
Copolymer and platinum ion assisted growth of functionalized gold nanonests
Jing-Cyuan
Yang
a,
Yi-Hsiang
Chang
a,
Wha-Tzong
Whang
a,
Chun-Hua
Chen
*a and
Ren-Jye
Wu
b
aDepartment of Materials Science and Engineering, National Chiao Tung University, Hsin-Chu, 30010, Taiwan, Republic of China. E-mail: ChunHuaChen@mail.nctu.edu.tw
bMaterial and Chemical Research Laboratories, Industrial Technology Research Institute, Hsin-Chu, 31040, Taiwan, Republic of China
First published on 6th November 2014
Abstract
This paper describes a facile low-toxicity synthesis of two different types of Au nanonests and one binary Au–Pt core–shell nanonest with high yield and excellent uniformity in size and morphology using block copolymer P123 (EO20PO70EO20) as a soft template and the main reductant in an ice water bath. The introduction of a suitable second metallic source, i.e. Pt ions in the present case, was proven to be a unique strategy for effectively governing the reduction sites of P123 for Au nucleation, which leads to significant changes especially in the crystallite size of the grown Au nanonests. The Au nanonests exhibited a distinct morphology-dependent surface plasmon resonance (SPR) that is red-shifted by ∼140 nm from the spherical particles wavelength, and significant surface-enhanced Raman scattering (SERS), allowing the rapid determination of rare malachite green oxalate (MGO).
Hollow-core metallic nanostructures having a high surface-area-to-volume ratio, structural rigidity and stability, internal storage space, and surface permeability have attracted growing interest as an important class of nanomaterials that exhibit unusual chemical and physical phenomena for a variety of potential applications such as catalysts, drug delivery vehicles, photonic crystals, and localized surface plasmon resonance (LSPR) or surface-enhanced Raman scattering (SERS) sensors.1–6
Among the relevant findings, hemispherical hollow structures, which typically display a container-like morphology, have been evolving into a new and interesting branch.7–11 The hemispherical geometry basically possesses advantages and possibilities similar to those considered for the spherical ones. But moreover, the particularly low morphological symmetry commonly leads to dissimilar anisotropic SPRs which are significantly geometry- and direction-dependent and complicated in comparison with the isotropic ones found in highly symmetrical objects.12,13 To date, the main strategy used to prepare the hemispherical hollow structures has focused on physical or chemical deposition of metallic materials on a variety of sacrificial templates.14–16 However, it still remains a challenge, mainly due to the difficulties in building suitable templates. The absence of effective synthesis strategies certainly limits the development of hemispherical hollow structures and the fundamental understanding of the corresponding anisotropic nano-optics.
In this work, we describe a completely new one-step copolymer-mediated strategy for controlled manufacturing of two types of mono-dispersed Au hemispherical nanoshells (denoted as nanonests) with tunable crystallite size and shell porosity and one distinct Au–Pt core–shell nanonest. It is worth pointing out that the present synthesis approach involves two key and essential concepts that have never been considered and reported before, namely creating hemispherical templates simply using block-copolymers and then simultaneously governing the nucleation sites of the formed soft templates by the addition of second metallic ions.
To realize the concepts, Pluronic P123 (PEO–PPO–PEO), one of the commercial tri-block copolymers, was introduced and expected to play multiple roles as a reductant, a protective agent, and also a morphology-tunable soft template in the present synthesis.17,18 Even though P123 has been experimentally confirmed to form isolated micelles, or lamellar, hexagonal, and cubic stacks, at specific concentration and temperature ranges in the aqueous solution,19 these self-assemblies obviously cannot be directly applied as hemispherical templates as needed. However, a slight aggregation of spherical micelles, which can be achieved by increasing the polymer concentration and lowering the temperature, as evidenced by our results, seems to be beneficial for forming hemisphere-like templates on top of the aggregates, which just look like a cluster of grapes. It is known that the hydrophilic PEO blocks in PEO–PPO–PEO form cavities to bind metal ions and then reduce them to metals through oxidation of their oxyethylene and oxypropylene groups.20–28
To adjust the Au nucleation sites of the formed soft templates, in addition to Au ions, we simultaneously introduce another metallic ion source, namely Pt ions, which will also be caught by the PEO blocks. However, in our experience, Pt ions cannot be reduced by PEO–PPO–PEO without any additional reductant. The nucleation sites will then be partially occupied by Pt ions, and therefore the Au nucleation sites as well as the crystal size and shell porosity of the fabricated Au nanonests will be effectively controlled.
Fig. 1(a) is the SEM image of the Au nanonests (I) (type I) formed at the temperature of an ice water bath in the absence of Pt ions (PtCl62−). Obviously, the Au nanonests (I) macroscopically exhibit similar nest-like morphologies with similar outer diameters of ∼300 nm and inner diameters of ∼150 nm. A closer look at the extremely porous nest shells confirms an assembly-like branching or granular structure comprising a number of irregular tiny nanocrystals. Almost the same structural characteristics can also be determined from the significant contrast between the lighter central part and the darker outer regions of the TEM images taken from two of the suspended Au nanonests (I) (see Fig. 1(b) or (c)). The broadened Au XRD peaks (see Fig. 1(f)) indicating the existence of tiny Au nanocrystals are consistent with the SEM and TEM observations. The average crystallite size of Au nanocrystals, nanonests (I) and (II) estimated using the Scherrer equation from a full-width at half-maximum (FWHM) of the (111) peak is 60, 15 and 20 nm, respectively. It is worth noting that such unique porous Au nanonests (I) can only be obtained at an approximate reduction temperature of 5 °C. When the reduction temperature increased from 5 °C to room temperature (25 °C), only faceted Au nanocrystals were found (see Fig. 1(d)). In contrast to the very tiny building blocks for the Au nanonests (I), the very large faceted Au nanocrystals led to rather sharp XRD peaks, as can be seen in Fig. 1(f). Fig. 1(e) shows the UV-visible absorption spectra of the prepared Au nanonests (I) and the faceted Au nanocrystals. Significantly, in addition to the sharper absorption peak at around 560 nm typical for spherical Au nanocrystals, a very broad absorption band from ∼700 nm to over 1100 nm (the maximum absorption is located at ∼850 nm) evidently originates from the nest-like morphology of the Au nanonests (I). Similar absorption spectra have also been reported previously for a variety of hollow Au spheres.29 The absorption spectrum recorded from the faceted Au nanocrystals is in good agreement with the calculated spectrum, in which Au solid spheres with a mean diameter of ∼150 nm suspended in water were assumed. As described above, Au and Pt ions should be simultaneously caught by the PEO blocks, but only Au can be reduced at such a low temperature (5 °C) in the absence of any reductants. Pt ions are thus used as caps to control the amount of nucleation sites for Au ions in the PEO–PPO–PEO tri-block polymers.
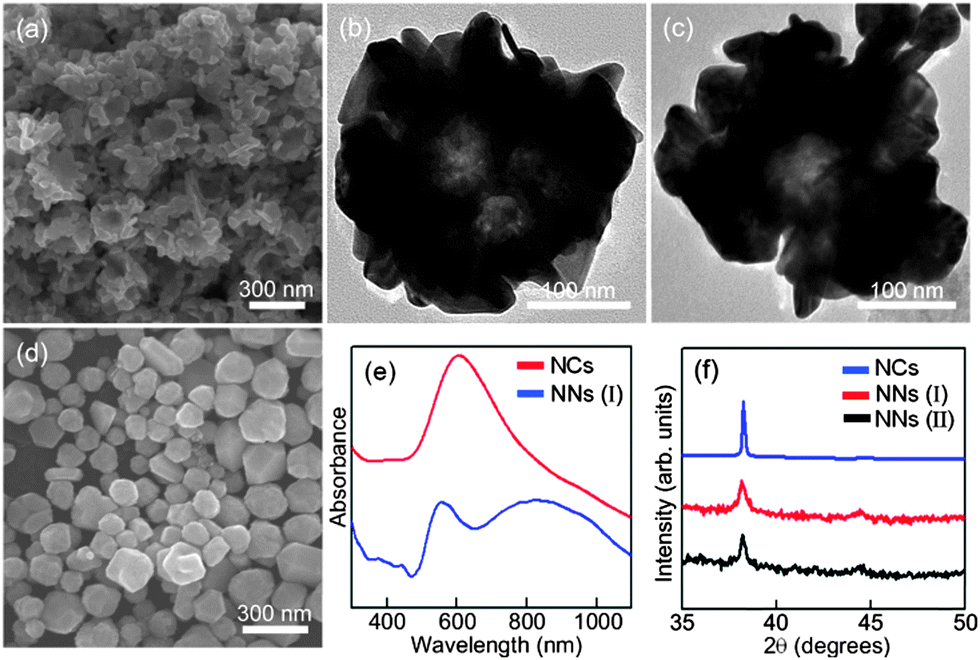 | ||
| Fig. 1 (a) The SEM and (b and c) TEM images of the Au nanonests (NNs) (I); (d) the SEM image of Au nanocrystals formed at room temperature; (e) the UV-visible spectra obtained from Au nanonests (I) and Au nanocrystals (NCs); (f) the XRD patterns of the prepared Au products. | ||
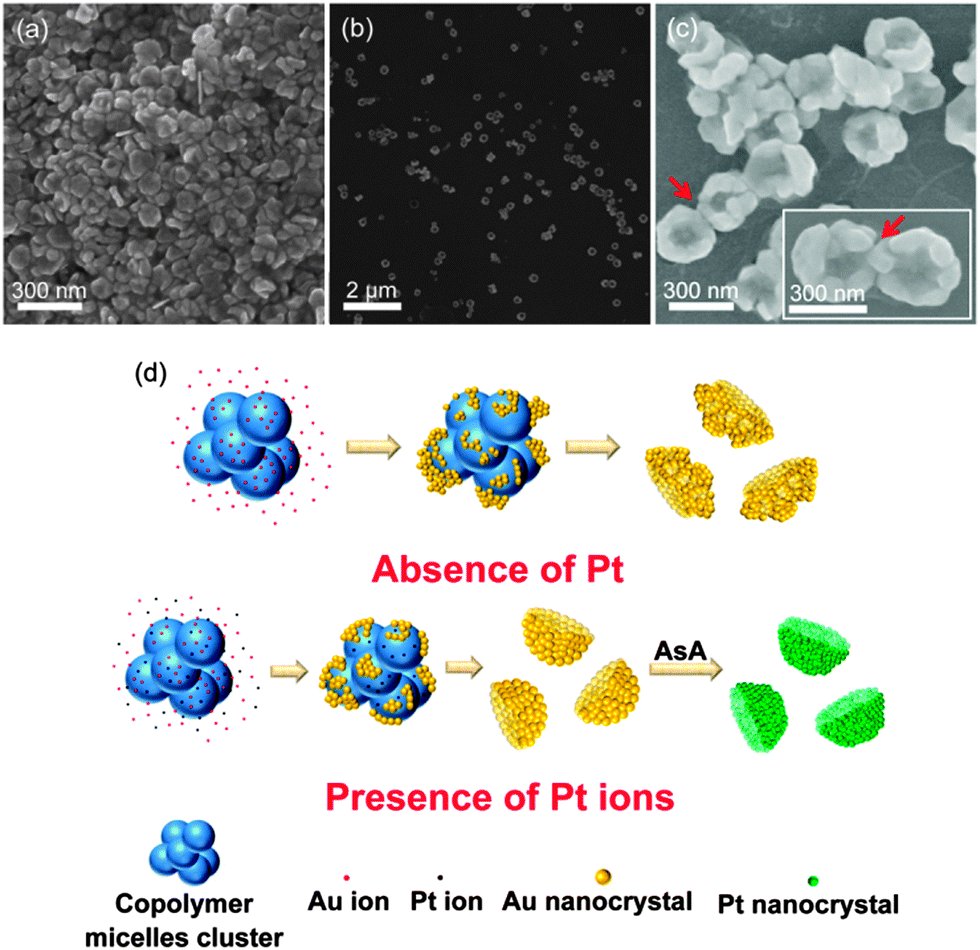
Fig. 2(a) and (b) respectively show the SEM and TEM images of the synthesized Au nanonests (II) (type II) in the presence of Pt ions during the reduction processes. The nest-like morphology and corresponding diameters basically remain like those mentioned in Fig. 1. As expected, the TEM energy-dispersive spectroscopy (EDS) (not shown here) confirms the absence of Pt in the Au nanonests (II), indicating that the introduced Pt ions were not reduced under the present synthesizing conditions. Taking a closer look at the shells of the Au nanonests (II) (see Fig. 2(a)), it is obvious that the addition of Pt ions indeed increases the scale of the Au building blocks but hardly affects the diameters of the Au nanonests (II). The corresponding XRD pattern (see Fig. 1(f)), having a relatively small full width at half maximum (FWHM), also confirms the larger grain size. In other words, the Au nanonests (II) prepared in the presence of Pt ions are constructed with fewer building blocks than those without Pt ions, and have a planar shell structure with slight branching. However, very few exceptions were also found in the present products which have highly branched surfaces (see Fig. 2(c)). Fig. 2(d) is an interesting TEM image showing some split seams in the bottom of one Au nanonest (II), which can be considered as direct evidence that the Au seeds simultaneously nucleate and grow until they meet each other, instead of shell by shell reduction occurring. The high-resolution TEM image and the corresponding selected area electron diffraction (SAED) pattern also obtained for the bottom of the Au nanonest (II) clearly confirm that the bottom shells are constructed with limited numbers of grains (see Fig. 2(e)). As can be seen in Fig. 2(f), the present Au nanonests (II) exhibit a very broad single absorption band at around 700 nm in contrast to the double absorption peaks shown in Fig. 1(e), mainly due to the lower presence of tiny Au nanocrystals attached to the surface of the Au nanonests (II) (the absorption peak at around 560 nm corresponds to the Au nanoparticles).
 | ||
| Fig. 2 (a) The SEM, (b–d) TEM, (e) HRTEM images and (f) UV-visible spectrum of the Au nanonests (II); (g) the SEM, (h) TEM and (i) HRTEM images of the Au–Pt core–shell nanonest. | ||
Here we also demonstrate a very special Au–Pt core–shell nanonest which can be produced by adding the reductant of ascorbic acid (AsA) to subsequently reduce the Pt ions on the surface of the formed Au nanonests (II) as displayed in Fig. 2(g) and (h). The lattice fringes shown in Fig. 2(i) correspond to {111} planes of Pt, indicating the successful reduction of tiny crystals of pure Pt. The induced optical properties as well as the resulting potential applications are considered as future research topics.
To fundamentally understand the growth mechanism of the Au nanonests (II), we sampled the Au production at different growth stages for SEM observations. Interestingly, the 30 min reduction (half of the designated regular reduction time of 60 min) leads to very uniform cap-like Au nanocrystals, as displayed in Fig. 3(a). From the slight sinking of the cap center, it is reasonable to believe that the Au caps should form the bottom of the regular Au nanonests (II), evidently suggesting a bottom-to-side growth mechanism. Fig. 3(b) shows the regular Au nanonests (II) obtained with a regular reaction time of 60 min at 5 °C as mentioned in Fig. 2. It is worth emphasizing again that the formed Au nanonests (II) are perfectly dispersed with rather similar sizes. As the reaction time increased to 120 min, the Au nanonests (II) started to aggregate to each other. The connection between the aggregated Au nanonests (II) seems to be rather solid and inseparable (see Fig. 3(c)). The longer reaction time would significantly increase the side thickness and thus lead to a connection between the neighboring Au nanonests (II). In fact we believe that the growth of the observed reaction-time-dependent morphologies fully followed the soft templates constructed by the PEO–PPO–PEO tri-block copolymer due to the extremely sharp edge of the internal spherical core shown in the Au nanonests (II) (see Fig. 2(a)).
 | ||
| Fig. 3 The SEM images of the Au nanonests formed at different growth stages: (a) 30 min, (b) 60 min, and (c) 120 min reduction time. (d) Schematic illustration of the growth mechanisms. | ||
It has been demonstrated that the micelle of PEO–PPO–PEO could act as a perfect template for the formation of hollow sulfide spheres.30 However, the isolated spherical micelles of P123 would not be the reasonable template or hollow Au spheres might form. In addition, according to the phase diagram of P12331 and considering the critical micelle concentration (cmc) and critical micelle temperature (cmt), P123 micelles should not appear in our system, mainly due to the very low reduction temperature of 5 °C. To reasonably explain the formed nest-like morphology, we consider that the P123 micelles should not only actually exist in our system, but aggregate to form a number of grape-like templates as proposed in Fig. 3(d). Such grape-like copolymer clusters would act as templates for the precipitation of gold due to the strong coordination interaction between the PEO block and gold ions.17 It is known that the equilibrium of the tri-block copolymer system will change upon addition of inorganic precursors and/or according to the preforming conditions, and a new competition among all species will be triggered until a new equilibrium is reached.32 In the present work, since we stirred the P123 polymer solution at room temperature for 60 min and then immediately transferred it to a reduction temperature of 5 °C for another 60 min, we believe that the micelles might just be kept at 5 °C without fully becoming unimers again. To clarify this point, before the precursors were introduced, we extended the stirring time to 3 days at 5 °C and confirmed that only pure Au nanoparticles could be found.
Morphological modifications of noble or transition metals have been proven to be very effective strategies and are a hot research topic in the field of SERS sensors.33–37 The present Au nanonests, having advantages resulting from the nest-shaped morphologies and high chemical activity, are thus considered as an excellent medium for enhancing SERS detection. In general, the main Raman characteristic peak of MGO, as marked by the dashed lines, is extremely weak even at such a very high MGO concentration (0.01 M). It is thus obviously difficult to provide reliable and reproducible data without further enhancement. Fig. 4(a) and (b) show the SERS spectra of MGO using the prepared Au nanonests (I) and (II), respectively. As expected, these two Au nanonests significantly enhance the Raman signals to a traceable level.
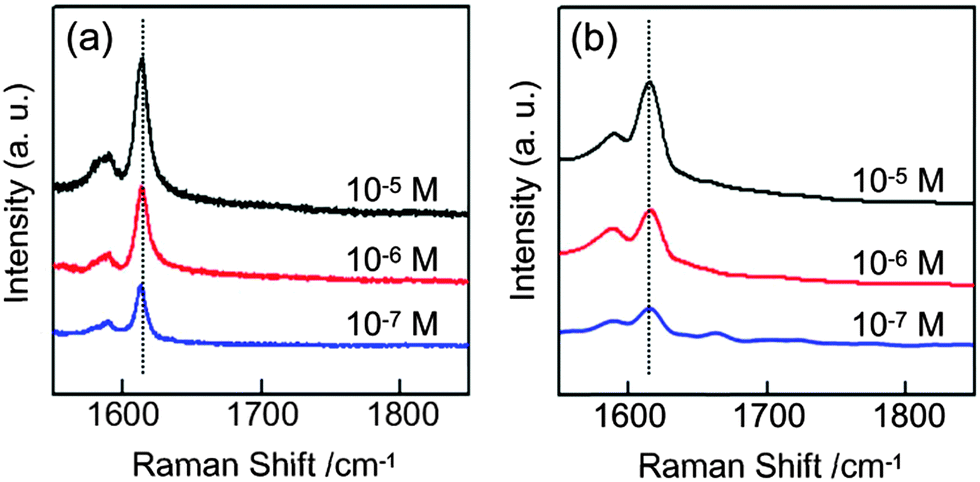 | ||
| Fig. 4 The SERS spectra of malachite green oxalate (MGO) respectively with (a) the Au nanonests (I) and (b) the Au nanonests (II). | ||
We successfully synthesized different types of Au nanonests using block-copolymer P123 as the soft template with a developed very unique strategy for effectively controlling the reduction sites for Au nucleation. The resulting obvious differences in the morphology, porosity and crystallite size of the formed Au nanonests evidently prove that the present strategy is not only a method to form a variety of hemispherical nanostructures, but a new concept for further developing novel nanostructures for both fundamental research and applications.
Experimental
The Pluronic amphiphilic P123 (EO20PO70EO20, Mw = 5800), AsA, and MGO (((C23H25N2)·(C2HO4))2·C2H2O4) were purchased from Aldrich. The metal ion precursors, hydrogen tetrachloroaureate(III) trihydrate (HAuCl4·3H2O) and hydrogen hexachloroplatinate(IV) hexahydrate (H2PtCl6·6H2O), were purchased from Acros. All the starting chemicals were used as received. An aqueous solution of P123 (44 g L−1) was prepared at room temperature and then cooled in a water-ice bath under vigorous stirring for an hour. Then, an aqueous solution of HAuCl4·3H2O (0.5 mL; 0.005 M) or a binary mixture of HAuCl4·3H2O (0.5 mL; 0.005 M) and H2PtCl6·6H2O (0.5 mL; 0.005 M) was respectively added to 4.5 mL of the P123 solution to give the designed molar ratio of metal ions to P123 for a one-hour reaction in the ice bath. The repeatedly washed metallic nanocups were subsequently thinly deposited on cleaned silicon substrates as the SERS sensors. The sensing experiments are demonstrated with the analyte molecules of aqueous MG from 10−5 to 10−7 M. The obtained products were characterized by field-emission scanning electron microscopy (FE-SEM, JEOL 6500), field-emission transmission electron microscopy (FE-TEM, JEOL-2100), and UV-visible spectroscopy (Thermo Scientific, Evolution 300). The SERS spectra were recorded using a Raman spectrometer (Horiba Jobin Yvon, LabRam HR model) equipped with a nitrogen-cooled CCD detector and a He–Ne laser (632.8 nm). Three scans were taken with 1 s accumulation time for each scan.Acknowledgements
Financial support from the National Science Council of the Republic of China under grant NSC 102-2221-E-009-175 and NSC 100-2221-E-009-023-MY3 is gratefully acknowledged.Notes and references
- H. Ataee-Esfahani, Y. Nemoto, L. Wang and Y. Yamauchi, Chem. Commun., 2011, 47, 3885 RSC.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2013, 135, 16762 CrossRef CAS.
- H. Ataee-Esfahani, J. Liu, M. Hu, N. Miyamoto, S. Tominaka, K. C. W. Wu and Y. Yamauchi, Small, 2013, 9, 1047 CrossRef CAS.
- D. Zhang, L. Qi, J. Ma and H. Cheng, Adv. Mater., 2002, 14, 1499 CrossRef CAS.
- C. Loo, A. Lin, L. Hirsch, M. H. Lee, J. Barton, N. Halas, J. West and R. Drezek, Technol. Cancer Res. Treat., 2004, 3, 33 CrossRef CAS.
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS.
- X. D. Wang, E. Graugnard, J. S. King, Z. L. Wang and C. J. Summers, Nano Lett., 2004, 4, 2223 CrossRef CAS.
- D. Jagadeesan, U. Mansoori, P. Mandal, A. Sundaresan and M. Eswaramoorthy, Angew. Chem., Int. Ed., 2008, 47, 7685 CrossRef CAS PubMed.
- X. Li, J. Peng, J. H. Kang, J. H. Choy, M. Steinhart, W. Knoll and D. H. Kim, Soft Matter, 2008, 4, 515 RSC.
- J. He, P. Zhang, J. Gong and Z. Nie, Chem. Commun., 2012, 48, 7344 RSC.
- Y. K. Mishra, R. Adelung, G. Kumar, M. Elbahri, S. Mohapatra, R. Singhal, A. Tripathi and D. K. Avasthi, Plasmonics, 2013, 8, 811 CrossRef CAS.
- N. A. Mirin and N. J. Halas, Nano Lett., 2009, 9, 1255 CrossRef CAS PubMed.
- J. Ye, N. Verellen, W. V. Roy, L. Lagae, G. Maes, G. Borghs and P. V. Dorpe, ACS Nano, 2010, 4, 1457 CrossRef CAS PubMed.
- J. C. Love, B. D. Gates, D. B. Wolf, K. E. Paul and G. M. Whitesides, Nano Lett., 2002, 2, 891 CrossRef CAS.
- C. Charnary, A. Lee, S. Q. Man, C. E. Moran, C. Radloff, R. K. Bradley and N. J. Halas, J. Phys. Chem. B, 2003, 107, 7327 CrossRef.
- J. Liu, A. I. Maaroof, L. Wieczorek and M. B. Cortie, Adv. Mater., 2005, 17, 1276 CrossRef CAS.
- T. Sakai and P. Alexandridis, J. Phys. Chem. B, 2005, 109, 7766 CrossRef CAS.
- S. Chen, C. Guo, G. H. Hu, J. Wang, J. H. Ma, X. F. Liang, L. Zheng and H. Z. Liu, Langmuir, 2006, 22, 9704 CrossRef CAS.
- P. Holmqvist, P. Alexandridis and B. Lindman, J. Phys. Chem. B, 1998, 102, 1149 CrossRef CAS.
- K. J. Liu, Macromolecules, 1968, 1, 308 CrossRef CAS.
- S. Yanagida, K. Takahashi and M. Okahara, Bull. Chem. Soc. Jpn., 1977, 50, 1386 CrossRef CAS.
- A. Warshawsky, R. Kalir, A. Deshe, H. Berkovitz and A. Patchornik, J. Am. Chem. Soc., 1979, 101, 4249 CrossRef CAS.
- K. Yokota, M. Matsumura, K. Yamaguchi and Y. Takada, Makromol. Chem., Rapid Commun., 1983, 4, 721 CrossRef CAS.
- M. D. Adams, P. W. Wade and R. D. Hancock, Talanta, 1990, 37, 875 CrossRef CAS.
- K. Ono and H. Honda, Macromolecules, 1992, 25, 6368 CrossRef CAS.
- A. M. Mathur and A. B. Scranton, Sep. Sci. Technol, 1995, 30, 1071 CrossRef CAS.
- B. J. Elliott, A. B. Scranton, J. H. Cameron and C. N. Bowman, Chem. Mater., 2000, 12, 633 CrossRef CAS.
- Y. Sakai, K. Ono, T. Hidaka, M. Takagi and R. W. Cattrall, Bull. Chem. Soc. Jpn., 2000, 73, 1165 CrossRef CAS.
- A. M. Schwartzberg, T. Y. Olson, C. E. Talley and J. Z. Zhang, J. Phys. Chem., 2006, 110, 19935 CrossRef CAS PubMed.
- Y. Ma, L. Qi, J. Ma and H. Cheng, Langmuir, 2003, 19, 4040 CrossRef CAS.
- G. Wanka, H. Hoffmann and W. Ulbricht, Macromolecules, 1994, 27, 4145 CrossRef CAS.
- C. S. Yang, D. D. Awschalom and G. D. Stucky, Chem. Mater., 2002, 14, 1277 CrossRef CAS.
- H. Liang, Z. Li, W. Wang, Y. Wu and H. Xu, Adv. Mater., 2009, 21, 4614 CrossRef CAS.
- T. Bhuvana and G. U. Kulkarni, Nanotechnology, 2009, 20, 045504 CrossRef CAS PubMed.
- C. Farcau and S. Astilean, J. Phys. Chem. C, 2010, 114, 11717 CAS.
- X. Liu, N. C. Linn, C. H. Sun and P. Jiang, Phys. Chem. Chem. Phys., 2010, 12, 1379 RSC.
- S. Boca, D. Rugina, A. Pintea, L. B. Tudoran and S. Astillean, Nanotechnology, 2011, 22, 055702 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |