
Microencapsulation of hexadecane by surface-initiated atom transfer radical polymerization on a Pickering stabilizer
Dezhong
Yin
*,
Jinjie
Liu
,
Wangchang
Geng
,
Baoliang
Zhang
and
Qiuyu
Zhang
School of Sciences, Northwestern Polytechnical University, Xi'an, China. E-mail: dezh_yin@nwpu.edu.cn; Tel: +86 02988431672
First published on 9th October 2014
Abstract
Hexadecane-loaded microcapsules with SiO2–poly(methyl methacrylate) (PMMA) hybrids as shells were prepared by surface-initiated atom transfer radical polymerization (SI-ATRP) in an oil-in-water Pickering emulsion. ATRP-initiators were introduced onto SiO2 particles by a homemade silane coupling reagent containing a 2-bromoisobutyrate group. SI-ATRP polymerization was conducted on Pickering stabilizers based on activators regenerated by electron transfer (ARGET). TGA results indicated that 80.3% of PMMA chains in the shell were covalently-bonded to SiO2, which was formed by SI-ATRP polymerization. The microcapsules show excellent potential for thermal energy storage, as evidenced by a high melting enthalpy of 161.7 J g−1.
Microcapsules have received considerable attention due to their particular structures and functions.1,2 A variety of interfacial related processes have been developed to fulfill the microencapsulation. Emulsion-based soft template-assisted approaches provide a good control over the microcapsule morphology and functionality.3,4 Particles can be irreversibly adsorbed at the oil–water interface,5 forming stable Pickering emulsions. The structure of Pickering emulsions makes them excellent templates to prepare microcapsules with inorganic–polymer hybrids as shells.6,7 These kinds of microcapsules exhibit special properties for particular applications. For example, Song8 showed that silver-incorporated microencapsulated phase change materials have better thermal stability than polymer-shelled microcapsules. Other research studies with incorporated carbon fibers,9 iron particles,10 and silica11 gave similar results.
It is crucial to confine the radical polymerization at oil/water interfaces for controlling the morphology of microcapsules. In traditional emulsion systems, reactive surfactants, such as polymerizable surfactants,12 amphiphilic transfer agents13 and amphiphilic initiators14 were employed to restrict the growing polymer to the interface. In surfactant-free Pickering emulsions, it is an exclusive choice to introduce radical initiators or chain transfer agents onto the surface of Pickering stabilizers. Following this strategy, Wang reported the use of initiator-modified silica nanoparticles as emulsifiers to confine the atom transfer radical polymerization (ATRP) of lightly cross-linked 2-hydroxyethylmethacrylate at the interface of a Pickering emulsion.15 Stöver coated LUDOX CL nanoparticles electrostatically with an anionic polymeric ATRP initiator for normal ATRP of water-soluble cross-linking monomers at the oil–water interface.16
Herein, we report ATRP for the fabrication of microcapsules with hybrid shells in a Pickering emulsion based on activators regenerated by electron transfer (ARGET). A silane coupling reagent containing a 2-bromoisobutyrate group (compound 2 in Fig. 1) was synthesized and employed directly to modify SiO2 to make SiO2 moderately hydrophilic and active for surface-initiated ATRP (SI-ATRP). Oil dissolved monomers were introduced in the Pickering emulsion. By the ARGET ATRP model using ascorbic acid (Vc) as a reducing agent, MMA was grafted on the surface of SiO2, leading to the formation of microcapsules with PMMA–SiO2 hybrids as shells. A complicated procedure to deoxygenate is shortened. Monomers and the catalyst (Cu+) were located in the dispersive and continuous phases, respectively, which is desirable to confine the ATRP at the interface. To the best of our knowledge, ARGET ATRP in a Pickering emulsion has not been demonstrated so far.
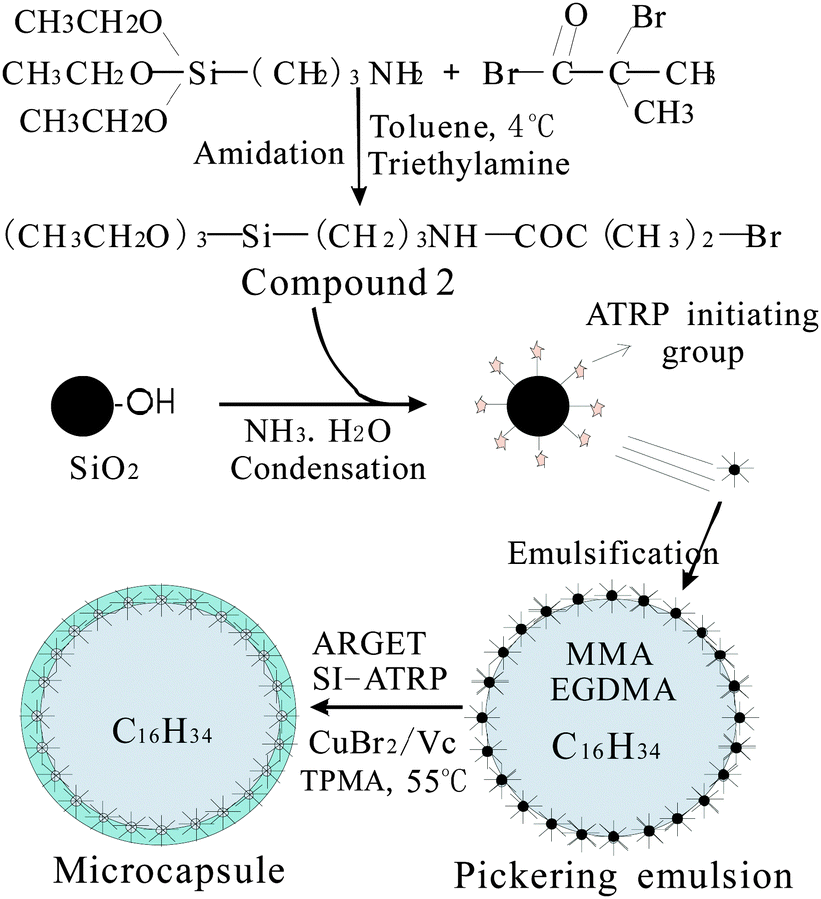 | ||
| Fig. 1 Scheme for immobilization of ATRP initiating groups and microencapsulation. | ||
Fig. 1 shows the overall scheme of microencapsulation. In the first step, we immobilized ATRP initiators on SiO2 particles. A silane coupling reagent containing a 2-bromoisobutyrate group (compound 2 in Fig. 1) was synthesized by amidation between 3-aminopropyl triethoxysilane (KH550) and 2-bromoisobutyryl bromide (BiBB) in anhydrous toluene using triethylamine as a deacid reagent. This amidation reaction is of high reactivity, and low temperature below 4 °C was maintained to control it, as commonly practiced in research studies.15 The reaction mixture was poured into a mortar containing silica particles and the powder was milled continuously. We optimized the amount of toluene to ensure that the silica particles can be soaked by the mixture, and as a result can be modified evenly by the silane coupling reagent. After evaporation of toluene, an ethanol–ammonia mixture was introduced similarly. Ammonia can catalyze the hydrolysis of the siloxane groups in KH550 into silanol groups and the condensation between silanol groups of hydrolyzed compound 2 and SiO2 particles. The modified SiO2 particles were washed with ethanol to remove the impurities. The average diameter of modified SiO2 was determined to be 70.8 nm using a laser particle size analyzer. By TGA analysis, the weight loss of modified SiO2 was estimated to be 7.14% at 600 °C. The molar mass of lost groups in compound 2 (–CH2–CH2–CH2–NH–CO–CBr(CH3)2) was 207 g mol−1. The surface density of ATRP initiators on SiO2 particles was calculated to be 0.345 mmol g−1 from these two data.
In the second step, an oil-in-water Pickering emulsion was stabilized solely by the modified SiO2, followed by ARGET ATRP polymerization to obtain microcapsules. SiO2 suspension, CuBr2 solution and tris(2-pyridylmethyl)amine(TPMA) were mixed as the water phase and monomers and hexadecane were mixed as the oil phase. After ultrasonic emulsification and displacing the air in the vessel with N2, ascorbic acid (Vc) was introduced to initiate ARGET ATRP at 55 °C for 48 hours. The mole ratio of the C–Br group/Cu2+/TPMA/Vc was fixed to be 0.103/0.0384/0.101/0.292. The majority of the Cu2+ and Cu+ complexes with TPMA were definitely located in the aqueous phase. So, a high amount of the catalyst was used to ensure a sufficient amount of the catalyst in the oil phase. With C–Br ATRP initiating groups on SiO2, polymer chains propagate from the SiO2 surface, producing microcapsules with covalently-bonded poly(methyl methacrylate) (PMMA)–SiO2 hybrid shells. A cross-linking structure was formed by ethyleneglycol dimethacrylate (EGDMA) between PMMA chains, which connected PMMA and SiO2 into whole shells. The microcapsules were filtrated and washed with deionized water exhaustedly, and volatilized at ambient temperature.
Four batches of microcapsules under different experimental conditions are summarized in Table 1. Due to abundant pyridyl groups, TPMA has suitable hydrophobicity to allocate part of its complexes to Cu(I) and Cu(II) in the oil phase. This is critical for a successful ATRP in emulsion. Monomers diffused to the interface, wherein they were initiated by the ATRP initiator on the SiO2 surface. The conversion of MMA for 48 h at 55 °C reached 75–81% (Table 1). This high conversion is ascribed to the high activity of the catalyst. Complexes of Cu+ with TPMA, a typical branched tetradentate ligand with nitrogen atoms in pyridine, produce the most active catalysts.17 Also, limited radical termination by oxygen in ARGET ATRP and suppressed disproportionation of Cu+ to form Cu and Cu2+ by Vc partially account for the high conversion.16
| No. | MMA (g) | EGDMA (g) | C16H34 (g) | Conversion of MMA (%) | Core content by TGA (%) | Bonded PMMA (%) | Core content by enthalpy (%) |
|---|---|---|---|---|---|---|---|
| 1 | 0.5 | 0 | 2 | 81.0 | 70.4 | 80.3 | 71.2 |
| 2 | 1.0 | 0 | 2 | 75.6 | 61.1 | 77.1 | 60.5 |
| 3 | 0.5 | 0.2 | 2 | 77.2 | 66.5 | — | 67.7 |
| 4 | 1.0 | 0.2 | 2 | 74.8 | 59.1 | — | 58.3 |
Fig. 2 shows an optical image of the emulsion and SEM images of the corresponding microcapsules. Almost all SiO2 particles were adsorbed at the interface of the emulsion, as evidenced by the fact that the aqueous phase of the emulsion was transparent (bottle shown in the inset of Fig. 2a). After polymerization, the individual spherical morphology of droplets was retained (Fig. 2b), indicating an excellent stability of the emulsion during polymerization. We soaked the microcapsules in ethanol for 48 h to dissolve the core material. The microcapsules without EGDMA collapsed and the sediment was separated for characterization of the shell material. The microcapsules with EGDMA as a cross-linker retain the particulate shape, even though they were distorted under high vacuum exerted during Au spraying and SEM detection (Fig. 2c). This indicated that the microcapsules have a core–shell structure with a single cavity and the SiO2 particles are connected together by cross-linkers (as shown in Fig. 1).
 | ||
| Fig. 2 Micrographs of (a) the emulsion, (b) microcapsule 3 and (c) ethanol-treated microcapsules. | ||
FT-IR spectra of modified SiO2, microcapsules, the shell material and SiO2 precipitation separated from microcapsule 2 are shown in Fig. 3. The spectrum of modified SiO2 (Fig. 3a) presents strong absorption peaks of amide groups at 1539 cm−1 and 1645 cm−1. This amide compound was formed exclusively by the reaction between KH550 and BiBB. The spectrum of microcapsules (Fig. 3b) presents characteristic peaks of hexadecane (C–H stretching at 2850–3000 cm−1), PMMA (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1726 cm−1) and SiO2 (Si–O–Si bending at 472 cm−1). The absorbance from 1726 cm−1 to 472 cm−1, as shown in the FT-IR spectrum of the shell material (Fig. 3c), is consistent with that shown in the spectrum of microcapsules, indicating that CO stretching originates from PMMA, rather than MMA. The peak of PMMA at 1726 cm−1 can be seen in the FT-IR spectrum of SiO2 precipitation (Fig. 3d). Because free PMMA was removed by THF, this PMMA was exclusively connected on the SiO2 surface and originated from the SI-ATRP of MMA.
O stretching at 1726 cm−1) and SiO2 (Si–O–Si bending at 472 cm−1). The absorbance from 1726 cm−1 to 472 cm−1, as shown in the FT-IR spectrum of the shell material (Fig. 3c), is consistent with that shown in the spectrum of microcapsules, indicating that CO stretching originates from PMMA, rather than MMA. The peak of PMMA at 1726 cm−1 can be seen in the FT-IR spectrum of SiO2 precipitation (Fig. 3d). Because free PMMA was removed by THF, this PMMA was exclusively connected on the SiO2 surface and originated from the SI-ATRP of MMA.
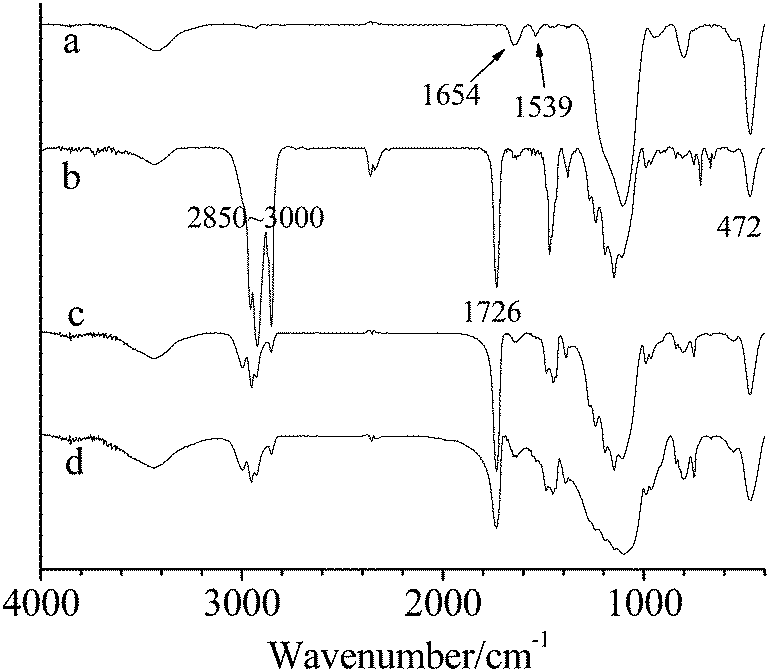 | ||
| Fig. 3 FTIR of (a) modified SiO2, (b) microcapsule 2, (c) the shell material and (d) SiO2 precipitation. | ||
Typical TGA curves of hexadecane, microcapsule 2, the shell material and SiO2 precipitation are shown in Fig. 4. By heating and flushing with N2, hexadecane evaporates at temperatures ranging from 120 to 220 °C (Fig. 4a). The shell material, a typical polymer, lost its weight at temperatures between 300 and 410 °C. The mass ratio of hexadecane/PMMA/SiO2 in microcapsule 2 is approximately 61.1/30.7/8.2 (Fig. 4b), a value consistent with the feed (as a ratio of 2/1/0.3). The core content of microcapsules was obtained according to the weight loss at 250 °C in the TGA curve, and the results are listed in Table 1. The weight loss of PMMA in SiO2 precipitation (Fig. 4d) verifies the existence of bonded PMMA on SiO2. Because the amount of SiO2 is consistent before and after THF treatment of the shell material, the percentage of bonded PMMA on SiO2 was calculated from the PMMA/SiO2 ratio obtained by TGA analysis.7 The results show that 80.3% and 77.1% of PMMA in microcapsule 1 and microcapsule 2 is bonded on SiO2, respectively.
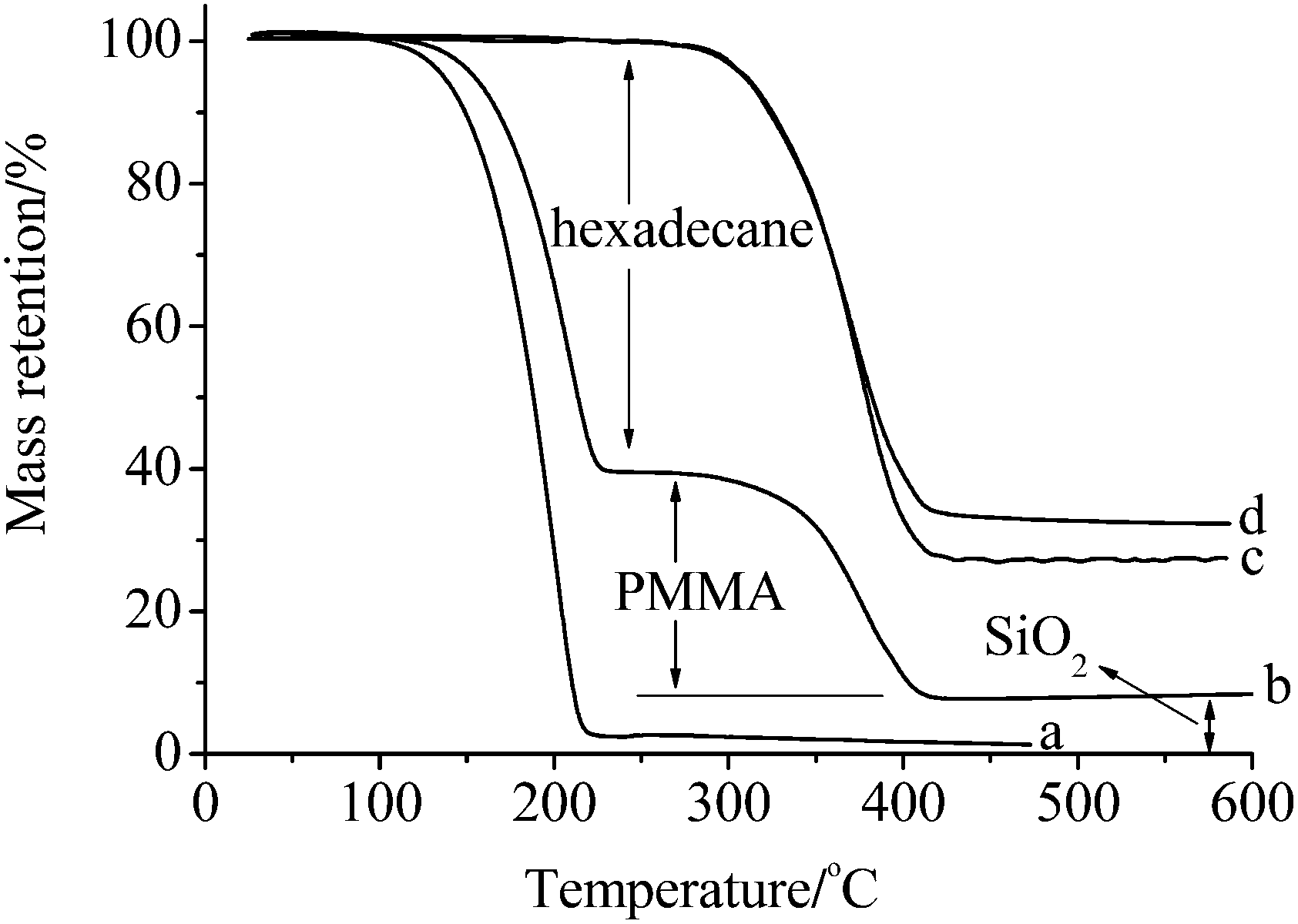 | ||
| Fig. 4 TGA curves of (a) hexadecane, (b) microcapsule 2, (c) the shell material and (d) SiO2 precipitation. | ||
Fig. 5 shows DSC curves of hexadecane and microcapsules. The melting thermogram of hexadecane reveals a well-defined exothermic peak at around 19.22 °C and a phase-change enthalpy of 227.2 J g−1. The suitable phase change temperature and high latent heat make hexadecane a predominant alternative in improving the thermal comfort of a fabric.18 After hexadecane was encapsulated, a complicated freezing process and supercooling to 3.3 °C were observed. The peak of microcapsules became wider than that of bulk hexadecane, which may be attributed to the low thermal conductivity of the polymer shell.19 The degree of super-cooling of hexadecane in microcapsules enhanced compared with that of bulk hexadecane, which indicates that the crystallization of an encapsulated liquid droplet was confined, and the number of nuclei in each droplet decreased compared to that of the bulk liquid droplet.20 The melting enthalpy of microcapsules ranges from 132.4 J g−1 to 161.7 J g−1. Core content values of microcapsules based on the enthalpy of microcapsules and bulk hexadecane, determined by methods reported elsewhere,4,20,21 are listed in Table 1. These results are well consistent with the recipes and the results obtained from TGA analysis, indicating a high efficiency of microencapsulation to the core material. The high enthalpy makes the microcapsules potential candidates for microencapsulated phase change materials.
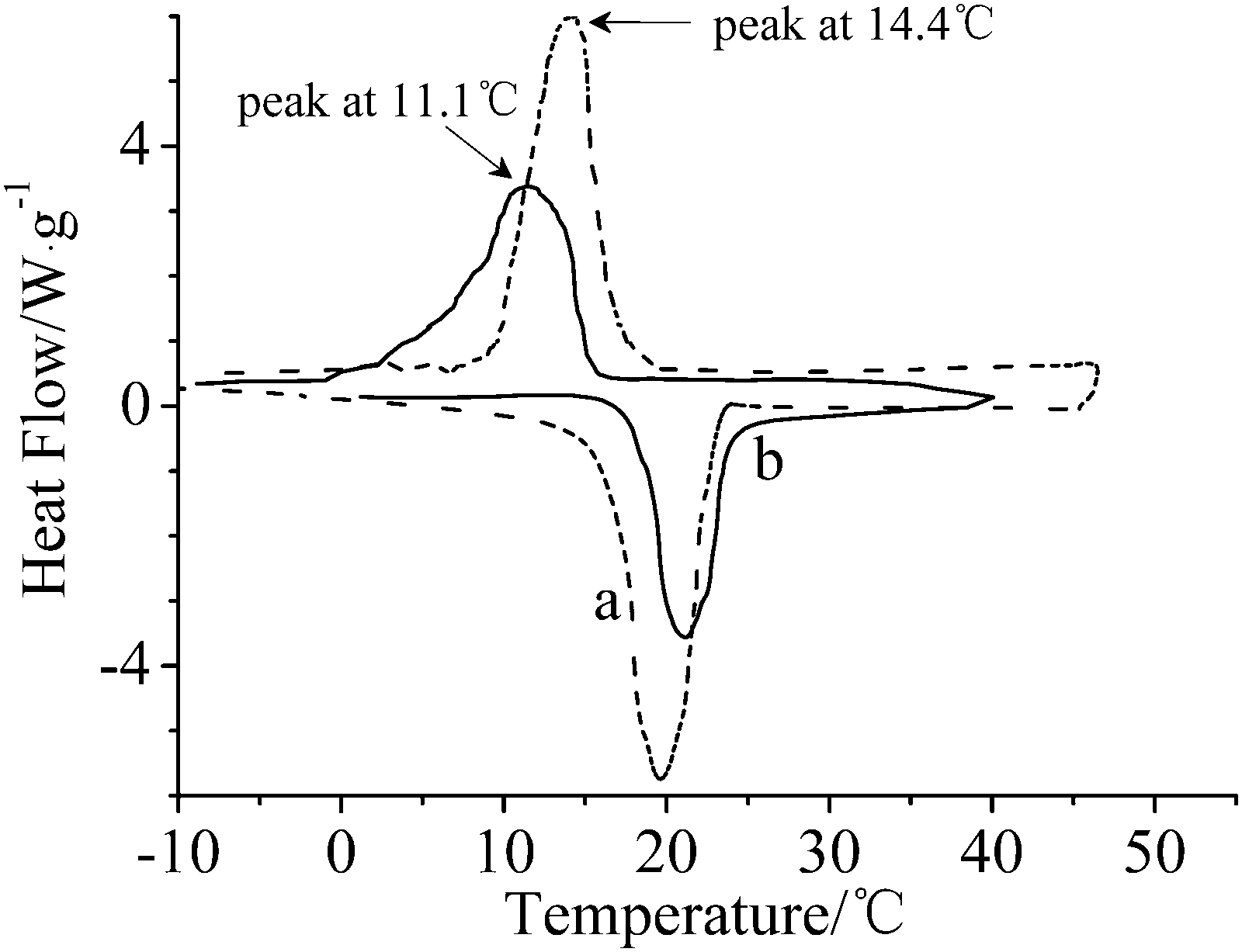 | ||
| Fig. 5 DSC curves of (a) hexadecane and (b) microcapsules. | ||
In conclusion, a microencapsulation method by ARGET ATRP on a Pickering stabilizer was developed. The procedure involves four successive steps, including the synthesis of a silane coupling agent containing ATRP initiating groups, modification of the SiO2 surface to make it active for ATRP, preparation of the oil-in-water Pickering emulsion and ARGET ATRP on a Pickering stabilizer. Conversion of 75–81% was achieved due to the high activity of the catalytic system BiBB/Cu2+/TPMA/Vc. ATRP polymerization of MMA was successfully confined at the oil/water interface, forming covalently-bonded PMMA–SiO2 hybrid shells by a strategy of grafting on the SiO2 surface. The FT-IR spectrum verified the existence of covalently-bonded PMMA on SiO2 and TGA analysis showed that proportion of bonded PMMA reaches 80.3%. The encapsulated hexadecane endows the microcapsules with properties which make them potential candidates for application in thermal storage, as evidenced by the DSC analysis on the phase change temperature, phase change enthalpy and core content of microcapsules.
Experimental
Anhydrous toluene (20 mL), KH550 (0.5 g, 2.26 mmol, jkchemical China) and triethylamine (1 g, 10 mmol) were introduced into a 100 mL three-neck flask. After cooling down to 4 °C, 10 mL of anhydrous toluene solution with 2-bromoisobutyryl bromide (0.62 g, 2.71 mmol, jkchemical China) was added dropwise. The reaction mixture was stirred for 2 h and then poured into a mortar containing 5 g silica particles (Wacker Silicones, Germany) and the powder was milled continuously. After evaporation of toluene, an ethanol–ammonia mixture was introduced similarly to catalyze the hydrolysis and the condensation between compound 2 and SiO2 particles. The modified SiO2 particles were washed with ethanol to remove the impurities and dispersed in Milli-Q water.In the second step, SiO2 suspension (10 mL, 3% of SiO2 content), CuBr2 solution and TPMA (TCI Japan) were mixed and treated ultrasonically. Separately, hexadecane and monomers (MMA and EGDMA, Sinopharm China, purified by a neutral alumina column before use) were mixed as the oil phase. Emulsification was carried out ultrasonically (200 w) for 1 min and the vessel was subsequently bubbled with N2 for 2 min to displace the air in the vessel. Then, ascorbic acid (Vc, Sinopharm China) was added into the vessel. The reaction mixture was sealed rapidly and placed in the roller reactor at 55 °C for 48 h. Microcapsules were filtrated and washed with deionized water exhaustedly, and volatilized at ambient temperature.
SiO2 in the shell was separated by eluting with ethanol and tetrahydrofuran (THF) and the proportion of PMMA covalently-bonded on SiO2 particles was determined, according to our previously reported method.7 A scanning electron microscope (JSM-6700F, Japan), a Fourier transformation infrared spectrometer (BRUKER Tensor 27, Germany), a thermal gravimetric analyzer (Q50, USA), a differential scanning calorimeter (DSC-2910, USA) and a laser particle size analyzer (Beckman LS13321, Germany) were employed to characterize the microcapsules. Conversion of MMA was determined by gas chromatography using an SE-54 cross-linked capillary column. The core material, hexadecane was chosen as an internal standard, because the amount of hexadecane was constant before and after polymerization. To prepare solution for GC testing, the emulsion or microcapsule was soaked with ethanol ultrasonically to extract hexadecane and unreacted monomers, followed by filtration and GC testing. Conversion was calculated from the ratio of peak area (PA) of MMA to C16H34 in the GC chromatogram of solution prepared from microcapsules and the emulsion, using eqn (1):
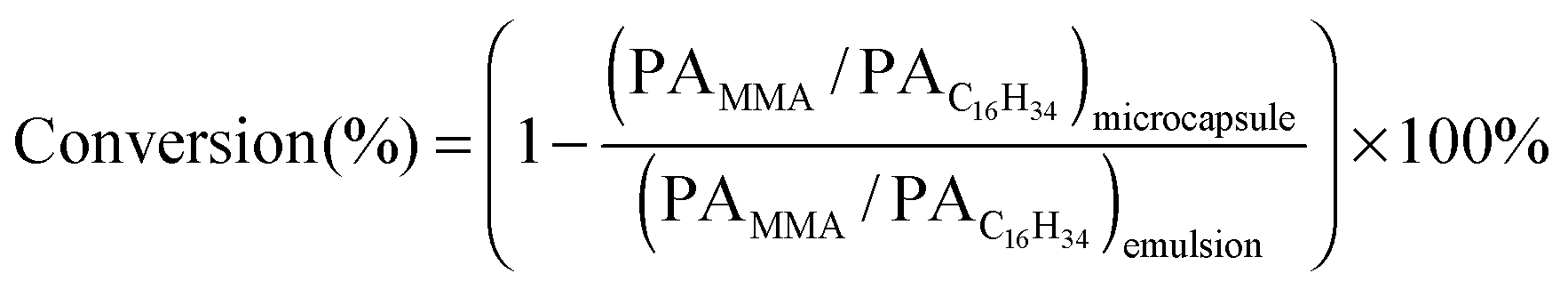 | (1) |
Acknowledgements
The support from the National Nature Science Foundation of China (51173147 and 51173146), the Natural Science Foundation of Shannxi Province (2014JM2038), and the NWPU Foundation for Fundamental Research (NWPU-FFR-JC20120250) is highly appreciated.Notes and references
- M. P. Neubauer, M. Poehlmann and A. Fery, Adv. Colloid Interface Sci., 2014, 207, 65 CrossRef CAS PubMed.
- L. Pastorino, S. Erokhina, F. C. Soumetz, P. Bianchini, O. Konovalov, A. Diaspro, C. Ruggiero and V. Erokhin, J. Colloid Interface Sci., 2011, 57, 56 CrossRef PubMed.
- L. B. Petrovic, V. J. Sovilj, J. M. Katona and J. L. Milanovic, J. Colloid Interface Sci., 2010, 342, 333 CrossRef CAS PubMed.
- D. Yin, L. Ma, J. Liu and Q. Zhang, Energy, 2014, 64, 575 CrossRef CAS PubMed.
- G. J. Lee, H. A. Son, J. W. Cho, S. K. Choi, H. T. Kim and J. W. Kim, J. Colloid Interface Sci., 2014, 413, 100 CrossRef CAS PubMed.
- S. Fujii, M. Okada, T. Nishimura, H. Maeda, T. Sugimoto, H. Hamasaki, T. Furuzono and Y. Nakamura, J. Colloid Interface Sci., 2012, 374, 1 CrossRef CAS PubMed.
- D. Yin, Q. Zhang, C. Yin, X. Zhao and H. Zhang, Polym. Adv. Technol., 2012, 23, 273 CrossRef CAS.
- Q. Song, Y. Li, J. Xing, J. Y. Hu and Y. Marcus, Polymer, 2007, 48, 3317 CrossRef CAS PubMed.
- S. Jegadheeswaran and S. D. Pohekar, Renewable Sustainable Energy Rev., 2009, 13, 2225 CrossRef CAS PubMed.
- Y. Xuan, Y. Huang and Q. Li, Chem. Phys. Lett., 2009, 479, 264 CrossRef CAS PubMed.
- C. C. Chang, Y. L. Tsai, J. J. Chiu and H. Chen, J. Appl. Polym. Sci., 2009, 112, 1850 CrossRef CAS.
- S. Voronov, A. Kohut, I. Tarnavchyk and A. Voronov, Curr. Opin. Colloid Interface Sci., 2014, 19, 95 CrossRef CAS PubMed.
- Y. Luo and H. Gu, Macromol. Rapid Commun., 2006, 27, 21 CrossRef CAS.
- W. Li and K. Matyjaszewski, Macromolecules, 2011, 44, 5578 CrossRef CAS.
- Y. Chen, C. Wang, J. Chen, X. Liu and Z. Tong, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1354 CrossRef CAS.
- J. Li, A. P. Hitchcock and H. D. H. Stöver, Langmuir, 2010, 26, 17926 CrossRef CAS PubMed.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953 CrossRef CAS.
- N. Sarier and E. Onder, Thermochim. Acta, 2007, 452, 14 CrossRef PubMed.
- W. Li, X. Zhang, X. Wang, G. Tang and H. Shi, Energy, 2012, 38, 249 CrossRef CAS PubMed.
- W. Li, G. L. Song, G. Y. Tang, X. D. Chu and S. D. Ma, Energy, 2011, 36, 785 CrossRef CAS PubMed.
- C. Alkan, A. Sarı, A. Karaipekli and O. Uzun, Sol. Energy Mater. Sol. Cells, 2009, 93, 143 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |