
Application of bis(oxazoline) in asymmetric β-amination of chalcones†
Tao
Deng
,
Hongjun
Wang
and
Chun
Cai
*
Chemical Engineering College, Nanjing University of Science & Technology, Nanjing, Jiangsu 210094, P. R. China. E-mail: c.cai@mail.njust.edu.cn
First published on 5th November 2014
Abstract
An effective enantioselective β-amination of chalcones with N-bromosuccinimide into β-imidoketones using a bis(oxazoline) ligand is described. A wide variety of β-imidoketone derivatives containing various functional groups can be obtained with high enantioselectivities. The products are highly valuable molecules regarding their vast applications as building blocks of drugs and biologically active compounds.
Over the past decade, tremendous progress has been achieved by employing different nitrogen nucleophiles and new acceptors as well as more efficient catalyst systems for carbon–nitrogen bond formation.1 Especially in the past several years, the development of efficient and conventional approaches2 (for example, the iminium-activated nucleophilic additions to α,β-unsaturated aldehydes3 and the Mannich-type reaction of bis(dialkylamino)boron enolates with aldehydes4), leading to chiral β-imidoketones and their derivatives has attracted much attention in organic synthesis, as the β-imidoketones obtained are highly valuable molecules regarding their vast applications as building blocks of drugs and biologically active compounds.5 Very recently, Han and co-workers developed an acid-functionalized ionic liquid as catalyst for hetero-Michael addition of nitrogen nucleophiles to α,β-unsaturated ketones.6 Brenna et al. reported that Ni(II) and Pd(II) pyridinyloxazolidine-compound catalyzed aza-Michael addition of aliphatic amines to α,β-unsaturated ketones7 is an efficient approach toward the synthesis of β-imidoketones.
Although a variety of methods have been reported, further development of asymmetric β-amination reactions still remains a hot topic. Therefore, the necessity to explore appropriate conditions to improve the ee is sometimes required. Among various subareas of the rapidly growing field of organocatalysis, the use of bis(oxazoline)-containing ligands including C2-symmetric bis(oxazoline) or aza-bis(oxazoline) turned out to be a powerful approach for the asymmetric synthesis of a great variety of highly enantioenriched organic compounds.8
With all of these precedents in mind, we were interested in exploring the enantioselectivity for the asymmetric β-amination reaction9 when NBS reacted as a nucleophilic nitrogen source with chalcones. Herein, we report the details of our studies and disclose improved enantiomeric ratios using an optimized bis(oxazoline) ligand.
Our initial studies were carried out with chalcone (1a) as the substrate and N-bromosuccinimide (NBS) as the nucleophilic nitrogen source under basic conditions. A variety of commonly used chiral ligands were examined (Fig. 1). Only modest ee values were generally obtained except for bis(oxazoline) (L3); good yield and ee value could be obtained with bis(oxazoline) (L3) (Table 1, entry 4). Decreasing L3 to 5 mol% led to a lower yield with obvious loss of ee (Table 1, entry 5).
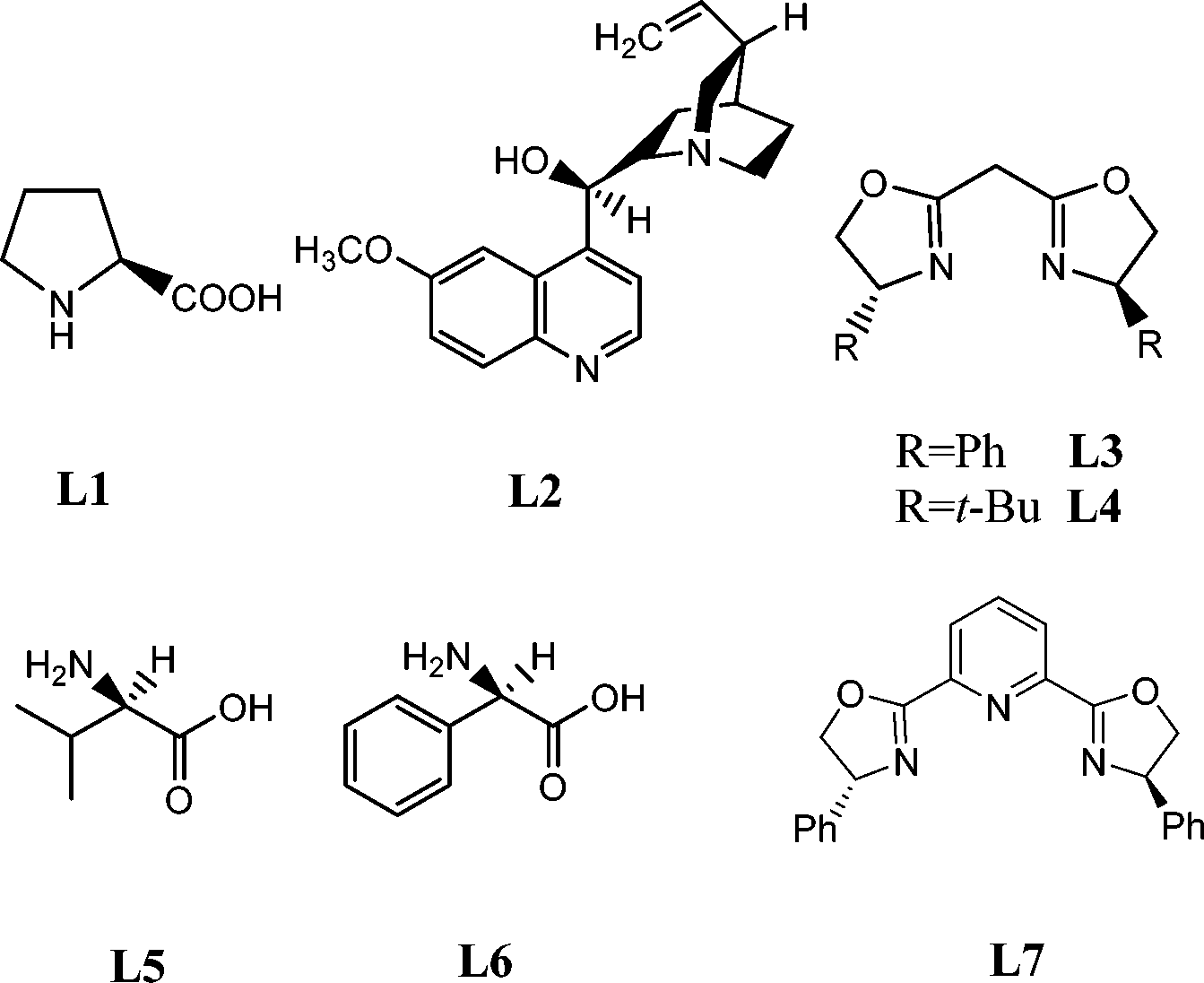 | ||
| Fig. 1 Selected examples of chiral ligands examined. | ||
|
|
|||
|---|---|---|---|
| Entry | Ligand | Yieldb (%) | eec (%) |
| a Reactions were carried out with 1a (1.0 mmol), NBS (1.2 mmol), ligand (0.1 mmol) and DBU (1.2 mmol) in MeCN (2.0 mL) for 15 h. b Determined by HPLC analysis. c The ee was checked by chiral HPLC using an Ultron ES-OVM column. d With L3 (0.05 mmol). | |||
| 1 | — | 69 | Racemic |
| 2 | L1 | 64 | 19 |
| 3 | L2 | 61 | 18 |
| 4 | L3 | 76 | 90 |
| 5d | L3 | 73 | 64 |
| 6 | L4 | 74 | 65 |
| 7 | L5 | 58 | 9 |
| 8 | L6 | 60 | 13 |
| 9 | L7 | 68 | 10 |
As shown in Table 2, no reaction occurred in MeCN at room temperature upon utilizing NaOH, K2CO3, Et3N, DABCO, and DMAP as bases (Table 2, entries 1–5). The reaction with pyridine (1.2 equiv.) as the base in MeCN gave 1-(3-oxo-1, 3-diphenylpropyl)-pyrrolidine-2, 5-dione (2a) in 39% yield (Table 2, entry 7). To our delight, DBU was found to be the most suitable base in terms of yield and enantioselectivity (Table 2, entry 6).
|
|
|||
|---|---|---|---|
| Entry | Base | Yieldb (%) | eec (%) |
| a Reactions were carried out with 1a (1.0 mmol), NBS (1.2 mmol), L3 (0.1 mmol) and base (1.2 mmol) in 2.0 mL MeCN for 15 h. b Determined by HPLC analysis. c The ee was checked by chiral HPLC using an Ultron ES-OVM column. | |||
| 1 | NaOH | n.r. | — |
| 2 | K2CO3 | n.r. | — |
| 3 | Et3N | n.r. | — |
| 4 | DABCO | n.r. | — |
| 5 | DMAP | n.r. | — |
| 6 | DBU | 76 | 90 |
| 7 | Pyridine | 39 | 71 |
The reaction was further optimized with respect to solvents (Table 3). When CH2Cl2, THF, EtOH, MeOH and DMF were selected as the solvents, the yields decreased (Table 3, entries 1–5). Additionally, no desired product was obtained with H2O as the solvent in the presence of TBAB (Table 3, entry 6). Among all the solvents tested, MeCN was the most efficient. 1-(3-Oxo-1, 3-diphenylpropyl)-pyrrolidine-2,5-dione (2a) was obtained in 76% yield with 90% ee using 10 mol% L3 in MeCN at room temperature (Table 3, entry 7). Other nucleophilic nitrogen sources were also examined (Table 3, entries 8 and 9). Moderate yield and ee were obtained with N-iodosuccinimide (NIS). However, no reaction was observed with N-chlorosuccinimide (NCS).
|
|
|||
|---|---|---|---|
| Entry | Solvent | Yieldb (%) | eec (%) |
| a Reactions were carried out with 1a (1.0 mmol), NBS (1.2 mmol), L3 (0.1 mmol) and DBU (1.2 mmol) in solvent (2.0 mL) for 15 h. b Determined by HPLC analysis. c The ee was checked by chiral HPLC using an Ultron ES-OVM column. d TBAB (0.05 mmol) was added. e With NIS (1.2 equiv.). f With NCS (1.2 equiv.). | |||
| 1 | CH2Cl2 | 57 | 51 |
| 2 | THF | 62 | 63 |
| 3 | EtOH | 33 | 73 |
| 4 | MeOH | 29 | 57 |
| 5 | DMF | 61 | 29 |
| 6d | H2O | n.r. | — |
| 7 | MeCN | 76 | 90 |
| 8e | MeCN | 54 | 84 |
| 9f | MeCN | n.r. | — |
The scope of this reaction was then evaluated with respect to substituted chalcones under the optimized conditions to form the corresponding β-imidoketones in 52–85% yield with 40–94% ee using 10 mol% L3 (Table 4, entries 1–15). The introduction of strong electron-donating groups (such as methoxy and methyl) at the para-position on the phenyl ring R1, led to a drop in yields and enantioselectivities (Table 4, entries 1, 12 and 13). When electron-withdrawing groups (such as para-bromo or trifluoromethyl groups) were introduced on the phenyl ring R1, lower enantioselectivities but higher yields were obtained (Table 4, entries 1, 14 and 15). However, the electron-donating groups (such as para-methyl or para-dimethylamino) on the phenyl ring R2 seemed to be less favorable in this protocol providing the corresponding products with moderate enantioselectivities (Table 4, entries 1, 5 and 6). Furthermore, with strong electron-withdrawing substituents (such as para-fluoro or para-nitro groups) on the phenyl ring R2, the reactions did not occur (Table 4, entries 2 and 4). It was notable that para-chloro substituents gave better ee in this reaction (Table 4, entry 3). Upon replacing the phenyl ring R2, with 1-naphthyl or other ortho-substituted phenyl on the phenyl ring R2, no desired products were obtained (Table 4, entries 7–9). This may be attributed to the effect of steric hindrance. Besides, this protocol could also be applied to heterocyclic chalcones as exemplified by (E)-1-phenyl-3-(pyridin-4-yl)prop-2-en-1-one in 67% yield and 64% ee (Table 4, entry 11). We further expanded the scope of this reaction to aliphatic chalcones. Unfortunately, we found that no desired product was obtained when a methyl substituent was introduced (R2 = Me) due to side reactions (Table 4, entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yieldb (%) | eec (%) |
| a Reactions were carried out with 1 (1.0 mmol), NBS (1.2 mmol), L3 (0.1 mmol) and DBU (1.2 mmol) in MeCN (2.0 mL) for 15 h. b Determined by HPLC analysis. c The ee was checked by chiral HPLC using an Ultron ES-OVM column. | |||||
| 1 | Ph | Ph | 2a | 76 | 90 |
| 2 | Ph | 4-FC6H4 | — | n.r. | — |
| 3 | Ph | 4-ClC6H4 | 2b | 83 | 94 |
| 4 | Ph | 4-NO2C6H4 | — | n.r. | — |
| 5 | Ph | 4-MeC6H4 | 2c | 73 | 80 |
| 6 | Ph | 4-NMe2C6H4 | 2d | 62 | 40 |
| 7 | Ph | 1-Naphthyl | — | n.r. | — |
| 8 | Ph | 2-ClC6H4 | — | n.r. | — |
| 9 | Ph | 2-BrC6H4 | — | n.r. | — |
| 10 | Ph | Me | — | n.r. | — |
| 11 | Ph | 4-Pyridinyl | 2e | 67 | 64 |
| 12 | 4-MeOC6H4 | Ph | 2f | 52 | 80 |
| 13 | 4-MeC6H4 | Ph | 2g | 71 | 76 |
| 14 | 4-CF3C6H4 | Ph | 2h | 79 | 66 |
| 15 | 4-BrC6H4 | Ph | 2i | 85 | 92 |
The reaction begins with the formation of intermediate A from NBS and DBU via halogen bond interaction. Then A transforms into a more electrophilic species B. Although the precise reaction mechanism is not clear, we proposed a possible plausible mechanism based on our work and on pertinent literature9 (Scheme 1).
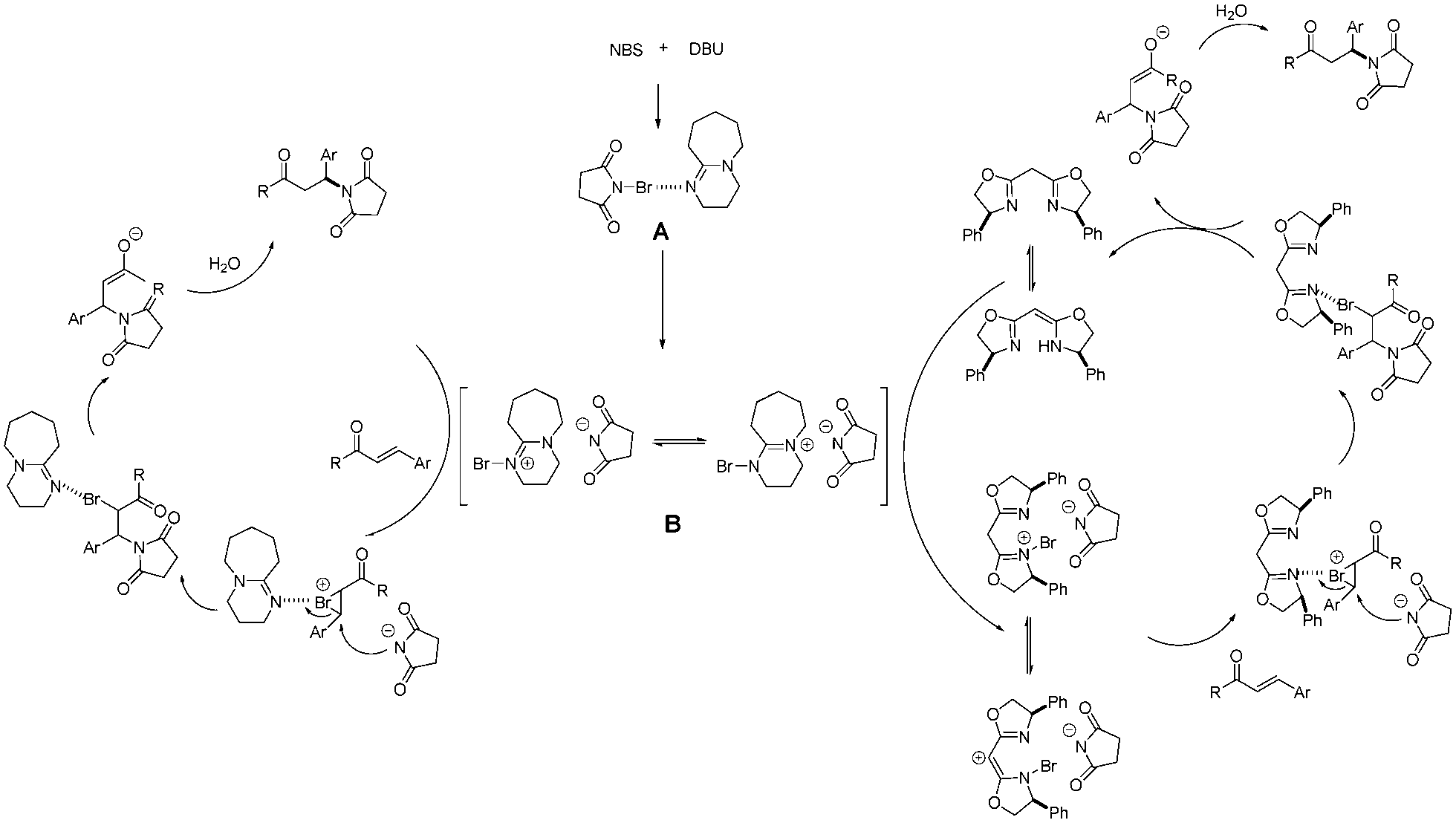 | ||
| Scheme 1 Plausible mechanism for the formation of β-aminoketones. | ||
In summary, we have developed an efficient enantioselective β-amination reaction of chalcones into β-imidoketones using NBS as nucleophilic nitrogen source and bis(oxazoline) as ligand. A wide variety of β-imidoketone derivatives with various functional groups were obtained in generally good yields with high enantioselectivities. Further transformations of these compounds provide access to useful intermediates with diverse functionality, such as building blocks of drugs and biologically active compounds.
Experimental section
General remarks
All reagents were purchased from commercial sources and used without treatment, unless otherwise indicated. The products were purified by column chromatography over silica gel. NMR spectra were recorded at 500 MHz using CDCl3 as the solvent. Elemental analysis was performed on a Vario EL III recorder. Mass spectra were obtained using an automated Fininigan TSQ Advantage mass spectrometer. Most of the products were known compounds and were identified by comparison of their physical and spectral data with those of authentic samples. The enantiomeric excess of the β-imidoketones was determined by HPLC on an Ultron ES-OVM column.Synthesis of chalcones (1a as an example)
A mixture of acetophenone (10 mmol) and benzaldehyde (1.1 equiv.) in anhydrous EtOH (15 mL) was stirred at room temperature for 5 min. Then, NaOH (3 equiv.) was added. The reaction mixture was stirred at room temperature overnight until aldehyde consumption. After that, HCl (10%) was added until neutrality. Then dichloromethane was added to dilute the reaction mixture. The organic layer was dried over anhydrous Na2SO4 and concentrated on Rotavapor under reduced pressure. Finally, the residue was purified by silica gel column chromatography to give 1a.A typical procedure for the β-amination of chalcones
A general procedure for the preparation of 2 (2a as an example): a mixture of chalcone 1a (208 mg, 1.0 mmol), L3 (30.6 mg, 0.1 mmol), and DBU (0.18 mL, 1.2 mmol) in MeCN (2.0 mL) was stirred at room temperature. NBS (213 mg, 1.2 mmol) was then added to the mixture. After starting material 1a was consumed as indicated by TLC, the reaction mixture was poured into water and then extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was washed with water (3 × 10 mL), dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography.Notes and references
- Recent examples: (a) X. F. Wang, J. An, X. X. Zhang, F. Tan, J. R. Xiao and W. J. Chen, Org. Lett., 2011, 13, 808 CrossRef CAS PubMed; (b) S. Fustero, S. Monteagudo, S. Flores and P. Barrio, Chem. – Eur. J., 2010, 16, 9835 CrossRef CAS PubMed; (c) Z. L. Yuan, Y. Wei and M. Shi, Eur. J. Org. Chem., 2010, 4088 CrossRef CAS; (d) S. Fustero, S. Monteagudo, S. Flores and P. Barrio, Org. Lett., 2010, 12, 5494 CrossRef CAS PubMed; (e) Y. F. Cai, X. H. Liu, Y. H. Hui, J. Jiang, W. T. Wang, W. L. Chen, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2010, 49, 6160 CrossRef CAS PubMed; (f) Y. F. Cai, X. H. Liu, J. Jiang, W. L. Chen, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2011, 133, 5636 CrossRef CAS PubMed; (g) W. Yang, H. X. He, Y. Gao and D. M. Du, Adv. Synth. Catal., 2013, 355, 3670 CrossRef CAS; (h) W. Yang and D. M. Du, Chem. Commun., 2013, 49, 8842 RSC.
- (a) J. Anthony, S. G. Burke, A. Davies and G. Christopher, Org. Biomol. Chem., 2004, 2, 1387 RSC; (b) L. Yang, L. W. Xu, W. Zhou, L. Li and C. G. Xia, Tetrahedron Lett., 2006, 47, 7723 CrossRef CAS PubMed; (c) M. Suginome, L. Uehlin and M. Murakami, J. Am. Chem. Soc., 2004, 126, 13196 CrossRef CAS PubMed; (d) A. Zarghia, S. A. Webb and S. Balalaieb, Eur. J. Org. Chem., 1998, 197 CrossRef.
- R. Appel, S. Chelli, T. Tokuyasu, K. Troshin and H. Mayr, J. Am. Chem. Soc., 2013, 135, 6579 CrossRef CAS PubMed.
- M. Suginome, L. Uehlin, A. Yamamoto and M. Murakami, Org. Lett., 2004, 6, 1167 CrossRef CAS PubMed.
- (a) S. A. Pishawikar and H. N. More, Int. J. Pharma Bio Sci., 2013, 4, 549 CAS; (b) D. Bhosle, S. Bharambe, N. Gairola and S. S. Dhaneshwar, Indian J. Pharm. Sci., 2006, 68, 286 CrossRef CAS PubMed; (c) S. N. Pandeya, D. Sriram and G. Nath, Sci. Pharm., 1999, 67, 10 Search PubMed; (d) S. N. Pandeya, V. S. Lakshmi and A. Pandeya, Indian J. Pharm. Sci., 2003, 65, 213 CAS; (e) J. V. D. Kamp and E. Mosettig, J. Am. Chem. Soc., 1936, 58, 1568 CrossRef.
- F. Han, L. Yang, L. Li and C. G. Xia, Org. Biomol. Chem., 2012, 10, 346 CAS.
- G. A. Ardizzoia, S. Brenna and B. Therrien, Dalton Trans., 2012, 41, 783 RSC.
- (a) M. Glos and O. Reiser, Org. Lett., 2000, 2, 2045 CrossRef CAS PubMed; (b) K. Lang, J. Park and S. Hong, J. Org. Chem., 2010, 75, 6424 CrossRef CAS PubMed; (c) S. A. Girard, T. Knauber and C. J. Li, Angew. Chem., 2014, 126, 76 CrossRef; (d) S. Gao, J. R. Chen, X. Q. Hu, H. G. Cheng, L. Q. Lu and W. J. Xiao, Adv. Synth. Catal., 2013, 355, 3539 CrossRef CAS; (e) K. Toribatake and H. Nishiyama, Angew. Chem., 2013, 125, 11217 CrossRef; (f) W. Dai, J. Li, B. Chen, G. S. Li, Y. Lv, L. Y. Wang and S. Gao, Org. Lett., 2013, 15, 5658 CrossRef CAS PubMed; (g) Z. M. Zhou, Z. H. Li, X. Y. Hao, X. Dong, X. Li, L. Dai, Y. Q. Liu, J. Zhang, H. F. Huang, X. Li and J. L. Wang, Green Chem., 2011, 13, 2963 RSC; (h) Z. M. Zhou, Z. H. Li, X. Y. Hao, J. Zhang, X. Dong, Y. Q. Liu, W. W. Sun, D. Cao and J. L. Wang, Org. Biomol. Chem., 2012, 10, 2113 RSC; (i) Z. H. Li, Z. M. Zhou, X. Y. Hao, J. Zhang, X. Dong, Y. Q. Liu, W. W. Sun and D. Cao, Appl. Catal., A, 2012, 28, 425–426 CrossRef PubMed; (j) S. F. Lu, D. M. Du, J. Xu and S. W. Zhang, J. Am. Chem. Soc., 2006, 128, 7418 CrossRef CAS PubMed; (k) H. Liu, S. F. Lu, J. Xu and D. M. Du, Chem. – Asian J., 2008, 3, 1111 CrossRef CAS PubMed.
- Y. Wei, S. X. Lin, F. S. Liang and J. P. Zhang, Org. Lett., 2013, 15, 852 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nj01660b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |