
Quick and easy access to N-Mannich bases of 1-isoindolinones by catalytic electroactivation of primary and secondary amines and tandem reaction with 2-formylbenzonitriles†
Laura
Palombi
*a,
Antonia
Di Mola
b and
Antonio
Massa
a
aDipartimento di Chimica e Biologia, Università di Salerno, Via Giovanni Paolo II, 84084 Fisciano (Sa), Italy. E-mail: lpalombi@unisa.it; Fax: +39-089-969603; Tel: +39-089-969575
bDipartimento di Scienze Farmaceutiche, Università di Salerno, Via Giovanni Paolo II, 84084 Fisciano (Sa), Italy
First published on 6th October 2014
Abstract
N-Mannich bases of 1-isoindolinones can be rapidly assembled by reacting 2-formylbenzonitriles with electroactivated amines on a Pt cathode, using a catalytic amount of electricity. Usefully, chiral amines allow the attainment of enantiopure N-Mannich bases by simple chromatographic separation.
The construction of heterocyclic cores and, at once, the introduction of several different functionalities on the selected molecular target is the subject of a large part of the current literature on organic synthesis. Nowadays, cascade and multicomponent reactions1 are the most common approaches to achieve this goal and are especially applied to the attainment of heterocyclic scaffolds with potential bio- and pharmacological activity. Due to their relevance in medicinal chemistry, in the last few years, we have become interested in the design and synthesis of 3-substituted isoindolinone derivatives,2 easily accessible by reacting 2-formylbenzonitriles with suitable carbon nucleophiles, using chemical3 or electrochemical4 catalytic systems. In particular, eco- and atom-economy, efficiency and readiness of the electrochemical approach5 prompted us to check if other useful nucleophiles could be electroactivated and used in a similar way. Intrigued by the possibility to introduce an exocyclic nitrogen atom on the third position of the isoindolinonic ring, we firstly focused our attention on amino compounds. In this work we wish to present our results concerning the tandem reaction of 2-formylbenzonitriles with electroactivated primary and secondary amines to afford N-Mannich bases 1, a class of compounds with easily conceivable versatility for a wide gamut of applications ranging from pharmacology, use as intermediates in drug synthesis, as well as ligands for catalytic purposes.6
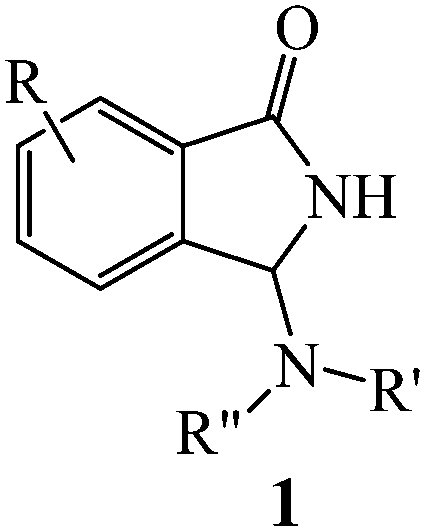
Despite their potential, a literature survey reveals that, after the pioneering study by R. Sato7 which shows the uncatalyzed access to compounds 1 when a large excess (more than 2000-fold molar excess) of amine is used, no more convenient catalyzed procedures have been reported starting from 2-formylbenzonitriles. However, other routes have been proposed (for example, the reaction of aldimines with isocyanates using a rhenium catalyst),8 demonstrating the interest for these kinds of heterocyclic scaffolds.9
Although simple amines are well-known nonreducible compounds at the Pt-cathode, previous investigations by one of the authors10 and others11 clearly demonstrated their indirect electrochemical activation in the addition reaction with weak electrophiles such as CO2 gas, via electrogenerated cyanomethyl anion or via CO2˙−. Taking into account these studies, benzylamine (2a), chosen as the model compound, has been electrolyzed in the cathodic side of a U-shaped two-compartment cell, in the presence of 2-formylbenzonitrile as an electrophile, under a variety of experimental conditions (Table 1). Interestingly, while pre-electrolyzed solutions of CH3CN/Et4N+BF4− and CH3CN/Et4N+BF4−/2-CNC6H4CHO (entries 1–2) were found to be completely ineffective as catalytic systems even after a long reaction time, N-Mannich base 1aa was obtained in very good yield by performing constant current electrolysis in the presence of 2a and 2-formylbenzonitrile 3a (entry 3). On the other hand, under the latter conditions, the current efficiency of the process is strikingly high, since a quantitative conversion of the starting materials is at once achieved by using only 2.5% of electricity. Although the electrochemical mechanism by which the reaction is initiated is not clear in detail, such a good turnover of the electron transfer process is highly desirable also in terms of atom-economy since, in parallel, it can drastically reduce the amount of the supporting electrolyte required to carry out the electrolysis.12
|
|
|||
|---|---|---|---|
| Entry | Q (e per molecule of 3a) | 3a Conversion | 1aa Yield |
| a A solution of CH3CN/Et4N+BF4− was pre-electrolyzed under galvanostatic conditions using a current quantity as reported in the table. 2a and 3a were added to the cathodic compartment after the current was switched off. b The electrolysis was performed in the presence of 3a. 2a was added after the current was switched off. c The electrolysis was performed in the presence of 2a and 3a. | |||
| 1a | 0.05 | — | — |
| 2b | 0.05 | — | — |
| 3c | 0.025 | 100% | 95% |
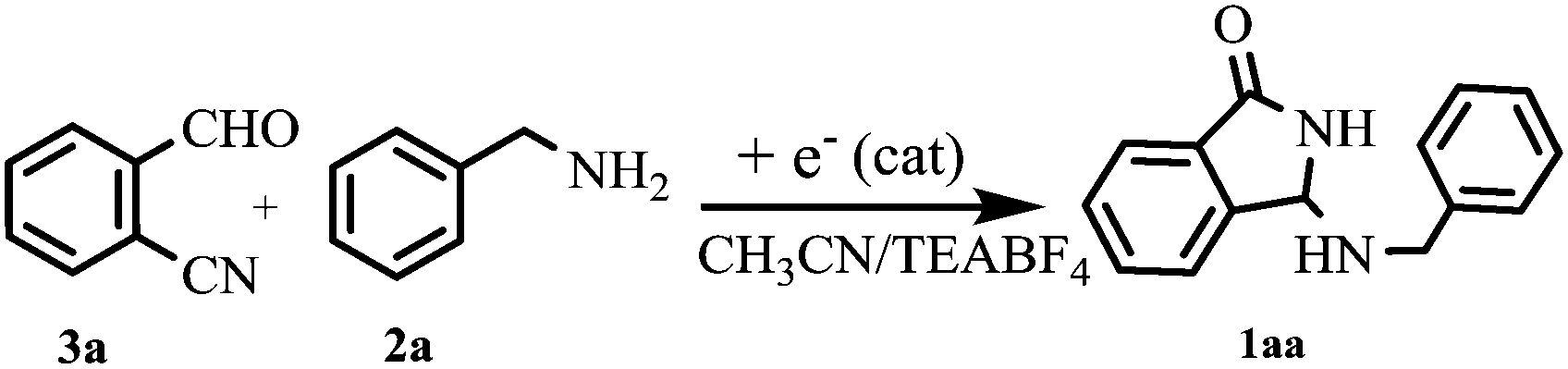
Taking into account these experiments, we hypothesized a reaction pathway which involves the electrochemically initiated deprotonation of the hemiaminal intermediate A, and the subsequent cascade of reactions (cyclization-rearrangement)7,13 as depicted in Fig. 1.
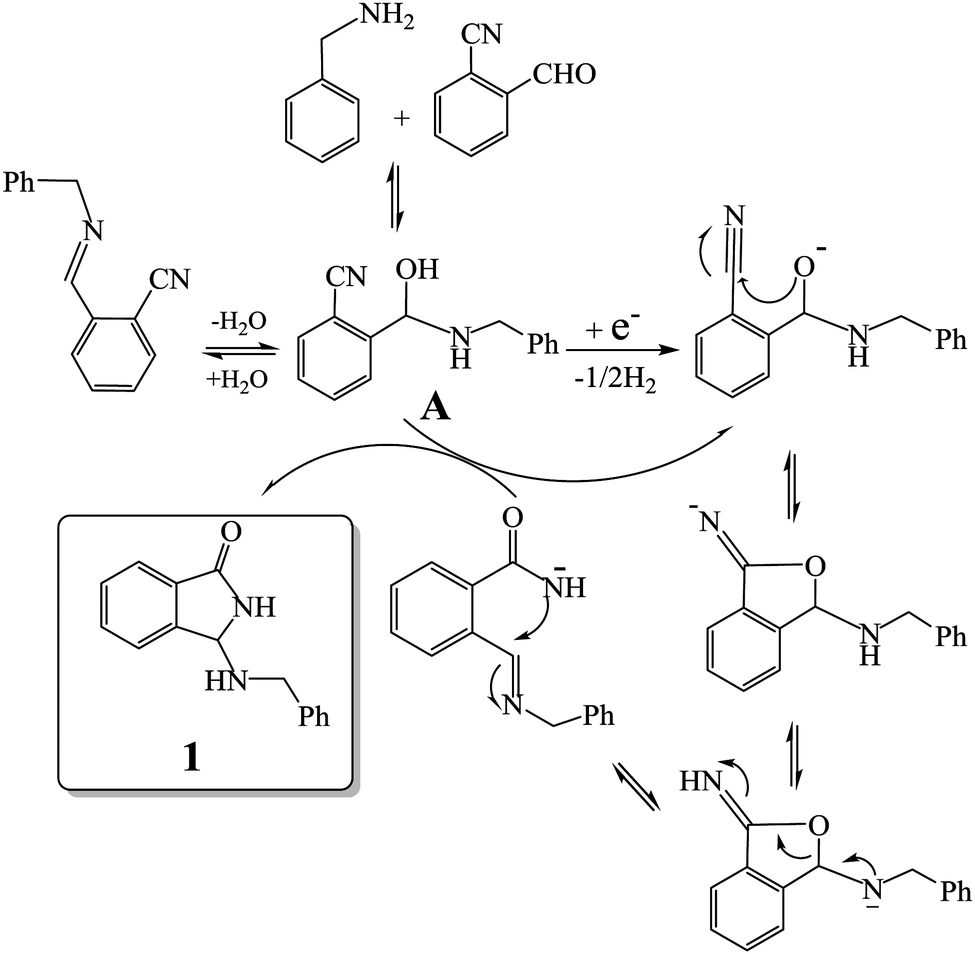 | ||
| Fig. 1 Proposed reaction pathway for the electrochemically initiated tandem reaction of amines with 2-formylbenzonitriles. | ||
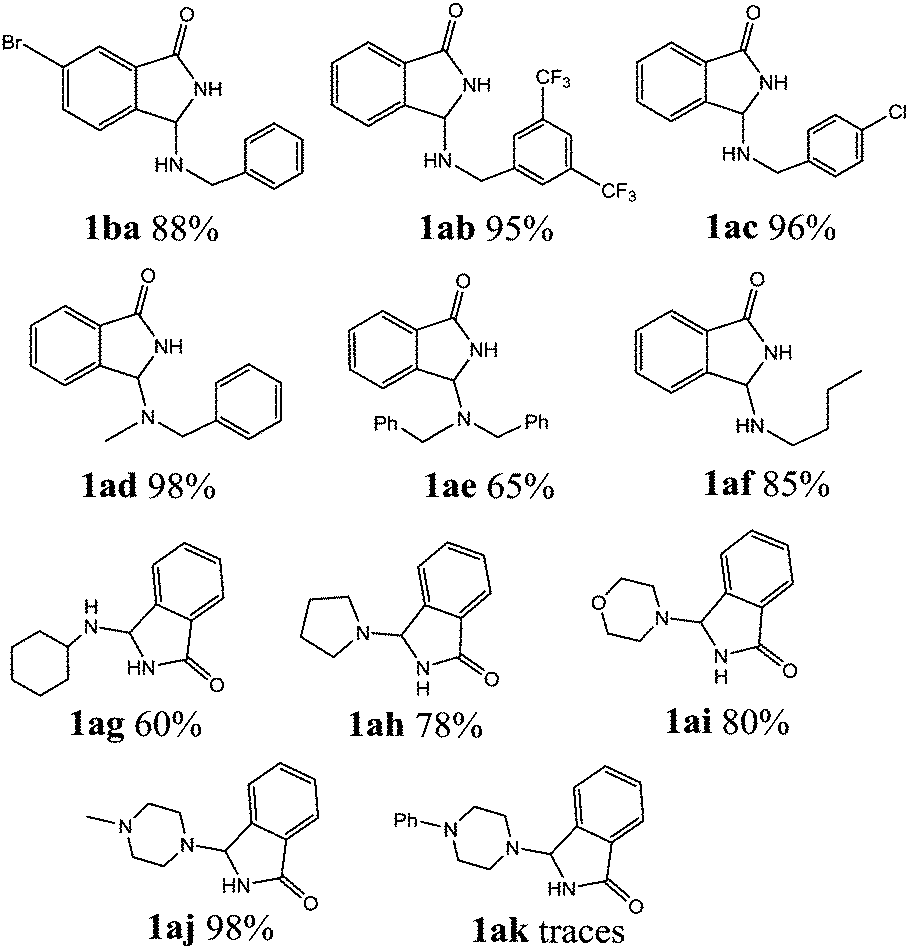
With the optimized set of conditions in hand, the electrochemically induced cascade reaction was investigated using different amine substrates 2a–k (Fig. 2). Table 2 summarizes the variety of N-Mannich bases accessible through the reaction of primary and secondary amines with 2-formylbenzonitriles (3a,b). The reaction was found to be general and successful with both primary and secondary alkyl- and benzyl-amines, as the products have been obtained in very good yield and complete regioselectivity was observed in almost all the cases. A slight decrease of the yield (as well as an increase of the reaction time) was observed only for amines 2e and 2g, probably because of the steric hindrance of these substrates. However, the reaction with amine 2k, which is scarcely soluble in the electrolysis solvent, did not occur, either by using a larger amount of current quantity (up to 1 electron per molecule) or by prolonging the reaction time.
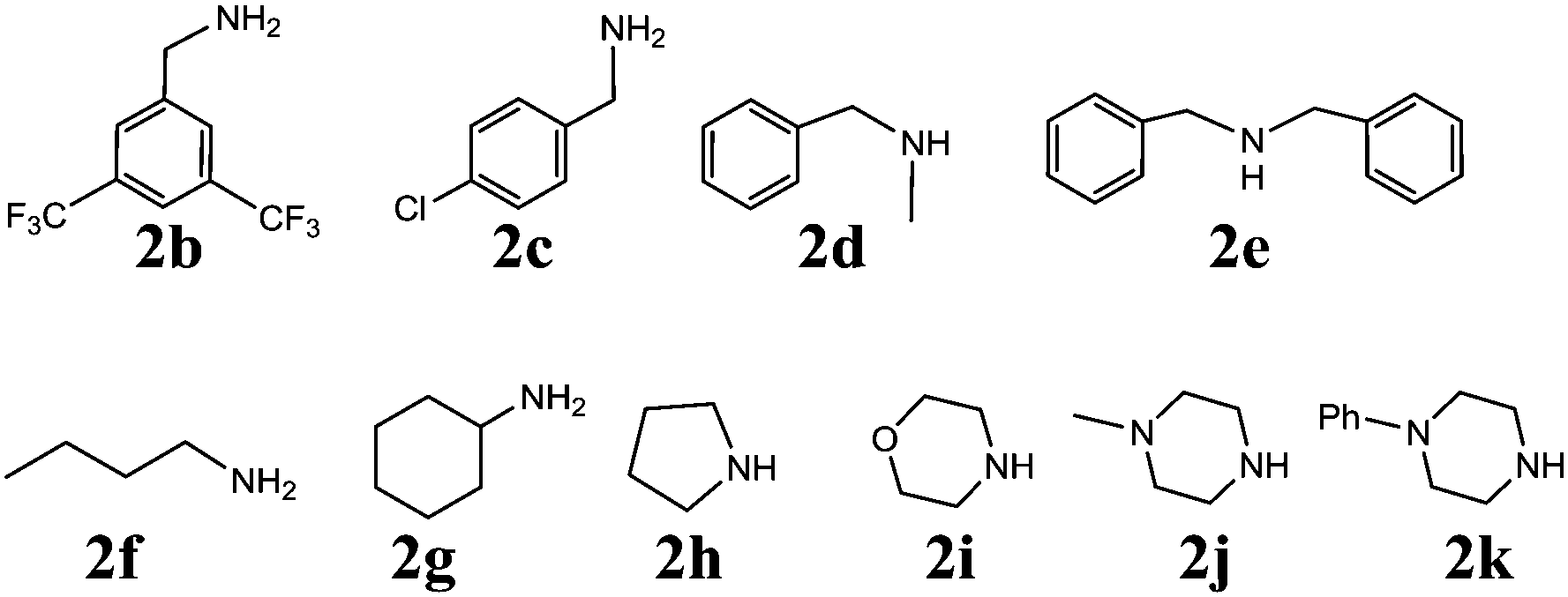 | ||
| Fig. 2 List of amines tested in the tandem reaction with 3a,b. | ||
|
|
|---|
|
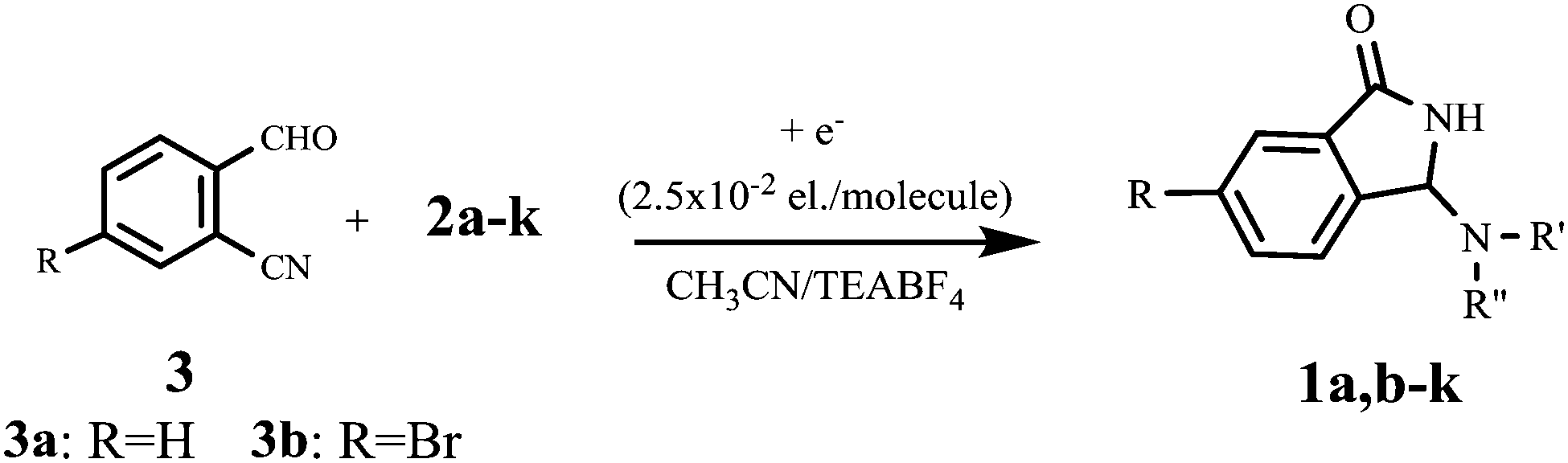
In view of the potential use of compounds 1 as chiral ligands or as pharmaceutical agents, in a follow-up of this investigation, we focused our attention on the possibility to obtain the newly created stereogenic center in the third position of the isoindolinonic ring in optically pure form. As, at this stage, we were not able to set up a suitable chiral environment that would allow their asymmetric synthesis under the electrochemical conditions, we tested chiral amines as resolving reagents (Fig. 3).
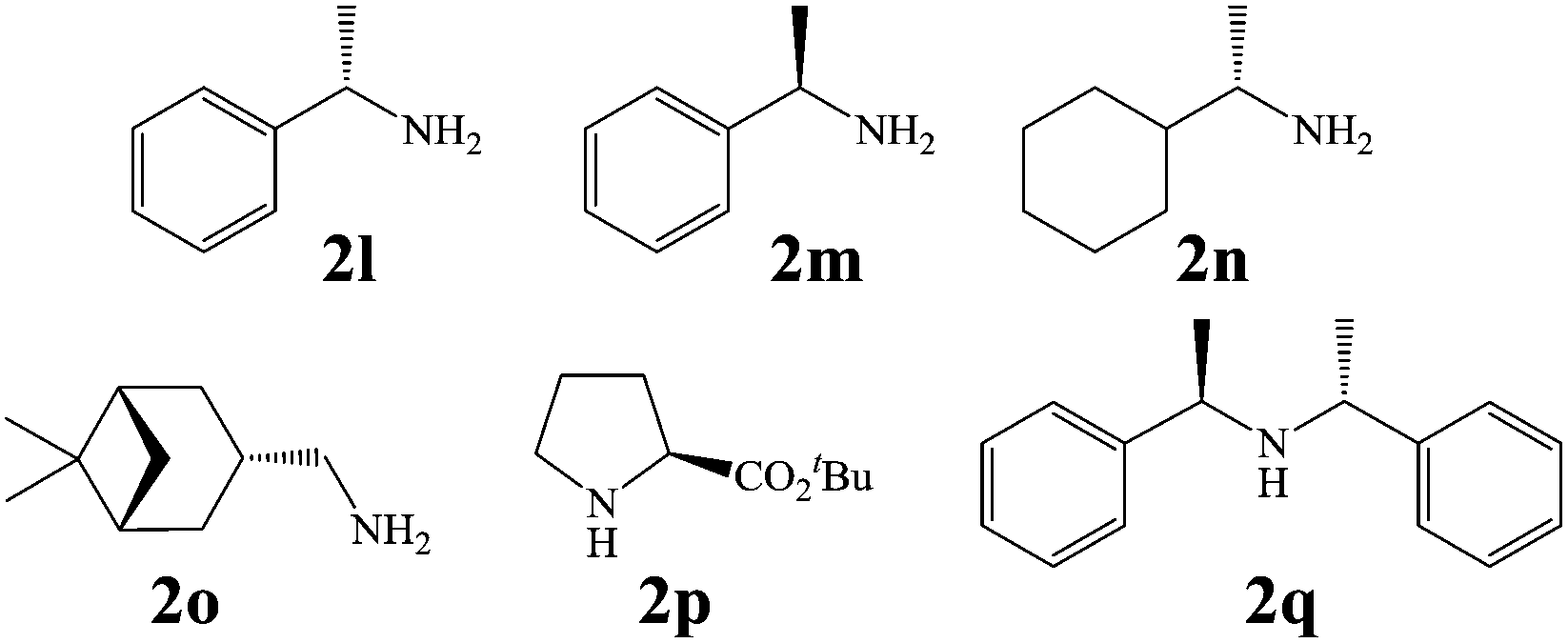 | ||
| Fig. 3 Chiral amines tested in the tandem reaction with 3a,b. | ||
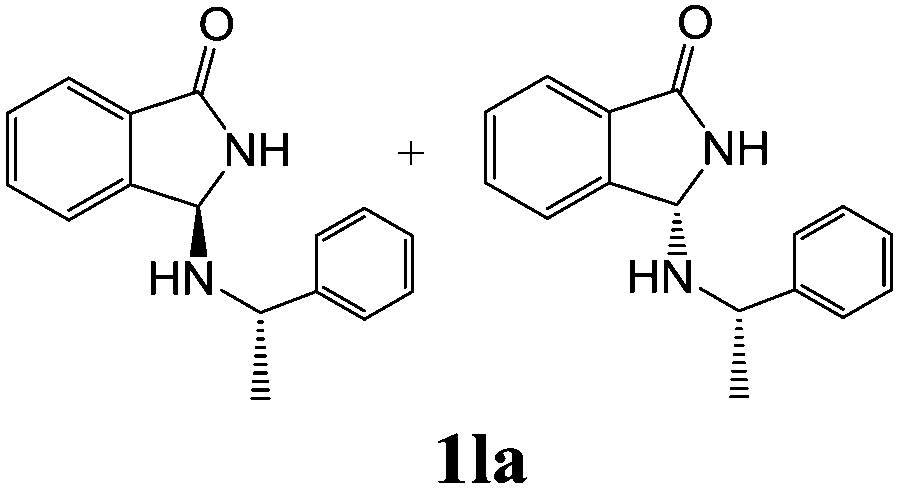
As shown in Table 3, with the exception of the bulky substrate 2q, chiral amines 2l–p provided the N-Mannich bases 1 with satisfactory to good yields, as epimeric mixtures. Though the diastereoselectivities observed were generally low, we were pleased to ascertain the possibility to access both the enantiopure diastereoisomers by simple separation on silica gel (entries 1–3 and 6).
| Entry | 2 | 1 | Yielda (%) | d.eb (%) |
|---|---|---|---|---|
| a Isolated yields refer to the mixture of diastereoisomers. b Diastereoisomeric excesses have been calculated by 1H-NMR analysis of the crude reaction mixtures. c 1H-NMR analysis of the crude mixture reveals a competitive reaction of formation of imine. | ||||
| 1 | 2l |
|
65 | — |
| 2 | 2m |
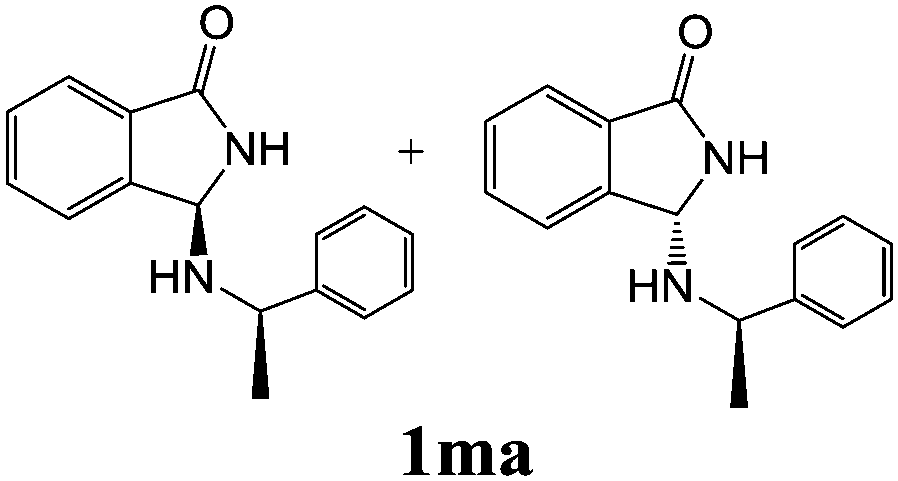
|
67 | — |
| 3 | 2m |
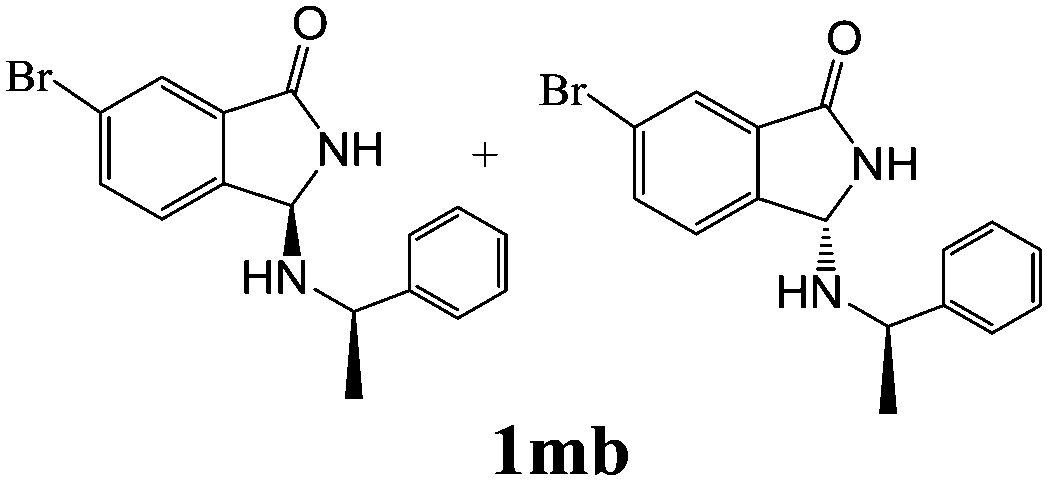
|
80 | — |
| 4c | 2n |
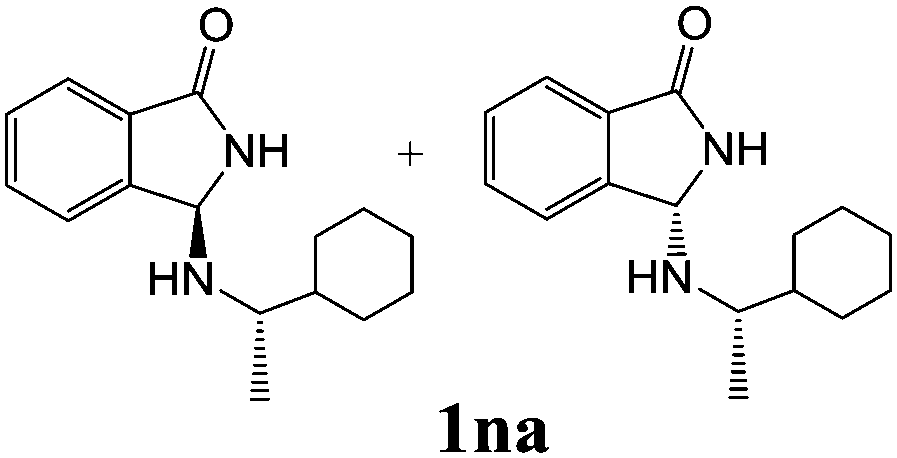
|
45 | 28 |
| 5c | 2o |
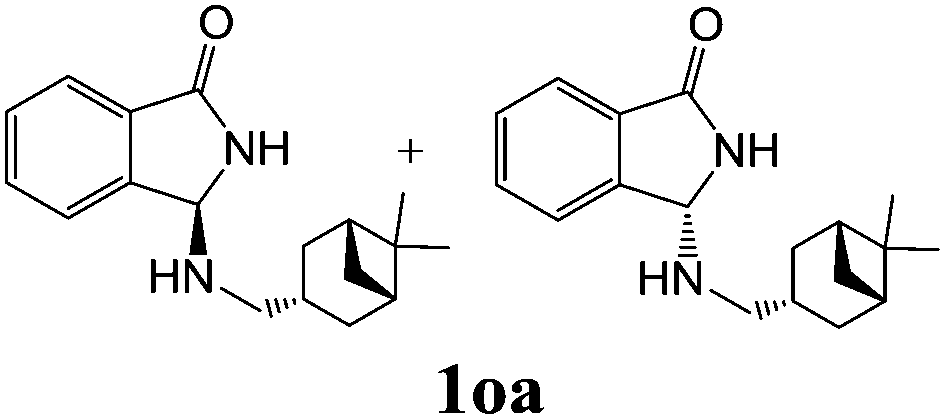
|
58 | — |
| 6 | 2p |
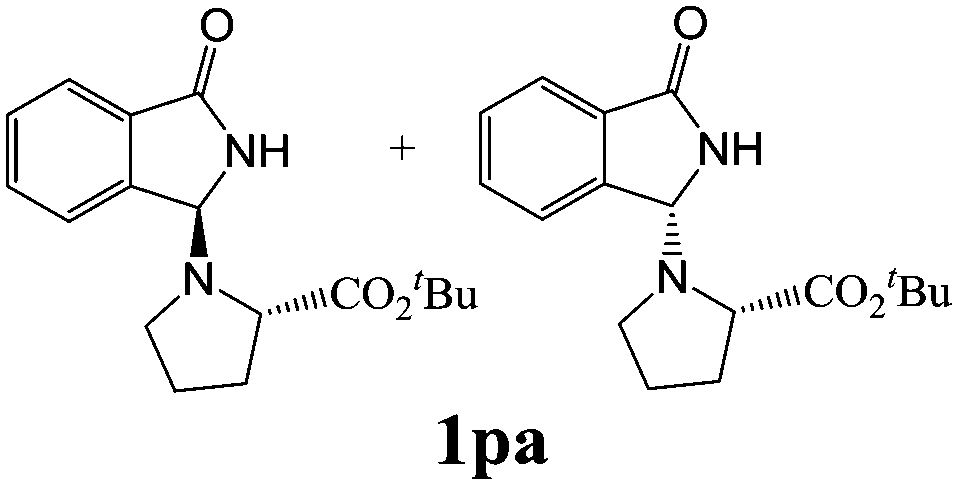
|
82 | 33 |
| 7 | 2q | — | — | — |
In summary, we have succeeded in the electrosynthesis of a series of N-Mannich bases of 1-isoindolinones, a class of heterocyclic compounds which may find applications in medicinal chemistry as well as ligands in catalysis. As postulated, cathodic electrolysis applying a catalytic amount of current under simple and handy galvanostatic conditions resulted in the electro-activation of primary and secondary alkyl- and benzyl-amines in the tandem reaction with 2-formylbenzonitriles. The desired products have been generally obtained in good yield and in a short reaction time, avoiding either metal or basic catalyst and, as a consequence, easy set-up and work-up procedures could be established. Furthermore, the use of chiral non-racemic amines as starting materials has allowed access to enantiopure 3-aminosubstituted isoindolinone derivatives 1 by simple chromatographic separation. We are currently working to develop an asymmetric version of this reaction.
Experimental
Typical experimental procedure for electro-induced synthesis of isoindolinones 1 in CH3CN: A solution of 2 (0.21 mmol) and 3 (0.2 mmol) in CH3CN/TEABF4 (0.4 ml/0.03 mmol) was electrolyzed at r.t., under galvanostatic conditions (8 mA, 0.025 electrons per molecule of 3), using a Hewlett Packard DC Power Supply Mod. E3612A. The experiments were carried out in the cathodic compartment of a U-divided glass cell separated through a porous G-4 glass plug. Platinum spirals (apparent area 1 cm2) were used as anode and cathode. In all the experiments the anolyte was constituted by a solution of 0.05 M TEABF4 in CH3CN.At the end of the electrolysis, TLC analyses showed disappearance of 3 and the reaction was in any case prolonged at r.t. under magnetic stirring for 2 h. The mixture was then concentrated in vacuum and directly purified by silica gel chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcEt = 2:1; CH3Cl:AcEt = 9:1 or CH3Cl:MeOH = 9:1).
:AcEt = 2:1; CH3Cl:AcEt = 9:1 or CH3Cl:MeOH = 9:1).
Notes and references
- (a) D. B. Ramachary and S. Jain, Org. Biomol. Chem., 2011, 9, 1277–1300 RSC; (b) W. Zhao and F.-E. Chen, Curr. Org. Synth., 2012, 9, 873 CrossRef CAS; (c) E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234 CrossRef CAS PubMed; (d) I. Akritopoulou-Zanze and S. W. Djuric, Top. Heterocycl. Chem., 2010, 25, 231 CAS; (e) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605 CrossRef CAS PubMed.
- (a) A. Di Mola, L. Palombi and A. Massa, Curr. Org. Chem., 2012, 16, 2302 CrossRef CAS; (b) A. Couture and P. Grandclaudon, Stud. Cercet. Stiint.: Chim. Ing. Chim., Biotehnol., Ind. Aliment., 2010, 11, 11 CAS.
- (a) C. Petronzi, S. Collarile, G. Croce, R. Filosa, P. De Caprariis, A. Peduto, L. Palombi, V. Intintoli, A. Di Mola and A. Massa, Eur. J. Org. Chem., 2012, 5357 CrossRef CAS; (b) V. More, R. Rohlmann, O. G. Mancheno, C. Petronzi, L. Palombi, A. De Rosa, A. Di Mola and A. Massa, RSC Adv., 2012, 2, 3592 RSC; (c) M. Perillo, A. Di Mola, R. Filosa, L. Palombi and A. Massa, RSC Adv., 2014, 4, 4239 RSC; (d) M. Angelin, M. Rahm, A. Fischer, T. Brinck and O. Ramstrom, J. Org. Chem., 2010, 75, 5882 CrossRef CAS PubMed; (e) M. Angelin, P. Vongvilai, A. Fischer and O. Ramstrom, Chem. Commun., 2008, 768 RSC.
- (a) L. Palombi, A. Di Mola, C. Vignes and A. Massa, Mol. Diversity, 2014, 18, 323–333 CrossRef CAS PubMed; (b) P. Antico, V. Capaccio, A. Di Mola, A. Massa and L. Palombi, Adv. Synth. Catal., 2012, 354, 1717 CrossRef CAS.
- (a) E. Dunach, M. J. Medeiros and S. Olivero, New J. Chem., 2006, 30, 1534 RSC; (b) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS PubMed.
- (a) S. N. Pandeya, V. S. Lakshmi and A. Pandey, Indian J. Pharm. Sci., 2003, 65, 213 CAS; (b) R. Bamnela and S. P. Shrivastava, Chem. Sci. Trans., 2012, 1, 431 CrossRef CAS; (c) L. Muruganandam and K. Balasubramanian, Chem. Sci. Rev. Lett., 2012, 1, 172 CAS.
- R. Sato, T. Senzaki, T. Goto and M. Saito, Chem. Lett., 1984, 1599 CrossRef CAS.
- Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed.
- K. Kobayashi, K. Nakagawa and T. Kozuki, Heterocycles, 2012, 85, 165 CrossRef CAS.
- M. Feroci, M. A. Casadei, M. Orsini, L. Palombi and A. Inesi, J. Org. Chem., 2003, 68, 1548 CrossRef CAS PubMed.
- D. F. Niu, L. Zhang, L. P. Xiao, Y. W. Luo and J. X. Lu, Appl. Organomet. Chem., 2007, 21, 941 CrossRef CAS.
- (a) L. Palombi, M. Feroci, M. Orsini and A. Inesi, Chem. Commun., 2004, 1846 RSC; (b) T. Caruso, M. Feroci, M. Orsini, A. Inesi, A. Scettri and L. Palombi, Adv. Synth. Catal., 2006, 348, 1942 CrossRef CAS.
- K. Kobayashi, K. Matsumoto, D. Nakamura, S. Fukamachi and H. Konishi, Helv. Chim. Acta, 2010, 93, 1048–1051 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental instrumentation and procedures, spectroscopic data, copies of 1H NMR and 13C NMR are provided. See DOI: 10.1039/c4nj01606h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |