
Environmentally benign synthesis of 4-aminoquinoline-2-ones using recyclable choline hydroxide†
Anita Kailas
Sanap
and
Ganapati Subray
Shankarling
*
Dyestuff Technology Department, Institute of Chemical Technology, N. P. Marg, Matunga, Mumbai-400019, India. E-mail: gsshankarling@gmail.com; Fax: +91-22-33611020; Tel: +91-22-33612708
First published on 2nd October 2014
Abstract
Greener synthesis of 4-aminoquinoline-2-ones was achieved by intramolecular cyclization of 2-cyanophenylamide derivatives using biodegradable and recyclable choline hydroxide (ChOH). The reaction proceeds rapidly and affords the corresponding 4-aminoquinoline-2-ones with a good to excellent yield. The protocol has the advantage of easy workup, high yields, and an environmentally benign methodology compared to other reported methods. The simplicity of this method makes it an interesting alternative to other approaches.
Introduction
4-Amino-2(1H)-quinolone scaffolds can be found in a large variety of biologically active compounds. 4-Amino-2(1H)-quinolone derivatives have attracted the attention of researchers not only because of their strong pain-relief activity but also due to their extremely weak acidic properties. Many non-narcotic analgesics are strong acids; even their salts can cause ulcers. There is no basis for such a side effect in the case of 4-amino-2(1H)-quinolone derivatives, at least, in such pronounced form. 4-Amino-3-phenylquinolin-2(1H)-one shows antagonist activity at the glycine site on the NMDA (N-methyl-D-aspartate) receptor and anticonvulsant activity.1 Additionally, 4-amino-3-benzimidazol-2-ylhydroquinolin-2-one was reported as a tyrosine kinase receptor and its analogues are reversible ATP competitive inhibitors of VEGFR-2, FGFR-1, and PDGFR with IC50 values <0.1 μM.2,3 Considering the significant applications of quinolones in the medicinal and agrochemical field, there has been tremendous interest in developing new efficient synthetic strategies which involve rapid functionalization and diversification of quinolones.Therefore, various synthetic methodologies have been reported to access these molecules as a backbone in their active moieties. Literature survey on the synthesis of 4-amino-3-substituted-2(1H)-quinolones did not show uniform reaction conditions. 4-Amino-1,2-dihydro-2-oxo-3-quinolinecarbonitrile was synthesized in five steps starting from isatoic anhydride and dimethyl malonate to give 4-hydroxyquinolone derivatives, which upon further reactions gave the title compound.4 Direct reaction of 2-aminobenzonitriles with dialkyl malonates gave a very low yield of the product.5 In an alternative approach, the intramolecular cyclization of malonyl ester amide of 2-aminobenzonitrile and 2-cyanophenyl-N-acetoacetamide was reported using tin(IV) chloride, sodium methoxide and sodium ethoxide to give a low yield of quinolone derivatives.5,6 Low yields were obtained in the cyclisation of the malonyl ester amide of 2-aminobenzonitrile and 2-cyanophenyl-N-acetoacetamide due to difficulty in coordinating both the β-dicarbonyl moiety and the cyano group of the same molecule, possibly due to geometric reasons.5 In some cases intramolecular cyclisation of corresponding amides of 2-aminobenzonitriles was reported using sodium hydride,7,8 potassium tert-butoxide,9 and LiHMDS.10 4-Amino-2(1H)-quinolones were synthesized from the reaction of 4-hydroxy-2-quinolones with benzylamine and the cleavage of the benzyl group by catalytic hydrogenation.11 Synthesis of 4-amino-2-quinolones was also reported using conventional amination of 4-hydroxy or 4-chloro derivatives with NH3 in a sealed tube12 or from the reaction of 4-hydroxyquinolones with benzyl ammonium chloride at high temperatures.13 Recently, Toche R. B. et al. reported the synthesis of ethyl-4-amino-2-oxo-1,2-dihydroquinoline-3-carboxylate by the reaction of 2-aminobenzonitrile with diethylmalonate using sodium ethoxide in long reaction times.14
These methods, however, suffer from one or another drawback, such as the use of metal mediated catalysis, a larger number of steps, strong basic conditions, longer reaction times, moisture-sensitive catalysts or expensive reagents, and low conversions, which limit their practical utility in organic synthesis. Our protocols describe a simple method for the synthesis of 4-amino-2(1H)-quinolones from their corresponding amides that were prepared from 2-aminobenzonitriles.
In recent years, the use of environmentally benign catalysts has received tremendous interest in different areas of organic synthesis.15 Choline hydroxide (ChOH) is an environmentally benign, easy to prepare base, which displays strong basicity. It has received considerable interest as an economical, easily available, and recyclable catalyst for various organic transformations. It was thought worthwhile to explore the catalytic potential of ChOH for this particular conversion. In view of the above and as a part of our ongoing program to develop viable protocols,16–22 we report herein a ChOH catalyzed, mild, simple, and efficient procedure for the construction of 4-amino-2-quinolones.
Experimental
Materials and equipment
All the solvents and chemicals were procured from S D Fine Chemicals (India) and were used without further purification. The reactions were monitored by TLC using 0.25 mm E-Merck silica gel 60 F254 precoated plates, which were visualized with UV light. 1H NMR spectra were recorded on a Bruker 400 MHz spectrometer, and chemical shifts are expressed in δ ppm using TMS as an internal standard. Mass spectral data were obtained using a micromass-Q-Tof (YA105) spectrometer. Infrared spectra were recorded on a Jasco-FT/IR 4100 LE ATR PRO450-S spectrometer.Preparation of choline hydroxide
To a solution of choline chloride in methanol, potassium hydroxide was added at room temperature. Then it was heated to 61 °C for 12 h with constant stirring. After cooling to room temperature, the reaction mixture was filtered to remove solid KCl and a solution was obtained. This was concentrated to remove methanol and used without further purification.General procedure for preparation of 3a–3h
To a solution of 2-aminobenzonitrile (1) (10 mmol) in dichloromethane (15 mL), 2 (10 mmol) was added at 0 °C. The mixture was stirred at room temperature for 1 h. After completion of the reaction, volatile components of the reaction mixture were removed under reduced pressure to give oil. It was quenched with ice cold water (10 mL) and the solution was extracted with ethyl acetate (25 mL × 3). The combined extracts were washed with saturated NaHCO3 (2 × 10 mL) and dried over Na2SO4. After removal of the solvent, the residue was purified by recrystallization in ethyl acetate to afford 3 as a pale yellow solid.General procedure for preparation of 5a–5e
To a solution of 2-aminobenzonitrile (1) (10 mmol) in dry xylene (15 mL), 4 (12 mmol) was added. The reaction mixture was refluxed for 4 h in a round-bottomed flask fitted with a Dean–Stark apparatus, kept in an oil bath. After completion of the reaction, it was cooled to 0 °C to let the products solidify and filtered. The crude product was washed with diethyl ether to afford 5 as a pale yellow solid.General procedure for preparation of choline hydroxide catalyzed synthesis of 4-aminoquinoline-2-ones (6a–6m)
Compound 3/5 (1 mmol) was dissolved in 3 mL of choline hydroxide at room temperature and then heated for appropriate time. After completion of reaction, cold water was added to the reaction mixture. The precipitated solid product (6) was filtered off and purified by column chromatography using hexane: ethyl acetate.Results and discussion
Preparation of 2-cyanophenylamides (3a–3h and 5a–5e)
In the first part, we carried out the reactions of 2-aminobenzonitriles with various acid chlorides like cyanoacetylchloride, phenyl acetyl chloride, monoethylmalonyl chloride, and 7-methylcoumarin-4-acetyl chloride to give corresponding amide derivatives (3a–3h) using the literature procedure23 (Scheme 1).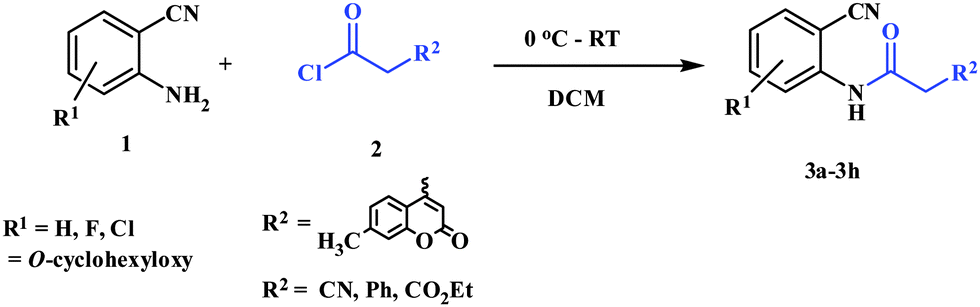 | ||
| Scheme 1 Synthesis of 3a–3h using 2-aminobenzoniriles and acid chlorides. | ||
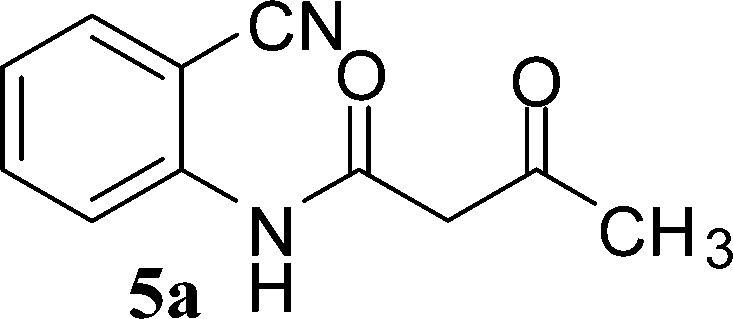
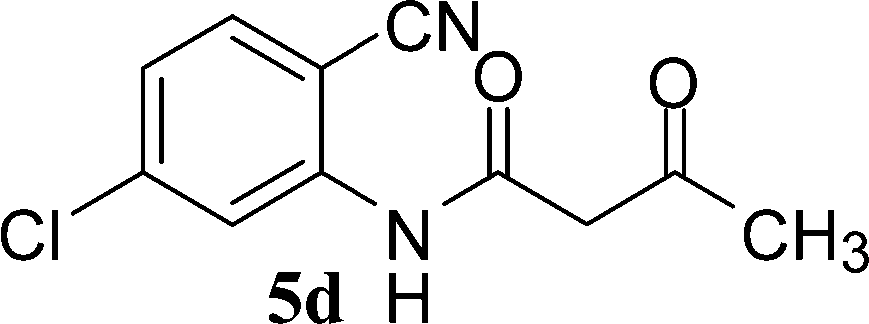
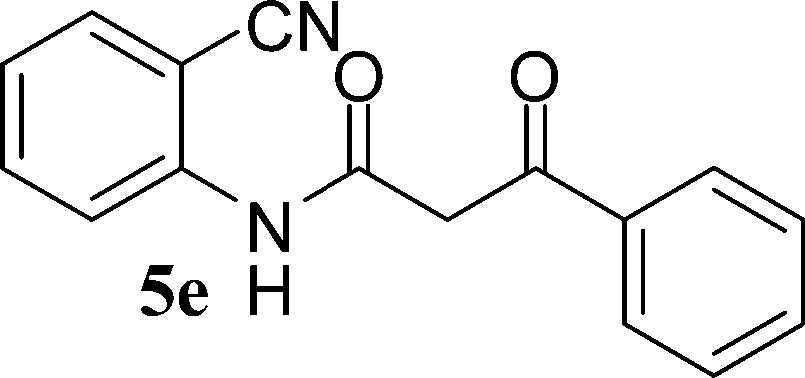
N-(2-Cyanophenyl)-3-oxobutanamides (5a–5d) and N-(2-cyanophenyl)-3-oxo-3-phenylpropanamide (5e) were prepared by the reaction of 2-aminobenzonitriles (1) with ethylacetoacetate and ethyl benzoylacetate in boiling xylene as per reported procedures24 (Scheme 2).
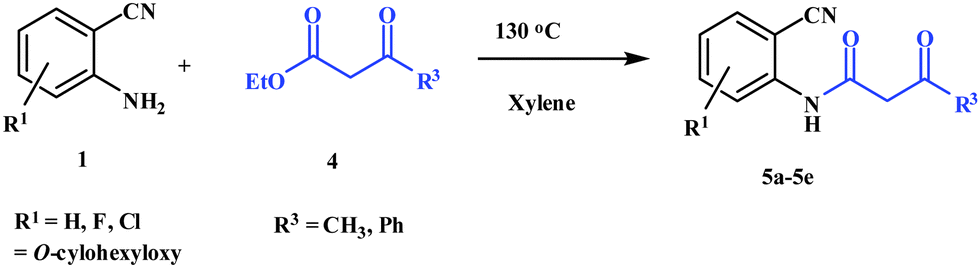 | ||
| Scheme 2 Synthesis of N-(2-cyanophenyl)-3-oxobutanamides (5a–5d) and N-(2-cyanophenyl)-3-oxo-3-phenylpropanamide (5e) using 2-aminobenzoniriles and β-ketoester. | ||
Optimization of reaction conditions
In order to optimize the reaction conditions for cyclization, the effects of different organic bases and solvents were investigated in detail for the reactions of 2-cyano-N-(2-cyanophenyl)acetamide (3a) and N-(2-cyanophenyl)-3-oxobutanamide (5a). The outcome is presented in Table 1. It is evident that no conversion into the product was obtained in the absence of a base even after a prolonged reaction time of 24 h (Table 1, entry 1). Deep eutectic mixtures (DES) such as choline chloride (ChCl): urea, ChCl: malonic acid, ChCl: oxalic acid have intrinsic advantages as green catalysts.16,17 Hence, attempts were made to study the model reactions using these catalysts, which resulted in no net reaction for ChCl: malonic acid/oxalic acid DES (Table 1, entries 2 and 3). Low yields of products were obtained in ChCl: urea DES (Table 1, entry 4). However, many other organic bases such as Et3N, piperidine, and pyridine promoted the reaction quite well for 3a in a short time as compared to 5a (Table 1, entries 5–7). Whereas inorganic bases such as sodium hydride, potassium hydroxide, sodium hydroxide, and potassium tert-butoxide provided a good yield of the product as compared to organic bases (Table 1, entries 8–11). Out of all the bases monitored, ChOH was concluded to be the best at 80 °C and gave products 6a and 6i in 83% and 80% yield respectively (Table 1, entry 12).|
|
|||||
|---|---|---|---|---|---|
| Entry | Base/solvent | For reactant 3aa | For reactant 5aa | ||
| Time (h) | Yieldf of 6a | Time (h) | Yieldf of 6i | ||
| a Reaction conditions: 3a/5a (1 mmol). b DES used (3 mL/80 °C). c Base used (2.5 equiv./reflux). d ChOH (3 mL/80 °C). e ChOH used (1 equiv./reflux). f Isolated yields, NR: no reaction. | |||||
| 1 | Without catalyst/reagent | 24 | NR | 24 | NR |
| 2b | ChCl: urea | 10 | 21 | 11 | 17 |
| 3b | ChCl: malonic acid | 10 | NR | 10 | NR |
| 4b | ChCl: oxalic acid | 10 | NR | 10 | NR |
| 5c | Et3N–ethanol | 4.5 | 48 | 24 | 36 |
| 6c | Piperidine–ethanol | 1.5 | 61 | 10 | 57 |
| 7c | Pyridine–THF | 2.5 | 47 | 11 | 42 |
| 8c | Sodium hydride–THF | 4 | 58 | 10 | 51 |
| 9c | Potassium tert-butoxide–THF | 6 | 62 | 9 | 54 |
| 10c | Sodium hydroxide–ethanol | 1.5 | 70 | 4.5 | 69 |
| 11c | Potassium hydroxide–ethanol | 1.5 | 72 | 4.5 | 63 |
| 12d | ChOH | 20 min | 83 | 50 min | 80 |
| 13e | Water | 12 | 30 | 12 | 21 |
| 14e | Ethanol | 1 | 63 | 4 | 64 |
| 15e | Acetonitrile | 1.5 | 61 | 6 | 67 |
| 16e | Tetrahydrofuran | 2 | 55 | 10 | 50 |
| 17e | Dichloromethane | 6 | 43 | 12 | 30 |
| 18e | Toluene | 2 | 49 | 2 | 51 |
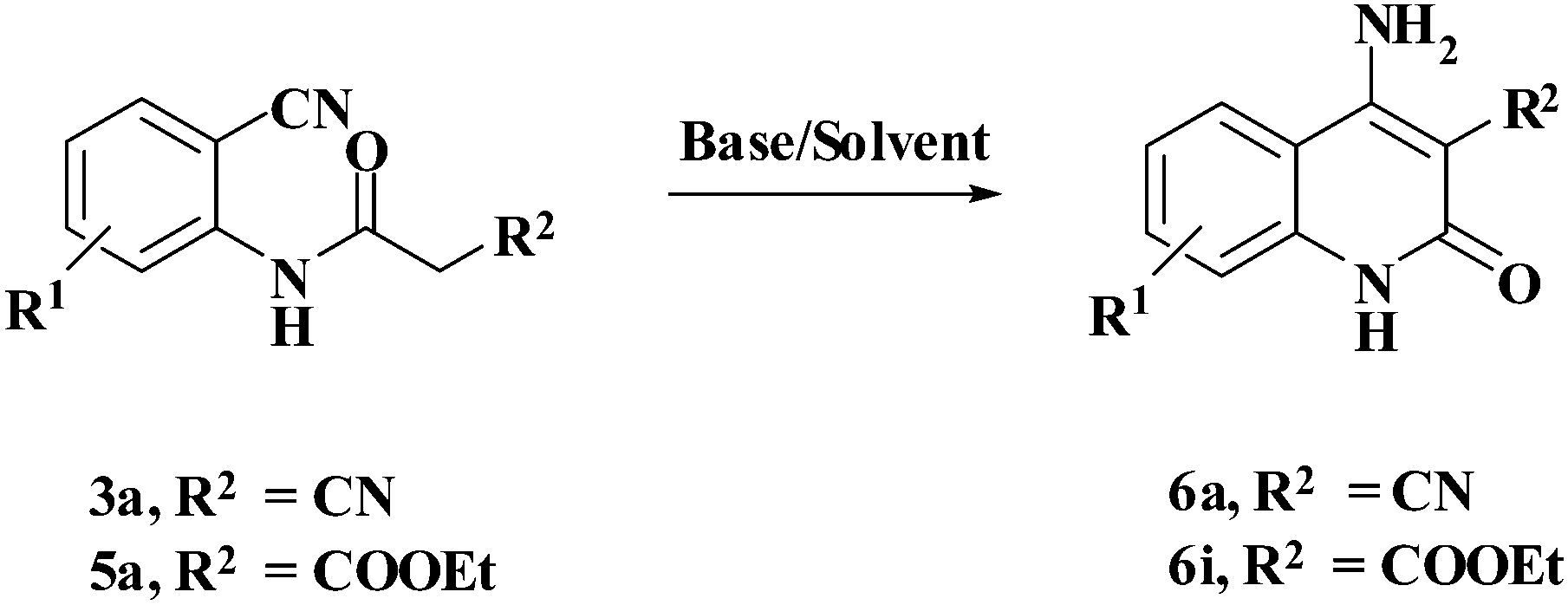
In order to screen the effect of the solvent, the model reactions were carried out using 1 equiv. of ChOH in different solvents under reflux conditions (Table 1, entries 13–18). It was observed that in polar solvents like ethanol and acetonitrile (Table 1, entries 14 and 15) good yields of the product were obtained in short reaction times as compared to non-polar solvents (Table 1, entries 16–18). In water, it gave a very low yield of the product under the same reaction conditions (Table 1, entry 13). As solvent-free synthesis has gained much current interest, it was considered important to investigate the reaction under these conditions as well. The best conversion was finally achieved under solvent-free conditions using ChOH at 80 °C (Table 1, entry 12).
Synthesis of 4-aminoquinoline-2-ones in choline hydroxide
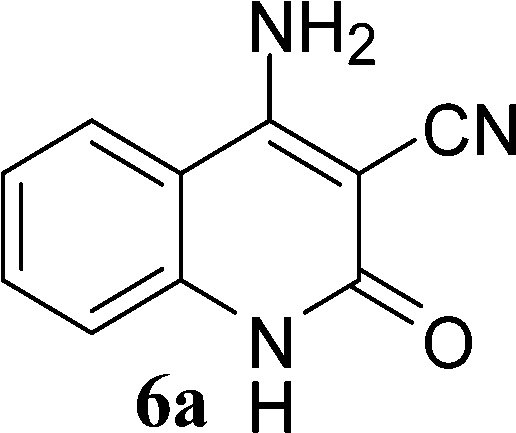
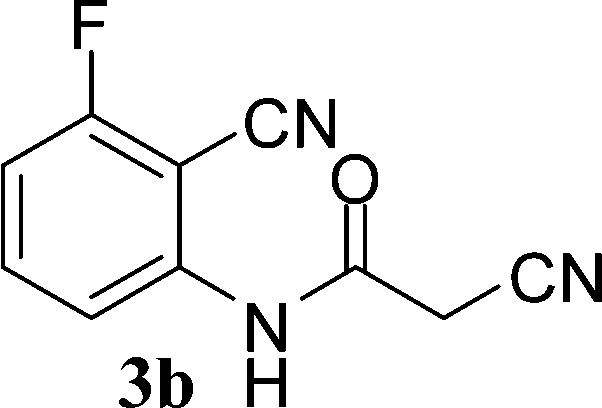
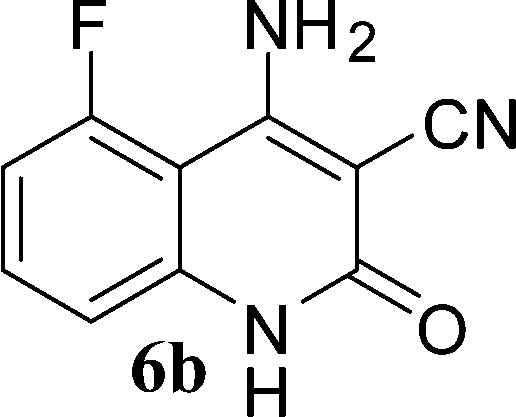
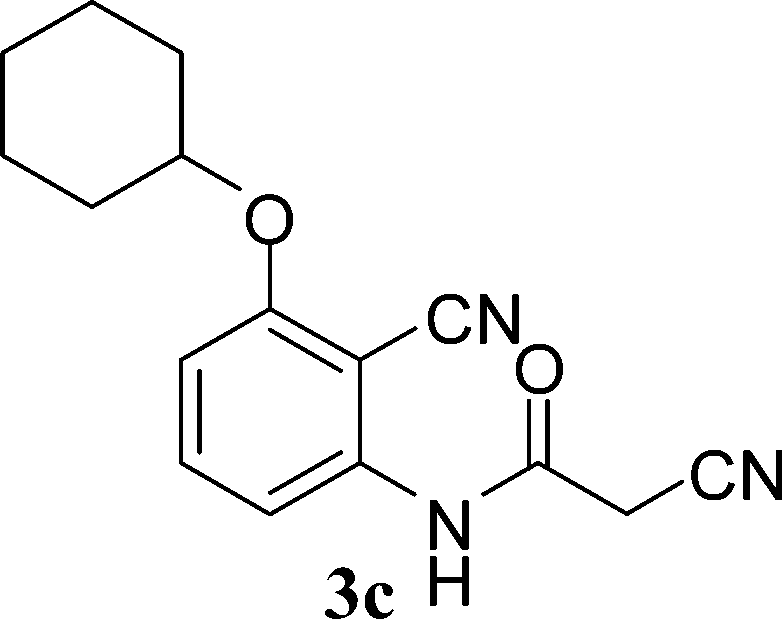
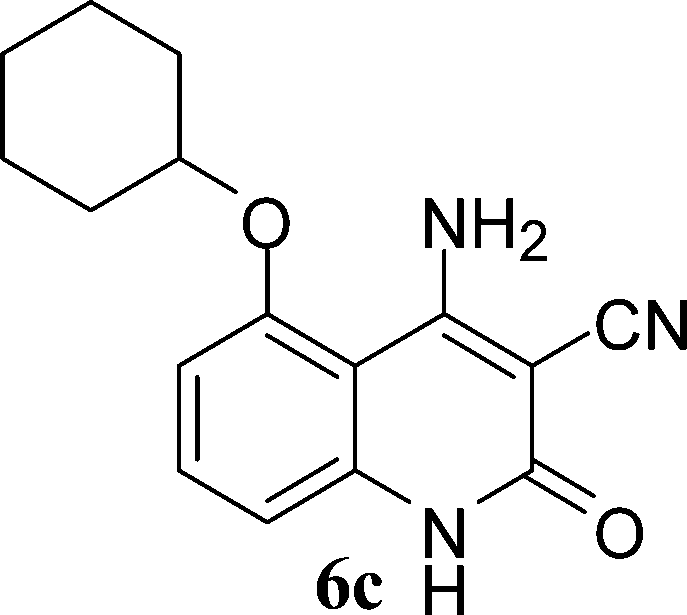
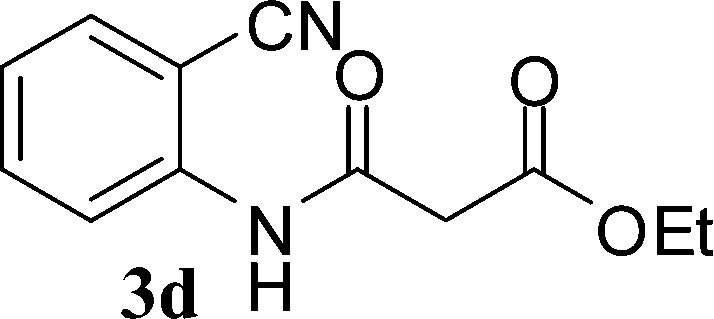
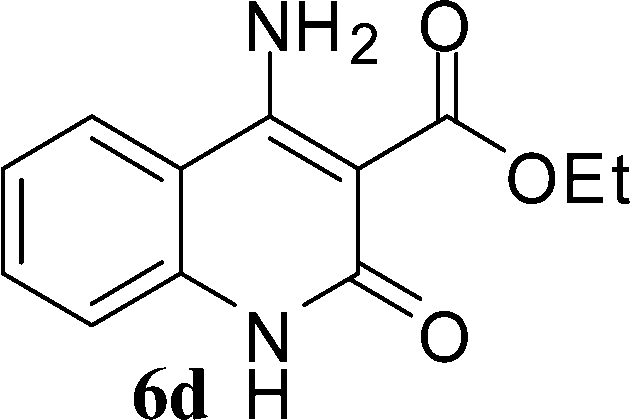
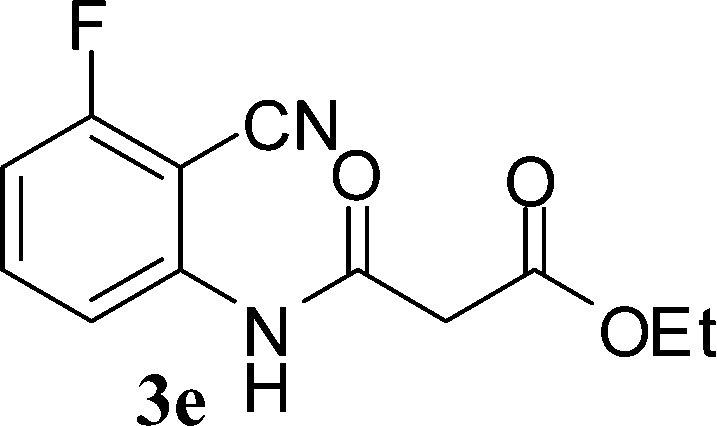
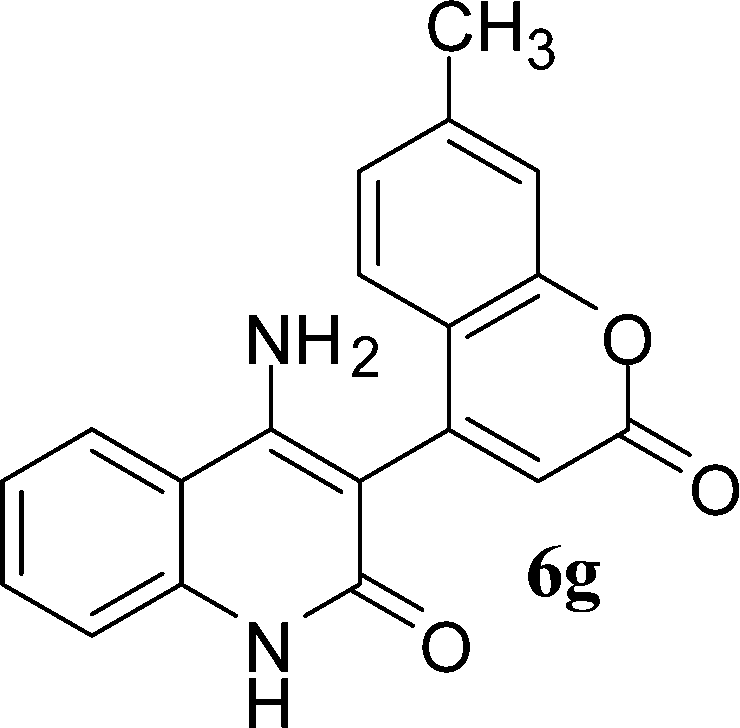
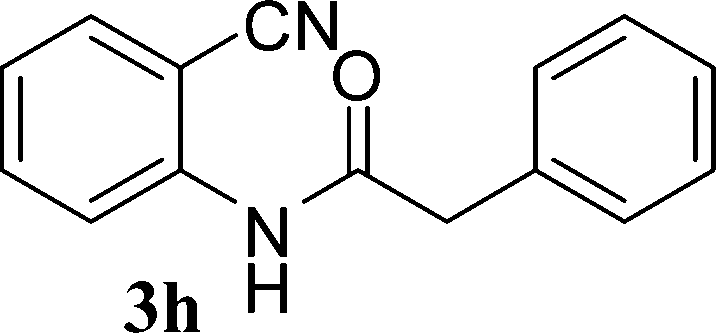
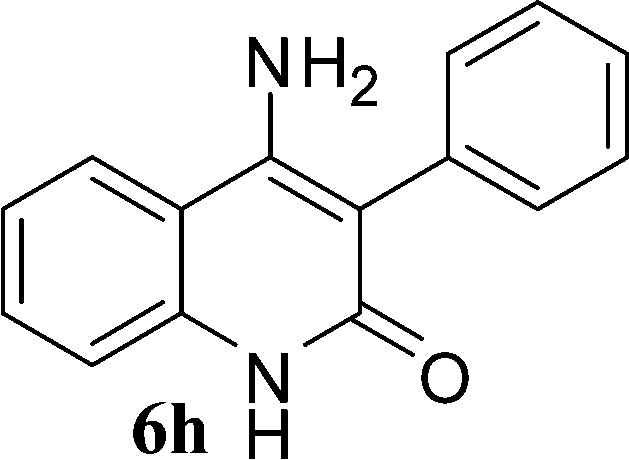
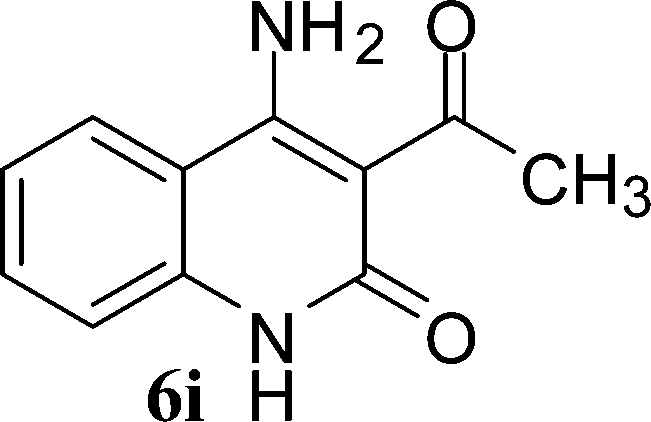
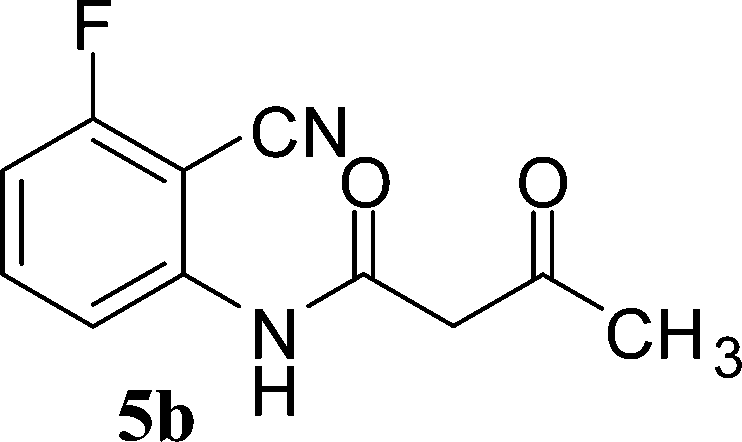
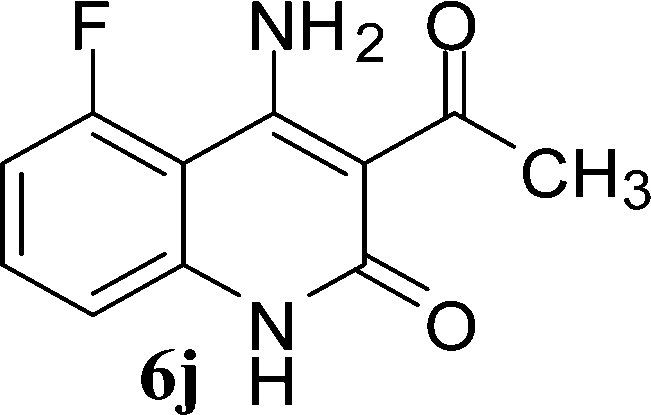
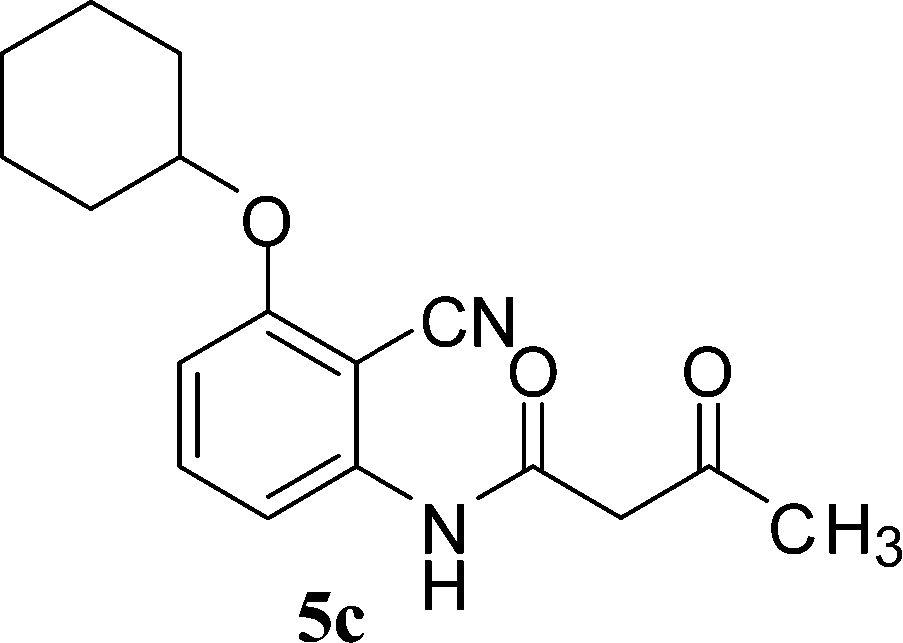
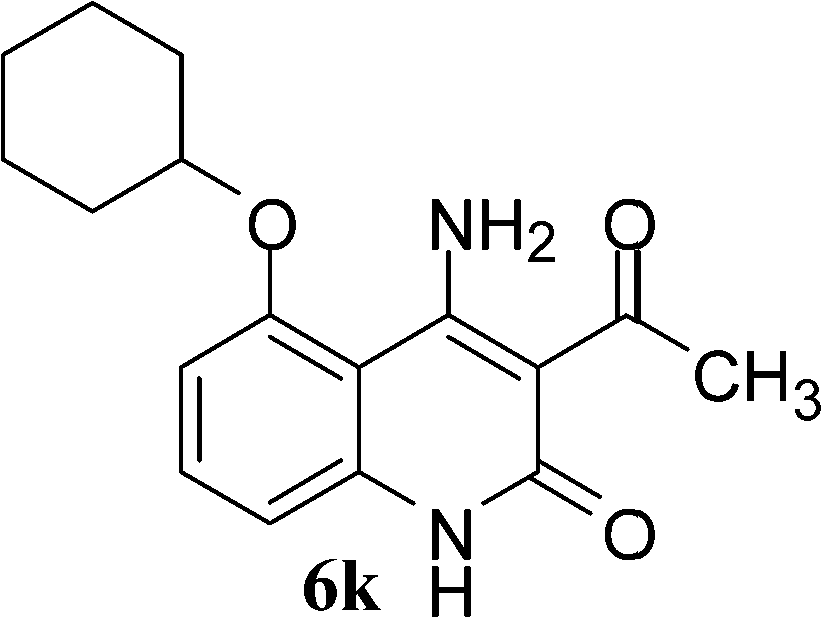
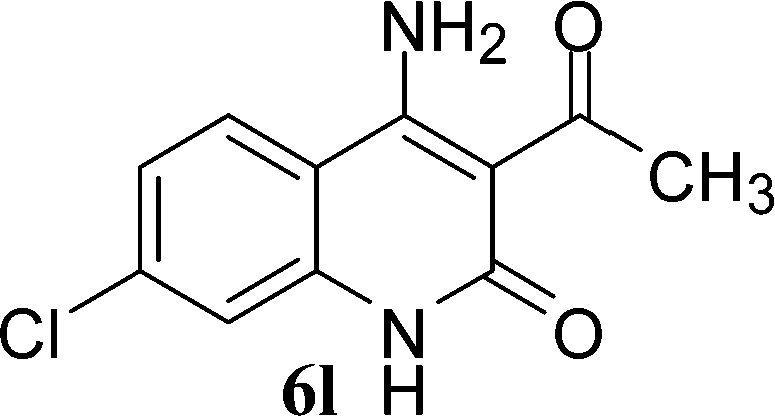
With the optimal conditions in hand, we extended our studies to a variety of substrates to evaluate the scope of this methodology and the results are presented in Table 2. The amides (3a–3h & 5a–5e) have an ArNHCOCH2R type of structure; whereas the Ar group consists of cyano substituents at the 2-position with respect to the NH2 group. The R1 group may be H/F/Cl/O-cyclohexyloxy; the R2 group may be CN/Ph/CO2Et/7-methylcoumarin-4-yl; and the R3 group may be CH3 or Ph. The substituent effect on the yield and the rate of cyclisation was determined (Scheme 3).
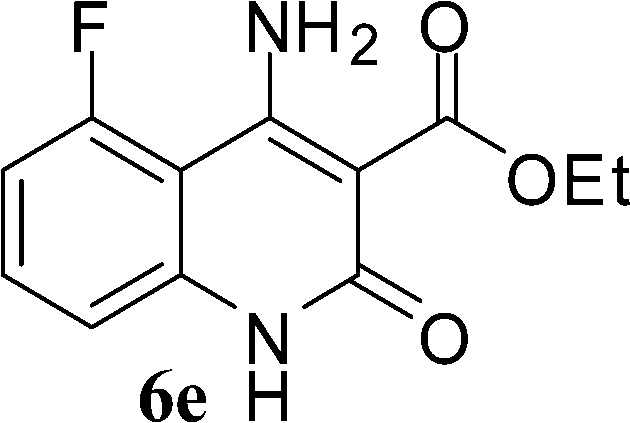
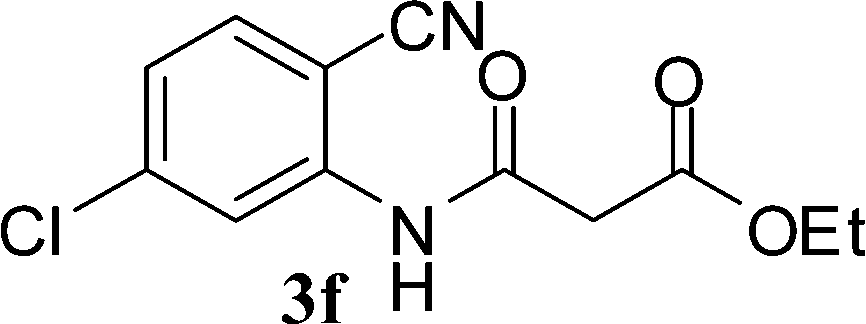
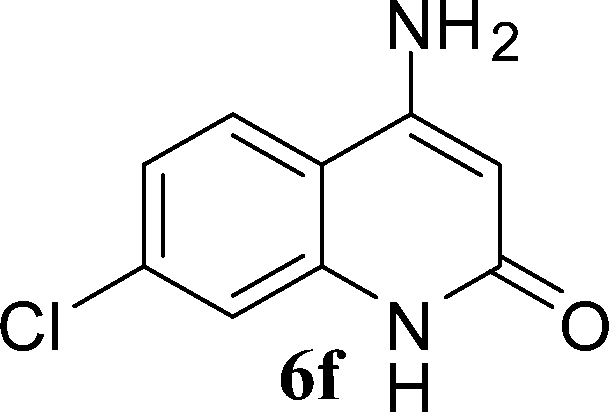
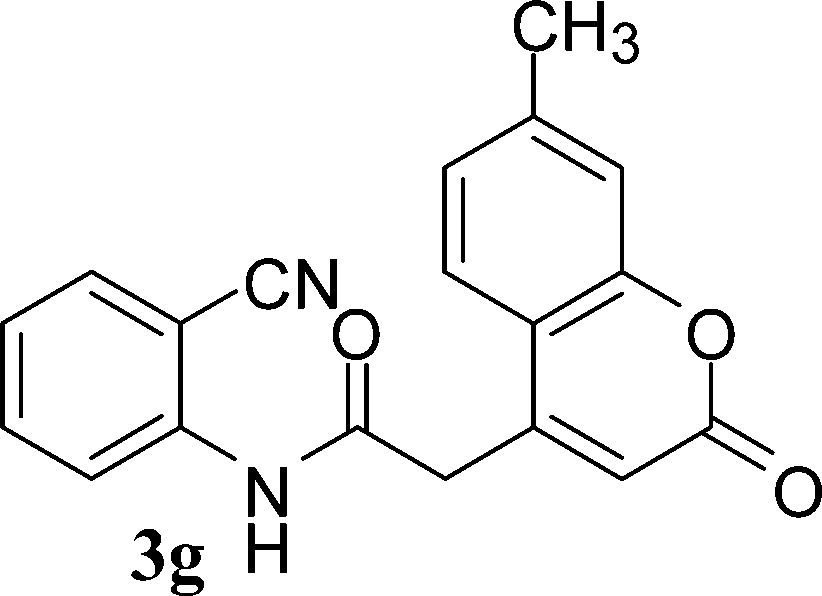
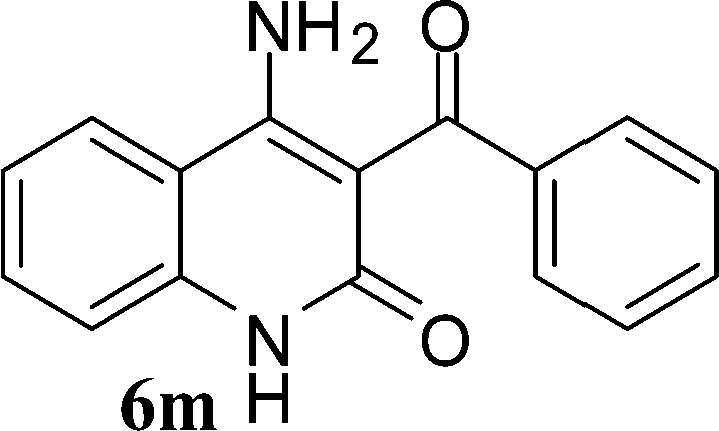
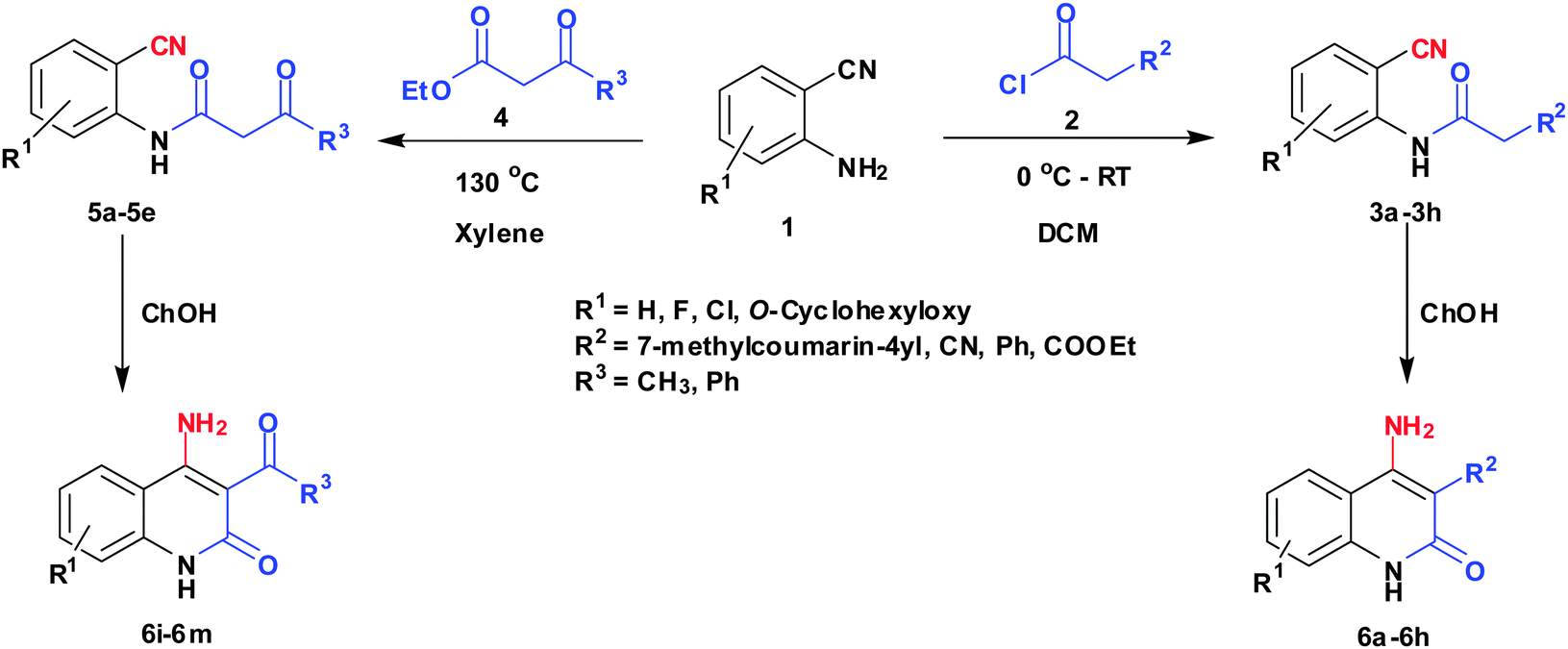 | ||
| Scheme 3 Synthesis of 4-aminoquinoline-2-ones in choline hydroxide. | ||
When R1 = H, then the rate of cyclisation was fast and a higher yield of the desired product (Table 2, entries 1, 4, 7–9 and 13) was observed as compared to other substituents (R1 = F, Cl and O-cyclohexyloxy). When R1 = F, Cl, decreased reactivity in the cyclization was due to their electron withdrawing effects. The longer reaction time required for the O-cyclohexyloxy group was due to its steric hindrance. Hence, more time was required for cyclisation of 3 giving a lesser yield of the 4-aminoquinoline-2-ones (Table 2, entries 2, 3, 5, 6, and 10–12). All the reactions were successful and gave a better yield of the products.
The time required for the cyclisation reaction also depends upon the nature of the R2/R3 group, as the electron withdrawing power of the R2/R3 group increases, the cyclisation reaction becomes faster. When R2 = CN, CO2Et (3a–3e), the reaction completed in a very short time and gave the best yield of 6a–6e (Table 2, entries 1–5). For the substrate 3f, we deliberately carried out the reaction for a longer time to see the effect of ChOH on ester functionality (R2 = CO2Et) (Table 2, entry 6). It was observed that hydrolysis and decarboxylation result in the formation of 6f rather than the desired product. Initially, the reaction was very rapid giving the corresponding cyclized product. After that ester was hydrolyzed to acid and then acid was decarboxylated to give 62% of 6f in 2 h.
In the case of 3g and 3h (R2 = Ar), carbanions are better stabilized by the aromatic ring thereby decreasing its nucleophilicity, which results in a longer reaction time (Table 2, entries 7 and 8). When R3 = CH3 or Ar (5a–5e), the reaction requires less time and gave a good yield of 6i–6m (Table 2, entries 9–13).
Recovery of ChOH
The recovery was very simple involving evaporation of water after isolation of the product by filtration. The recycled ChOH was used for the next batch with minimal loss of activity till four consecutive runs (Fig. 1).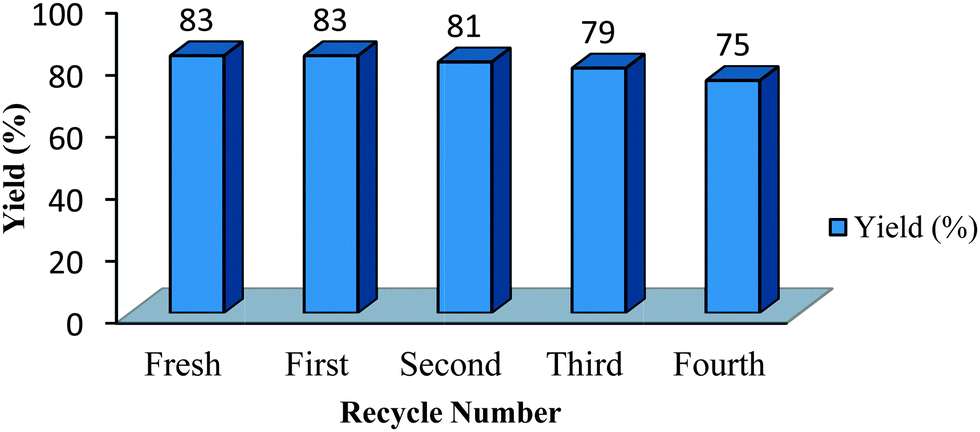 | ||
| Fig. 1 Recyclability studies for product 6a. | ||
FT-IR analysis
The compound having the covalent bond absorbs various frequencies of electromagnetic radiation in the infrared region of the electromagnetic spectrum. The infrared spectrum is used to determine structural information about a molecule. ChOH before the reaction and after the reaction was characterized using ATR-FTIR spectroscopy. The absorption peak at around 3365 cm−1 is characteristic of O–H stretching vibration. The bands at 1236 and 1087 cm−1 result from the C–N and C–O stretching. Also bands at 888 and 628 cm−1 are due to the C–H bending vibration. There is no change observed in the IR spectra of ChOH before the reaction and after four runs (Fig. 2).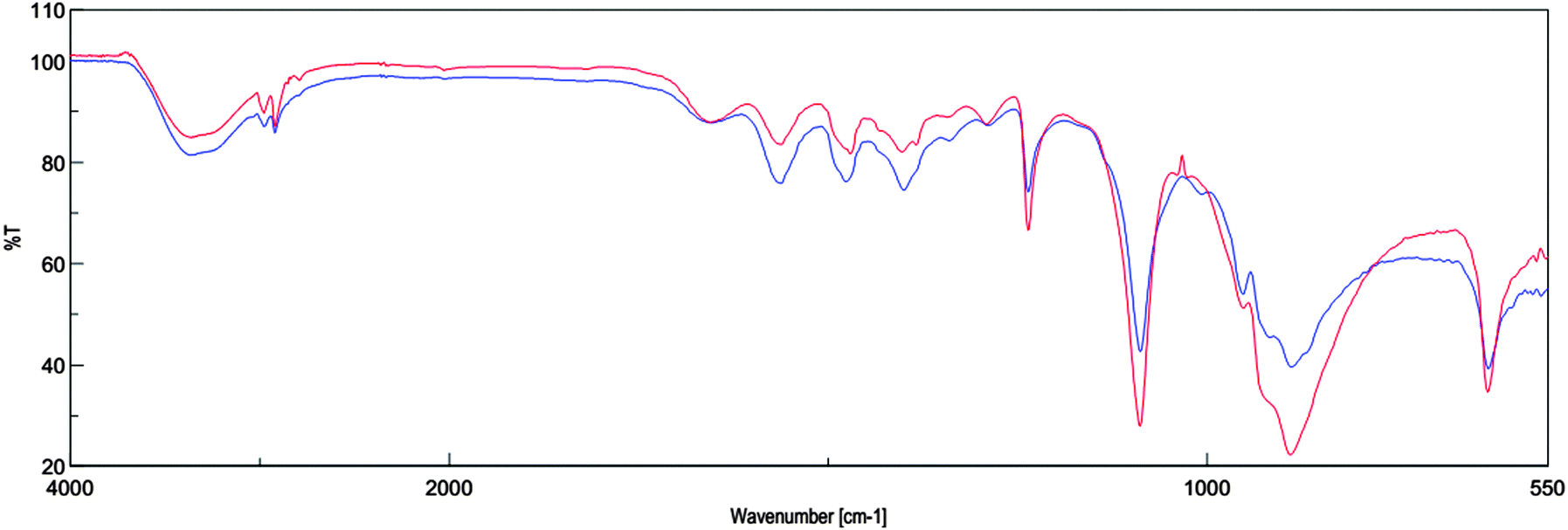 | ||
| Fig. 2 FT-IR spectra of ChOH before reaction (lower blue curve) and ChOH after four runs (upper red curve). | ||
Proposed mechanism
Although the mechanism illustrating the role of such ChOH in 4-aminoquinoline-2-ones synthesis is yet to be confirmed, we suggest a mechanism that highlights the probable role of ChOH in synthesis. The hydroxide ion of ChOH forms hydrogen bonding with acidic protons of amide (Fig. 3). ChOH assists in improving reactivity and finally promoting cyclization to form the 4-aminoquinoline-2-ones core.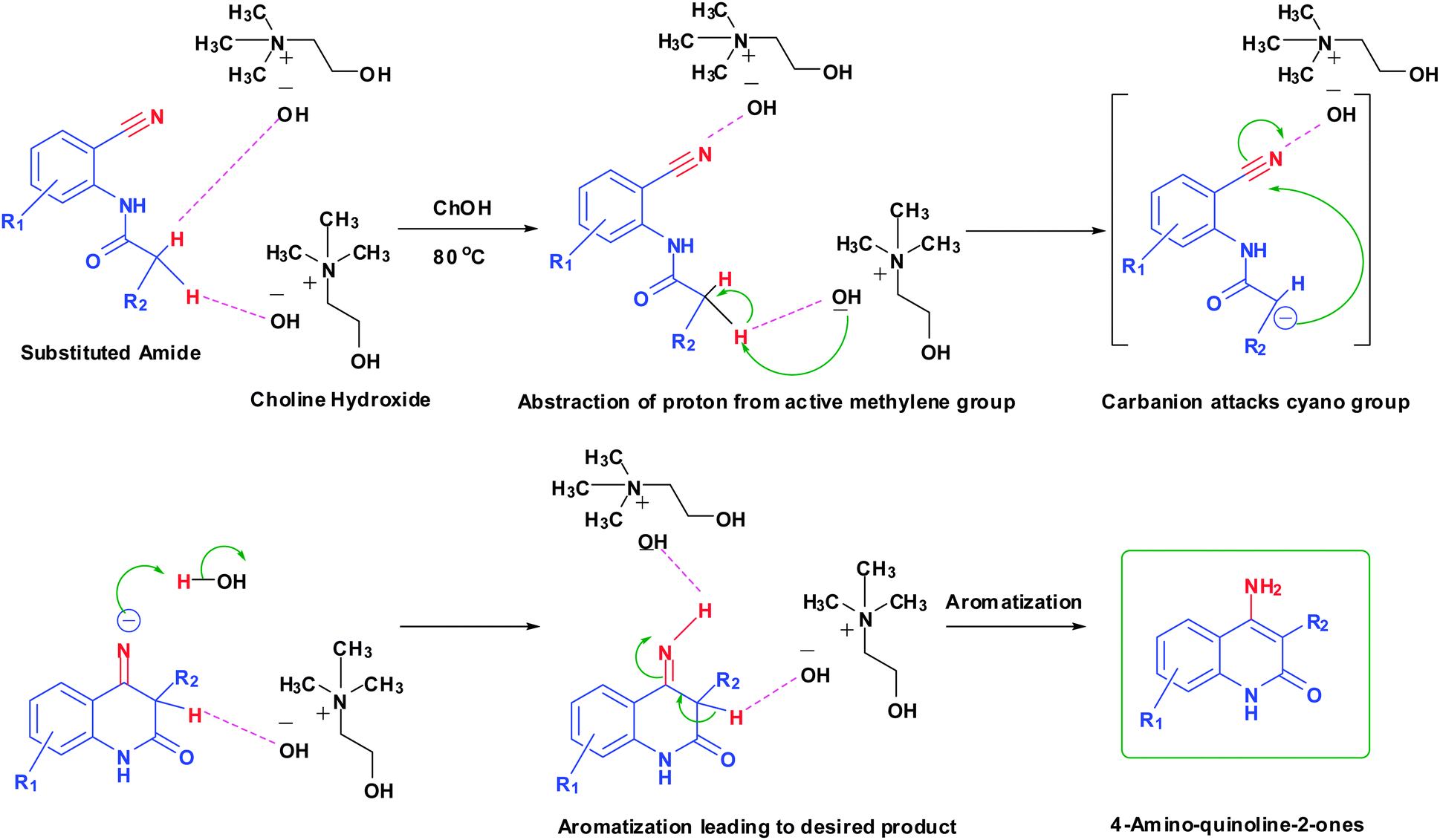 | ||
| Fig. 3 Mechanism proposing the role of ChOH in synthesis of 4-aminoquinoline-2-one derivatives. | ||
Conclusion
In this study, we have described an efficient and practical methodology for the synthesis of 4-aminoquinoline-2-ones in ChOH. Moreover, the influence of the substituent type (–CN, –COCH3, –COPh, –CO2Et, –Ph, 7-methylcoumarin-4-yl substituents) and the position on the rate of cyclisation of amides to 4-aminoquinoline-2-ones was investigated. When R1 = H, then the rate of cyclisation was fast as compared to other substituents (R1 = F, Cl and O-cyclohexyloxy). The time required for the cyclisation reaction depends upon the nature of the R2/R3 group. The electron withdrawing cyano group increases the cyclisation reaction rate and gave a high yield of the product (6a–6c) and when R2/R3 = Ar (3g and 3h), the reaction required longer time. This may be due to stabilization of carbanions by the benzene ring. FTIR spectra of fresh ChOH and recycled ChOH remain unchanged. Hence, ChOH can be recycled and reused. These features will facilitate this procedure to find extensive applications in the field of organic synthesis.Acknowledgements
A.K.S. is thankful to the Institute of Chemical Technology and UGC-CAS for providing financial assistance and IIRBS-Kottayam for spectral analysis.Notes and references
- R. W. Carling, P. D. Leeson, K. W. Moore, C. R. Moyes, M. Duncton, M. L. Hudson, R. Baker, A. C. Foster, S. Grimwood, J. A. Kemp, G. R. Marshall, M. D. Tricklebank and K. L. Saywell, J. Med. Chem., 1997, 40, 754–765 CrossRef CAS PubMed.
- P. A. Renhowe, S. Pecchi, C. M. Shafer, T. D. Machajewski, E. M. Jazan, C. Taylor, W. Antonios-McCrea, C. M. McBride, K. Frazier, M. Wiesmann, G. R. Lapointe, P. H. Feucht, R. L. Warne, C. C. Heise, D. Menezes, K. Aardalen, H. Ye, M. He, V. Le, J. Vora, J. M. Jansen, M. E. Wernette-Hammond and A. L. Harris, J. Med. Chem., 2009, 52, 278–292 CrossRef CAS PubMed.
- P. A. Renhowe, S. Pecchi, T. D. Machajewski, C. M. Shafer, E. M. Jazan, C. Taylor, W. B. McCrea, C. M. McBride, E. Jazan, M. E. Wernette-Hammond and A. L. Harris, WO Pat., 0222598A1, 2002 Search PubMed.
- C. Tachdjian, D. S. Karanewsky, X.-Q. Tang, X. Li, F. Zhang, G. Servant, Q. Chen, V. Darmohusodo, R. Fine, J. R. Fotsing, J. R. Hammaker, X. Kang, R. D. A. Kimmich, B. Klebansky, H. Liu, G. Petrovic, M. Rinnova, S. Adamski-Werner, J. Yamamoto, H. Zhang, A. Zlotnik, M. Zoller and K. Zoller, US Pat., 20110224155A, 2011 Search PubMed.
- A. C. Veronese, R. Callegari and S. A. A. Salah, Tetrahedron Lett., 1990, 31, 3485–3488 CrossRef CAS.
- A. C. Veronese, R. Callegari and C. F. Morelli, Tetrahedron, 1995, 51, 12277–12284 CrossRef CAS.
- A. Walser, G. Silverman, T. Flynn and R. I. Fryer, J. Heterocycl. Chem., 1975, 12, 351–358 CrossRef CAS.
- M. Rowley, J. J. Kulagowski, A. P. Watt, D. Rathbone, G. I. Stevenson, R. W. Carling, R. Baker, G. R. Marshall, J. A. Kemp, A. C. Foster, S. Grimwood, R. Hargreaves, C. Hurley, K. L. Saywell, M. D. Tricklebank and P. D. Leeson, J. Med. Chem., 1997, 40, 4053–4068 CrossRef CAS PubMed.
- M. Rehwald, K. Gewald, H. Lankaun and K. Unverferth, Heterocycles, 1997, 45, 483–492 CrossRef CAS PubMed.
- E. J. Paul, P. A. Renhowe, S. Pecchi, T. D. Machajewski, C. M. Shafer, C. Taylor, W. R. McCrea Jr., C. M. McBride and E. Jazan, US Pat., 2003028018A1, 2003 Search PubMed.
- W. Steinschifter and W. Stadlbauer, J. Prakt. Chem./Chem.-Ztg., 1994, 336, 311–318 CrossRef CAS.
- W. Stadlbauer and T. Kappe, Monatsh. Chem., 1982, 113, 751–760 CrossRef CAS.
- T. K. W. Stadlbauer, Synthesis, 1981, 833–834 CrossRef.
- R. B. Toche, R. A. Janrao, S. M. Bagul, S. P. Patil, B. P. Pagar and P. S. Nikam, J. Fluoresc., 2011, 21, 1617–1624 CrossRef CAS PubMed.
- C. Ruß and B. König, Green Chem., 2012, 14, 2969–2982 RSC.
- Y. A. Sonawane, S. B. Phadtare, B. N. Borse, A. R. Jagtap and G. S. Shankarling, Org. Lett., 2010, 12, 1456–1459 CrossRef CAS PubMed.
- P. M. Pawar, K. J. Jarag and G. S. Shankarling, Green Chem., 2011, 13, 2130–2134 RSC.
- H. R. Lobo, B. S. Singh and G. S. Shankarling, Catal. Commun., 2012, 27, 179–183 CrossRef CAS PubMed.
- S. B. Phadtare, K. J. Jarag and G. S. Shankarling, Dyes Pigm., 2013, 97, 105–112 CrossRef CAS PubMed.
- A. K. Sanap and G. S. Shankarling, Catal. Commun., 2014, 49, 58–62 CrossRef CAS PubMed.
- U. N. Yadav and G. S. Shankarling, J. Mol. Liq., 2014, 19, 137–141 CrossRef PubMed.
- A. K. Sanap and G. S. Shankarling, RSC Adv., 2014, 4, 34938–34943 RSC.
- J. Shao, X. Huang, S. Wang, B. Liu and B. Xu, Tetrahedron, 2012, 68, 573–579 CrossRef CAS PubMed.
- K. Chakraborty and C. Devakumar, J. Agric. Food Chem., 2006, 54, 6800–6808 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nj01281j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |