
Synthesis, characterization, and physical properties of a poly(acrylamide-co-4-cyanobiphenyl-4′-oxyundecylacrylate)
Tahseen
Kamal
and
Soo-young
Park
*
Department of Polymer Science & Engineering, Kyungpook National University, #1370 Sangyuk-dong, Buk-gu, Daegu 702-701, Korea. E-mail: psy@knu.ac.kr
First published on 14th October 2014
Abstract
A series of copolymers containing 4-cyanobiphenyl-4′-oxyundecylacrylate (LC11) as the hydrophobic liquid crystal (LC) unit and acrylamide (AAm) as the hydrophilic unit were synthesized by free radical polymerization. 1H NMR spectra of the copolymers confirmed the successful copolymerization. The PAAm-co-PLC11 (75/25), (80/20), and (85/15) copolymers showed swelling behavior in water and formed hydrogels because of the hydrophilic AAm units in the copolymer chains while the LC11 units formed cross-linking junctions by organizing into microstructures. The organization of the LC11 units into microstructures was revealed by a polarized optical microscopy (POM) study of the copolymer hydrogels. The copolymers with LC11 ratios higher than 25 mol% were too hydrophobic to swell in water to form hydrogels. This might be because of the molecular weight of the LC11 units, which is several fold higher than AAm. The copolymers with less than 15 mol% LC11 were so hydrophilic that water dissolved them. The POM and wide angle X-ray scattering (WAXS) studies of the copolymer fiber revealed that the LC11 units were aligned with the mesogens' direction, which is perpendicular to the fiber axis.
Introduction
A hydrogel is a type of cross-linked polymer network holding a large amount of water in the network interspaces. Hydrogels are increasingly attracting the interest of academic and industrial researchers due to their unique properties.1–3 They are prepared primarily from hydrophilic polymers containing either chemically or physically cross-linked junctions. Chemically cross-linked networks have permanent junctions and the degree of water swelling in these cross-linked systems is mainly dependent on the cross-linking densities. On the other hand, physical network systems have transient junctions that arise from either polymer chain entanglements or physical links. The physical interactions capable of constructing and maintaining a gel network system include hydrogen bonding,4 crystallization,5 hydrophobic interaction,6 inclusion complexation,7 ionic complexation,8 van der Waals interactions,9 and interpolymer complexation.10 Only a few reported hydrogels rely totally on physically cross-linked systems. One example is an ionically cross-linked triblock copolymer hydrogel;11 another example is a poly(vinyl alcohol) (PVA) hydrogel made by the repeated freezing–thawing method, in which some parts of the PVA chains form crystallites held by very strong hydrogen bonds.12 Similarly, Osada et al. reported on copolymer hydrogels containing acrylic acid monomer units as hydrophilic parts and n-octadecyl acrylate,13 hexadecyl acrylate,14 and 16-acryloyloxy-hexadecanoic acid15 as hydrophobic units.Recently, materials scientists have struggled to create anisotropic and birefringent hydrogel systems.16–20 Although several effective approaches have shown promise for inducing anisotropy in gels, including the use of an electrical field,21 inclusion of ionic liquid moieties,16 and utilization of mechanical stretching18 or shear force from flow,22 all these systems exclude common liquid crystals (LCs), which are anisotropic molecules and well known for their birefringent properties. The coupling of order with fluidity makes LCs very interesting materials as their orientation-dependent properties can be influenced by readily accessible stimuli.23
In this paper, 4-cyanobiphenyl-4′-oxyundecylacrylate (LC11) was copolymerized with an acrylamide monomer (AAm) by free radical polymerization. The hydrophilic AAm units were hydrated whereas the LC11, being hydrophobic, organized into birefringent structures during swelling experiments.
Experimental
Copolymerization of AAm and LC11
The copolymers were synthesized via free radical polymerization. Firstly, the LC11 monomer was synthesized according to our previous method. The copolymer synthesis was performed in a 50 mL Schlenk flask equipped with a mechanical stirrer and a nitrogen inlet. The two monomers AAm and LC11 (Table 1) were dissolved in 7 mL of THF at 25 °C and purged with nitrogen for at least 1 h. Polymerization was initiated by adding AIBN (dissolved in 1 mL of degassed THF) to the monomer mixture and was carried out for a given time at 65 °C, in a constant gas stream, and at constant stirring at 330 rpm. After polymerization, the reaction mixture was charged into a clean beaker containing 60 mL of methanol for precipitating the polymer. The polymer was purified three times by repeated precipitation from THF and methanol.| Product name | Result (Mn and PDI) |
|---|---|
| PAAm-co-PLC11 (90/10) | 5.2 K and 1.4 |
| PAAm-co-PLC11 (85/15) | 6.2 K and 1.5 |
| PAAm-co-PLC11 (80/20) | 6.5 K and 1.2 |
| PAAm-co-PLC11 (75/25) | 5.9 K and 1.2 |
| PAAm-co-PLC11 (65/35) | 5.8 K and 1.4 |
| PAAm-co-PLC11 (50/50) | 4.3 K and 1.2 |
The sample composition in mole percentage is indicated in the sample name; for example, PAAm-co-PLC11 (90/10) (Table 1), where the first number in the parentheses denotes the mole percentage of the AAm monomer and the second number gives the percentage of LC11 in the monomer feed. The numbers indicate the mole percentages that were used for the synthesis.
Characterization
The molar masses and molar mass distributions were determined by gel permeation chromatography (GPC) using a PLgel Organic GPC column and an RI750F refractive index detector (Young Lin Instrument Co., Acme 9000, Korea). THF was used as the eluent with a flow rate of 0.8 mL min−1 and calibration was carried out using polystyrene standards (Aldrich Chemical Co., USA). The 1H NMR spectra were obtained at ambient temperature using a 400 MHz Bruker spectrometer (Germany) and using deuterated chloroform (CDCl3) as the solvent for the LC11 monomer and random copolymers. The optical images were obtained through a polarized optical microscope (Samwon, LSP-13, Korea) equipped with crossed polarizers. Hydrogel samples were sandwiched between two glass slides and viewed in the cross polar states. The swelling ratio of the copolymers was determined by measuring the volume of the samples. Wide angle X-ray scattering patterns were recorded on phosphor image plates (Perkin Elmer, Cyclone) using a Statton camera. An Anton-Parr X-ray generator operated at 40 kV and 40 mA produced Cu Kα radiation, which was monochromatized using a flat monochromator (Huber model 151).Results and discussion
Fig. 1a and b show the 1H NMR spectra of PAAm-co-PLC11 (65/35) and PAAm-co-PLC11 (50/50). The rest of the copolymers with feed molar ratios of 75/25, 80/20, 85/15, and 90/10 were insoluble in CDCl3. However, other copolymers with different compositions had the same synthesis procedure so that the same chemical structure is expected. The characteristic NMR peaks in the corresponding spectra of copolymers were assigned to the protons in their chemical structures. The peaks from vinyl protons in the δ = 5.6–6.4 ppm range disappeared, confirming the successful copolymerization. The backbone protons of the AAm parts in the 1H NMR spectra of the copolymers had a chemical shift at 1.64 ppm, which overlapped the protons from LC11. Thus, it was not possible to determine the exact composition with NMR spectroscopy because of overlapping.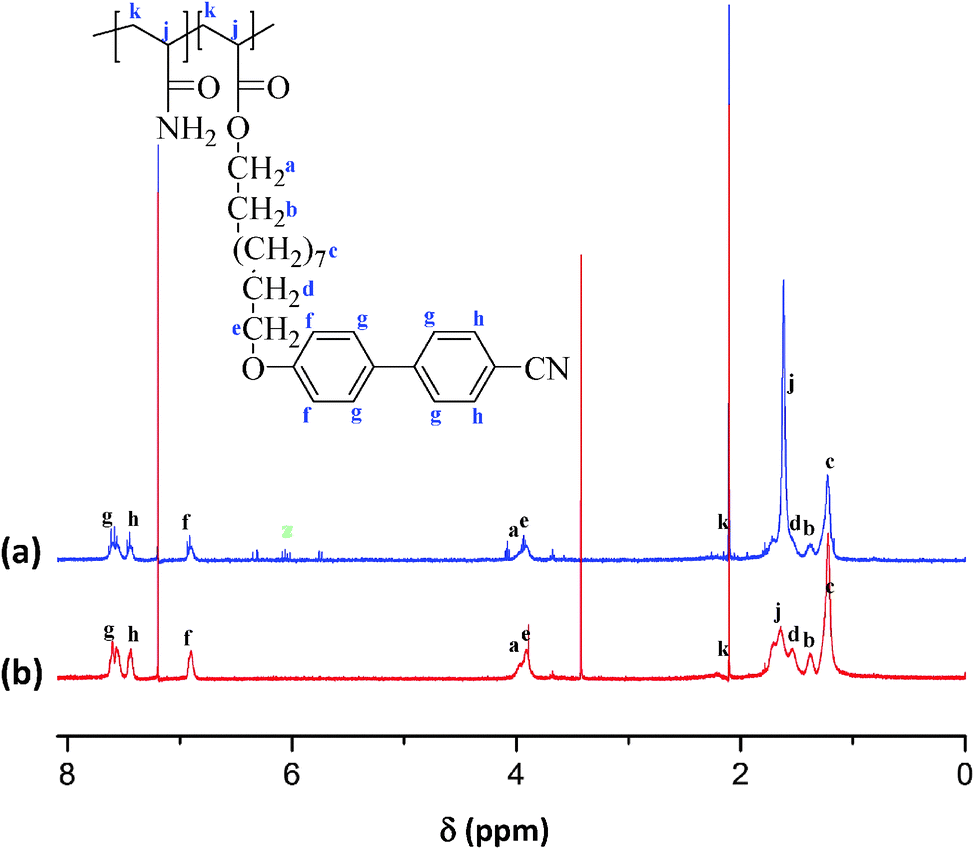 | ||
| Fig. 1 1H NMR spectra of (a) PAAm-co-PLC11 (65/35), and (b) PAAm-co-PLC11 (50/50). Peaks at z are due to the remaining monomer. | ||
Fig. 2a and b show the digital photographs of all the Table 1 copolymers in DI water taken immediately after their placement in water and after 12 h, respectively. PAAm-co-PLC11 (50/50) and (65/35) remained unchanged in water after 12 h, whereas (75/25), (80/20), and (90/10) swelled. The degree of swelling was dependent on the AAm composition in the copolymers (Fig. 2c). Copolymer (90/10) was so hydrophilic that it dissolved in water just by slight shaking.
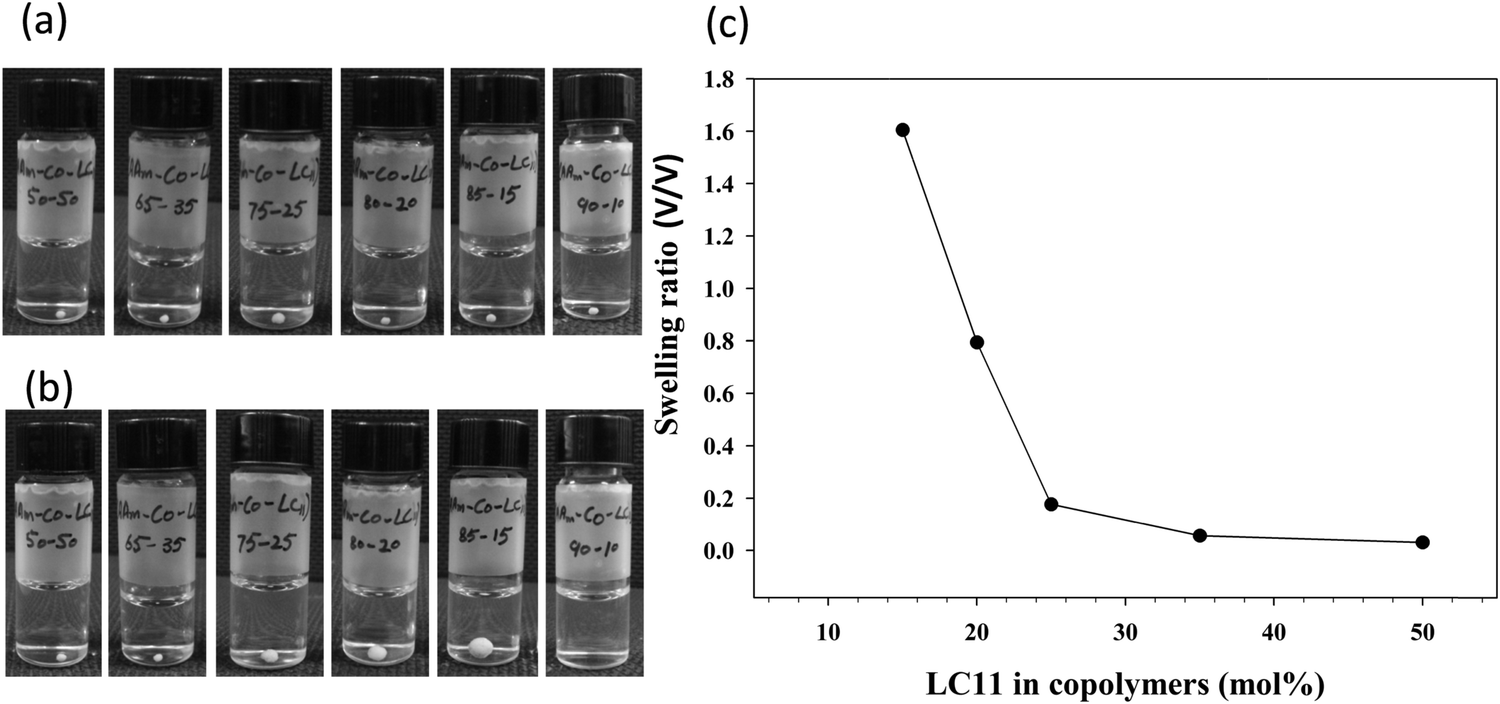 | ||
| Fig. 2 Digital photographs of compact spherical copolymer samples in (a) water and (b) after 12 h of swelling. (c) The corresponding swelling ratio vs. LC11 mol% in PAAm-co-PLC11. | ||
Fig. 3a–c show the POM images of the PAAm-co-PLC11 (75/25), (80/20), and (85/15) hydrogels, respectively, in the cross polar state. All these hydrogels had bright spots whose number and size decreased with a decrease in the LC11 content in the copolymer.24 The mesomorphic behaviors of the LC11 monomer and the PLC11 homopolymer have been examined by other researchers. They found that these materials exist in solid crystalline states at room temperature and exhibit smectic ordering at certain high temperatures.25 Thus, the bright spots in the hydrogels were due to the microstructure of the LC11 units obtained from the different PAAm-co-PLC11 chains, which formed the physical cross-linking junctions. The remaining black area represents the swollen amorphous PAAm part.
 | ||
| Fig. 3 POM images of (a) PAAm-co-PLC11 (75/25), (b) PAAm-co-PLC11 (80/20), and (c) PAAm-co-PLC11 (85/15) in the cross polar state; all scale bars are 100 μm. | ||
Because LC monomers were used in the synthesized copolymers that have anisotropic and alignment properties, additionally, we tested the copolymers for investigating the alignment properties of the LCs by fabricating the fiber. The PAAm-co-PLC11 (80/20) fiber was prepared by melt spinning at 70 °C. Fig. 4a shows an optical image of the fiber in bright mode. When viewed under crossed polarizers with the fiber axis at an angle of 45° to each polarizer, the fiber appeared to be birefringent (Fig. 4b). The birefringence of the fiber disappeared when the fiber was rotated such that its axis was parallel to either of the polarizers, indicating that the copolymer chains and the mesogens were aligned during the spinning process. To get molecular level details, WAXS was performed on this fiber. Fig. 4d shows the WAXS 2-dimensional (2D) pattern of the fiber with its axis vertical to the X-ray beam. A single broad peak appeared at 2θ = 20.4°. The X-ray scattering of the 2D pattern was concentrated in the meridional direction, which is clear from the azimuthal scan of this peak (Fig. 4e). The peak at 2θ = 20.4° might be due to the mesogen stacking and the splitting of this peak into two arcs in the meridional direction, suggesting that these mesogens were aligned perpendicular to the fiber direction.
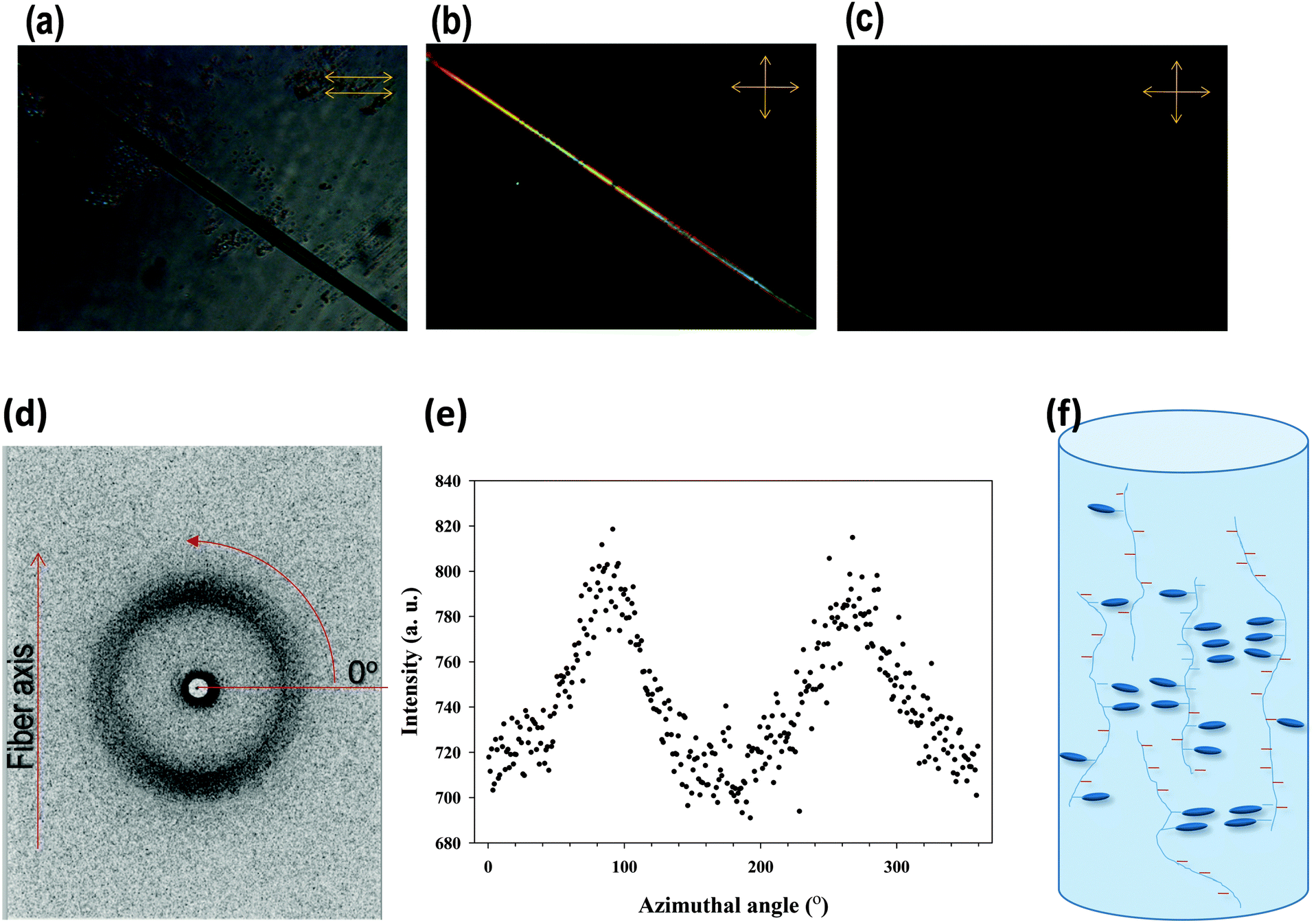 | ||
| Fig. 4 POM images of the PAAm-co-PLC11 (80/20) fiber in (a) bright mode, (b) the cross polar state with the fiber axis at an angle of 45° to either polarizer, and (c) with the fiber axis parallel to one of the polarizers. (d) 2D WAXS pattern of the fiber, (e) the intensity distribution of the peak at 2θ = 20.4° along the azimuthal angle and (f) the deduced molecular level structure of the copolymer fiber. | ||
Conclusions
A series of copolymers having LC11 as hydrophobic and AAm as hydrophilic units were prepared by free radical polymerization. The 1H NMR spectra of the copolymers confirmed successful copolymerization. The PAAm-co-PLC11 (75/25), (80/20), and (85/15) copolymers showed swelling behavior in water and formed hydrogels because of the hydrophilic AAm units in the copolymer chains while the LC11 units played a cross-linking role by forming microstructures. The polymers with LC11 ratios higher than 25 mol% were too hydrophobic to swell in water to form hydrogels and those with less than 15 mol% were so hydrophilic that water dissolved them. The POM and WAXS studies of the copolymer fiber revealed that the LC11 units were aligned with the mesogens' direction, which is perpendicular to the fiber axis.Acknowledgements
This research was supported by Kyungpook National University Research Fund, 2014.References
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- K. T. Nguyen and J. L. West, Biomaterials, 2002, 23, 4307–4314 CrossRef CAS.
- Y. Osada and J.-P. Gong, Adv. Mater. (Weinheim, Ger.), 1998, 10, 827–837 CrossRef CAS.
- H. Gao, N. Wang, X. Hu, W. Nan, Y. Han and W. Liu, Macromol. Rapid Commun., 2013, 34, 63–68 CrossRef CAS PubMed.
- L. Zhang, J. Zhao, J. Zhu, C. He and H. Wang, Soft Matter, 2012, 8, 10439–10447 RSC.
- J. Hao and R. A. Weiss, Macromolecules, 2011, 44, 9390–9398 CrossRef CAS.
- Y. Wang, L. Zhou, G. Sun, J. Xue, Z. Jia, X. Zhu and D. Yan, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1114–1120 CrossRef CAS.
- X. Feng and R. Pelton, Macromolecules, 2007, 40, 1624–1630 CrossRef CAS.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- D. Myung, D. Waters, M. Wiseman, P.-E. Duhamel, J. Noolandi, C. N. Ta and C. W. Frank, Polym. Adv. Technol., 2008, 19, 647–657 CrossRef CAS PubMed.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater. (Weinheim, Ger.), 2011, 23, 2327–2331 CrossRef CAS PubMed.
- C. M. Hassan and N. A. Peppas, Macromolecules, 2000, 33, 2472–2479 CrossRef CAS.
- A. Matsuda, J. I. Sato, H. Yasunaga and Y. Osada, Macromolecules, 1994, 27, 7695–7698 CrossRef CAS.
- A. Matsuda, J. P. Gong and Y. Osada, Polym. Gels Networks, 1998, 6, 307–317 CrossRef CAS.
- A. Matsuda, Y. Katayama, T. Kaneko, J. P. Gong and Y. Osada, J. Mol. Struct., 2000, 554, 91–97 CrossRef CAS.
- D. Batra, D. N. T. Hay and M. A. Firestone, Chem. Mater., 2007, 19, 4423–4431 CrossRef CAS.
- Z. L. Wu, T. Kurokawa, D. Sawada, J. Hu, H. Furukawa and J. P. Gong, Macromolecules, 2011, 44, 3535–3541 CrossRef CAS.
- Z. L. Wu, D. Sawada, T. Kurokawa, A. Kakugo, W. Yang, H. Furukawa and J. P. Gong, Macromolecules, 2011, 44, 3542–3547 CrossRef CAS.
- Z. L. Wu, T. Kurokawa, S. Liang, H. Furukawa and J. P. Gong, J. Am. Chem. Soc., 2010, 132, 10064–10069 CrossRef CAS PubMed.
- F. A. Aouada, M. R. de Moura, A. F. Rubira, E. C. Muniz, P. R. G. Fernandes, H. Mukai, A. C. F. da Silveira and R. Itri, Eur. Polym. J., 2006, 42, 2781–2790 CrossRef CAS PubMed.
- M. Yoshio, Y. Shoji, Y. Tochigi, Y. Nishikawa and T. Kato, J. Am. Chem. Soc., 2009, 131, 6763–6767 CrossRef CAS PubMed.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed.
- T. Kamal and S.-Y. Park, Chem. Commun. (Cambridge, U. K.), 2014, 50, 2030–2033 RSC.
- M. D. Kempe, J. A. Kornfield, C. K. Ober and S. D. Smith, Macromolecules, 2004, 37, 3569–3575 CrossRef CAS.
- V. P. Shibaev, S. G. Kostromin and N. A. Plate, Eur. Polym. J., 1982, 18, 651–659 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |