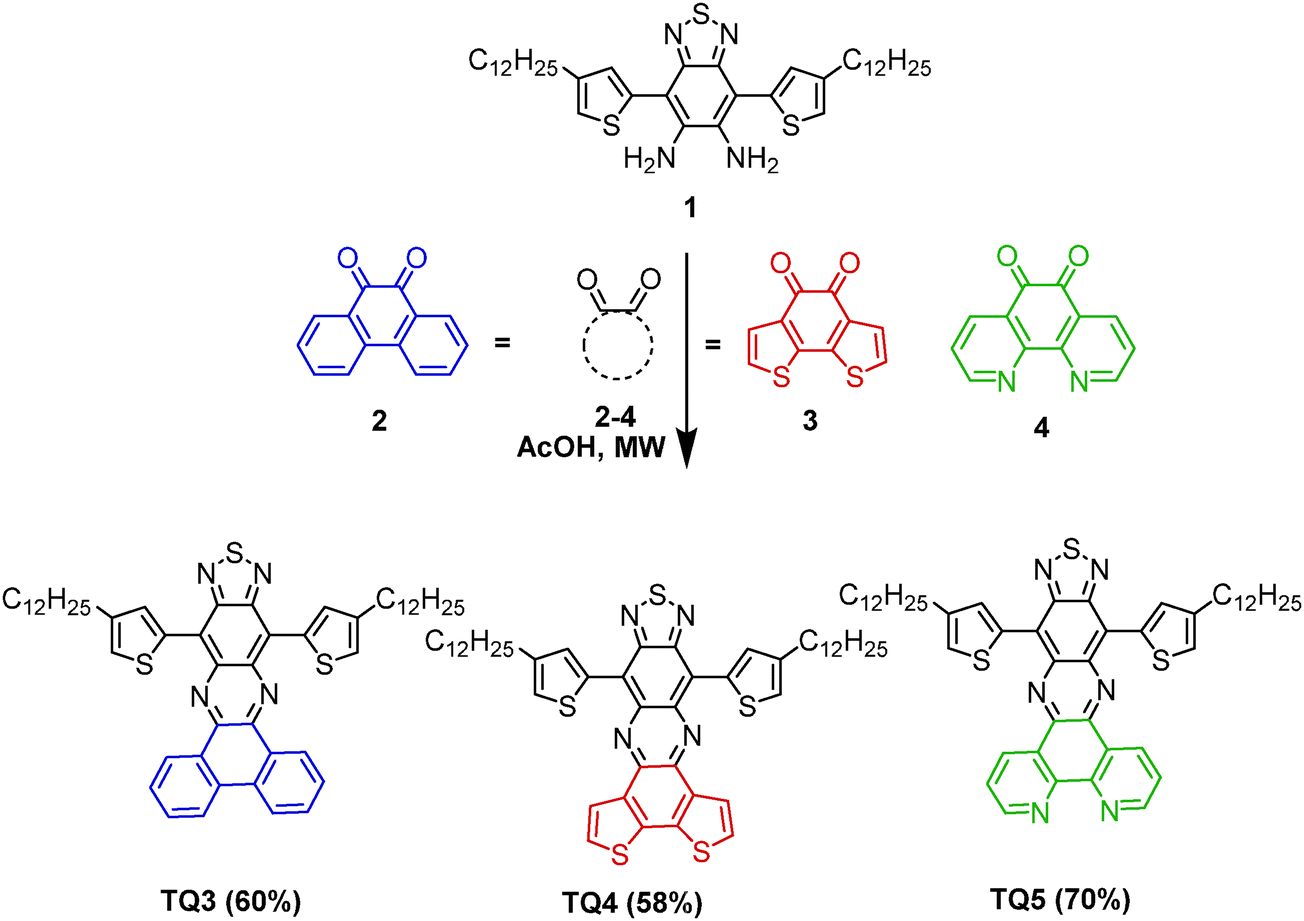 | ||
| Scheme 2 Synthesis of the fused-ring TQ derivatives TQ3–TQ5. | ||
2. Experimental
2.1. Materials and measurements
All chemicals and reagents were used as received from commercial sources without further purification. Unless stated otherwise, chemical reactions were carried out under ambient atmosphere. The microwave (MW) assisted synthesis was carried out in a CEM Discover™ system, in a 35 mL closed vial, controlling reaction temperatures, pressures, and stirring speeds.Proton nuclear magnetic resonance (1H-NMR) spectra, carbon nuclear magnetic resonance (13C-NMR) spectra were recorded on a Bruker AMX 500 NMR instrument (500 and 75 MHz, respectively) or on a Bruker AMX 250 NMR instrument (250 and 62.5 MHz, respectively) with trimethylsilane as internal standard. High-resolution MALDI-TOF mass spectra were recorded on a Bruker Reflex II-TOF Spectrometer using a 337 nm nitrogen laser with tetracyanochinodimethan (TCNQ) as matrix. The UV-Vis absorption measurements were performed on a Perkin-Elmer Lambda 15 spectrophotometer, and PL measurements on a SPEX Fluorolog 2 type F212 steady-state fluorometer.
The 2D wide angle X-ray scattering (2DWAXS) measurements were performed by means of a solid anode X-ray tube (Siemens Kristalloflex X-ray source, copper anode X-ray tube operated at 35 kV and 40 mA), Osmic confocal MaxFlux optics, X-ray beam with pinhole collimation and a MAR345 image plate detector. The samples were prepared as a thin filament of 0.7 mm in diameter via filament extrusion. For the measurements, the samples were positioned perpendicular to the incident X-ray beam and scattering intensity was detected on a 2D image plate (MAR345) with a pixel size of 100 μm (3450 × 3450 pixels). Data analysis was performed using the Datasqueeze 3.0.0 software.
OFETs were fabricated by employing the bottom-gate, bottom-contact architecture. The 230 nm thick SiO2 dielectric (capacitance = 14.6 nF cm−2) covering the highly doped Si acting as the gate electrode was functionalized with hexamethyldisilazane (HMDS) to minimize interfacial trapping sites. The compounds were deposited by drop-casting of 5 mg mL−1 in chloroform solution on FET substrates at room temperature, followed by annealing at 120 °C for 60 min. The source and drain electrodes consist of 10 nm of high work function (ITO) adhesive layer coated with 30 nm of Au. An interdigitated electrode configuration with typical length L = 5–20 μm and width W = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μm (WL−1 = 500–4000) was used. All the electrical measurements (using Keithley 4200 SCS) were performed in a glove box under nitrogen atmosphere.
000 μm (WL−1 = 500–4000) was used. All the electrical measurements (using Keithley 4200 SCS) were performed in a glove box under nitrogen atmosphere.
2.2. General synthetic procedures
3. Results and discussion
3.1. Synthesis
Following the typical synthetic procedure of the conventional TQ derivatives, the fused-ring TQ analogues (TQ3–TQ5) were obtained via the condensation of the 4,7-substituted benzo[c][1,2,5]thiadiazole-5,6-diamine (1)10 with a corresponding polycyclic α-dione (Scheme 2). The required diamine could be easily obtained from its corresponding 4,7-substituted 5,6-dinitrobenzo[c][1,2,5]thiadiazole.10 While 9,10-phenanthroquinone (2) and 1,10-phenanthroline-5,6-dione (4) are commercially available, benzo[2,1-b:3,4-b′]bithiophene-7,8-quinone (3) could be easily synthesized following a procedure previously described.11 Based on the beneficial effects pointed out in our earlier studies,3b the condensation was performed under MW irradiation reducing the reaction time to a few hours. Hence, the functionalized TQ analogues with an enforced planarity were isolated as green solids in 60% to 70% yield for TQ3–TQ5, respectively. All the compounds were thermally stable as investigated with thermogravimetric analysis (TGA) under N2 with a heating rate of 10 °C min−1. The temperatures with 5% weight loss for TQ3–TQ5 were 414, 426, and 418 °C, respectively (Fig. S1, ESI†). The differential scanning calorimetry (DSC) was also measured, and shown in Fig. S2 (ESI†). The DSC curves of three compounds didn't show any phase transition in the range from 25 to 350 °C.3.2. Photophysical and electrochemical properties
The absorption and emission spectra of TQ3–TQ5 were recorded in dilute toluene solution (c = 10−5 M, Fig. 1) and the extracted photophysical data are summarized in Table 1. For the sake of comparison, relevant data corresponding to TQ1–TQ2 are listed as well. The absorption profile of the fused-ring TQ analogues TQ3–TQ5 was composed of three absorption bands. All three compounds presented strong π–π* transitions at higher energies, between 300 and 380 nm. Between 380 and 550 nm multiple bands of close energetic spacing were observed which could be attributed to various vibrational components of the same π–π* transition. While for TQ4 this transition was more pronounced, in the case of TQ3 and TQ5 it was significantly reduced in intensity. The low energy transition resulting in a broad absorption band between 550 and 850 nm, possesses charge transfer characteristics as clearly shown by DFT calculations (Fig. 3). Further TD-DFT-SCF calculations (Fig. S4b, ESI†) well reproduced the long wavelength absorption, originating from the HOMO and LUMO tansitions. The long wavelength absorption maximum (λmax) was bathochromically shifted from TQ3 (λmax = 717 nm), TQ5 (λmax = 730 nm), and to TQ4 (λmax = 755 nm). From all three fused-ring TQ analogues, TQ3 (Eg = 1.50 eV) revealed the highest and TQ4 (Eg = 1.44 eV) the lowest optical energy gap, as calculated from the onset of the long wavelength absorption band. This is not surprising, considering that TQ3–TQ5 differ only in the fused-ring attached to the TQ core, therefore it is expected that extending the π-conjugation via the benzo[2,1-b:3,4-b′]bithiophene (TQ4) should result in a lower HOMO–LUMO energy difference. That TQ5 has a smaller optical gap than TQ3 in toluene solution can be ascribed to the more electron-poor phenanthroline group which stabilizes the LUMO more than the HOMO and lowers the gap.1 The differences between the characteristics of TQ1–TQ2 and the fused-ring TQ3–TQ5 analogues could thus be assigned to the extended planar central TQ segment with better delocalization.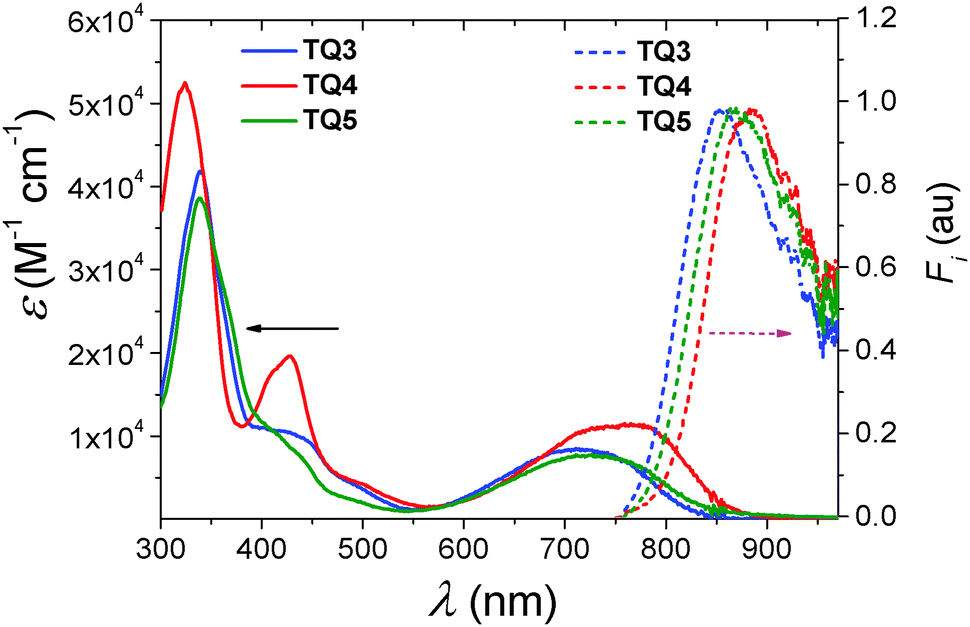 | ||
| Fig. 1 UV-Vis absorption (solid line) and fluorescence (dashed lines with the excitation wavelength at 750 nm) spectra of compounds TQ3–TQ5 in toluene solutions (c = 10−5 M). | ||
| Compd | λ absmax (nm) | λ emmax (nm) | λ absmax (nm) | E g (eV) | EAc (eV) | IP (eV) | LUMOe (eV) | E calc. (eV) |
|---|---|---|---|---|---|---|---|---|
| a In toluene (1 × 10−5 M). b Spin coated on a glass substrate from a 5 mg mL−1 toluene. c 0.1 M of n-Bu4NPF6, in DCM, glassy carbon electrode, scan rate 50 mV s−1. Calculated from EEA/IP = −(ERed/Ox,onset − E(1/2)Fc+/Fc + 4.8) eV, wherein, the E(1/2)Fc+/Fc = 0.54 eV (measured in our setup). d Calculated from Eopt − EA. e DFT quantum mechanical calculations B3LYP/6-311G(d). f Reported in the literature.7 | ||||||||
| TQ1 | 620 | N/A | 695 | 1.70 | −3.43 | −5.11d | −3.27 | 1.92 |
| TQ2 | 650 | N/A | 713 | 1.65 | −3.52 | −5.08d | −3.23 | 1.86 |
| TQ3 | 717 | 853 | 750 | 1.50 | −3.81 | −5.01c | −3.44 | 1.74 |
| TQ4 | 774 | 884 | 776 | 1.44 | −3.84 | −5.05c | −3.56 | 1.60 |
| TQ5 | 730 | 867 | 759 | 1.49 | −3.88 | −5.07c | −3.62 | 1.70 |
The UV-Vis absorptions of thin-films of the target compounds were examined as well (see Fig. S3, ESI†). The λabsmax of TQ3–TQ5 located at 750 nm, 776 nm and 759 nm, respectively. The absorption maxima of TQ3 and TQ5 in film showed evident red-shift compared to those in solution (see Table 1), suggesting the formation of more aggregated structures in the solid state. Surprisingly, for TQ4 only a tiny red-shift of 2 nm was found providing no major difference between the film and solution.
All three compounds revealed a single broad emission in the NIR region (Fig. 1). Within the series TQ3–TQ5, the emission maximum (λemmax) reflected essentially identical trend as λabsmax (Table 1). The extension of the π-conjugation via phenanthrene (TQ3) resulted in a higher energy emission at λem = 854 nm. Introducing the electron deficient phenanthroline moiety in the TQ core (TQ5) yielded a 13 nm bathochromic shift of λem to 867 nm. Furthermore, the incorporation of the benzo[2,1-b:3,4-b′]bithiophene into the TQ analogue (TQ4) led to a further red-shift of the λem to 884 nm.
Cyclic voltammetry (CV) measurements were carried out in order to estimate the electron affinities (EAs) and ionization potentials (IPs) of TQ3–TQ5. The corresponding data are collected in Table 1. All three compounds exhibit two reversible reduction peaks and one irreversible oxidation peak (Fig. 2). Going from TQ3, TQ4 to TQ5 the fused-ring TQ derivatives became easier to be reduced. Conversely, the EA values, calculated from the onset of the first reduction peak, showed a reverse trend. Phenanthrene containing TQ3 had the highest EA value (−3.81 eV), while TQ4, with the benzo[2,1-b:3,4-b′]bithiophene extension, had a lower EA value of −3.84 eV. In comparison with TQ3, the additional nitrogens of TQ5 can significantly stabilize its LUMO level,12 yielding the lowest electron affinity of −3.88 eV. Hence, the EA values were significantly lowered compared to the structurally closely related TQ1–TQ2 (−3.43 and −3.52 eV), which can be attributed to the better π-electron delocalization and conjugation within the extended planarized backbone. Additionally, the IP of three compounds estimated from the onset of first oxidation peak is −5.01, −5.05 and −5.07 eV for TQ3, TQ4 and TQ5, respectively.
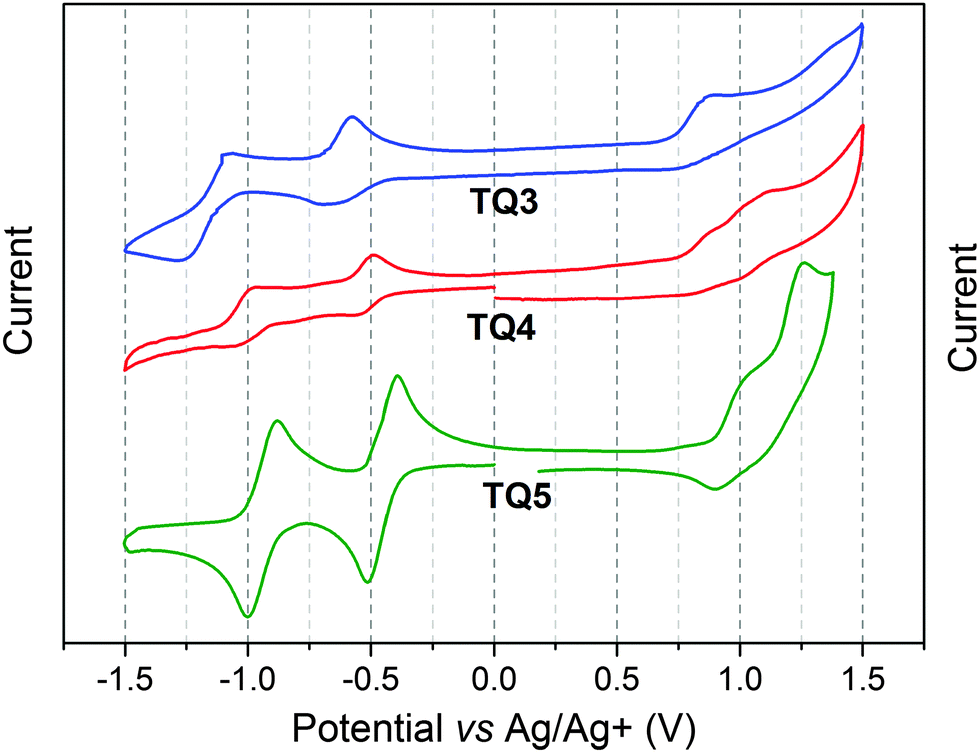 | ||
| Fig. 2 Cyclic voltammograms of compounds TQ3–TQ5 in DCM at 50 mV s−1, 0.1 M of n-Bu4NPF6, glassy carbon electrode, mV vs. Ag/Ag+. | ||
3.3. Quantum chemistry calculations
Quantum mechanical calculations (density functional theory (DFT) using B3LYP functional with 6-311G(d) basis set) were carried out to explore the geometric and electronic properties of the presented molecules (Fig. 3). As typical for functionalized TQ derivatives, the HOMOs reside on the thiophenes anchoring the TQ segment and the phenyl incorporated in the TQ core to which they are connected. Similarly, the LUMOs of TQ3–TQ5 are mainly localized on the ring extended TQ core. The calculated energy levels were in decent agreement with the experimentally determined values as also the calculated optical transitions (TD-DFT scf, Fig. S4a and b, ESI,†Table 1). Hence, the calculations come to support the fine tuning of the electronic properties through extension of the π-conjugation via enforced planarization, making TQ3–TQ5 attractive acceptor building blocks for organic electronics.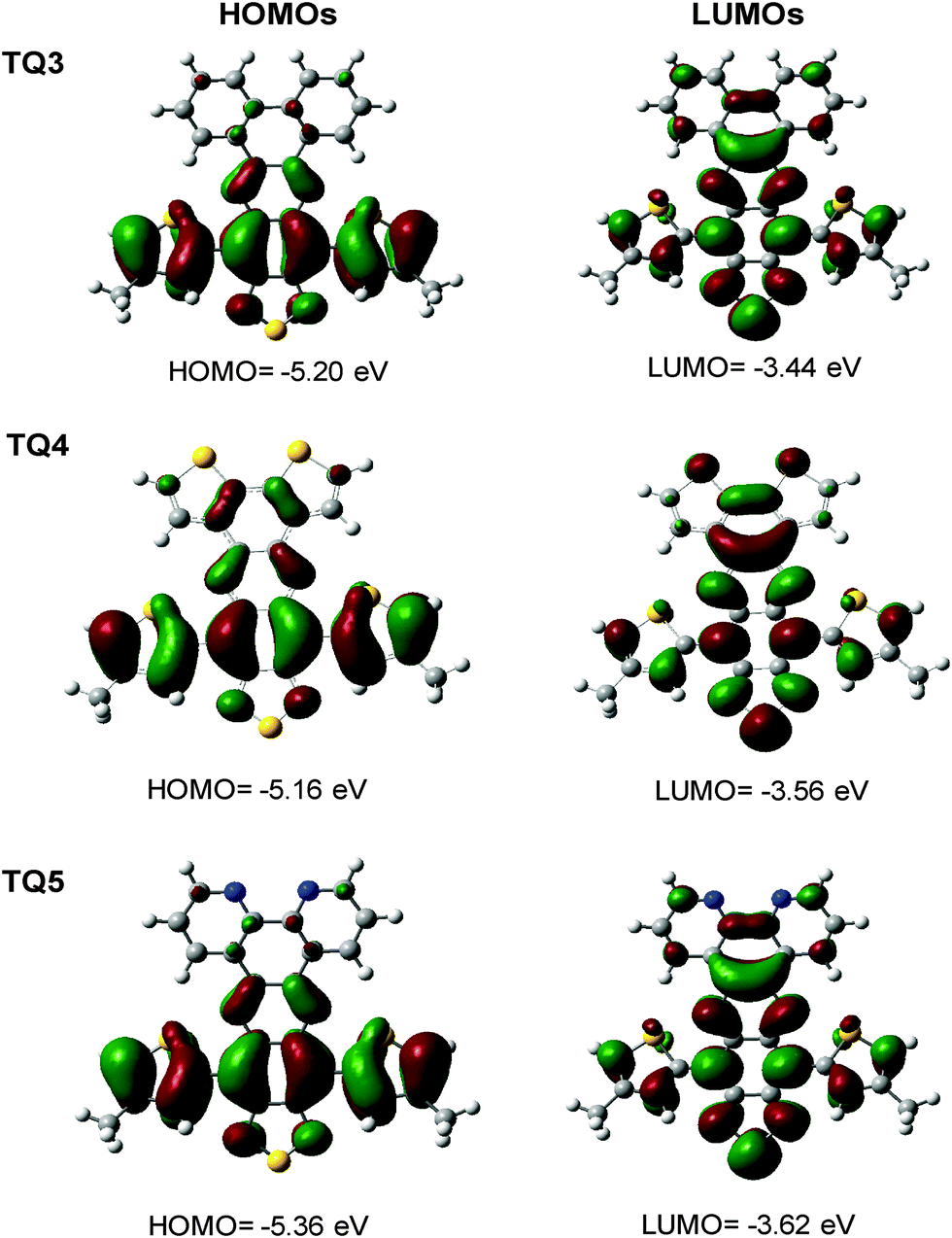 | ||
| Fig. 3 Electronic density contour calculated for the frontier molecular orbitals of TQ3–TQ5. | ||
3.4. Charge-carrier transport and solid-state organization
The charge carrier transport was determined in thin film bottom gate, bottom contact field-effect transistors. The compounds were drop-cast from a 5 mg mL−1 chloroform solution on top of the hexamethyldisilazane surface-modified SiO2 dielectric and afterwards annealed at 120 °C. A field-effect response was obtained for TQ5 with an electron mobility of 5 × 10−6 cm2 V−1 s−1 (Fig. 4a), However, TQ3 and TQ4 were not observed any field-effect characteristics. To better understand the differences in device performance between the compounds, the supramolecular bulk organization of TQ3–TQ5 was investigated on extruded fibers by two-dimensional wide-angle X-ray scattering (2DWAXS). All three compounds revealed a columnar organization in which the molecules are packed on top of each other (Fig. 5). Interestingly, within this series a discotic liquid crystalline phase was found only for TQ5, while TQ3 and TQ4 are crystalline as evident from the high number of reflections (Fig. 5a and b). At 110 °C the positions of the equatorial reflections in the pattern of TQ5 indicated a hexagonal arrangement of the columns with a lattice of 4.28 nm (Fig. 4b and Fig. S8, ESI†). Meridional scattering intensities at the wide-angle range are characteristic of non-tilted molecules with a π-stacking distance of 0.37 nm. Cooling back the sample to 30 °C decreases slightly the hexagonal parameter to 4.17 nm and induces a minor tilting of ca. 15° of the building blocks towards the columnar axis as evident from the double meridional reflection (Fig. 5c). In contrast to crystalline TQ3 and TQ4, the device performance of TQ5 was improved by self-healing during annealing the compound in the liquid crystalline phase and reducing in this way grain boundaries which are serious charge carrier traps.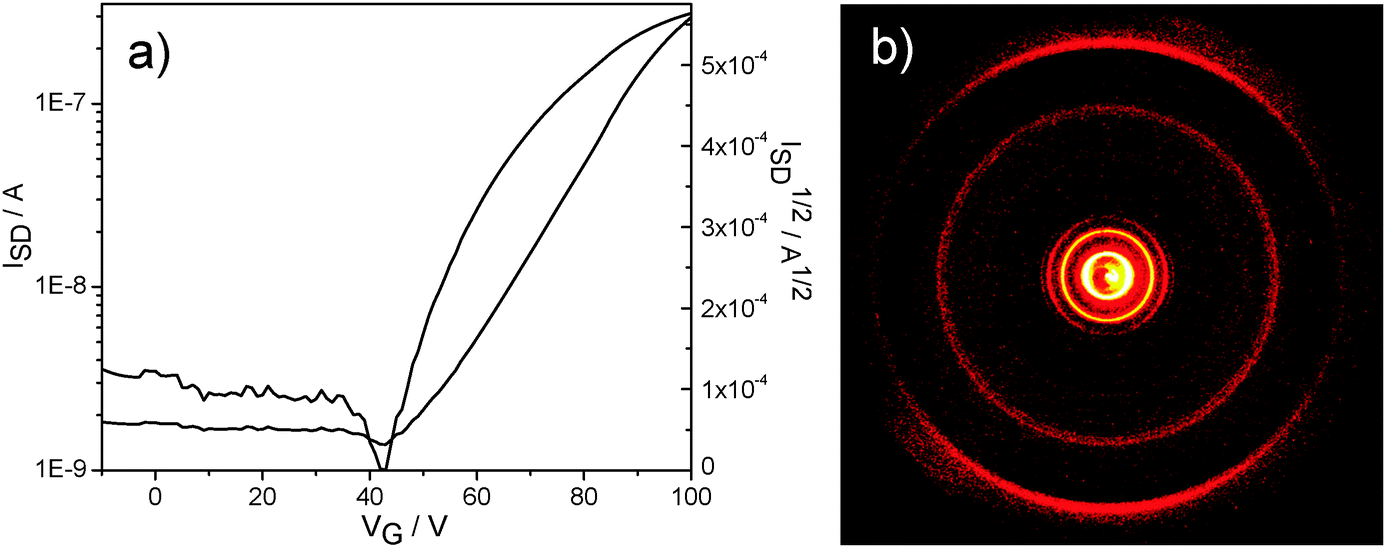 | ||
| Fig. 4 (a) Transistor transfer plot and (b) 2DWAXS at 100 °C of TQ5. | ||
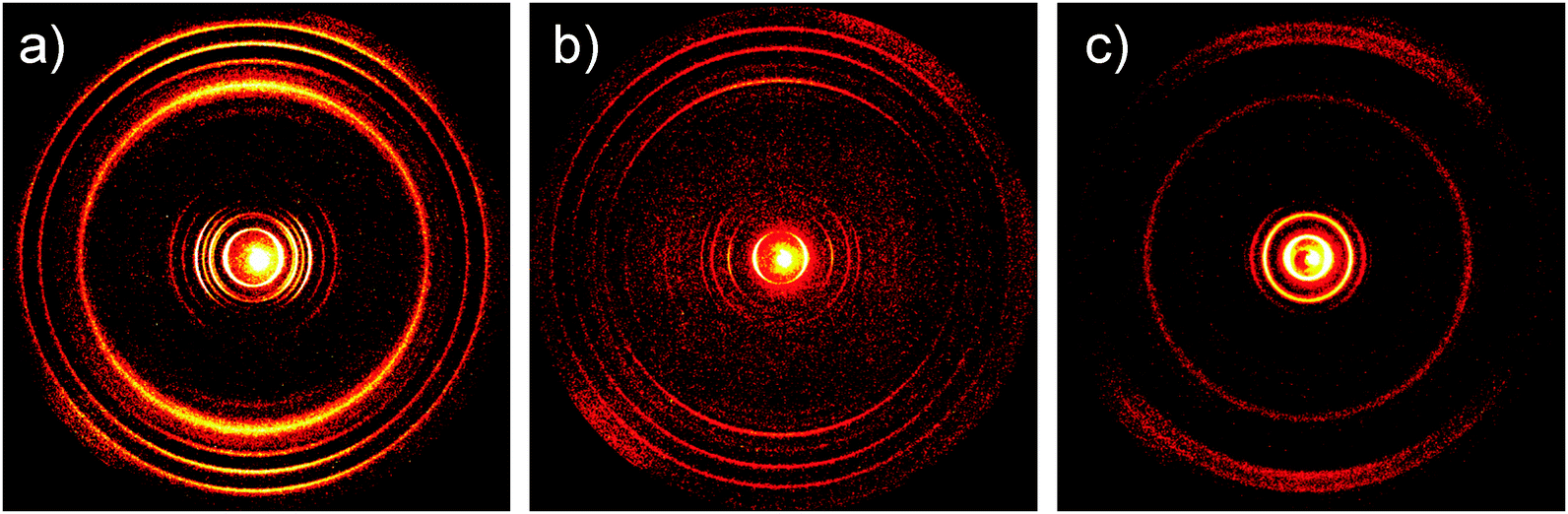 | ||
| Fig. 5 2DWAXS patterns of (a) TQ3, (b) TQ4 and (c) TQ5 recorded at 30 °C. | ||
4. Conclusions
In summary, three TQ derivatives with an extended planarized backbone were designed and synthesized. Their molecular optoelectronic properties were significantly affected by changing the fused aromatic ring in the TQ core. The extended π-conjugation led to much lower optical energy gaps and higher electron affinities than their non planarized counterparts. The lowest HOMO–LUMO gap was found for TQ4, the lowest EA value for TQ5 containing the electron deficient phenanthroline, which showed some electron mobilities and LC ordering. These three derivatives are valuable candidates as NIR absorbers and emitters. Even more the phenanthrene containing TQ3 showed useful implementation as phototransistor to be further optimized.13 The benzodithiophene derivative TQ4 allows polymerization along the condensed thiophenes,14 which may be interesting for photovoltaics, and the phenanthroline condensed TQ5 is a very important ligand for metal complexations towards triplet emitters and may be applied as sensing reagent for metal ions. Furthermore, these findings promote these derivatives as strong acceptor building blocks for the construction of conjugated polymers for organic electronics applications.Acknowledgements
S.Z. wants to thank the Chinese Academy of Sciences for a scholarship and his project leader and supervisor Hao Pang, Bing Liao from the Key Laboratory of Cellulose Chemistry, Guangzhou Institute of Chemistry, Chinese Academy of Sciences, Guangzhou 510650, People's Republic of China. C. A. gratefully acknowledges the China Scholarship Council (CSC) for offering a 3 years Scholarship. X.G. is grateful for an Alexander von Humboldt fellowship. Financial support by the SFB TR49 and the European Union within the EC FP7 ONE-P Project 212311 is gratefully acknowledged.References
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570 CrossRef CAS.
- (a) X. Wang, E. Perzon, W. Mammo, F. Oswald, S. Admassie, N.-K. Persson, F. Langa, M. Anderson and O. Inganäs, Thin Solid Films, 2006, 511, 576 CrossRef PubMed; (b) E. Perzon, F. Zhang, M. Andersson, W. Mammo, O. Inganäs and M. R. Andersson, Adv. Mater., 2007, 19, 3308 CrossRef CAS PubMed.
- (a) M. X. Chen, X. Crispin, E. Perzon, M. R. Andersson, T. Pullerits, M. Andersson, O. Inganäs and M. Berggren, Appl. Phys. Lett., 2005, 87, 252105 CrossRef PubMed; (b) T. Dallos, D. Beckmann, G. Brunklaus and M. Baumgarten, J. Am. Chem. Soc., 2011, 133, 13898 CrossRef CAS PubMed.
- M. X. Chen, E. Perzon, N. Robisson, S. K. M. Jönsson, M. R. Andersson, M. Fahlman and M. Berggren, Synth. Met., 2004, 146, 233 CrossRef CAS PubMed.
- J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS PubMed.
- S. C. Rasmussen, R. L. Schwiderski and M. E. Mulholland, Chem. Commun., 2011, 47, 11394 RSC.
- H. Li, T. L. Tam, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 46 CrossRef CAS PubMed.
- (a) C. Qin, Y. Fu, C.-H. Chui, A.-W. Kan, Z. Xie, L. Wang and W.-Y. Wong, Macromol. Rapid Commun., 2011, 32, 1472 CrossRef CAS PubMed; (b) J. Hai, G. Shi, J. Yu, E. Zhu, L. Bian, W. i. Ma and W. Tang, New J. Chem., 2014, 38, 4816 RSC.
- (a) K. Müllen and G. Wegner, Electronic Materials: The Oligomer Approach, Wiley-VCH, Weinheim, Germany, 1998 Search PubMed; (b) B. P. Karsten, L. Viani, J. Gierschner, J. Cornil and R. A. J. Janssen, J. Phys. Chem. A, 2009, 113, 10343 CrossRef CAS PubMed; (c) T. Dallos, M. Hamburger and M. Baumgarten, Org. Lett., 2011, 13, 1936 CrossRef CAS PubMed; (d) J. P. Nietfeld, R. L. Schwiderski, T. P. Gonnella and S. C. Rasmussen, J. Org. Chem., 2011, 76, 6383 CrossRef CAS PubMed.
- (a) A. P. Zoombelt, M. Fonrodona, M. M. Wienk, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Org. Lett., 2009, 11, 903 CrossRef CAS PubMed; (b) E. Perzon, X. Wang, S. Admassie, O. Inganäs and M. R. Andersson, Polymer, 2007, 47, 4261 CrossRef PubMed.
- A. Meyer, E. Sigmund, F. Luppertz, G. Schnakenburg, I. Gadaczek, T. Bredow, S.-S. Jester and S. Höger, Beilstein J. Org. Chem., 2010, 6, 1180 CrossRef CAS PubMed.
- J. P. Nietfeld, R. L. Schwiderski, T. P. Gonnella and S. C. Rasmussen, J. Org. Chem., 2011, 76, 6383 CrossRef CAS PubMed.
- M. Li, C. An, T. Marscalek, X. Guo, Y. Long, H. Yin, C. Gu, M. Baumgarten, W. Pisula and K. Müllen, Chem. Mater., 2015, 27, 2218 CrossRef CAS.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, MALDI-TOF, IR, MW reaction profiles, and 2D-WAXS integration. See DOI: 10.1039/c5nj00517e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |