
A mononuclear cobalt(III) complex and its catecholase activity†
Merry
Mitra
a,
Pallepogu
Raghavaiah‡
b and
Rajarshi
Ghosh
*a
aDepartment of Chemistry, The University of Burdwan, Burdwan 713 104, India. E-mail: rajarshi_chem@yahoo.co.in; Fax: +91-342-2530452; Tel: +91 342 2533913 ext: 424
bSchool of Chemistry, University of Hyderabad, Hyderabad 500 046, India
First published on 2nd October 2014
Abstract
The structural analysis of a cobalt(III) complex [Co(HL)2](OAc)·H2O (1) [H2L = N-(2-hydroxyethyl)-3-methoxysalicylaldimine] reveals a tridentate chelation behaviour of the ligand H2L having a distorted octahedral coordination environment around the cobalt(III) center with a CoN2O4 chromophore. 1 behaves as an effective catalyst towards the oxidation of 3,5-di-tert-butylcatechol in different solvents, viz. dichloromethane (DCM), methanol (MeOH) and acetonitrile (MeCN) to its corresponding quinone derivative in aerial oxygen. The reaction follows Michaelis–Menten enzymatic reaction kinetics with turnover numbers (Kcat), 1.46 × 103, 1.21 × 103 and 2.16 × 103 h−1 in DCM, MeOH and MeCN, respectively.
Introduction
In a plant system, the catalysis of the oxidation of o-diphenol (catechol) to corresponding quinone coupled with the 2e/2H+ reduction of oxygen to water in the presence of molecular oxygen is known as catecholase activity. The resulting quinones auto-polymerize to give brown pigments, which are responsible to defend the damages caused by pathogens and insects to plants. The crystal structure of the met form of the enzyme catechol oxidase, also known as o-diphenol oxidase, contains two hydroxobridged, which are strongly antiferromagnetically coupled with copper(II) centres in their active site. Each copper(II) centre is coordinated to three histidine nitrogens and adopts a trigonal pyramidal environment with one nitrogen in the apical site. Since the elucidation of its crystal structure,1 several reports of dicopper(II) complexes have been found2–5 to correlate to the structure–function relationship of this biocatalytic reaction. Moreover, different monocopper(II),6 manganese(III),6e,7 nickel(II),8 nickel(II)–manganese(II),9 Fe(III),10 cobalt(II/III)11 and zinc(II)12 compounds are available, which show catecholase activity. All these indicate that the exploration of the possibility of catecholase activity by new types of species with different ligand environment, and different metal ions along with their different oxidation state(s) and nuclearity, which can mimic the native enzyme, deserves special importance. The exact structure–property correlation for catecholase activity is yet to be discovered, indicating the necessity of modeling catecholase active complexes. Here, in this endeavor, we report the synthesis and characterization of a mononuclear cobalt(III) complex (1),13 with an N,O donor Schiff base ligand and its catalytic activity towards the oxidation of a catechol derivative to its corresponding quinone in different solvents.Experimental
Materials
High purity o-vanillin (Aldrich, UK), 2-aminoethanol (Aldrich, UK), cobalt(II) acetate tetrahydrate (Aldrich, UK), 3,5-di-tert-butylcatechol (Aldrich, UK) and all other solvents were purchased from the respective companies, and used as received. Solvents were dried according to the standard procedure and distilled prior to use.The ligand H2L [H2L = N-(2-hydroxyethyl)-3-methoxysalicylaldimine] was prepared using a reported procedure.14O-vanillin (0.3043 g, 2 mmol) was heated under reflux with 2-aminoethanol (0.1222 g, 2 mmol) in 30 ml dehydrated ethanol. After 2 h, the reaction solution was evaporated under reduced pressure to yield a yellow coloured solid, which was dried under vacuum and stored over CaCl2 for subsequent use.
For the catecholase activity study, 1 × 10−4 mol dm−3 solution of 1 (0.0005 g) was treated with 1 × 10−2 mol dm−3 (100 equivalents) of 3,5-DTBC (0.0222 g) under aerobic conditions.
Physical measurements
Elemental analyses (carbon, hydrogen and nitrogen) were performed on a Perkin-Elmer 2400 CHNS/O elemental analyzer. UV-Vis and IR spectra (KBr discs, 4000–300 cm−1) were recorded using a Shimadzu UV-Vis 2450 spectrophotometer and Perkin-Elmer FT-IR model RX1 spectrometer, respectively. The H1 NMR spectral data were collected in CDCl3 on a Bruker 400 MHz spectrometer. Mass spectrometric data were collected on Xevo G2 Q TOF mass spectrometer.Preparation of 1
A methanolic solution (5 cm3) of Co(OAc)2·4H2O (0.0623 g, 0.25 mmol) was added dropwise to a stirring solution of H2L (0.0244 g, 0.125 mmol) in DCM (10 cm3). The brown solution with reddish tinge was filtered and the supernatant liquid was kept in air for slow evaporation. The product was obtained as a square deep brown solid.Yield: (based on metal salt) 0.1126 g (86.22%). Anal. calc. for C22H27N2O9Co (1): C, 50.58; H, 5.21; N, 5.36; found: C, 50.32; H, 4.90; N, 4.92. Selected IR bands (KBr pellet, cm−1): 3461 (s), 1655 (s), 1648 (s), 973 (s). UV-Vis (λ, nm): 252, 390, 495 and 751.
X-ray diffraction study
Single crystals of 1 suitable for X-ray crystallographic analysis was selected following an examination under a microscope. Diffraction data were collected at 293(2) K on a Bruker SMART APEX CCD diffractometer using Mo-Kα radiation (λ = 0.71073 Å) and the crystal was identified to belong to P21/c space group. The crystal data and refinement details are listed in Table 1. The structure was solved by direct methods, and the structure solution and refinement were based on |F|2. The final differences Fourier map showed the maximum and minimum peak heights at 0.575 and −0.364 e Å−3 with no chemical significance. All the calculations were carried out using SHELXL-97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and were refined using SHELSL-97.15 All the figures have been generated using ORTEP-32.16
15 and were refined using SHELSL-97.15 All the figures have been generated using ORTEP-32.16
| Empirical formula | C22H27N2O9Co |
| Formula weight | 522.39 |
| T (K) | 293(2) |
| Wavelength (Å) | 0.71073 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Unit cell dimensions | |
| a (Å) | 15.959(5) |
| b (Å) | 11.194(3) |
| c (Å) | 13.281(4) |
| α (°) | 90.00 |
| β (°) | 100.678(5) |
| γ (°) | 90.00 |
| V (Å3) | 2331.4(12) |
| Z | 4 |
| D calc (mg m−3) | 1.488 |
| Absorption coefficient (mm−1) | 0.791 |
| F(000) | 1088 |
| Crystal size (mm3) | 0.36 × 0.21 × 0.09 |
| Theta range for data collection (°) | 1.30–25.97 |
| Index ranges | −19 ≤ h ≤ 19, −13 ≤ k ≤ 13, −16 ≤ l ≤ 16 |
| Reflections collected | 23447 |
| Independent reflections | 4563 [Rint = 0.0356] |
| Completeness to theta | 99.8% (θ = 25.97) |
| Absorption correction | Multi-scan |
| T max and Tmin | 0.9322 and 0.7638 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4563/2/318 |
| Goodness-of-fit (GOF) on F2 | 1.063 |
| Final R indices [l > 2σ(l)] | R 1 = 0.0556, wR2 = 0.1456 |
| R indices (all data) | R 1 = 0.0672, wR2 = 0.1542 |
| Largest difference in peak and hole (e Å−3) | 0.575, −0.364 |
Results and discussion
Synthesis and formulation
Complex 1 was synthesized by the addition of methanolic solution of Co(II) acetate tetrahydrate into the dichloromethane solution of the ligand H2L.The compound was characterized using elemental analysis, IR, UV and NMR spectroscopy, and single crystal X-ray crystallography. The IR spectrum of 1 shows relatively intense peaks around 1590–1600 cm−1 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching frequency and weak bands in the range of 2980–2900 cm−1 due to the aliphatic C–H stretching.
N stretching frequency and weak bands in the range of 2980–2900 cm−1 due to the aliphatic C–H stretching.
X-ray structure
The molecular structure of 1 is shown in Fig. 1. X-ray crystallography reveals (Table 1) that the hexa-coordination around the Co(III) centre is completed by the N and O donor centres from each of the organic ligand frameworks. | ||
| Fig. 1 ORTEP diagram of 1 with 20% ellipsoid probability plot. | ||
The diamagnetic behavior (Fig. S1; ESI†) as well as comparison of the bond angle–bond distance parameter (Table 2) of the complex with other reported ones confirms the oxidation state of the metal as +III. The phenolic OH in the ligand framework gets deprotonated13 because of its acidity being higher than alcoholic OH, thus the monocationic [Co(HL)2]+ is being formed. The positive charge of the complex is neutralized by a crystallized acetate anion. The geometry around the metal(III) centre is best described to be a distorted octahedron. Bond angle and bond distance data (Table 2) reveal that the phenolic oxygen O1 and the alcoholic oxygen O2 are at the axial position of the octahedron. The rest N1, N2 (imine nitrogens) and O3 (phenolic oxygen), O4 (alcoholic) are at axial position.
| Bond lengths | |||
| Co(1)–O(1) | 1.856(2) | Co(1)–O(4) | 1.952(3) |
| Co(1)–O(2) | 1.945(3) | Co(1)–N(1) | 1.891(3) |
| Co(1)–O(3) | 1.875(2) | Co(1)–N(2) | 1.887(3) |
| Bond angles | |||
| O(1)–Co(1)–O(2) | 178.46(11) | O(2)–Co(1)–N(2) | 90.07(11) |
| O(1)–Co(1)–O(3) | 91.56(11) | O(3)–Co(1)–O(4) | 177.08(11) |
| O(1)–Co(1)–O(4) | 91.28(12) | O(3)–Co(1)–N(1) | 89.47(11) |
| O(1)–Co(1)–N(1) | 95.11(14) | O(3)–Co(1)–N(2) | 95.12(10) |
| O(1)–Co(1)–N(2) | 89.62(10) | N(1)–Co(1)–O(4) | 90.93(12) |
| O(2)–Co(1)–O(3) | 89.97(10) | N(1)–Co(1)–N(2) | 173.33(13) |
| O(2)–Co(1)–O(4) | 87.18(12) | N(2)–Co(1)–O(4) | 84.25(11) |
| O(2)–Co(1)–N(1) | 85.08(14) | ||
Catecholase activity of 1: spectrophotometric study
To study the catecholase activity of the complex 1; 3,5-DTBC with two bulky t-butyl substituents on the ring and low quinone-catechol reduction potential was selected to be the substrate. This makes it easily oxidized to the corresponding o-quinone, 3,5-DTBQ, which is highly stable and shows a maximum absorption at 401 nm in DCM. Solution of 1 was treated with 100 equivalents of 3,5-DTBC under aerobic conditions. The repetitive UV-Vis spectral scan was recorded in pure DCM (Fig. 2). Spectral bands at 751, 495, 390 and 252 nm appeared in the electronic spectrum of complex 1, whereas 3,5-DTBC showed a single band at 282 nm. After the addition of 3,5-DTBC, the time dependent spectral scan showed very smooth increase of the quinone band at 401 nm, as reported by Krebs et al.,17 which indicated the formation of the respective quinone derivative, 3,5-DTBQ, which was purified by column chromatography. The product was isolated in high yield (71.1%) by the slow evaporation of the eluant and was identified by H1 NMR spectroscopy (Fig. S2; ESI†). H1 NMR (CDCl3, 400 MHz): δH = 1.16 (s, 9H), 1.20 (s, 9H), 6.15 (d, J = 2.4 Hz, 1H), 6.86 (d, J = 2.4 Hz, 1H).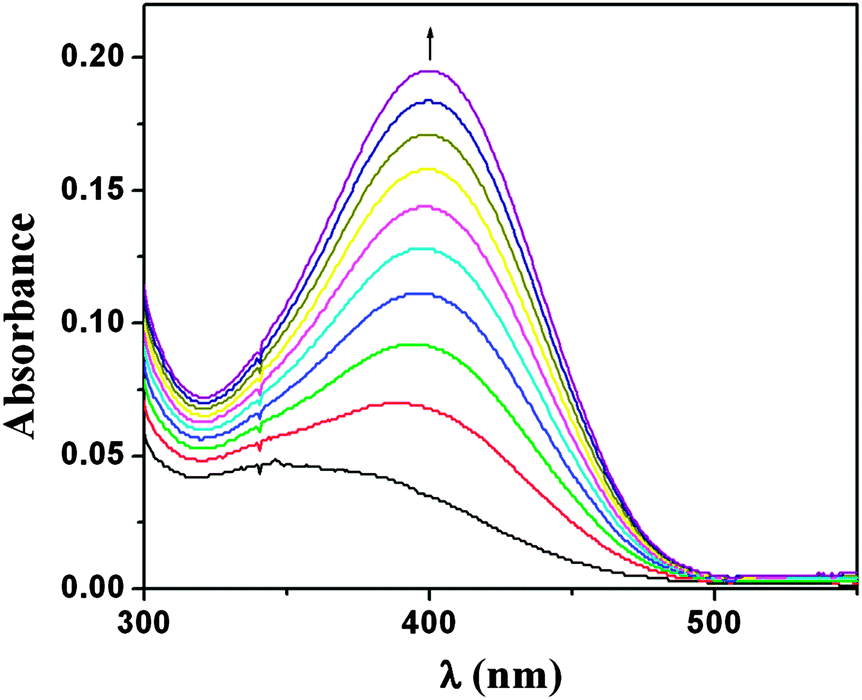 | ||
| Fig. 2 Change in spectral pattern of complex 1 in DCM after reaction with 3,5-DTBC, after observing the reaction for 4 h. | ||
To find out the comparative reaction velocity between 3,5-DTBC and 1, the reaction kinetics between 1 and 3,5-DTBC were studied by observing the time dependent change in absorbance at a wavelength of 401 nm, which is characteristic of 3,5-DTBQ in DCM. The colour of the solution gradually turned deep brown indicative of the gradual conversion of 3,5-DTBC to 3,5-DTBQ. The difference in absorbance ΔA, at 401 nm, was plotted against time to obtain the initial rate for that particular catalyst to substrate concentration ratio (Fig. 3). A first-order catalytic reaction was observed with an initial rate of 6.2 × 10−3 min−1. The reaction followed a pseudo first order kinetics in oxygen-saturated solvent medium.
 | ||
| Fig. 3 A plot of the difference in absorbance (ΔA) vs. time to evaluate the initial rate of the catalytic oxidation of 3,5-DTBC by 1 in DCM. | ||
The catecholase activity of complex 1 was similarly studied in MeOH and MeCN media. In MeOH and MeCN also, 3,5-DTBQ shows maximum absorption at 401 nm (Fig. 4 and 5). 3,5-DTBQ obtained in each medium was purified by column chromatography with yields 67.8% in MeOH and 76.5% in MeCN. It was further characterized by determining its melting point (∼110 °C), which agreed well with that reported in the literature.18 The reaction kinetics was studied by observing the time dependent change in absorbance at a wavelength of 401 nm for catalysis in MeOH as well as in MeCN. The difference in absorbance ΔA at this particular wavelength, were plotted against time to obtain the initial rate of the reaction. A first-order catalytic reaction is observed in both the solvents, where the initial rates are found to be 8.98 × 10−4 min−1 and 1.09 × 10−3 min−1 in MeOH and MeCN, respectively (Fig. S3 and S4; ESI†).
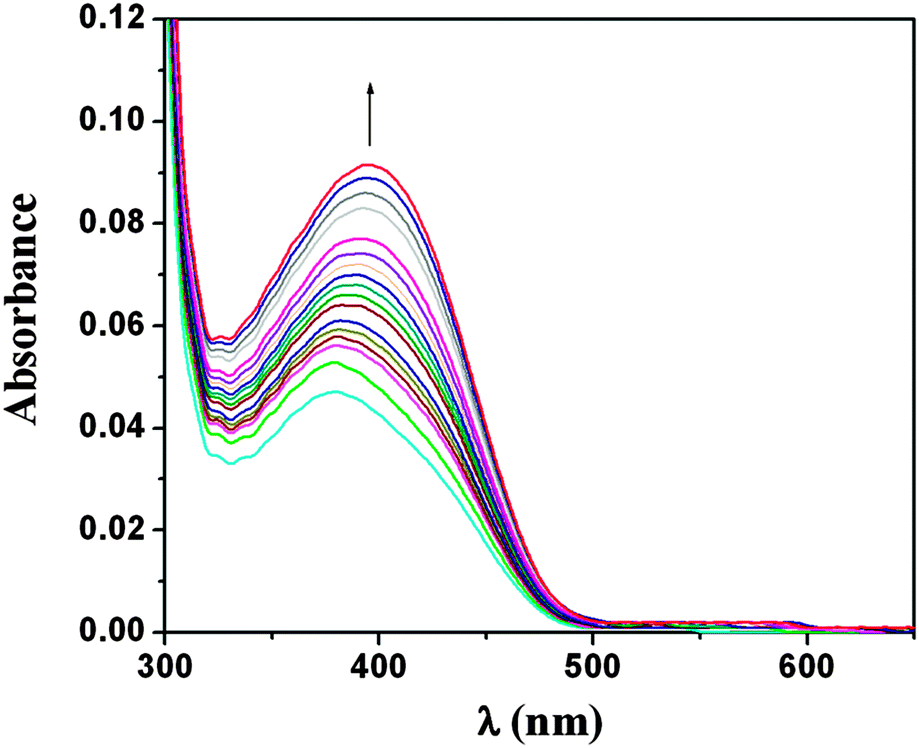 | ||
| Fig. 4 Change in spectral pattern of complex 1 after reaction with 3,5-DTBC, after observing the reaction for 6 h in MeOH. | ||
 | ||
| Fig. 5 Change in the spectral pattern of complex 1 in MeCN after reaction with 3,5-DTBC, after observing the reaction for 6 h. | ||
Enzyme kinetics study
Enzymatic kinetic experiments were performed UV-Vis spectrophotometrically, thermostated at 25 °C for complex 1 and the substrate 3,5-DTBC in DCM, MeOH and MeCN. 0.04 ml of the complex solution, with a constant concentration of 1 × 10−4 M, was added to 2 ml of 3,5-DTBC of a particular concentration (varying its concentration from 1 × 10−3 M to 1 × 10−2 M) to achieve the ultimate concentration of the complex as 1 × 10−4 M. The conversion of 3,5-DTBC to 3,5-DTBQ was monitored with time at a wavelength of 401 nm for solutions in DCM, MeOH and MeCN. The rate for each concentration of the substrate was determined by the initial rate method.The rate versus concentration of substrate data were analyzed on the basis of Michaelis–Menten approach of enzymatic kinetics to get the Lineweaver–Burk (double reciprocal) plot as well as the values of the various kinetic parameters Vmax, KM and Kcat. The observed rate vs. [substrate] plot in DCM solution as well as Lineweaver–Burk plot is given in Fig. 6.
 | ||
| Fig. 6 Plot of rate vs. [substrate] in the presence of 1 in DCM; inset: Lineweaver–Burk plot. | ||
Similar plots in MeOH and MeCN are given in Fig. S5 and S6 of ESI.† The kinetic parameters are listed in Table 3. The turnover numbers (Kcat) are 1.46 × 103, 1.21 × 103 and 2.16 × 103 h−1 in DCM, MeOH and MeCN, respectively.
| Solvent | V max (M s−1) | Std. error | K M (M) | Std. error | K cat (h−1) |
|---|---|---|---|---|---|
| DCM | 4.06 × 10−5 | 5.71 × 10−6 | 1.25 × 10−3 | 2.29 × 10−4 | 1.46 × 103 |
| MeOH | 3.36 × 10−5 | 3.39 × 10−6 | 7.38 × 10−4 | 5.97 × 10−5 | 1.21 × 103 |
| MeCN | 5.99 × 10−5 | 1.98 × 10−5 | 4.90 × 10−3 | 2.36 × 10−3 | 2.16 × 103 |
Reaction mechanism
The catalytic process follows a two-step mechanistic pathway. This is evident from the rate plot (Fig. S7; ESI†). The first step, with a lesser rate constant value, is the rate determining step. Probably, in this step, the 1:1 adduct of catechol and the cobalt complex is formed. To obtain a mechanistic inference of the catecholase activity and to get an idea about the complex-substrate intermediate, we recorded an ESI-MS spectrum (Fig. S8; ESI†) of a 1:100 mixture of complex 1 and 3,5-DTBC of mixing them together. The signal at m/z = 196 is due to the formation of the protonated ligand [(L2)H]+. 3,5-DTBC can be indicated by the peak at m/z = 221. The peak at m/z = 243 can be assigned to sodium aggregate of quinone [3,5-DTBQ-Na]+. The aqueous complex [Co(L)2(H2O)]+ exhibits a peak at m/z = 463. The formation of a sodium aggregate of the species 1a (Scheme 1) is identified by the peak at m/z = 354. The intermediate Co(III) complex is reduced to Co(II) by the catechol derivative, and 3,5-DTBC itself gets oxidised to quinone in the presence of oxygen. The oxygen that takes part in this process is converted to H2O2. H2O2 thus liberated was identified and characterized spectrophotometrically (S1; ESI†).19
 | ||
| Scheme 1 | ||
Conclusions
In conclusion, we have synthesized and structurally characterized one monometallic cobalt(III) complex (1) with an N,O donor Schiff base ligand. The catalytic property of 1 has been kinetically investigated for the aerobic oxidation of 3,5-DTBC to 3,5-DTBQ in DCM, MeOH and MeCN, which reveals that the catalytic reaction follows a first order reaction pathway. The turnover numbers of 1 are 1.46 × 103, 1.21 × 103 and 2.16 × 103 h−1 in DCM, MeOH and MeCN, respectively, which are considerably greater than those reported in recent times. In a recent report by Mohanta et al., the turnover numbers are 39 h−1, 40 h−1, and 48 h−1 in DMF, and 167 h−1 and 215 h−1 in MeCN for Cu(II) complexes.4b The same group reported a mixed valence Co(III/II) complex with turnover numbers 482.16 h−1 and 45.38 h−1 in MeCN and MeOH, respectively.11 Rajak et al. reported two Cu(II) complexes, which show a turnover rate of about 29 and 37 h−1.4e A turnover rate of 28 h−1 of a Cu(II) complex is reported by Neves et al.5b Ghosh et al. reported three Ni(II) complexes with turnover numbers 64.1, 51.1 and 81.7 h−1 in MeCN,8 and three heterometallic Ni(II)–Mn(II) complexes with their turnover rates ranging from 25.8 to 104.5 h−1.9 This indicates that 1 is a better and more effective model for catecholase activity than the recently reported ones, though, to the best of our knowledge, the most active catalyst4a reported to date exhibits a turnover number of 3.24 × 104 h−1. Therefore, by comparing all these data it can be concluded that the reported complex (1) is considerably an efficient catalyst and has an appreciable turnover rate in various solvents. Moreover, 1 being a mononuclear complex with non-copper centre is mimicking an enzyme with a dicopper active site.Acknowledgements
Financial support by the Department of Science & Technology, New Delhi, India (F. No. SR/FT/CS-83/2010 dt. 11-02-2011) is gratefully acknowledged by RG. Generous help in magnetic data collection and mass spectrometric analysis respectively by Prof. T. Mallah, Université Paris Sud, France and Prof. M. Ali, Jadavpur University, India are also acknowledged. MM is thankful to The University of Burdwan for her research fellowship.Notes and references
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084 CrossRef CAS PubMed.
- (a) M. Merkel, N. Möller, M. Piacenza, S. Grimme, A. Rompel and B. Krebs, Chem. – Eur. J., 2005, 11, 1201 CrossRef CAS PubMed; (b) J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793 RSC; (c) B. Sreenivasulu, F. Zhao, S. Gao and J. J. Vittal, Eur. J. Inorg. Chem., 2006, 2656 CrossRef CAS; (d) C.-T. Yang, M. Vetrichelvan, M. Yang, B. Moubaraki, K. S. Murrey and J. J. Vittal, Dalton Trans., 2004, 113 RSC.
- (a) I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814 RSC; (b) I. A. Koval, K. Selmeczi, C. Belle, C. Philouze, E. Saint-Aman, I. Gautier-Luneau, A. M. Schuitema, M. van Vliet, P. Gamez, O. Roubeau, M. Lüken, B. Krebs, M. Lutz, A. L. Spek, J.-L. Pierre and J. Reedijk, Chem. – Eur. J., 2006, 12, 6138 CrossRef CAS PubMed.
- (a) K. S. Banu, T. Chattopadhyay, A. Banerjee, S. Bhattacharya, E. Suresh, M. Nethaji, E. Zangrando and D. Das, Inorg. Chem., 2008, 47, 7083 CrossRef CAS PubMed; (b) S. Majumder, S. Sarkar, S. Sasmal, E. Carolina Sãnudo and S. S. Mohanta, Inorg. Chem., 2011, 50, 7540 CrossRef CAS PubMed; (c) A. Biswas, L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Inorg. Chem., 2012, 51, 10111 CrossRef CAS PubMed; (d) S. Mandal, J. Mukherjee, F. Lloret and R. Mukherjee, Inorg. Chem., 2012, 51, 13148 CrossRef CAS PubMed; (e) A. Banerjee, S. Sarkar, D. Chopra, E. Colacio and K. K. Rajak, Inorg. Chem., 2008, 47, 4023 CrossRef CAS PubMed.
- (a) P. Comba, B. Martin, A. Muruganantham and J. Straub, Inorg. Chem., 2012, 51, 9214 CrossRef CAS PubMed; (b) A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki and E. Schwingel, Inorg. Chem., 2002, 41, 1788 CrossRef CAS PubMed; (c) S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. L. Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526 CrossRef CAS; (d) S.-C. Cheng and H.-H. Wei, Inorg. Chim. Acta, 2002, 340, 105 CrossRef CAS.
- (a) A. L. Abuhijleh, J. Pollitte and C. Woods, Inorg. Chim. Acta, 1994, 215, 131 CrossRef CAS; (b) A. L. Abuhijleh, C. Woods, E. Bogas and G. L. Guenniou, Inorg. Chim. Acta, 1992, 195, 67 CrossRef CAS; (c) M. R. Malachowski, M. G. Davidson and J. N. Hoffman, Inorg. Chim. Acta, 1989, 157, 91 CrossRef CAS; (d) M. R. Malachowski and M. G. Davidson, Inorg. Chim. Acta, 1989, 162, 199 CrossRef CAS; (e) M. Mitra, A. K. Maji, B. K. Ghosh, G. Kaur, A. Roy Choudhury, C.-H. Lin, J. Ribas and R. Ghosh, Polyhedron, 2013, 61, 15 CrossRef CAS PubMed.
- (a) P. Kar, R. Haldar, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2012, 51, 4265 CrossRef CAS PubMed; (b) S. Mukherjee, T. Weyhermüller, E. Bothe, K. Wieghardt and P. Chaudhuri, Dalton Trans., 2004, 3842 RSC; (c) K. S. Banu, T. Chattopadhyay, A. Banerjee, M. Mukherjee, S. Bhattacharya, G. K. Patra, E. Zangrando and D. Das, Dalton Trans., 2009, 8755 RSC.
- A. Biswas, L. K. Das, M. G. B. Drew, G. Aromí, P. Gamez and A. Ghosh, Inorg. Chem., 2012, 51, 7993 CrossRef CAS PubMed.
- P. Seth, L. K. Das, M. G. B. Drew and A. Ghosh, Eur. J. Inorg. Chem., 2012, 2232 CrossRef CAS.
- (a) L. I. Simándi, T. M. Simándi, Z. May and G. Besenyei, Coord. Chem. Rev., 2003, 245, 85 CrossRef; (b) T. Megyes, Z. May, G. Schubert, T. Grósz, L. I. Simándi and T. Radnai, Inorg. Chim. Acta, 2006, 359, 2329 CrossRef CAS PubMed; (c) Z. May, L. I. Simándi and A. J. Vértes, J. Mol. Catal. A: Chem., 2007, 266, 239 CrossRef CAS PubMed; (d) S.-I. Lo, J.-W. Lu, W.-J. Chen, S.-R. Wang, H.-H. Wei and M. Katada, Inorg. Chim. Acta, 2009, 362, 4699 CrossRef CAS PubMed; (e) M. Mitra, A. K. Maji, B. K. Ghosh, P. Raghavaiah, J. Ribas and R. Ghosh, Polyhedron, 2014, 67, 19 CrossRef CAS PubMed.
- S. Majumder, S. Mondal, P. Lemonie and S. S. Mohanta, Dalton Trans., 2013, 42, 4561 RSC.
- A. Guha, T. Chattopadhyay, N. D. Paul, M. Mukherjee, S. Goswami, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2012, 51, 8750 CrossRef CAS PubMed.
- S. Yamada, Y. Kuge and K. Yamanouchi, Bull. Chem. Soc. Jpn., 1970, 43, 406 CrossRef CAS.
- (a) S. Hazra, R. Koner, P. Lemoine, E. Carolina Sañudo and S. Mohanta, Eur. J. Inorg. Chem., 2009, 3458 CrossRef CAS; (b) R. Koner, S. Hazra, M. Fleck, A. Jana, C. R. Lucas and S. Mohanta, Eur. J. Inorg. Chem., 2009, 4982 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, ORTEP-32 for Windows, University of Glasgow, Scotland, 1998 Search PubMed.
- F. Zippel, F. Ahlers, R. Werner, W. Haase, H.-F. Nolting and B. Krebs, Inorg. Chem., 1996, 35, 3409 CrossRef CAS PubMed.
- S. Tsuruya, S.-I. Yanai and M. Masai, Inorg. Chem., 1986, 25, 141 CrossRef CAS.
- (a) A. I. Vogel, Textbook of quantitative inorganic analysis, Longmans, Green and Co. Ltd, London, 3rd edn, 1961, p. 366 Search PubMed; (b) A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki and E. Schwingel, Inorg. Chem., 2002, 41, 1788 CrossRef CAS PubMed; (c) E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Magnetic and spectroscopic plots are included here. CCDC 918017. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01587h |
| ‡ Present address: Department of Chemistry, Dr. Harisingh Gour University, Sagar 470 003, India. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |