
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of an ionic liquid on the rate of reaction at a phosphorus centre†
Bradley J.
Butler
and
Jason B.
Harper
*
School of Chemistry, University of New South Wales, UNSW Sydney 2052, Australia. E-mail: j.harper@unsw.edu.au
First published on 16th October 2014
Abstract
The effect of the ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide on the ethanolysis of diethyl chlorophosphate was investigated. The addition of the salt increases the rate constant, though the extent of this increase was dependent on the mole fraction used and the organic co-solvent. Temperature-dependent kinetic analyses were used to demonstrate that the rate enhancement was as a result of a decrease in the activation enthalpy offsetting a decrease in the activation entropy. Altering the salt used showed the importance of the balance of these enthalpic and entropic effects.
Introduction
Recently there has been an increase in the investigation of the use of ionic liquids as alternatives to traditional molecular solvents.1–5 Ionic liquids have many desirable properties as solvents such as low volatility6 and the potential to alter the solvent properties based on their components.6–9 However, in order for ionic liquids to be used widely as solvents, an understanding of how they affect reaction outcome is required.2There are a growing number of studies on how ionic liquids affect reaction outcomes, such as reaction rate and selectivity, for examples see the work of Welton,10–20 Chiappe15,21–31 and D'Anna32–40 along with that from our own group.2,5,7,41–54 These investigations have typically been limited, particularly for kinetic studies, to well-defined organic processes focusing on reactions at carbon centres such as the reaction of benzyl halides with pyridine44,49,51,52 and the methanolysis of 3-chloro-3,7-dimethyloctane.41,44,49,52 Thus, while there is a growing understanding as to the effect of ionic liquids on reactions at carbon centres, these is little in the way of investigation at alternate centres such as phosphorus. Reactions at phosphorus centres are important industrially in the preparation of plasticisers55 and pesticides,56 along with on the smaller scale such as the preparation of oligonucleotides.57
While there have been some studies into phosphorus chemistry in ionic liquids, they have been primarily qualitative in nature (i.e. no kinetic analyses undertaken), focusing on the stability of the phosphorus compounds in these solvents and the ability of ionic liquids to prevent unwanted side reactions such as hydrolysis. Initial work58 demonstrated the different activity of water in a range of ionic liquids and highlighted the importance of the nature of the water present. In a subsequent study,59 the same group considered the reaction of phosphorus trichloride with isopropyl amine in either one of a variety of hydrophobic ionic liquids or the molecular solvent hexane; when carried out in the ionic liquids there was no hydrolysis of phosphorous reagent (cf. a significant extent in the hexane case) and a greater extent of conversion to the desired product. Subsequently, the synthetic utility of this type of reaction control has been demonstrated.60,61
A study where kinetic data was obtained involved reaction of Paroxon with piperidine in a series of ionic liquids.62 It was noted that the ionic liquids affected both the rate constant of reaction of the phosphorus species and the product distribution; in the example used nucleophilic substitution at the phosphorus centre (SN2@P), an aliphatic carbon centre (SN2@C) and at an aromatic carbon centre (SNAr) were all possible. Whilst of interest in noting the different reactivity in ionic liquids relative to molecular solvents (and between ionic liquids), no activation parameter data was determined so the origins of the rate changes in terms of entropy and enthalpy of activation were not discussed. Further, no dependence of reaction outcome on the proportion of ionic liquid in the reaction mixture was considered; this has been shown to be significant in previous reports.5,41,42,51,54
The work described herein aims to consider the effects of an ionic liquid on the rate of a reaction at a phosphorus centre and to correlate this with what is known about how ionic liquids affect reactions at carbon centres.
The reaction chosen was the ethanolysis of diethyl chlorophosphate (Scheme 1), which proceeds through a bimolecular substitution mechanism.63 Diethyl chlorophosphate is a commonly used analogue for both pesticides and nerve agents due to it having comparable reactivity while being of a much low volatility64 and therefore safer to handle.65–67 The ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([bmim][(CF3SO2)2N]) was chosen as it is well studied and allows direct comparisons with previous work where this ionic liquid was also used.7,41,42,44,51,53
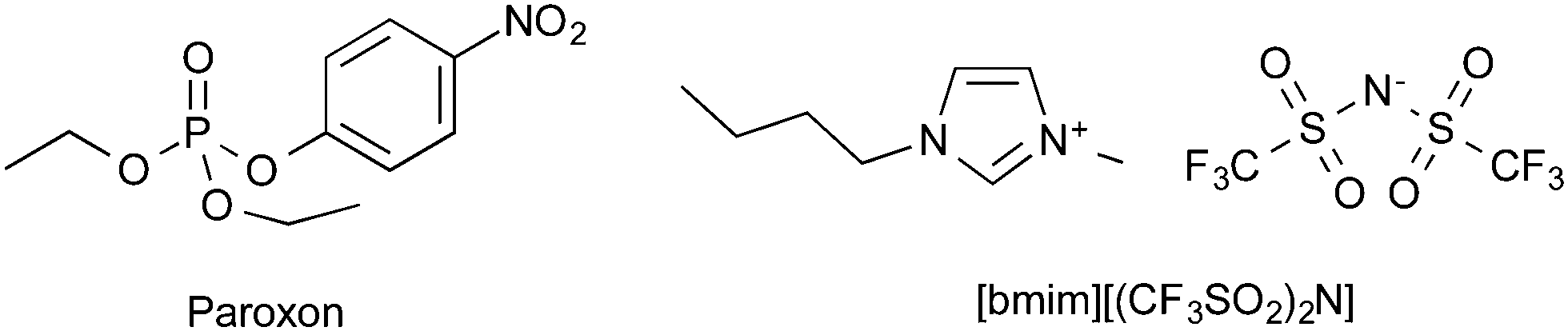
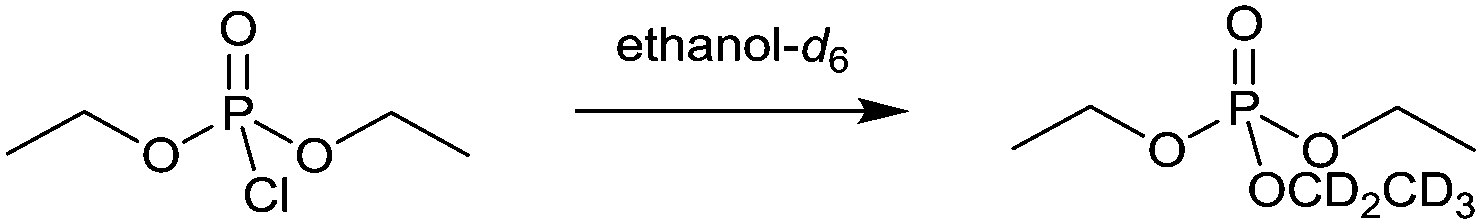 | ||
| Scheme 1 Ethanolysis of diethyl chlorophosphate by ethanol-d6 to afford the partially deuterated triethylphosphate. | ||
Experimental
Diethyl chlorophosphate, lithium bis(trifluoromethylsulfonyl)-imide and acetonitrile were used as received without further purification. Ethanol-d6 and acetonitrile were stored over molecular sieves. The ionic liquid [bmim][(CF3SO2)2N] was synthesised from [bmim][Br] via an anion metathesis.68 The ionic liquid [bmim][Cl] was synthesised through a modification of a literature method.69 All salts were dried to constant weight at 80 °C under reduced pressure. Karl Fischer analysis was used to determine the water content of solvents used (ethanol, 1000 ppm; acetonitrile, <200 ppm; [bmim][(CF3SO2)2N], <100 ppm). 1H NMR spectra were obtained on either a Bruker Avance 400 spectrometer (400 MHz), Bruker Avance 500 spectrometer (500 MHz) or a Bruker Avance 600 spectrometer (600 MHz) with the resulting spectra processed using MestReNova (Mestrelab Research S.L.). Results were found to be reproducible between different spectrometers.Kinetic analyses were carried out in solutions containing diethyl chlorophosphate (ca. 0.3 mol L−1) at a given temperature and at a given mole fraction of salt. For the cases where the co-solvent was ethanol-d6 it also served as reagent. In cases where the co-solvent was acetonitrile, ethanol-d6 was also present at a concentration of ca. 1.0 mol L−1. The reaction was followed using 1H NMR spectroscopy‡ with spectra recorded at regular intervals with a minimum of 100 spectra recorded to a minimum of one half life. For cases where the observed rate constant was <8 × 10−6 s−1in situ monitoring was impractical and the sample was held at the desired temperature in a water bath. Spectra were acquired periodically and initial rates determined up to ca. 10% conversion.
For cases where a large excess of nucleophile was used, the extent of conversion was determined through monitoring the change in the integration either of the signal at δ ca. 4.3 due to the CH2 protons of the diethyl chlorophosphate or of the signal at δ ca. 4.1 corresponding to the CH2 protons of the resultant triethylphosphate. From these integrations, pseudo-first order rate constants and further, the corresponding second order rate constants could be calculated. In the case of second order reactions, the relative concentrations of the diethyl chlorophosphate and the triethylphosphate were determined through comparison of signal at δ ca. 4.3 allowing the absolute concentrations to be calculated from the initial concentration of the reagent; from these and the concentration of the nucleophile, the second order rate constant was determined. The bimolecular form of the Eyring equation70 was utilised to calculate the activation parameters.
Results and discussion
Effect of the mole fraction of ionic liquid on rate of reaction
Initial studies focused on the effect of the change in concentration of the ionic liquid on the rate constant of the ethanolysis of diethyl chlorophosphate. As seen in previous studies, the relation between ionic liquid concentration and rate constant can be non-linear5,41,51,54 therefore an investigation into the optimal concentration of an ionic liquid for rate constant enhancement was performed.There are potential competing reactions to that shown in Scheme 1; both hydrolysis due to adventitious water and nucleophilic substitution at a methylene group (SN2@C) are possible. The former process would generate the diester diethyl hydrogen phosphate, while the latter would generate a phosphochloridate that may react further with ethanol to generate the same diester. Under all of the reaction conditions described here only a single byproduct (identified through spiking experiments to be the diester) was observed, and in negligible amounts (with the exception of the chloride salt, discussed further below), using either 1H or 31P NMR spectroscopy (for representative spectra, see ESI†).
[bmim][(CF3SO2)2N] in ethanol
In this case, the mole fraction of the ionic liquid [bmim][(CF3SO2)2N] was controlled by altering the ratio of [bmim][(CF3SO2)2N] and ethanol-d6 with the concentration of the diethyl chlorophosphate staying constant. This resulted in the ethanol-d6 being both a reactant and solvent. This was advantageous as the higher concentrations of ethanol provided pseudo-first order conditions along with rapid reaction times.As represented in Fig. 1 there is a clear trend in the rate constant for the reaction shown in Scheme 1 as the mole fraction of the ionic liquid in the reaction mixture changes. Initially, the rate constant for the process increases up to a maximum value at ca. χIL 0.3, corresponding to a ca. four-fold (Table 1) increase from the case in ethanol (χIL 0). As the mole fraction increases further, there is a decrease in the rate constant up until the maximum mole fraction used (χIL 0.72); at this point there is still an increase in rate constant compared to the molecular solvent case (of ca. two-fold).
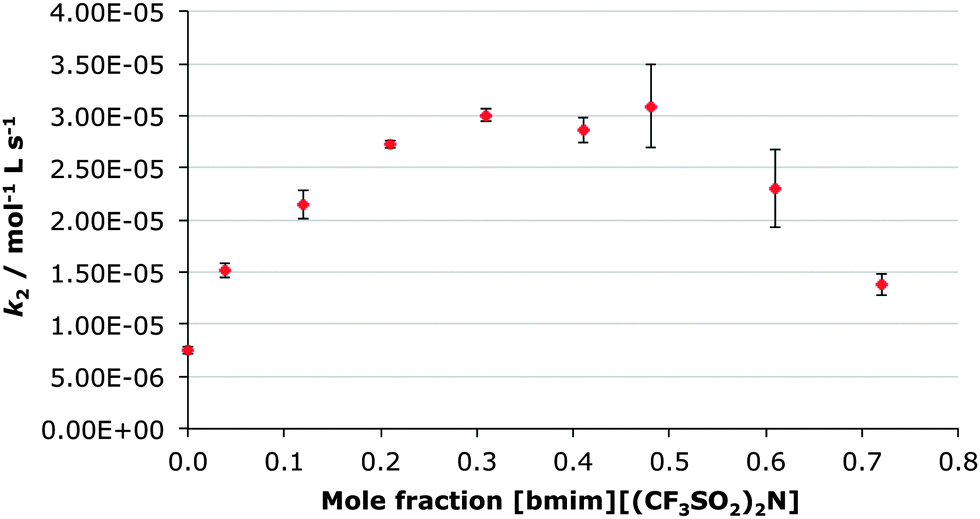 | ||
| Fig. 1 The bimolecular rate constant (k2) for the ethanolysis of diethyl chlorophosphate at different mole fractions of [bmim][(CF3SO2)2N] in ethanol-d6 at 298.2 K. Uncertainties are the standard error of the mean derived from at least three replicate measurements at each mole fraction. | ||
This trend does not exactly parallel those trends observed previously for reactions at carbon centres. Arguably the closer comparison mechanistically – the reaction of benzyl halides with pyridine in acetonitrile proceeding through a bimolecular mechanism – showed an increase in the rate constant with proportion of ionic liquid concentration to a maximum at χIL 0.77 followed by a slight decrease to χIL 0.85.51 The extent of rate enhancement was similar to that described here, with the maximum enhancement being ca. 4.5 times.
The alternate comparison is with the SN1 methanolysis of 3-chloro-3,7-dimethyloctane in mixtures of methanol and [bmim][(CF3SO2)2N].41 In that case, an initial increase in rate constant with proportion of ionic liquid, followed by a decrease as more ionic liquid was added, was also observed. In that case, however, the maximum effect occurred at a much lower proportion of ionic liquid – a relative rate enhancement of ca. two-fold at χIL 0.02.
It is interesting to note that this trend of rate constant enhancement has similar elements to those previous studies, particularly an increase in rate constant to a given mole fraction of ionic liquid followed by a decrease. In this case, the concentration at which the maximum effect occurs is different to those described previously, lying approximately halfway between the two cases. This suggests that the mechanism of reaction is not the only variable that must be considered when analysing the effect of ionic liquid concentration on the rate of a particular reaction.
[bmim][(CF3SO2)2N] in acetonitrile and ethanol
In order to investigate any effect the molecular solvent may have on the effect of the ionic liquid on the rate constant in the above reaction, an alternate molecular solvent, acetonitrile, was introduced. This solvent was chosen in order to change the nature of the molecular solvent (from protic ethanol to aprotic acetonitrile) while keeping the apparent polarity similar to both [bmim][(CF3SO2)2N] and ethanol.17 By keeping the concentration of the ethanol low and constant (ca. 1 mol L−1), any effect it may have could be minimised. The mole fraction of [bmim][(CF3SO2)2N] in this case was controlled by altering the ratio of [bmim][(CF3SO2)2N]) and acetonitrile.Unlike the previous case, which was carried out under pseudo-first order conditions, the low concentration of ethanol used meant that kinetic analysis had to take into account the change in concentration of this reagent. As ethanol-d6 is NMR silent the change in concentration of this reagent had to be calculated utilising the known starting concentration and using the assumption that any absolute change in concentration of the diethyl chlorophosphate was equivalent to the absolute change in concentration of the ethanol-d6. This, and the need to determine the concentrations of multiple species, complicated analysis in this case.
As seen in Fig. 2, the general trend of an increase in rate constant to a maximum at χILca. 0.3 followed by a decrease, mirrors that seen in the previous case of [bmim][(CF3SO2)2N] in ethanol-d6 (Fig. 1). However the actual extent of rate constant enhancement is greater (Table 2), with a ca. sixteen-fold increase. This is primarily due to the rate constant in the χIL 0 case being markedly lower in acetonitrile ((1.4 ± 0.2) × 10−6 mol L−1 s−1) than in ethanol-d6 ((7.5 ± 0.3) × 10−6 mol L−1 s−1). This is most likely due to the relative natures of ethanol and acetonitrile with a larger rate constant in a protic solvent likely due to more effective stabilisation of the charge-separated transition state. The most interesting observation here is that the effect from the presence of the [bmim][(CF3SO2)2N] is much greater than any affect caused by changing the molecular co-solvent.
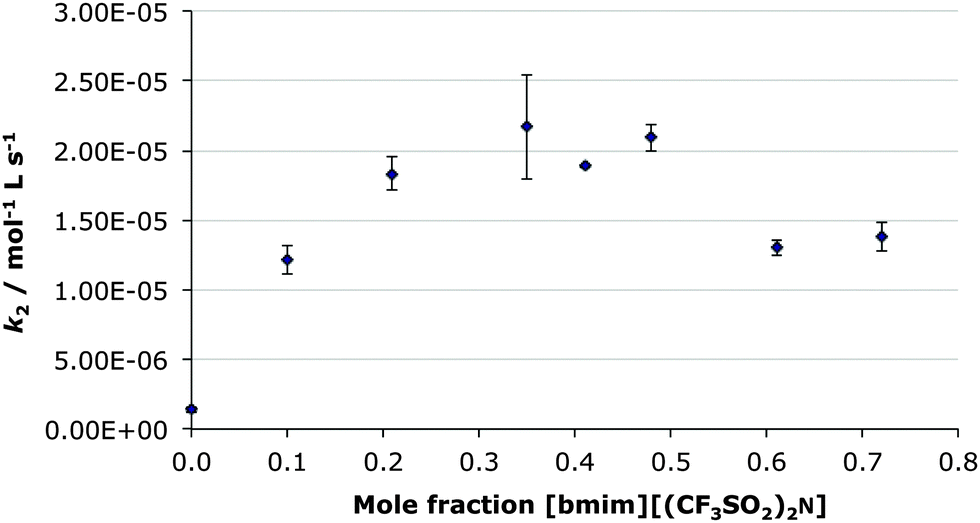 | ||
| Fig. 2 The bimolecular rate constant (k2) for the ethanolysis of diethyl chlorophosphate at different mole fractions of [bmim][(CF3SO2)2N] in acetonitrile at 298.2 K. Uncertainties are the standard error of the mean derived from at least three replicate measurements at each mole fraction. | ||
Activation parameters for the reaction
In order to better understand the origin of the effect of the ionic liquid [bmim][(CF3SO2)2N] on the rate constant for the reaction shown in Scheme 1, temperature dependent kinetic analyses were carried out in order to obtain the activation parameters for the reaction. The changes in both enthalpy and entropy of activation were investigated to provide insight into the interaction of the solvents with both the starting materials and the transition state of the reaction.[bmim][(CF3SO2)2N] in ethanol
The rate constant for the reaction was obtained over a range of temperatures and at three different mole fractions of χIL – 0, 0.3 and 0.7. The particular mole fractions were selected as these correspond to no ionic liquid, the mole fraction of ionic liquid that gave the largest rate constant, and the maximum mole fraction of ionic liquid used. An Eyring plot (see ESI†) allowed determination of the activation parameters ΔH‡ and ΔS‡ (Table 3).70 The equivalent was attempted with acetonitrile as a co-solvent as per the mole fraction dependence study however due to the large errors in the resultant Eyring plot, the results were unable to provide any insight (see ESI†).At χIL 0, the activation enthalpy was positive and the activation entropy large and negative, as would be expected for a non-spontaneous reaction that proceeds through an associative mechanism. Upon moving to χIL 0.3, there is a marked decrease in both the activation parameters. A decrease in the activation enthalpy corresponds to a decrease in the activation energy (an enthalpic benefit) whilst a decrease in the activation enthalpy results in an increase in the activation energy (an entropic cost). Increasing the proportion of ionic liquid further does not significantly affect the activation parameters significantly.
These data are consistent with the observed dependencies of the rate constant of the reaction on the proportion of the ionic liquid [bmim][(CF3SO2)2N] in the reaction mixture. At low proportions of [bmim][(CF3SO2)2N], the decrease in the activation enthalpy outweighs the decrease in the activation entropy; the result is a decrease in the activation energy and an increase in the rate constant. Whilst the uncertainties in the activation parameters negate detailed interpretation, it is likely that the balance of enthalpic and entropic effects changes slightly with the latter dominating and the rate constant decreasing at high mole fractions of ionic liquid.
It is worth considering the microscopic origins of the observed changes in the activation parameters. The decrease in both parameters suggests either a stabilisation of the transition state through interaction with the ionic liquid (which also involves some ordering) or decreased organisation about and stabilisation of the starting materials in the presence of the salt. Given the charge development in the transition state, the former is considered more reasonable.
The outcomes of this reaction can be compared to the previously discussed SN2 Menschutkin reactions of a benzyl halide with pyridine.7,44,49 In those cases, rather than an enthalpic benefit causing the rate constant enhancement, an increase in the activation entropy was observed on moving to ionic liquid solvent; there is also corresponding increase in the activation enthalpy. This is consistent with organisation of the solvent about the starting material being more significant in the ionic liquid case; this interaction (which was shown to be between the cation and the lone pair of the nucleophile49 is not present in the transition state. Therefore, while mechanistically similar, the cause of the rate constant increase in the ethanolysis of diethyl phosphate on addition of ionic liquid is not the same.
In comparing the effects of an ionic liquid on the activation parameters of the reaction shown in Scheme 1 with other systems, the SN1 methanolysis of 3-chloro-3,7-dimethyloctane is affected by [bmim][(CF3SO2)2N] in a similar fashion with decreases in both activation parameters observed.42 The origin of the similarities in outcomes is unclear though stabilisation of developing charge in the transition state likely dominates the interaction with the comparatively less interacting nucleophile in the phosphate ester case.
The presence of highly coordinating ions
In order to investigate the effect that a highly coordinating ion may have, the ionic liquid was substituted with either lithium bis(trifluoromethylsulfonyl)imide or [bmim][Cl]. The corresponding increase in coordinating ability in the cation or the anion, respectively, was used to determine possible sources of interaction present in the parent ionic liquid [bmim][(CF3SO2)2N]. Again ethanol was chosen as the solvent as it provided reasonable solubility for both lithium bis(trifluoromethylsulfonyl)imide and [bmim][Cl] and allowed for pseudo-first order kinetics to be utilised while not affecting the overall trend as seen previously.Effect of the mole fraction of the salts on rate of reaction
In this case the [bmim] cation was replaced with the much more coordinating lithium cation through preparation of a range of solutions of lithium bis(trifluoromethylsulfonyl)imide in ethanol-d6 up to χsalt 0.24 and measuring the rate constant at each concentration (Fig. 3). Subsequently, the bis(trifluoromethylsulfonyl)imide anion was replaced with the much more coordinating chloride anion by creating a range of solution of [bmim][Cl] in ethanol-d6 up to χsalt 0.45 and measuring the rate constant at each concentration (Fig. 4). The mole fraction range used was limited by the solubilities of the salts in ethanol. Whilst no byproducts were observed in the case of the lithium salt, up to 10% of the corresponding diester was observed in the chloride salt cases. This is likely due to the presence of adventitious water (due to the hygroscopic salt) however this did not affect the acquisition of kinetic data.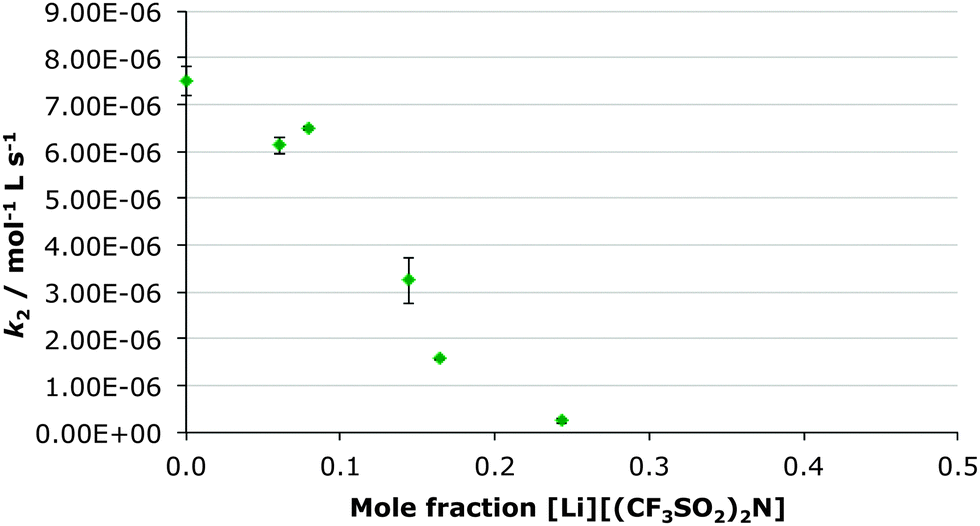 | ||
| Fig. 3 The bimolecular rate constant (k2) for the ethanolysis of diethyl chlorophosphate at different mole fractions of lithium bis(trifluoromethylsulfonyl)imide in ethanol-d6 at 298.2 K. Uncertainties are the standard error of the mean derived from at least three replicate measurements at each mole fraction. | ||
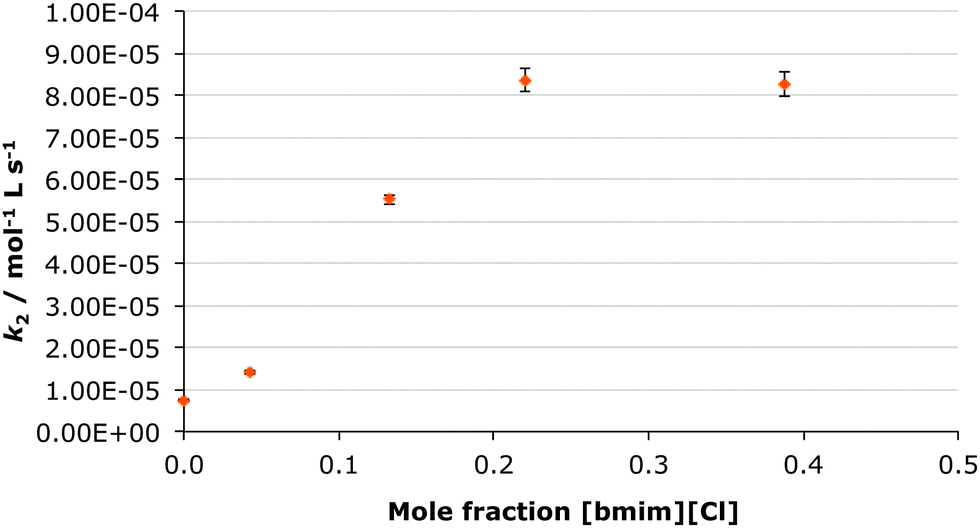 | ||
| Fig. 4 The bimolecular rate constant (k2) for the ethanolysis of diethyl chlorophosphate at different mole fractions of [bmim][Cl] in ethanol-d6 at 298.2 K. Uncertainties are the standard error of the mean derived from at least three replicate measurements at each mole fraction. | ||
While previously there was an increase in rate constant on increasing proportion of the ionic liquid in the reaction mixture, when lithium bis(trifluoromethylsulfonyl)imide was added, there is a significant decrease in the rate constant of the reaction of ca. thirty-fold at χsalt 0.24 (Table 4). Therefore any positive effects of the parent [bmim][(CF3SO2)2N] are negated. In contrast, when the chloride salt was added, a large increase in the rate constant for ethanolysis of diethyl chlorophosphate was observed, the effect was greater than in the [bmim][(CF3SO2)2N] case (Table 4).
| χ salt |
k
2/10−6 mol L−1 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
k
2/k2(ethanol)b |
|---|---|---|
| a Uncertainties quoted are the standard error of the mean derived from at least three replicate measurements at each mole fraction. b Uncertainties are compounded from rate data. | ||
| 0 | 7.5 ± 0.3 | 1 |
| 0.24 [Li][(CF3SO2)2N] | 0.26 ± 0.06 | 0.035 ± 0.008 |
| 0.22 [bmim][Cl] | 83 ± 5 | 11.0 ± 0.8 |
Activation parameters for the reaction
In order to better understand the origin of this change in effect, temperature dependent kinetic analyses were again carried out in order to obtain the activation parameters (Table 5). To facilitate direct comparison, all reactions were performed at a mole fraction ca. 0.2 (once again, the Eyring plot is presented in the ESI†).The first thing to note directly is that the activation parameters are markedly different in the presence of the lithium salt. The increase in the activation enthalpy (the large uncertainty in the activation entropy makes further discussion on that point difficult) suggests either increased stabilisation of the starting materials or decreased interaction of the solvent mixture with the incipient charges in the transition state. The former seems unlikely (given the charge development in the transition state) and hence this change is likely the result of coordination of the oxophilic lithium71 with the nucleophile.
The [bmim][Cl] case however is more enlightening. Here, the change in activation parameters is again an enthalpic benefit with an entropic cost much like the [bmim][(CF3SO2)2N] case. The extent of these changes however is markedly different with a marginally lower decrease in enthalpy of activation and a much lower decrease in the entropy of activation. This change in enthalpic benefit/entropic cost balance results in a much greater rate constant enhancement. Of most interest here is the fact that any interactions between the salt and the transition state are reduced in the [bmim][Cl] case when compared to the [bmim][(CF3SO2)2N] case. This may suggest preferential ion pairing of the [bmim] cation with the chloride anion, reducing interactions with the transition state. It is important to note that rate constant enhancement is not proportional to the level of interaction, rather the balance of enthalpic and entropic effects is most important; this has been seen previously for reaction at carbon centres.54 Altering the makeup of both the cation and anion in an effort to alter potential interactions in solution is clearly of great interest.
Conclusions
The effect of the ionic liquid [bmim][(CF3SO2)2N] on the rate constant of the ethanolysis of diethyl chlorophosphate was found to be highly dependent on the mole fraction of the salt in solution. This change in rate constant was found to be due to a decrease in both the activation parameters for the reaction due to interactions of the components of the salt [bmim][(CF3SO2)2N] with the transition state. Through the use of highly coordinating ions, it was found that the level of this interaction and the balance of enthalpic benefit and entropic cost are critical in affecting the rate constant for the reaction.Acknowledgements
BJB acknowledges the support of the Australian government through the receipt of an Australian Postgraduate Award. JBH acknowledges financial support from the Australian Research Council Discovery Project Funding Scheme (Project DP130102331).Notes and references
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- J. B. Harper and M. N. Kobrak, Mini-Rev. Org. Chem., 2006, 3, 253 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- H. M. Yau, S. T. Keaveney, B. J. Butler, E. E. L. Tanner, M. S. Guerry, S. R. D. George, M. H. Dunn, A. K. Croft and J. B. Harper, Pure Appl. Chem., 2013, 85, 1979–1990 CrossRef CAS.
- K. Seddon, Kinet. Catal., 1995, 37, 693–697 Search PubMed.
- E. E. L. Tanner, H. M. Yau, R. R. Hawker, A. K. Croft and J. B. Harper, Org. Biomol. Chem., 2013, 11, 6170–6175 CAS.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- J. G. Huddleston and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- N. L. Lancaster, T. Welton and G. B. Young, J. Chem. Soc., Perkin Trans. 2, 2001, 2267–2270 RSC.
- N. L. Lancaster, P. A. Salter, T. Welton and G. B. Young, J. Org. Chem., 2002, 67, 8855–8861 CrossRef CAS PubMed.
- L. Crowhurst, N. L. Lancaster, J. M. Pérez Arlandis and T. Welton, J. Am. Chem. Soc., 2004, 126, 11549–11555 CrossRef CAS PubMed.
- N. L. Lancaster and T. Welton, J. Org. Chem., 2004, 69, 5986–5992 CrossRef CAS PubMed.
- L. Crowhurst, R. Falcone, N. L. Lancaster, V. Llopis-Mestre and T. Welton, J. Org. Chem., 2006, 71, 8847–8853 CrossRef CAS PubMed.
- R. Bini, C. Chiappe, V. L. Mestre, C. S. Pomelli and T. Welton, Org. Biomol. Chem., 2008, 6, 2522–2529 CAS.
- G. Ranieri, J. P. Hallett and T. Welton, Ind. Eng. Chem. Res., 2008, 47, 638–644 CrossRef CAS.
- M. Y. Lui, L. Crowhurst, J. P. Hallett, P. A. Hunt, H. Niedermeyer and T. Welton, Chem. Sci., 2011, 2, 1491–1496 RSC.
- I. Skarmoutsos, D. Dellis, R. P. Matthews, T. Welton and P. A. Hunt, J. Phys. Chem. B, 2012, 116, 4921–4933 CrossRef CAS PubMed.
- H. Niedermeyer, C. Ashworth, A. Brandt, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2013, 15, 11566–11578 RSC.
- L. Chen, M. Sharifzadeh, N. Mac Dowell, T. Welton, N. Shah and J. P. Hallett, Green Chem., 2014, 16, 3098–3106 RSC.
- C. Chiappe, D. Capraro, V. Conte and D. Pieraccini, Org. Lett., 2001, 3, 1061–1063 CrossRef CAS PubMed.
- C. Chiappe, V. Conte and D. Pieraccini, Eur. J. Org. Chem., 2002, 2831–2837 CrossRef CAS.
- C. Chiappe, D. Pieraccini and P. Saullo, J. Org. Chem., 2003, 68, 6710–6715 CrossRef CAS PubMed.
- C. Chiappe and D. Pieraccini, J. Org. Chem., 2004, 69, 6059–6064 CrossRef CAS PubMed.
- R. Bini, C. Chiappe, E. Marmugi and D. Pieraccini, Chem. Commun., 2006, 897–899 RSC.
- R. Bini, C. Chiappe, C. S. Pomelli and B. Parisi, J. Org. Chem., 2009, 74, 8522–8530 CrossRef CAS PubMed.
- C. Chiappe, M. Malvaldi and C. S. Pomelli, Green Chem., 2010, 12, 1330–1339 RSC.
- A. Cerda-Monje, A. Aizman, R. A. Tapia, C. Chiappe and R. Contreras, Phys. Chem. Chem. Phys., 2012, 14, 10041–10049 RSC.
- C. Chiappe, A. Sanzone, D. Mendola, F. Castiglione, A. Famulari, G. Raos and A. Mele, J. Phys. Chem. B, 2012, 117, 668–676 CrossRef PubMed.
- C. Chiappe and C. S. Pomelli, Phys. Chem. Chem. Phys., 2013, 15, 412–423 RSC.
- C. Chiappe, S. Rajamani and F. D'Andrea, Green Chem., 2013, 15, 137–143 RSC.
- F. D'Anna, V. Frenna, R. Noto, V. Pace and D. Spinelli, J. Org. Chem., 2006, 71, 9637–9642 CrossRef PubMed.
- F. D'Anna, V. Frenna, R. Noto, V. Pace and D. Spinelli, J. Org. Chem., 2006, 71, 5144–5150 CrossRef PubMed.
- F. D'Anna, V. Frenna, V. Pace and R. Noto, Tetrahedron, 2006, 62, 1690–1698 CrossRef PubMed.
- F. D'Anna, V. Frenna, S. La Marca, R. Noto, V. Pace and D. Spinelli, Tetrahedron, 2008, 64, 672–680 CrossRef PubMed.
- F. D'Anna, S. La Marca, P. Lo Meo and R. Noto, Chem. – Eur. J., 2009, 15, 7896–7902 CrossRef PubMed.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, Eur. J. Org. Chem., 2011, 5681–5689 CrossRef.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, ChemPhysChem, 2012, 13, 1877–1884 CrossRef PubMed.
- F. D'Anna, H. Q. Nimal Gunaratne, G. Lazzara, R. Noto, C. Rizzo and K. R. Seddon, Org. Biomol. Chem., 2013, 11, 5836–5846 Search PubMed.
- F. D'Anna, S. Marullo, P. Vitale, C. Rizzo, P. Lo Meo and R. Noto, Appl. Catal., A, 2014, 482, 287–293 CrossRef PubMed.
- B. Y. W. Man, J. M. Hook and J. B. Harper, Tetrahedron Lett., 2005, 46, 7641–7645 CrossRef CAS PubMed.
- H. M. Yau, S. Barnes, J. M. Hook, T. G. Youngs, A. K. Croft and J. B. Harper, Chem. Commun., 2008, 3576–3578 RSC.
- H. M. Yau, S. J. Chan, S. R. D. George, J. M. Hook, A. K. Croft and J. B. Harper, Molecules, 2009, 14, 2521–2534 CrossRef CAS PubMed.
- H. M. Yau, A. G. Howe, J. M. Hook, A. K. Croft and J. B. Harper, Org. Biomol. Chem., 2009, 7, 3572–3575 CAS.
- C. E. Rosella and J. B. Harper, Tetrahedron Lett., 2009, 50, 992–994 CrossRef CAS PubMed.
- S. R. D. George, G. L. Edwards and J. B. Harper, Org. Biomol. Chem., 2010, 8, 5354–5358 CAS.
- S. G. Jones, H. M. Yau, E. Davies, J. M. Hook, T. G. A. Youngs, J. B. Harper and A. K. Croft, Phys. Chem. Chem. Phys., 2010, 12, 1873–1878 RSC.
- M. R. Gyton, M. L. Cole and J. B. Harper, Chem. Commun., 2011, 47, 9200–9202 RSC.
- H. M. Yau, A. K. Croft and J. B. Harper, Faraday Discuss., 2012, 154, 365–371 RSC.
- M. H. Dunn, M. L. Cole and J. B. Harper, RSC Adv., 2012, 2, 10160–10162 RSC.
- S. T. Keaveney and J. B. Harper, RSC Adv., 2013, 3, 15698–15704 RSC.
- E. E. L. Tanner, R. R. Hawker, H. M. Yau, A. K. Croft and J. B. Harper, Org. Biomol. Chem., 2013, 11, 7516–7521 CAS.
- S. T. Keaveney, D. V. Francis, W. Cao, R. S. Haines and J. B. Harper, Aust. J. Chem. DOI:10.1071/CH14117.
- S. T. Keaveney, K. S. Schaffarczyk McHale, R. S. Haines and J. B. Harper, Org. Biomol. Chem., 2014, 12, 7092–7099 CAS.
- S. M. Kent, US Pat., 2595310, 1952 Search PubMed.
- J. M. Deming, US Pat., 3408442, 1968 Search PubMed.
- W. J. Stec, US Pat., 5856465, 1999 Search PubMed.
- E. Amigues, C. Hardacre, G. Keane, M. Migaud and M. O'Neill, Chem. Commun., 2006, 72–74 RSC.
- E. J. Amigues, C. Hardacre, G. Keane and M. E. Migaud, Green Chem., 2008, 10, 660 RSC.
- E. J. Amigues, C. Hardacre, G. Keane, M. E. Migaud and W. R. Pitner, Green Chem., 2009, 11, 1391–1396 RSC.
- K. Crossey, C. Hardacre, M. E. Migaud and S. E. Norman, RSC Adv., 2012, 2, 2988–2993 RSC.
- P. Pavez, D. Millán, J. I. Morales, E. A. Castro, C. A. López and J. G. Santos, J. Org. Chem., 2013, 78, 9670–9676 CrossRef CAS PubMed.
- R. F. Hudson and L. Keay, J. Chem. Soc., 1960, 1859–1864 RSC.
- Diethyl chlorophosphate, Version 5.2 [Online], Sigma-Aldrich Pty. Ltd: Castle Hill, NSW, Australia, April 16, 2013, http://www.sigmaaldrich.com/catalog/product/aldrich/d91632?lang=en®ion=AU, accessed July 4, 2014.
- T. J. Dale and J. Rebek, Angew. Chem., Int. Ed., 2009, 48, 7850–7852 CrossRef CAS PubMed.
- T. J. Dale and J. Rebek, J. Am. Chem. Soc., 2006, 128, 4500–4501 CrossRef CAS PubMed.
- S. Bencic-Nagale, T. Sternfeld and D. R. Walt, J. Am. Chem. Soc., 2006, 128, 5041–5048 CrossRef CAS PubMed.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192–5200 RSC.
- P. Nockemann, K. Binnemans and K. Driesen, Chem. Phys. Lett., 2005, 415, 131–136 CrossRef CAS PubMed.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS PubMed.
- U. Olsher, R. M. Izatt, J. S. Bradshaw and N. K. Dalley, Chem. Rev., 1991, 91, 137–164 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Representative NMR spectra demonstrating the extent of byproduct formation. Kinetic data on which Fig. 1–4 and Tables 1, 2 and 4 are based. Kinetic data and Eyring plots from which the data in Tables 3 and 5 are derived. Kinetic data, Eyring plot and activation parameters for the ethanolysis of diethyl chlorophosphate in acetonitrile. See DOI: 10.1039/c4nj01224k |
| ‡ 31P NMR spectroscopy was considered, particularly in order to identify any formation of byproducts. The overlap of signals in such spectra for many of the solvents systems used meant that 31P NMR spectroscopy was subsequently not used for kinetic analysis. For more details, see ESI.† |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |