
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect preparation and conversion of copper hydroxide-based monolithic xerogels with hierarchical pores†
Shotaro
Fukumoto
,
Kazuki
Nakanishi
* and
Kazuyoshi
Kanamori
Department of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: kazuki@kuchem.kyoto-u.ac.jp; Fax: +81 757 532 925; Tel: +81 757 532 925
First published on 4th June 2015
Abstract
Copper hydroxide-based monolithic xerogels with controlled hierarchical pores have been prepared directly by a sol–gel process from an ionic precursor, CuCl2·2H2O. Propylene oxide acts as a gelation inducer by increasing pH homogeneously in a reaction solution. Poly(acrylamide) is utilized not only to control macroscopic phase separation but also to support the network physically. Glycerol contributes to the formation of monolithic gels by suppressing the growth of copper hydroxide crystals. An appropriate starting composition leads to co-continuous gel skeletons and macropores. Although the as-dried gels were amorphous, post-treatments (calcination and solvothermal treatment) formed metallic copper and copper oxides (CuO and Cu2O) without the collapse of the monolithic form and macrostructure.
Introduction
Porous materials are used in various applications such as catalysis, adsorption, separation, electrodes, and insulation, and play a crucial role in the development of materials for a sustainable society.1–4 Although several material forms such as particles and films are widely studied, monolithic porous materials are attracting increasing attention due to their easy handling and potential for, for example, separation media5,6 and efficient column-type contact devices such as monolithic flow-through reactors.7,8 Recently, there have been many preparation methods of monolithic porous materials such as nanocasting,9 foaming,10,11 replication/sacrificial templating,11 and sol–gel processing accompanied by phase separation.5,12 A typical nanocasting method uses a hard silica template and the silica has to be removed under severe conditions using reagents such as a strong base and hydrofluoric acid.9 A sol–gel process accompanied by phase separation, which has been developed in sol–gel systems derived from alkoxysilanes, enables the spontaneous formation of macroporous monoliths and there is no need to use harmful reagents or a template. In this method, controlled phase separation is achieved in the course of hydrolysis and polycondensation of a precursor and a well-defined co-continuous macroporous structure can be obtained by freezing the temporally coarsening structure during spinodal decomposition by a sol–gel transition.12 Macroporous morphology can be tuned predominantly via the concentration of a phase separation inducer, e.g. water-soluble polymers and surfactants. Mesopores, which increase the surface area, can be added to macroporous skeletons by a dissolution–reprecipitation-based post-treatment13 (Ostwald ripening) and/or soft templating using a structure-directing agent.14 In addition, micropores can also be tailored by selecting microporous frameworks such as activated carbons and zeolites.15–17For the more rational design of porous materials with desired chemical compositions, furthermore, diversity of derivatives would be another key point. In other words, the parent material should be transformed into different compounds/composites by post-treatment such as solvothermal treatment and calcination in a controlled atmosphere. We have reported metal (oxy)hydroxide-based porous monoliths (the metal species include titanium,18,19 aluminum,20 zirconium,21 iron,22 nickel,23 and chromium24) that can be converted into oxygen-deficient metal oxides,25,26 elemental metals, metal carbides, metal nitrides, and their composites with carbons. Many of these metal oxide-related materials have been obtained by an extension of an epoxide-mediated sol–gel process from ionic precursors,27–29 rather than via highly reactive metal alkoxide-derived systems. An epoxide-mediated sol–gel process utilizes an irreversible epoxide opening reaction, which consumes protons in a solution to swiftly and uniformly raise the solution pH. Hydrolysis and condensation of metal aquo cations occur simultaneously and metal (oxy)hydroxide networks are formed as a result.27–29 Further development of this strategy would allow us to prepare macroporous monoliths of various trivalent metal oxides, divalent metal oxides and mixed metal oxides. In comparison to previous works, however, it is increasingly difficult to prepare copper-based monolithic gels, because coordination polyhedra of divalent cations are less stable than those of trivalent cations30 and it is hard to control the rate of precipitation–sedimentation against that of the formation of a monolithic gel network.
Copper-based materials have been utilized in various applications such as catalysts31–33 and semiconductors.34 Control over the valence state and composition of copper-based materials is particularly important in practical applications, including the abovementioned examples. Porous copper oxide materials reported in previous works35 are presumed to be aggregates of precipitates rather than homogeneously crosslinked monoliths (after being used as a catalyst, centrifugation was required to separate the material, which was probably due to its very low mechanical strength) and their macropores are uncontrolled. In this work, we report the direct preparation of porous copper hydroxide-based monolithic xerogels with controlled macro/mesopores and their conversion to copper-based materials with various valence states from Cu(0) to Cu(II). The copper hydroxide-based porous monolithic materials were successfully prepared with the aid of organic additives that prevent undesirable crystallization and reinforce the network.22–24 The effects of changes in the starting composition and the role of one of the additives (glycerol) in gel morphology and crystal growth have been investigated in detail. Heat treatment in oxidative and inert atmospheres at different temperatures has been found to yield different crystalline phases, e.g. metallic copper and copper oxides (CuO and Cu2O). In addition, solvothermal treatment in ethylene glycol has also caused reduction to metallic copper under milder conditions. Pore properties and monolithicity are also influenced by these post-treatment processes.
Experimental
Synthesis
Copper(II) chloride dihydrate (CuCl2·2H2O: Sigma-Aldrich Co., USA, 98%) was used as the copper source. Distilled water, ethanol (Kishida Chemical, Japan, ≥99.5%), and glycerol (GLY: Kishida Chemical, Japan, ≥99.0%) were used as the solvent mixture. Polyacrylamide (PAAm: Sigma-Aldrich Co., USA, 50 wt% in water) with an average molecular weight of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 was used to control macroscopic phase separation. Propylene oxide (PO: Sigma-Aldrich Co., USA, ≥99%) was added to promote gelation by enhancing the hydrolysis of copper aquo ions. To wash the as-prepared samples, 2-propanol (IPA: Kishida Chemical, Japan, ≥99.0%) was used. Ethylene glycol (EG: Kishida Chemical, Japan, ≥99.5%) was used for solvothermal treatment.
000 was used to control macroscopic phase separation. Propylene oxide (PO: Sigma-Aldrich Co., USA, ≥99%) was added to promote gelation by enhancing the hydrolysis of copper aquo ions. To wash the as-prepared samples, 2-propanol (IPA: Kishida Chemical, Japan, ≥99.0%) was used. Ethylene glycol (EG: Kishida Chemical, Japan, ≥99.5%) was used for solvothermal treatment.
Sample gels were prepared with the starting compositions listed in Table 1. Firstly, 1.53 g CuCl2·2H2O and WPAAm g aqueous PAAm solution were dissolved in a mixture of Vwater mL distilled water, Vethanol mL ethanol and VGLY mL glycerol. Then, 1.47 mL PO was added under stirring at ambient temperature (25 °C). After stirring for 1 min, the homogeneous solution thus prepared was sealed and kept at 30 °C for gelation. The wet gels that were typically obtained after 10–15 min were aged at 30 °C for 24 h, immersed in IPA at 60 °C for 24 h three times and then dried by evaporation at 60 °C.
| Sample ID | CuCl2·2H2O/g | Watera/mL (total) | Ethanol/mL | GLY/mL | PAAmb/g (absolute) | PO/mL |
|---|---|---|---|---|---|---|
| a PAAm is 50 wt% in water, so the total volume of water is Vwater + WPAAm × 1/2, assuming the density of water is ∼1 g cm−3. b PAAm is 50 wt% in water, so the absolute weight of PAAm is WPAAm × 1/2. | ||||||
| C1 | 1.53 | 1.1 | 0.3 | 2.4 | 0.5 | 1.47 |
| C2 | 1.53 | 1.1 | 0.3 | 2.4 | 0.6 | 1.47 |
| C3 | 1.53 | 1.1 | 0.3 | 2.4 | 0.7 | 1.47 |
| C4 | 1.53 | 1.1 | 0.3 | 2.4 | 0.8 | 1.47 |
| C5 | 1.53 | 1.1 | 0.3 | 2.4 | 0.9 | 1.47 |
| C6 | 1.53 | 1.2 | 0.3 | 2.4 | 0.6 | 1.47 |
| C7 | 1.53 | 1.2 | 0.3 | 2.4 | 0 | 1.47 |
| C8 | 1.53 | 3.6 | 0.3 | 0 | 0.6 | 1.47 |
| C9 | 1.53 | 3.6 | 0.3 | 0 | 0 | 1.47 |
Some samples were calcined at different temperatures for 4 h at a heating rate of 4 °C min−1 in an air or argon stream at a rate of 1 L min−1 in an electric furnace. Some samples that were calcined in argon were further calcined at 400 °C for 4 h at a heating rate of 4 °C min−1 in air to remove remnant carbons.
Some wet gel samples that were immersed in ethylene glycol after gelation were subjected to solvothermal treatment in an autoclave heated to 180 °C for various durations up to 6 h.
Characterizations
The morphology of the dried gels and heat-treated gels was observed by a scanning electron microscope (SEM: JSM-6060, JEOL Ltd., Japan, with Pt coating). Thermogravimetry-differential thermal analysis (TG-DTA: Thermo Plus TG 8120, Rigaku Corporation, Japan) up to 600 °C was performed at a heating rate of 5 °C min−1 while continuously supplying air at a rate of 100 mL min−1. Molecular-level information on the sample was obtained by Fourier transform infrared spectroscopy (FT-IR: IR Affinity-1, Shimadzu Corp., Japan) using the conventional pellet technique with potassium bromide (KBr); spectra were collected with 100 scans over the wavenumber range 400–4000 cm−1 at a resolution of 2 cm−1. X-ray diffraction (XRD) analysis was carried out with a RINT system (2θ from 10° to 90°, 1.6 kW, Cu Kα: λ = 0.154 nm, RINT-Ultima III, Rigaku Corp., Japan). Micro/mesopores were characterized by the nitrogen adsorption–desorption technique (BELSORP-mini II, Bel Japan Inc., Japan). Before measurement, each sample was degassed at 80 °C under vacuum for more than 4 h. Surface areas were obtained by the Brunauer–Emmett–Teller (BET) method.Results and discussion
Effect of PAAm on gel morphology
The starting solutions with the compositions listed in Table 1 were homogeneous, transparent and green in color. The addition of PO quickly resulted in the formation of copper hydroxide-based gels (∼13 min for C3),36,37 whereas no gelation was observed without addition of PO.Fig. 1(a) shows the appearance of a dried gel and Fig. 1(b)–(f) show SEM images of dried gels prepared with varying values of WPAAm. The total volume of water, (Vwater + WPAAm × 1/2, assuming the density of water is ∼1 g cm−3), was set at 1.1 mL for C1 to C5.
 | ||
| Fig. 1 Appearance of the resulting dried gel (a), SEM images of samples prepared with varying starting compositions: (b) C1, (c) C2, (d) C3, (e) C4, and (f) C5. Detailed starting compositions are listed in Table 1. | ||
In the absence of PAAm, no gelation was observed but dispersed precipitates were formed. On the other hand, with PAAm monolithic gels were formed and their morphologies varied depending on the value of WPAAm. In C1, the gel skeleton exhibits a co-continuous structure, which consists of similar-sized globular units (Fig. 1(b)), whereas the appropriate value of WPAAm leads to co-continuous gel skeletons with smoother surfaces (C2, Fig. 1(c)). A further increase in WPAAm makes the size of macropores smaller (C3, Fig. 1(d) and C4, Fig. 1(e)), which means PAAm suppresses the progress of phase separation. The addition of an excessive amount of PAAm resulted in morphology with isolated macropores (C5, Fig. 1(f)). Moreover, with an increasing amount of PAAm, the volume fraction of macropore skeletons are increased, which indicates that PAAm is preferentially incorporated in the skeletons.
For the purpose of clarifying the role of PAAm, we carried out thermal analysis and molecular-level structural analysis, by which the distribution of PAAm between the gel phase and the solvent phase was determined. TG-DTA curves of gels prepared with a smaller amount of PAAm (C1) and a larger amount (C5) are shown in Fig. 2(a). The curves show that the weight loss of the gel exhibits a reasonable correlation with the amount of PAAm contained in the starting composition. Exothermic peaks around 200 °C and 350 °C are ascribed to the combustion of glycerol and PAAm, respectively.22,38 The FT-IR spectrum of C6 is shown in Fig. 2(b). The presence of PAAm in the gel skeletons is confirmed by the peaks that are derived from the primary amide group of PAAm: deformation vibrations of –NH groups (1200 cm−1), stretching vibrations of C–N bonds (1450 cm−1), bending vibrations of N–H bonds (1604 cm−1), stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups (1658 cm−1), and stretching vibrations of N–H bonds (3186 cm−1).39–42
O groups (1658 cm−1), and stretching vibrations of N–H bonds (3186 cm−1).39–42
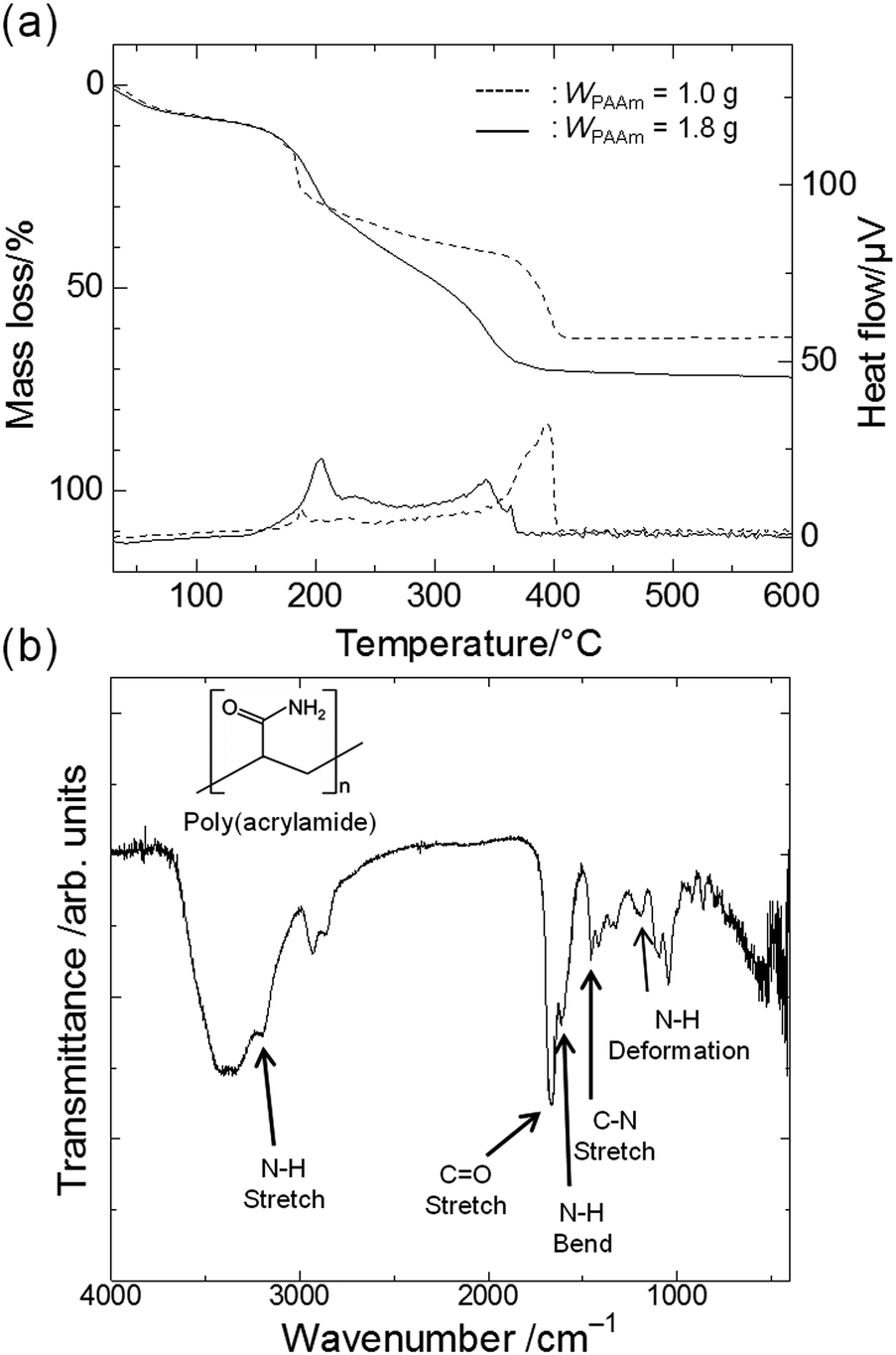 | ||
| Fig. 2 (a) TG-DTA curves of the gel phase of dried samples prepared with a smaller amount of PAAm (C1, dashed lines) and a larger amount of PAAm (C5, solid lines). (b) FT-IR spectrum of C6. | ||
The homogeneous solution separates into two phases, one rich in a PAAm–copper(II) hydroxide-based composite (gel phase) and the other rich in the solvent mixture (solvent phase), and the sol–gel transition arrests the transient phase-separating structure.12 Both PAAm and copper(II) hydroxide can interact with each other strongly, by hydrogen bonds between amide groups and hydroxyl groups or by coordinate bonds between nitrogen atoms in amide groups and copper ions.43 After removal of the solvent by evaporative drying, the spaces occupied by the solvent phase become macropores. It was previously reported that in iron(III) hydroxide systems PAAm acts not only to control phase separation but also to physically support the network.22 Also in the present case, PAAm plays a double role, similarly to the case in iron(III) hydroxide systems.
Effect of glycerol on crystallization
Fig. 3(a) shows XRD patterns of the samples prepared with glycerol and PAAm (C6, i), with glycerol and without PAAm (C7, ii), without glycerol and with PAAm (C8, iii), and without glycerol or PAAm (C9, iv). C6 is a monolithic gel with co-continuous macropores (Fig. 4c(i)). The total volume of solvent, (Vwater + WPAAm × 1/2 + VGLY), was set at 3.6 mL for C6 to C9.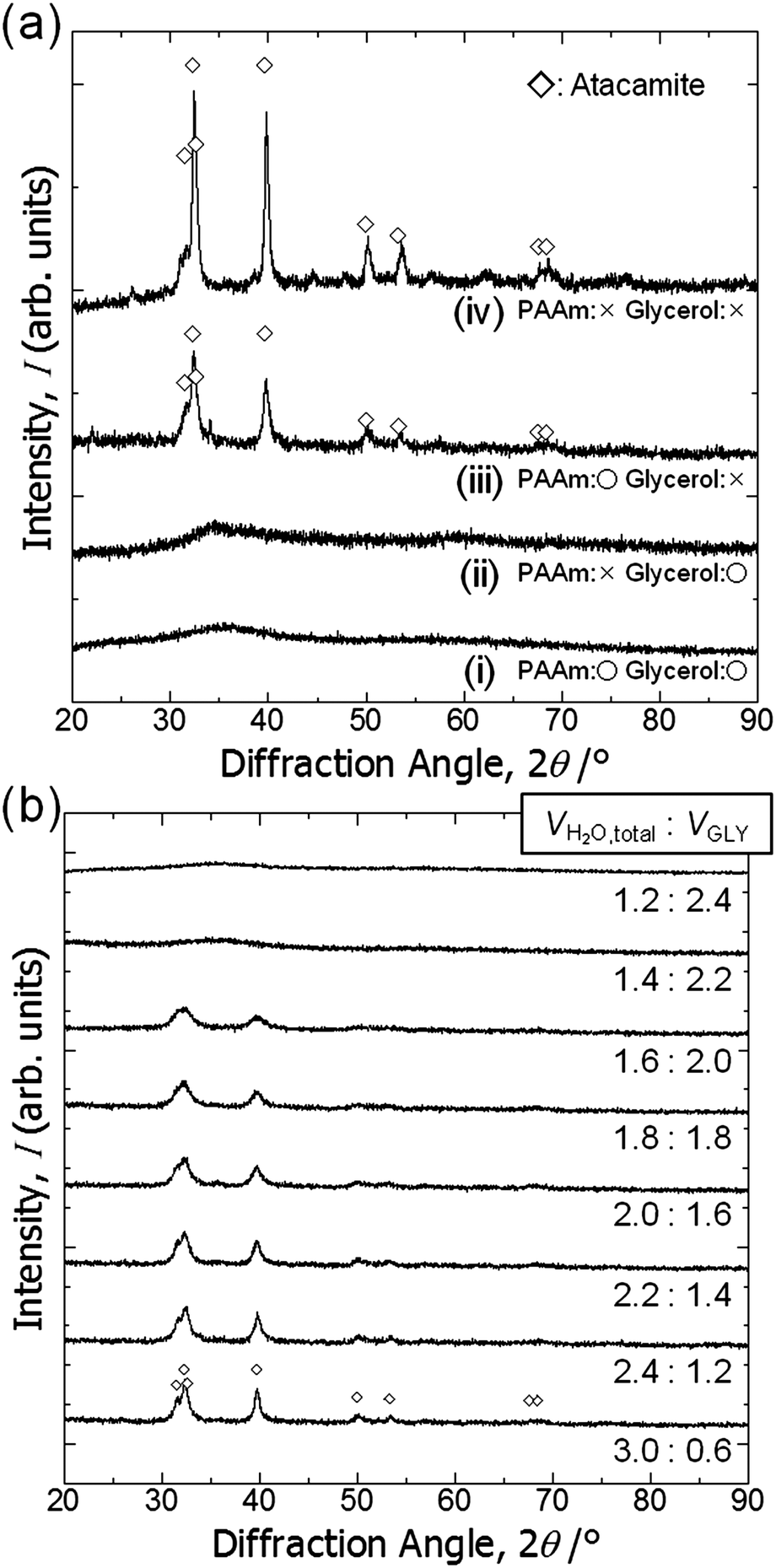 | ||
| Fig. 3 (a) X-ray diffraction patterns of the samples prepared with or without glycerol and PAAm: (i) C6, (ii) C7, (iii) C8, and (iv) C9. (b) X-ray diffraction patterns of the samples prepared with varying VH2O:VGLY ratios. | ||
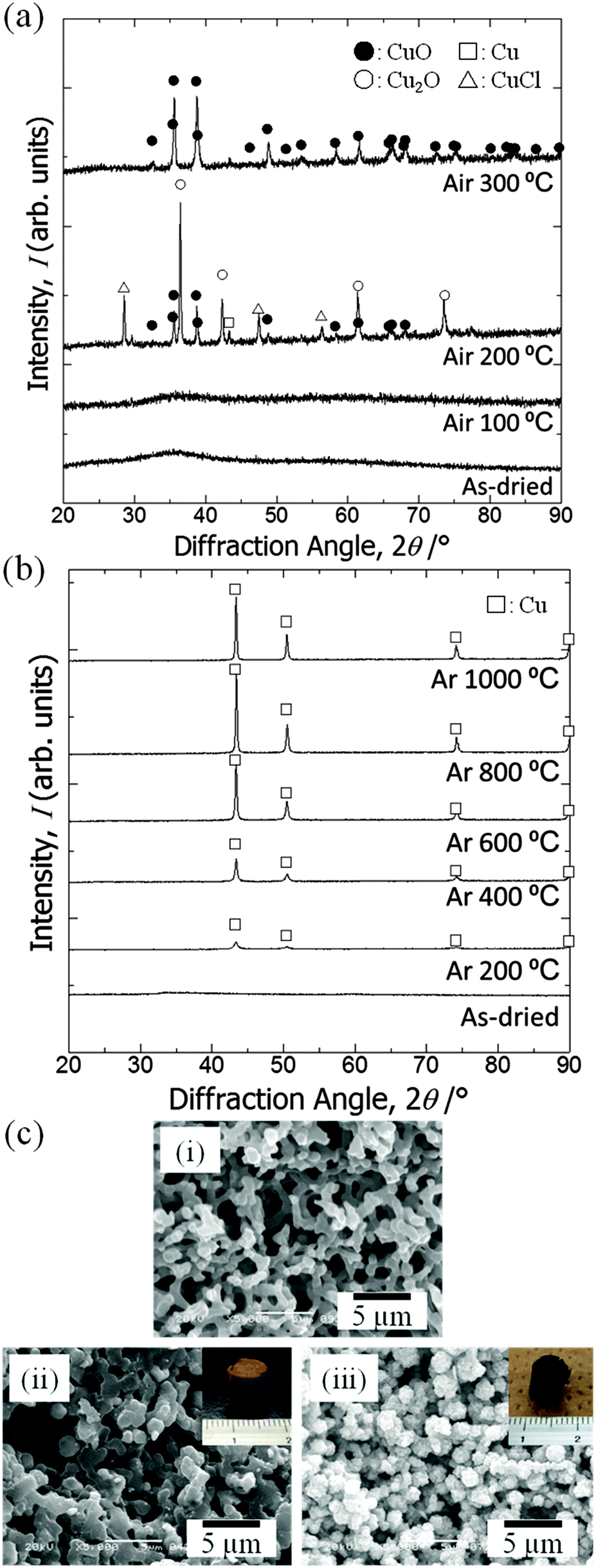 | ||
| Fig. 4 X-ray diffraction patterns of the samples: (a) as-dried and calcined in air at different temperatures, (b) as-dried and calcined in argon at different temperatures. Symbols indicate open squares: Cu [PDF #00-004-0836], open triangles: CuCl [PDF #01-081-1841], closed circles: CuO [PDF #00-048-1548], and open circles: Cu2O [PDF #01-073-6371], respectively. (c) SEM images of the samples: (i) as-dried, (ii) calcined in argon at 800 °C and (iii) calcined in argon at 800 °C and successively in air at 400 °C. Insets show the appearance of each sample. | ||
Regardless of the presence of PAAm, no obvious diffraction peaks are found for the samples prepared with glycerol. It should be pointed out that the crystalline phase is atacamite (Cu2(OH)3Cl) [PDF #00-023-0948], which suggests a considerable amount of Cl− is incorporated in the original gel structure. This means that some chloride anions are coordinated to copper cations and do not react with PO. Fig. 3(b) shows XRD patterns of samples prepared with varying ratios of Vwater and VGLY, setting the total volume of solvents, (VH2O + WPAAm × 1/2 + VGLY), constant at 3.6 mL. With an increase in VGLY the diffraction peaks change into peaks with lower intensity and larger width and no obvious peaks are found for samples prepared with more than 2.2 mL glycerol, which indicates that glycerol prevents crystal growth. Samples prepared with less than 2.0 mL glycerol are precipitates but do not form a monolith, whereas with more than 2.2 mL glycerol monolithic wet gels have been obtained. In addition, atacamite is known to form plate-like crystals, which means that the suppression of crystal growth by glycerol allows the network to expand in three dimensions and monolithic gels are formed. Zhao et al. also reported the suppression of crystal growth in magnetites by polyols, which can be attributed to limited dissolution–reprecipitation and binding of polyols on the crystal surface.44
Crystal phase transition by calcination
XRD patterns of samples calcined at varying calcination temperatures in air are shown in Fig. 4(a). Calcination was performed on the gel prepared with the starting composition C6. The as-dried gel exhibits no obvious diffraction peaks, which indicates that it only contains amorphous and/or nanocrystalline phases that are undetectable by XRD. After calcination in air at 100 °C the gel remains amorphous, whereas at 200 °C diffraction peaks from mixed phases appear and at 300 °C peaks ascribed to copper(II) oxide become evident. After calcination at 200 °C or 300 °C the monolithic form was not preserved. We assume that the burn-off of PAAm, which had firmly supported the gel skeletons via strong interactions, has resulted in collapse of the monolithic form of the gel samples. Fig. 4(b) shows XRD patterns of samples calcined at varying temperatures in argon. Again, calcination was performed on C6 gels. At 200 °C a metallic copper phase is recognized.45 As the calcination temperature is increased, the peaks become sharper and stronger until 800 °C. At 1000 °C, i.e. near the melting point of copper, growth of peaks is no longer seen, which is probably due to the partial melting of grown copper crystals. Although calcination in argon causes overall shrinkage, complete burn-off of PAAm is prevented and instead carbonized materials are left within the skeleton. Because of the uniform distribution of carbon in the skeletons, the monolithic form and microstructure have been preserved (Fig. 4c(ii)).22–24 Carbons that are derived from the organic species not only act to support the network but also as a reducing agent, which leads to the metallic copper phase.45 The absence of bulk electrical conductivity in the metallic copper–carbon composite thus prepared indicates that the copper phase exists in a non-percolating form. In order to confirm the morphological and compositional changes, we calcined samples first in argon at 800 °C, followed by in air at 400 °C to burn off carbons. The gel skeletons are considered to be densified after shrinkage during calcination in argon, which is accompanied by the collapse of mesopores (Fig. 5). As a result, the monolithic form has been preserved (Fig. 4c(iii)) in the sample, which consists of copper(II) oxide (Fig. S1, ESI†). In this case, almost all carbon was burnt off (Fig. S2, ESI†). By successive heat treatment, macroporous monoliths of copper–carbon composite and copper(II) oxide can be selectively prepared.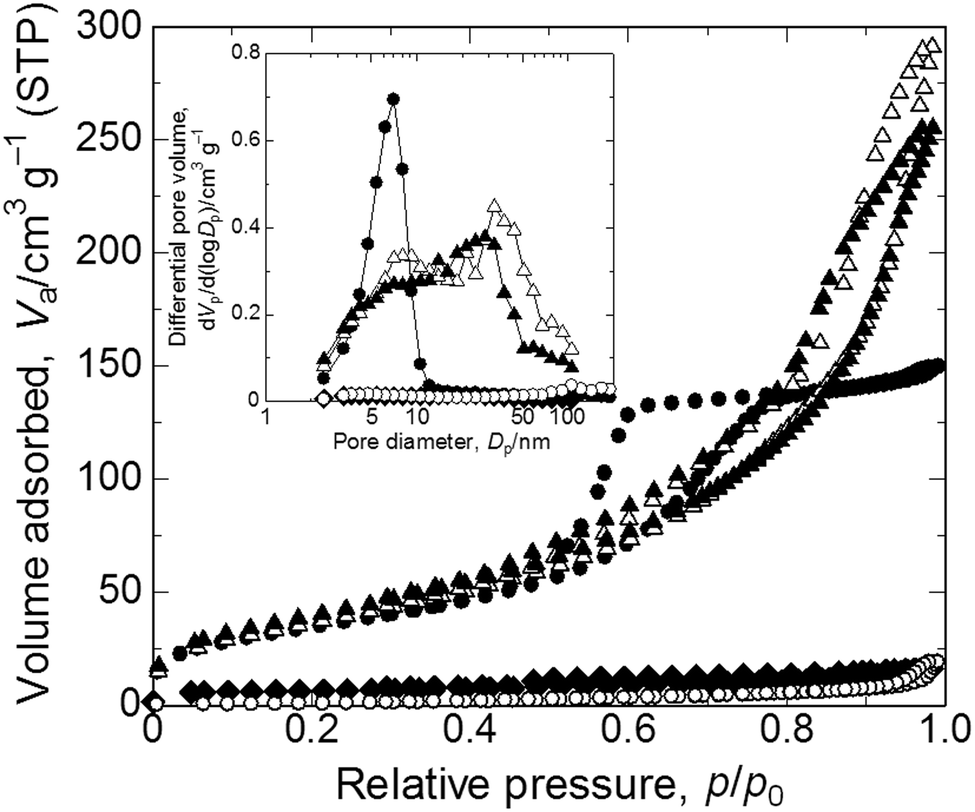 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms of the as-dried and heat-treated samples. Symbols indicate closed circles: as-dried, closed rhombuses: calcined in argon at 800 °C, open circles: calcined in argon at 800 °C followed by in air at 400 °C, open triangles: solvothermally treated for 2 h, and closed triangles: solvothermally treated for 6 h. Inset shows BJH pore distributions. Refer to Table 2 for details of pore parameters. | ||
| Symbol | Sample ID | Mesopore size/nm | S BET/m2 g−1 | Pore volume/cm3 g−1 | Constituent elements | Microstructure |
|---|---|---|---|---|---|---|
| ● | C6 | 7 | 127 | 0.24 | Amorphous atacamite/PAAm | Macro–mesoporous |
| ◆ | Ar800 | — | 21 | 0.02 | Copper(0)/carbon | Macroporous |
| ○ | Ar800 + Air400 | — | 5 | 0.03 | Copper(II) oxide | Macroporous |
| △ | EG2 | 33 | 138 | 0.46 | Copper(I) oxide/PAAm | Macro–mesoporous |
| ▲ | EG6 | 28 | 149 | 0.40 | Copper(0)/PAAm | Macro–mesoporous |
Solvothermal treatment
As samples calcined in argon at 200 °C contain a copper(0) phase, we assumed that copper ions in the as-dried samples are in a state that is easily reduced. We then tried solvothermal treatment, which is a milder reducing process,46–48 in ethylene glycol as the reducing agent to obtain macroporous metallic Cu and Cu2O monoliths.49,50 As a result, we could not only preserve the monolithic form and microstructure (Fig. 6(a)) but also obtain reduced states of copper, depending on the duration of the treatment (Fig. 6(b)). Unlike in calcination, because apparent shrinkage does not occur, mesopores of the samples are well preserved (Fig. 5). This is therefore a superior process for obtaining hierarchically macro- and mesoporous copper(I) oxide–PAAm composites and metallic copper–PAAm composite gels.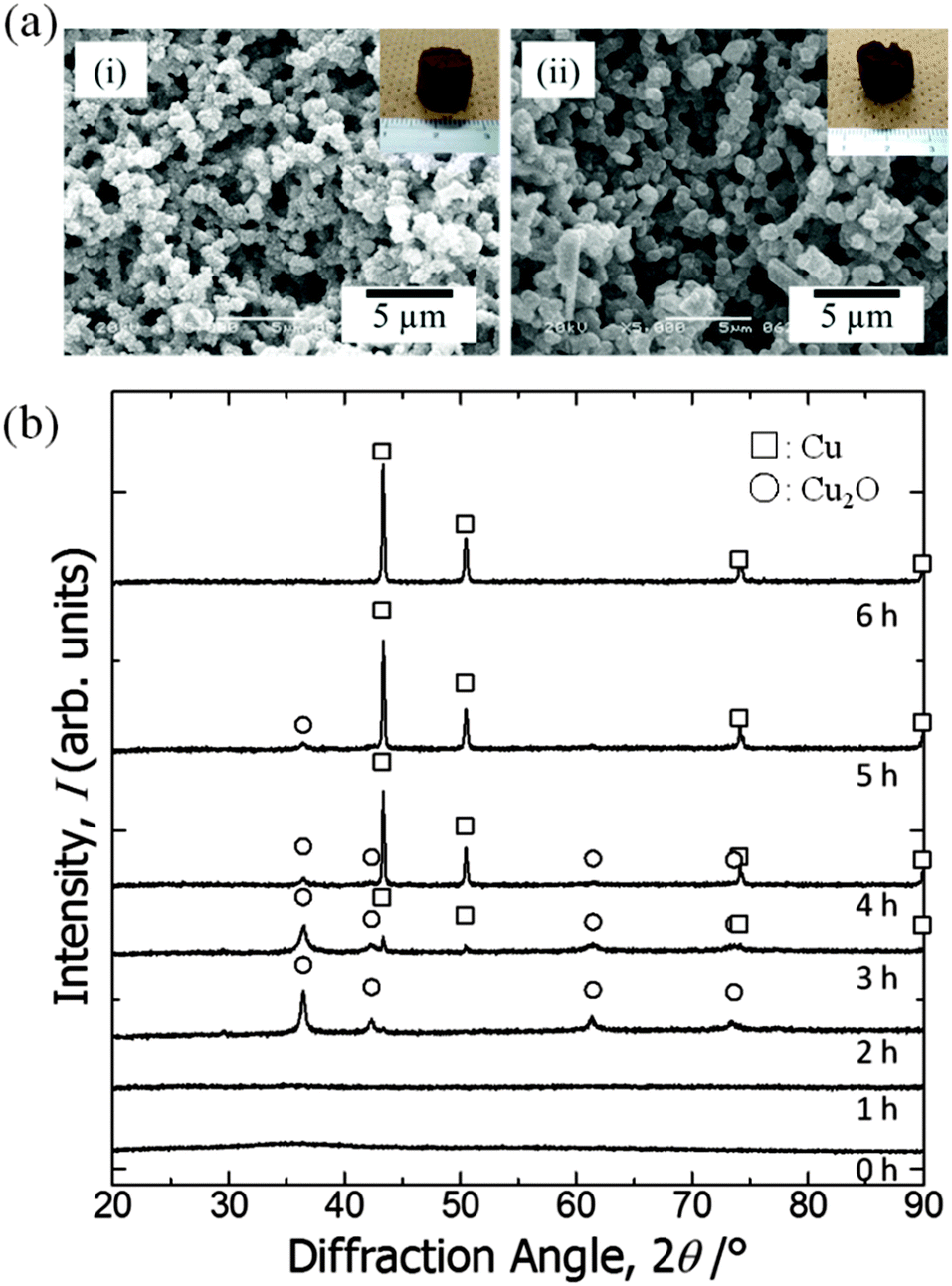 | ||
| Fig. 6 (a) SEM images of the samples treated in solvothermal conditions in ethylene glycol for various durations: (i) 2 h (EG2 in Table 2) and (ii) 6 h (EG6). Insets in (i) and (ii) show the appearance of the samples. (b) X-ray diffraction patterns of the samples: as-dried and solvothermally treated in ethylene glycol for various durations. Symbols indicate open squares: Cu and open circles: Cu2O. | ||
Conclusions
Copper-based monolithic xerogels with macropores have been prepared using a copper salt as a precursor in an epoxide-mediated sol–gel process accompanied by phase separation. Glycerol in the starting solution is incorporated in the gel skeletons, and it suppresses the crystallization–precipitation of the copper hydroxide-based network and poly(acrylamide) (PAAm) is also incorporated in the gel skeletons and aids not only in controlling phase separation but also in physically supporting the network. Appropriate starting compositions allow the concurrent phase separation and gelation, which leads to the formation of macroporous gels with co-continuous structure. By calcination in air, a copper(II) oxide phase appears, whereas the monolithic form collapses. By calcination in argon, copper(0)/carbon macroporous monoliths have been obtained. The as-dried gels are reduced and a metallic copper phase appears while retaining the microstructure and monolithic form, whereas mesopores collapse due to shrinkage. Successive calcination in air of the samples calcined in argon results in crystallization to form copper(II) oxide while retaining the macroporous morphology and monolithic form (CuO macroporous monoliths). By solvothermal treatment in ethylene glycol, copper(0)/PAAm and copper(I) oxide/PAAm macro/mesoporous monoliths have been obtained while preserving most of the mesopores and monolithic form. Another important transformation of the as-dried gels into porous coordination polymers/metal–organic frameworks (PCPs/MOFs) is reported elsewhere, in parallel with the present paper.51 The present comprehensive strategy for producing monolithic porous materials with different copper-based phases ensures different applications such as in catalysis, electrochemical devices and separations.Notes and references
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481 RSC.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696 RSC.
- S. L. Candelaria, Y. Shao, W. Zhou, X. Li, J. Xiao, J. G. Zhang, Y. Wang, J. Liu, J. Li and G. Cao, Nano Energy, 2012, 1, 195 CrossRef CAS PubMed.
- K. Nakanishi and N. Tanaka, Acc. Chem. Res., 2007, 40, 863 CrossRef CAS PubMed.
- F. Svec, J. Chromatogr. A, 2010, 1217, 902 CrossRef CAS PubMed.
- G. Jas and A. Kirschning, Chem. – Eur. J., 2003, 9, 5708 CrossRef CAS PubMed.
- A. Sachse, A. Galarneau, F. Fajula, F. Di Renzo, P. Creux and B. Coq, Microporous Mesoporous Mater., 2011, 140, 58 CrossRef CAS PubMed.
- A. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793 CrossRef CAS PubMed.
- N. Brun, S. Ungureanu, H. Deleuze and R. Backov, Chem. Soc. Rev., 2011, 40, 771 RSC.
- A. R. Studart, U. T. Gonzenbach, E. Tervoort and L. J. Gauckler, J. Am. Ceram. Soc., 2006, 89, 1771 CrossRef CAS PubMed.
- K. Nakanishi, J. Porous Mater., 1997, 4, 67 CrossRef CAS.
- K. Nakanishi, R. Takahashi, T. Nagakane, K. Kitayama, N. Koheiya, H. Shikata and N. Soga, J. Sol-Gel Sci. Technol., 2000, 17, 191 CrossRef CAS.
- T. Amatani, K. Nakanishi, K. Hirao and T. Kodaira, Chem. Mater., 2005, 17, 2114 CrossRef CAS.
- Y. Tokudome, K. Nakanishi, S. Kosaka, A. Kariya, H. Kaji and T. Hanada, Microporous Mesoporous Mater., 2010, 132, 538 CrossRef CAS PubMed.
- G. Hasegawa, K. Kanamori, K. Nakanishi and T. Hanada, Carbon, 2010, 48, 1757 CrossRef CAS PubMed.
- G. Hasegawa, K. Kanamori, K. Nakanishi and T. Hanada, Chem. Commun., 2010, 46, 8037 RSC.
- J. Konishi, K. Fujita, K. Nakanishi and K. Hirao, Chem. Mater., 2006, 18, 6069 CrossRef CAS.
- G. Hasegawa, K. Kanamori, K. Nakanishi and T. Hanada, J. Am. Ceram. Soc., 2010, 93, 3110 CrossRef CAS PubMed.
- Y. Tokudome, K. Fujita, K. Nakanishi, K. Miura and K. Hirao, Chem. Mater., 2007, 19, 3393 CrossRef CAS.
- J. Konishi, K. Fujita, S. Oiwa, K. Nakanishi and K. Hirao, Chem. Mater., 2008, 20, 2165 CrossRef CAS.
- Y. Kido, K. Nakanishi, A. Miyasaka and K. Kanamori, Chem. Mater., 2012, 24, 2071 CrossRef CAS.
- Y. Kido, K. Nakanishi, N. Okumura and K. Kanamori, Microporous Mesoporous Mater., 2013, 176, 64 CrossRef CAS PubMed.
- Y. Kido, G. Hasegawa, K. Kanamori and K. Nakanishi, J. Mater. Chem. A, 2014, 2, 745 CAS.
- A. Kitada, G. Hasegawa, Y. Kobayashi, K. Kanamori, K. Nakanishi and H. Kageyama, J. Am. Chem. Soc., 2012, 134, 10894 CrossRef CAS PubMed.
- A. Kitada, G. Hasegawa, Y. Kobayashi, K. Miyazaki, T. Abe, K. Kanamori, K. Nakanishi and H. Kageyama, RSC Adv., 2013, 3, 7205 RSC.
- H. Itoh, T. Tabata, M. Kokitsu, N. Okazaki, Y. Imizu and A. Tada, J. Ceram. Soc. Jpn., 1993, 101, 1081 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher, Jr., J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher, Jr., L. W. Hrubesh and R. L. Simpson, J. Non-Cryst. Solids, 2001, 285, 22 CrossRef CAS.
- P. W. Atkins, T. L. Overton, J. P. Rourke, M. T. Weller and F. A. Armstrong, Inorganic Chemistry, Oxford University Press, Oxford, 2010, p. 123 Search PubMed.
- K. L. Hohn and Y. C. Lin, ChemSusChem, 2009, 2, 927 CrossRef CAS PubMed.
- S. Sá, H. Silva, L. Brandão, J. M. Sousa and A. Mendes, Appl. Catal., B, 2010, 99, 43 CrossRef PubMed.
- S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778 CrossRef CAS PubMed.
- E. Fortunato, P. Barquinha and R. Martins, Adv. Mater., 2012, 24, 2945 CrossRef CAS PubMed.
- G. A. Naikoo, R. A. Dar and F. Khan, J. Mater. Chem. A, 2014, 2, 11792 CAS.
- Y. Liu, W. Ren and H. Cui, Micro Nano Lett., 2011, 6, 823 CAS.
- A. Du, B. Zhou, J. Shen, S. Xiao, Z. Zhang, C. Liu and M. Zhang, J. Non-Cryst. Solids, 2009, 355, 175 CrossRef CAS PubMed.
- M. Tutas, M. Saglam, M. Yuksel and C. Guler, Thermochim. Acta, 1987, 111, 121 CrossRef CAS.
- S. Rajendran, B. V. Apparao and N. Palaniswamy, Electrochim. Acta, 1998, 44, 533 CrossRef CAS.
- H. Kasgöz, S. Özgümüş and M. Orbay, Polymer, 2003, 44, 1785 CrossRef.
- J. F. Zhu, Y. J. Zhu, M. G. Ma, L. X. Yang and L. Gao, J. Phys. Chem. C, 2007, 111, 3920 CAS.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Infrared Spectrometry: Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., New York, 2005, pp. 99–101 Search PubMed.
- B. L. Rivas, E. D. Pereira and I. Moreno-Villoslada, Prog. Polym. Sci., 2003, 28, 173 CrossRef CAS.
- L. Zhao, H. Zhang, Y. Xing, S. Song, S. Yu, W. Shi, X. Guo, J. Yang, Y. Lei and F. Cao, Chem. Mater., 2008, 20, 198 CrossRef CAS.
- B. V. L’vov, Thermochim. Acta, 2000, 360, 109 CrossRef.
- Y. Zheng, L. Zheng, Y. Zhan, X. Lin, Q. Zheng and K. Wei, Inorg. Chem., 2007, 46, 6980 CrossRef CAS PubMed.
- C. Nethravathi and M. Rajamathi, Carbon, 2008, 46, 1994 CrossRef CAS PubMed.
- X. M. Chen and M. L. Tong, Acc. Chem. Res., 2007, 40, 162 CrossRef CAS PubMed.
- S. J. Chen, X. T. Chen, Z. Xue, L. H. Li and X. Z. You, J. Cryst. Growth, 2002, 246, 169 CrossRef CAS.
- M. Wei, N. Lun, X. Ma and S. Wen, Mater. Lett., 2007, 61, 2147 CrossRef CAS PubMed.
- N. Moitra, S. Fukumoto, J. Reboul, K. Sumida, Y. Zhu, K. Nakanishi, S. Kitagawa, S. Furukawa and K. Kanamori, Chem. Commun., 2015, 51, 3511 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction patterns of the samples calcined in argon at 800 °C followed by calcination in air at 400 °C and TG-DTA curves of the sample calcined in argon at 800 °C and one calcined in argon at 800 °C followed by calcination in air at 400 °C. See DOI: 10.1039/c5nj00479a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |