
Alkane oxidation with peroxides catalyzed by cage-like copper(II) silsesquioxanes†‡
Mikhail M.
Vinogradov
ab,
Yuriy N.
Kozlov
c,
Alexey N.
Bilyachenko
a,
Dmytro S.
Nesterov
b,
Lidia S.
Shul'pina
a,
Yan V.
Zubavichus
ad,
Armando J. L.
Pombeiro
b,
Mikhail M.
Levitsky
a,
Alexey I.
Yalymov
a and
Georgiy B.
Shul'pin
*c
aNesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ulitsa Vavilova, dom 28, Moscow 119991, Russia
bCentro de Química Estrutural, Complexo I, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
cSemenov Institute of Chemical Physics, Russian Academy of Sciences, ulitsa Kosygina, dom 4, Moscow 119991, Russia. E-mail: Shulpin@chph.ras.ru; gbsh@mail.ru; Fax: +7 499 1376130; Fax: +7 495 6512191; Tel: +7 495 9397317
dNational Research Center “Kurchatov Institute”, pl. Akad. Kurchatova, dom 1, Moscow 123098, Russia
First published on 22nd September 2014
Abstract
Isomeric cage-like tetracopper(II) silsesquioxane complexes [(PhSiO1.5)12(CuO)4(NaO0.5)4] (1a), [(PhSiO1.5)6(CuO)4(NaO0.5)4(PhSiO1.5)6] (1b) and binuclear complex [(PhSiO1.5)10(CuO)2(NaO0.5)2] (2) have been studied by various methods. These compounds can be considered as models of some multinuclear copper-containing enzymes. Compounds 1a and 2 are good pre-catalysts for the alkane oxygenation with hydrogen peroxide in air in an acetonitrile solution. Thus, the 1a-catalyzed reaction with cyclohexane at 60 °C gave mainly cyclohexyl hydroperoxide in 17% yield (turnover number, TON, was 190 after 230 min and initial turnover frequency, TOF, was 100 h−1). The alkyl hydroperoxide partly decomposes in the course of the reaction to afford the corresponding ketone and alcohol. The effective activation energy for the cyclohexane oxygenation catalyzed by compounds 1a and 2 is 16 ± 2 and 17 ± 2 kcal mol−1, respectively. Selectivity parameters measured in the oxidation of linear and branched alkanes and the kinetic analysis revealed that the oxidizing species in the reaction is the hydroxyl radical. The analysis of the dependence of the initial reaction rate on the initial concentration of cyclohexane led to a conclusion that hydroxyl radicals attack the cyclohexane molecules in proximity to the copper reaction centers. The oxidations of saturated hydrocarbons with tert-butylhydroperoxide (TBHP) catalyzed by complexes 1a and 2 exhibit unusual selectivity parameters which are due to the steric hindrance created by bulky silsesquioxane ligands surrounding copper reactive centers. Thus, the methylene groups in n-octane have different reactivities: the regioselectivity parameter for the oxidation with TBHP catalyzed by 1a is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.5:8:7. Furthermore, in the oxidation of methylcyclohexane the position 2 relative to the methyl group of this substrate is noticeably less reactive than the corresponding positions 3 and 4. Finally, the oxidation of trans-1,2-dimethylcyclohexane with TBHP catalyzed by complexes 1a and 2 proceeds stereoselectively with the inversion of configuration. The 1a-catalyzed reaction of cyclohexane with H216O2 in an atmosphere of 18O2 gives cyclohexyl hydroperoxide containing up to 50% of 18O. The small amount of cyclohexanone, produced along with cyclohexyl hydroperoxide, is 18O-free and is generated apparently via a mechanism which does not include hydroxyl radicals and incorporation of molecular oxygen from the atmosphere.
:10.5:8:7. Furthermore, in the oxidation of methylcyclohexane the position 2 relative to the methyl group of this substrate is noticeably less reactive than the corresponding positions 3 and 4. Finally, the oxidation of trans-1,2-dimethylcyclohexane with TBHP catalyzed by complexes 1a and 2 proceeds stereoselectively with the inversion of configuration. The 1a-catalyzed reaction of cyclohexane with H216O2 in an atmosphere of 18O2 gives cyclohexyl hydroperoxide containing up to 50% of 18O. The small amount of cyclohexanone, produced along with cyclohexyl hydroperoxide, is 18O-free and is generated apparently via a mechanism which does not include hydroxyl radicals and incorporation of molecular oxygen from the atmosphere.
1. Introduction
Various transition metal complexes are able to activate C–H bonds in alkanes and arenes. In particular, soluble mono and polynuclear copper compounds are good pre-catalysts for oxidation reactions of hydrocarbons with molecular oxygen and peroxides.1 Hydrogen peroxide, tert-butyl hydroperoxide (TBHP), and peroxyacetic acid are typically employed in the oxidation of saturated and aromatic hydrocarbons.2,3 The development of novel metal complex catalysts has been inspired by the action of some copper-containing enzymes and especially particulate methane monooxygenase (pMMO), which bears a polynuclear copper fragment and oxidizes alkanes including methane under very mild conditions.4,5 The reactions of copper ions with H2O2 typically result in the production of either hydroxyl radicals or Cu(III) derivatives.6Recently some of us reported7 the first examples of the oxidation of benzene and 1-phenylethanol with H2O2 or TBHP catalyzed by new bi- and tetranuclear copper(II) silsesquioxanes (for the synthesis and structural features of metallasilsesquioxanes, see selected reviews8). Only two papers have been devoted to the oxygenation of alkanes catalyzed by iron silsesquioxanes.9 As a continuation of our studies on copper derivatives we have performed herein a further study of two isomeric tetracopper(II) compounds, a “Globule”-like [(PhSiO1.5)12(CuO)4(NaO0.5)4] (1a) and “Sandwich”-like [(PhSiO1.5)6(CuO)4(NaO0.5)4(PhSiO1.5)6] (1b) (Fig. 1) derivatives as well as a dicopper(II) [(PhSiO1.5)10(CuO)2(NaO0.5)2] (2) complex with the structure of a “Cooling tower” (Fig. 2). These compounds were used for the first time as pre-catalysts in the oxidation of saturated hydrocarbons with hydrogen peroxide and tert-butyl hydroperoxide. A kinetic analysis of these reactions has also been performed.
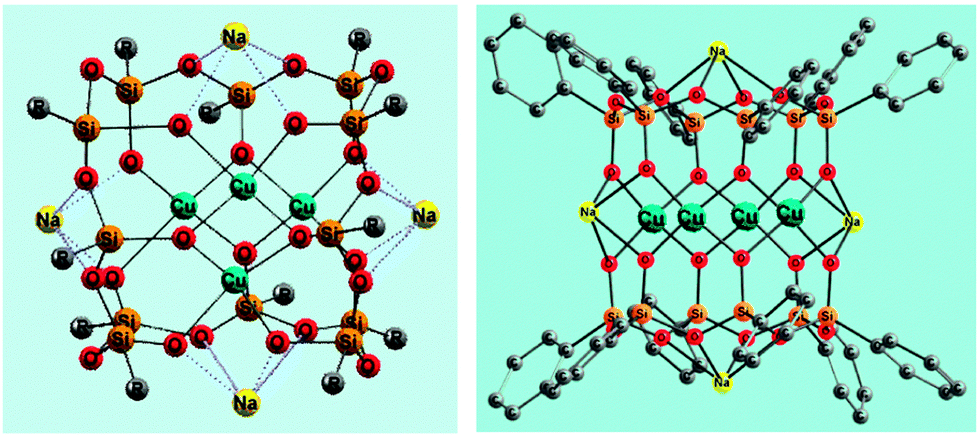 | ||
| Fig. 1 Structures of tetracopper(II) derivatives 1a7b (CCDC 920381), [(PhSiO1.5)12(CuO)4(NaO0.5)4] (left), and 1b7b (CCDC 931312), [(PhSiO1.5)6(CuO)4(NaO0.5)4(PhSiO1.5)6] (right). Grey balls R in 1a are phenyl substituents. Solvating molecules of 1-butanol (in the case of 1a) and 1,4-dioxane (in the case of 1b) are omitted for clarity. | ||
 | ||
| Fig. 2 Structure of dinuclear complex 27a (CCDC 931703), [(PhSiO1.5)10(CuO)2(NaO0.5)2]. Grey balls are phenyl substituents. Solvating molecules of ethanol are omitted for clarity. | ||
2. Results and discussion
2.1. Local-structure study of copper complexes by EXAFS
The local structure around Cu atoms in the compounds 1a, 1b and 2 was also elucidated by means of EXAFS spectroscopy. Some amounts (∼70 mg) of compounds 1a, 1b and 2 were studied by EXAFS in order to illustrate the identity of structures (in terms of Cu-ions surrounding) of bulky polycrystalline samples of catalysts and known X-ray results (obtained for monocrystals). The XANES spectra for the complexes are consistent with that of Cu2+ ions in a slightly distorted square-planar coordination by oxygen atoms. Experimental and best-fit theoretical Fourier Transforms (FTs) of EXAFS spectra are shown in Fig. 3. The dominant maxima in the FTs correspond to the Cu–O first coordination sphere, whereas the second distinct peaks are due to a superposition of Cu⋯Cu, Cu⋯Na, and Cu⋯Si contributions. Interatomic distances obtained by the non-linear curve-fitting procedure are summarized in Table 1, being quite close to expected values from respective X-ray crystallographic data. | ||
| Fig. 3 Fourier transforms of Cu K-edge EXAFS spectra for Cu complexes 1a, 1b, and 2. | ||
| Complex | Coordination sphere | Coordination number | Interatomic distance (Å) |
|---|---|---|---|
| 1a | Cu–O1 | 2 | 1.89 |
| Cu–O2 | 2 | 1.99 | |
| Cu⋯Cu | 1 | 3.02 | |
| Cu⋯Na | 1 | 3.25 | |
| Cu⋯Si | 4 | 3.20 | |
| 1b | Cu–O1 | 4 | 1.93 |
| Cu–O2 | 0.5 | 2.41 | |
| Cu⋯Cu | 1 | 2.95 | |
| Cu⋯Na | 1 | 3.02 | |
| Cu⋯Si | 4 | 3.21 | |
| 2 | Cu–O | 4 | 1.93 |
| Cu⋯Cu | 1 | 3.07 | |
| Cu⋯Na | 1 | 3.14 | |
| Cu⋯Si | 4 | 3.19 | |
2.2. Main features of the alkane oxidation
We studied the oxidation of alkanes in an acetonitrile solution with hydrogen peroxide catalyzed by complex 1a (see Fig. 1) as well as complex 2 (Fig. 2). Since complex 1b turned out to be not very stable and active in the oxidation of benzene,7b we did not use this compound in the alkane oxidations. The reactions occur in the presence of nitric acid. Examples of the kinetic curves for the oxidation of cyclohexane are shown in Fig. 4. The oxygenation of cyclohexane gives rise to the formation of the corresponding alkyl hydroperoxide, ROOH, as the main primary product. To demonstrate the formation of alkyl hydroperoxide in this oxidation and to estimate its concentration in the course of the reaction we used a simple method developed earlier by Shul'pin.10 If an excess of solid PPh3 is added to the sample of the reaction solution before the GC analysis, the alkyl hydroperoxide present is completely reduced to the corresponding alcohol. By comparing the GC concentrations of the alcohol and ketone measured before and after reduction with PPh3 we can estimate the real concentrations of the three products (alkyl hydroperoxide, ketone and alcohol) present in the reaction solution.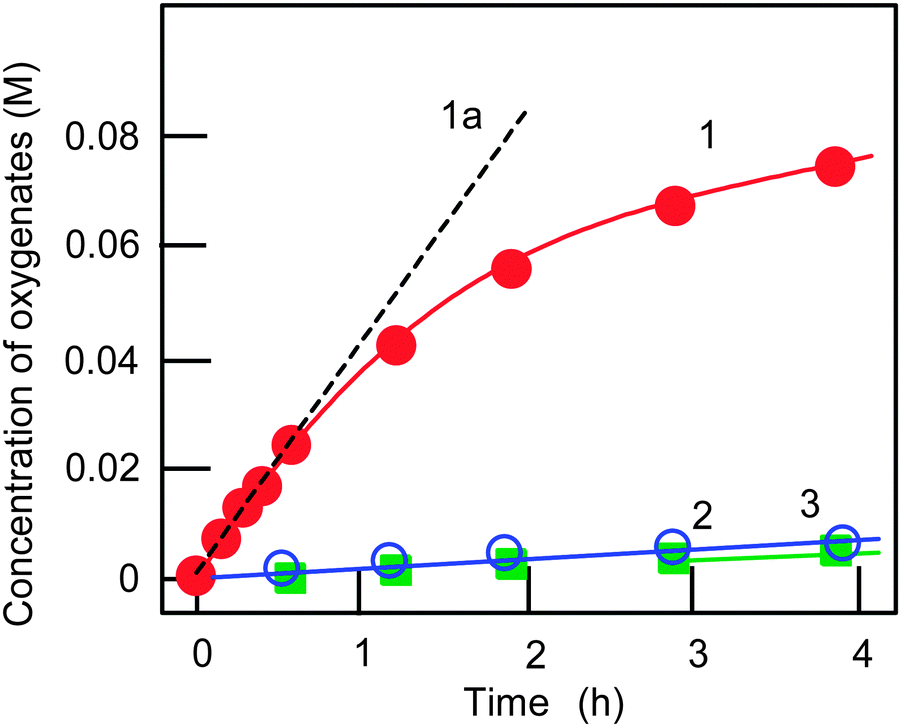 | ||
| Fig. 4 Kinetic curves of accumulation of oxygenates (cyclohexyl hydroperoxide, curve 1; cyclohexanol, curve 2; cyclohexanone, curve 3) in the oxidation of cyclohexane with H2O2 catalyzed by complex 1a. Conditions: [1a]0 = 4.1 × 10−4 M, [cyclohexane]0 = 0.46 M, [HNO3] = 0.4 M, [H2O2]0 = 1.0 M (50% aqueous), [H2O]total = 2.65 M, solvent MeCN, 60 °C. Concentrations of the three oxygenated products (cyclohexyl hydroperoxide, cyclohexanol and cyclohexanone) were calculated by comparing the concentrations of cyclohexanol and cyclohexanone measured by GC before and after reduction of samples with PPh3 (for this method, see ref. 5 and Experimental section). The initial oxidation rate W0 was determined from the slope of tangent (dotted straight line 1a) to the kinetic curve 1. Yield of oxygenates was 17% and TON was 190 after 230 min. Initial TOF (line 1a) was 100 h−1. | ||
Using cyclohexane as a model substrate we studied dependences of the initial reaction rate W0 (based on the sum of cyclohexanol and cyclohexanone concentrations measured after reduction of the reaction sample with PPh3) on the initial concentration of each reactant at fixed concentrations of all other components of the reaction solution. These dependences are shown in Fig. 5–8. The dependence of W0 on the initial concentration of H2O2 (straight line) is shown in Fig. 7A. Addition of water enhances the initial reaction rate and the dependence has a maximum (Fig. 7B).
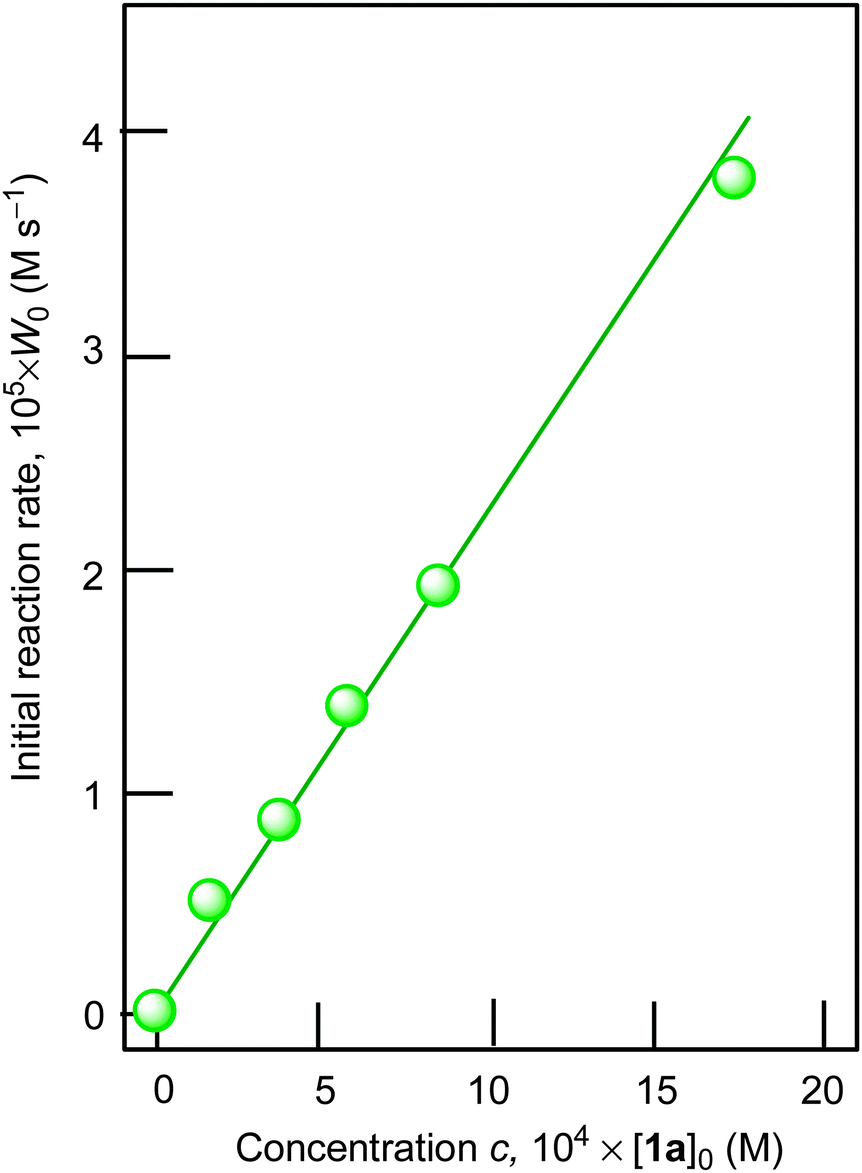 | ||
| Fig. 5 Dependence of cyclohexane oxidation rate W0 on initial concentration of catalyst 1a in the oxidation of cyclohexane with H2O2 (1.0 M) in the presence of HNO3 (0.4 M) ([cyclohexane]0 = 0.46 M, solvent MeCN, 60 °C). For the original kinetic curves, see Fig. S1 (ESI‡). | ||
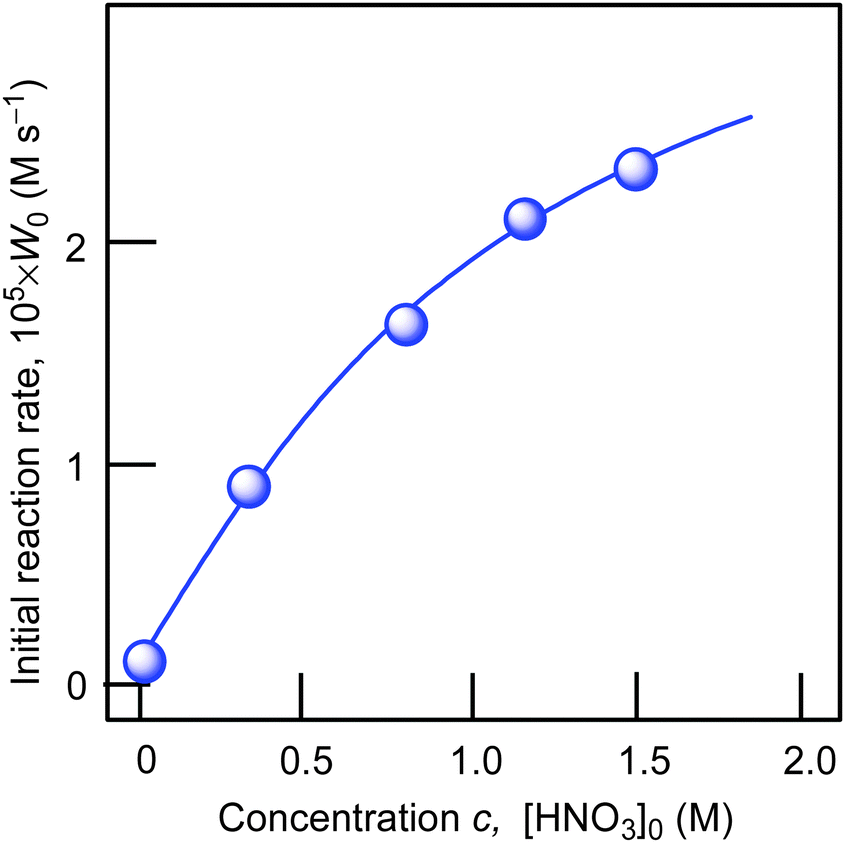 | ||
| Fig. 6 Dependence of initial reaction rate W0 on concentration of added nitric acid in the cyclohexane oxidation. Conditions: [1a]0 = 4.1 × 10−4 M, [cyclohexane]0 = 0.46 M, [H2O2]0 = 1.0 M (50% aqueous), [H2O]total = 2.65 M, solvent MeCN, 60 °C. For the original kinetic curves, see Fig. S2 (ESI‡). The initial rate W0 was determined (as shown in Fig. 4) from the slope of tangent to the kinetic curve of accumulation of the sum of cyclohexyl hydroperoxide, cyclohexanone and cyclohexanol (in order to obtain the value of concentration of all products we measured the concentration of the sum cyclohexanol + cyclohexanone after reduction of the sample with PPh3). | ||
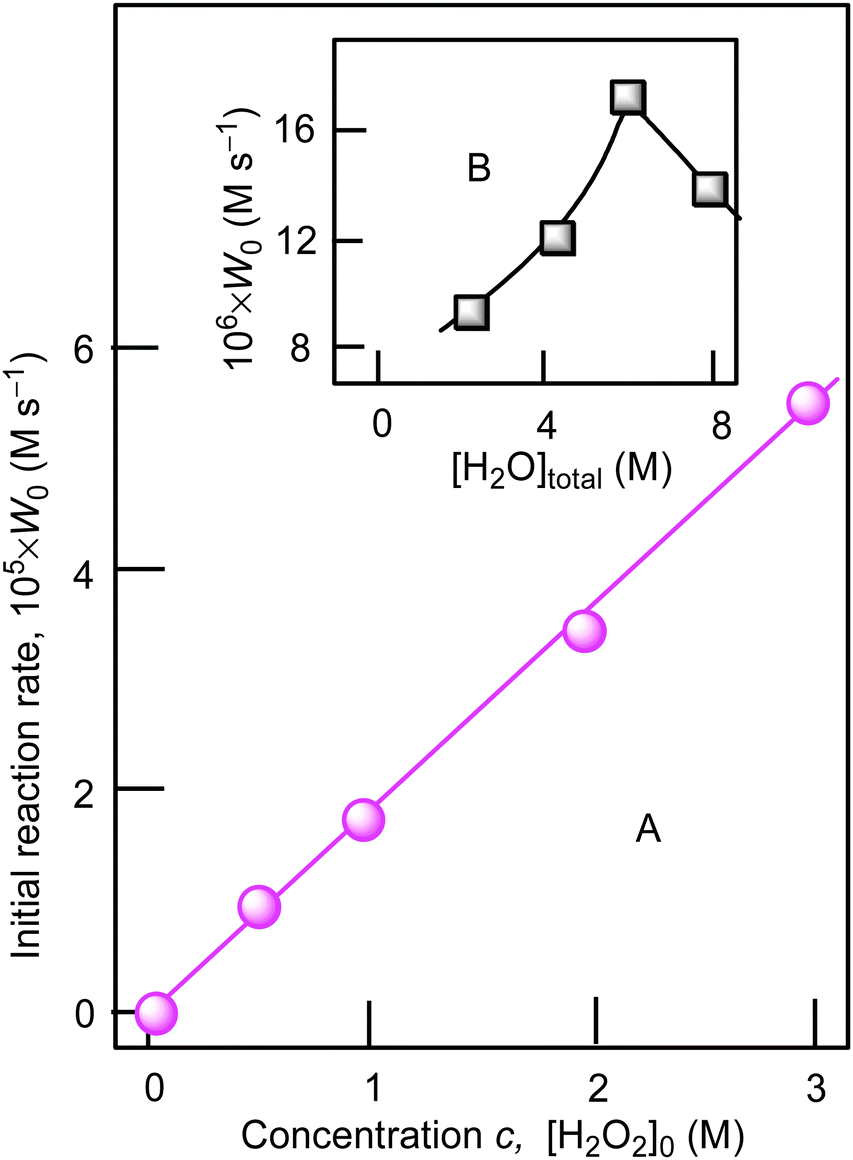 | ||
| Fig. 7 Graph A: dependence of initial reaction rate W0 on initial concentration of hydrogen peroxide (50% aqueous was used) in the cyclohexane oxidation. Conditions: [1a]0 = 4.1 × 10−4 M, [cyclohexane]0 = 0.46 M, [HNO3]0 = 0.4 M, solvent MeCN, 60 °C. For the original kinetic curves, see Fig. S3 (ESI‡). The concentration of water in the reaction was maintained constant [H2O]total = const = 2.65 M, by adding necessary amounts of H2O. Graph B: dependence of initial reaction rate W0 on total concentration of water at [H2O2]0 = 1.0 M. Concentrations of the products (cyclohexanol and cyclohexanone) were measured after reduction with PPh3. | ||
 | ||
| Fig. 8 Graph A: dependence of oxidation rate W0 on initial concentration of cyclohexane in the oxidation of cyclohexane with H2O2. Conditions: [1a]0 = 4.1 × 10−4 M, [H2O2]0 = 1.0 M (50% aqueous), [H2O]total = 2.65 M, [HNO3]0 = 0.4 M, solvent MeCN, 60 °C. For the original kinetic curves, see Fig. S4 (ESI‡). Concentrations of the products (cyclohexanol and cyclohexanone) were measured after reduction with PPh3. Graph B: linearization of dependence presented in graph A using coordinates 1/[cyclohexane]0 − 1/W0. | ||
We carried out the oxidation of cyclohexane at different temperatures catalyzed by complex 1a (Fig. S5, ESI‡). The estimated effective activation energy is 16 ± 2 kcal mol−1. (Fig. S6, ESI‡). For the reaction catalyzed by complex 2, Ea = 17 ± 2 kcal mol−1 (Fig. S7 and S8, ESI‡). It is interesting that the addition of benzene as a potential radical trap practically does not affect the initial rate of cyclohexane oxidation (Fig. S9, ESI‡), a phenomenon that remains difficult to explain. A similar behaviour was observed in the oxidation of cyclohexane with the H2O2-vanadate ion-pyrazine-2-carboxylic acid reagent, which also operates with the participation of hydroxyl radicals (see below, Section 2.4; for this system, see ref. 11).
2.3. Selectivity in the alkane oxidation with H2O2 and TBHP
In order to determine the nature of the alkane oxidizing species we measured the selectivity parameters in oxidations of certain alkanes with H2O2 catalyzed by compounds 1a and 2 (Table 2, entries 1 and 2). These values can be compared with the parameters determined previously12 for other systems which are also given in Table 2 for comparison (see also the discussion of various selectivity parameters collected in Table S3 (ESI‡) from the paper12m which has been unwittingly lost in ref. 12m). It can be seen that these parameters are close to the selectivities determined previously for the vanadium, iron, osmium, nickel, rhenium and aluminum-based systems generating free hydroxyl radicals (entries 5–28) and noticeably lower than parameters determined for the oxidation systems operating either without the participation of reactive hydroxyl radicals or in narrow cages (entries 29–36). The oxidation of cis-1,2-DMCH and trans-1,2-DMCH with the 1/H2O2 and 2/H2O2 systems proceeds non-stereoselectively, similarly to other oxidizing systems shown in entries 5–28.| Entry | Oxidizing system | C(1):C(2):C(3):C(4) |
1°:2°:3° |
trans:cis | Ref. | |
|---|---|---|---|---|---|---|
| n-Heptane | MCH | c-1,2-DMCH | t-1,2-DMCH | |||
|
a All parameters were measured after reduction of the reaction mixtures with triphenylphosphine before GC analysis and calculated based on the ratios of isomeric alcohols. Parameter C(1):C(2):C(3):C(4) is the relative normalized (taking into account the number of hydrogen atoms at each carbon) reactivities of hydrogen atoms at carbons 1, 2, 3 and 4 of the chain of n-heptane. Parameter 1°:2°:3° is the relative normalized reactivities of hydrogen atoms at primary, secondary and tertiary carbons of methylcyclohexane (MCH). Parameter trans/cis is the ratio of isomers of tert-alcohols with mutual trans- and cis-orientation of the methyl groups formed in the oxidation of cis- and trans-1,2-dimethylcyclohexane (DMCH).
b Abbreviations. MCH, c-1,2-DMCH and t-1,2-DMCH are methylcyclohexane, cis-1,2-dimethylcyclohexane and trans-1,2-dimethylcyclohexane, respectively. Symbol hν means UV irradiation. PCA is pyrazine-2-carboxylic acid. Vanadatrane is oxovanadium(V) triethanolaminate. V is [{VO(OEt)(EtOH)}2L] where H4L is bis(2-hydroxybenzylidene)terephthalohydrazide. Fe2(HPTB) is complex [Fe2(HPTB)(μ-OH)(NO3)2](NO3)2, HPTB = N,N,N′,N′-tetrakis(2-benzimidazolylmethyl)-2-hydroxo-1,3-diaminopropane. Cp2Fe is ferrocene. Fe2 is binuclear complex [Fe2(N3O-L1)2(μ-O)(μ-OOCCH3)]+, where L1 = 1-carboxymethyl-4,7-dimethyl-1,4,7-triazacyclononane. Fe4 is tetranuclear complex [Fe4(N3O2-L)4(μ-O)2]4+ with ligand N3O2-L1. Cp2*Os is decamethylosmocene. Os is complex (2,3-η-1,4-diphenylbut-2-en-1,4-dione)undecacarbonyl triangulotriosmium. The salt Ni(ClO4)2 was used in combination with L2 = 1,4,7-trimethyl-1,4,7-triazacyclononane. Complex Re is cis-(Cl,Cl)-[Re(p-NC6H4CH3)Cl2(ind-3-COO)(PPh3)]·2MeOH (where ind-3-COOH is indazole-3-carboxylic acid). Complex [Co4Fe2OSae8]·4DMF·H2O, where H2Sae = salicylidene-2-ethanolamine. Cu4 is tetracopper(II) triethanolaminate complex [O⊂Cu4{N(CH2CH2O)3}4(BOH)4][BF4]2. Mn is complex [Mn2(R-LMe2R)2(μ-O)2]3+ where R-LMe2R = (R)-1-(2-hydroxypropyl)-4,7-dimethyl-1,4,7-triazacyclononane. Oxone is 2KHSO5·KHSO4·K2SO4. In the complex Cu(H3L3)(NCS), ligand H4L3 is N,N,N′,N′-tetrakis-(2-hydroxyethyl)ethylenediamine. Ti-MMM-2 is a heterogeneous Ti-containing catalyst. Complex [Mn2L22O3]2+ is a binuclear manganese derivative, where L2 = 1,4,7-trimethyl-1,4,7-triazacyclononane. L4 is tetradentate amine N,N-bis(2-pyridylmethylene)-1,4-diaminodiphenyl ether. m-CPBA is metachloroperoxybenzoic acid. TS-1 is a heterogeneous titanosilicalite catalyst.
c
n-Octane was used instead of n-heptane (see Fig. S10, ESI).
d In the presence of pyridine trans:cis = 0.62.
e In the presence of pyridine trans:cis = 0.35.
|
||||||
| 1 | 1a/H2O2/HNO3 | 1:3.5:3.5:3.2c |
1:5:14 |
1.1 | 0.8 | This work |
| 2 | 2/H2O2/HNO3 | 1:5:14 |
0.65 | 0.66 | This work | |
| 3 | 1a/TBHP | 1:10.5:8:7c |
1:10:60 |
0.65 | 0.40 | This work |
| 4 | 2/TBHP | 1:12:93 |
0.8d | 0.53e | This work | |
| 5 | hν/H2O2 | 1:7:6:7 |
0.9 | 12a | ||
| 6 | (n-Bu4N)VO3/PCA/H2O2 | 1:9:7:7 |
1:6:18 |
0.75 | 0.8 | 12a–c |
| 7 | Vanadatrane/PCA/H2O2 | 1:5.5:6:5 |
1:4:10 |
0.7 | 0.75 | 12c |
| 8 | “V”/PCA/H2O2 | 1:5:4:3 |
1:5:14 |
0.7 | 0.6 | 12d |
| 9 | (n-Bu4N)VO3/HClO4/H2O2 | 1:6:6:6 |
12a | |||
| 10 | (n-Bu4N)VO3/H2SO4/H2O2 | 1:7:7:6 |
1:7:26 |
0.9 | 0.9 | 12e |
| 11 | FeSO4/H2O2 | 1:5:5:4.5 |
1:3:6 |
1.3 | 1.2 | 12a,f |
| 12 | Fe(ClO4)3/H2O2 | 1:9:9 |
1:7:43 |
12a,f | ||
| 13 | Fe2(HPTB)/PCA/H2O2 | 1:6:6:5 |
1:6:13 |
12g | ||
| 14 | Cp2Fe/PCA/H2O2 | 1:7:7:6 |
1:10:33 |
0.8 | 0.8 | 12h,i |
| 15 | “Fe2”/H2O2 | 1:10:10:6 |
1.6 | 1.2 | 12j | |
| 16 | “Fe4”/H2O2 | 1:15:14:11 |
0.9 | 1.3 | 12j | |
| 17 | (OC)3Fe(μ-PhS)2Fe(CO)3/PCA/py/H2O2 | 1:6.5:6.5:6 |
1:11:29 |
0.9 | 1.0 | 12k |
| 18 | Cp2*Os/py/H2O2 | 1:7:7:7 |
1:8:23 |
1.0 | 0.9 | 12l |
| 19 | Os3(CO)12/py/H2O2 | 1:4:4:4 |
1:5:11 |
0.85 | 10d, 12m | |
| 20 | Os3(CO)12/H2O2 | 1:5:5:5 |
1:6:14 |
10d, 12m | ||
| 21 | “Os”/H2O2 | 1:5.5:5:4.5 |
1:4:10 |
0.9 | 12n | |
| 22 | OsCl3/py/H2O2 | 1:12:10:3.5 |
12o | |||
| 23 | Ni(ClO4)4/L2/H2O2 | 1:1:7:6 |
1:7:15 |
12p | ||
| 24 | Al(NO3)3/H2O2 | 1:5:5:5 |
1:6:23 |
0.8 | 0.8 | 12q |
| 24 | “Re”/H2O2 | 1:6:6:5 |
1:6:19 |
0.9 | 0.9 | 12r |
| 25 | [Co4Fe2OSae8]/HNO3/H2O2 | 1:7:7:6 |
1:7:20 |
0.85 | 0.85 | 12s |
| 26 | “Cu4”/CF3COOH/H2O2 | 1:8:7:5.5 |
1:5:14 |
0.8 | 0.8 | 12t |
| 27 | TS-1/NaOH/MeCN/H2O2 | 1:8:8:8 |
1:6:21 |
0.85 | 0.95 | 12u |
| 28 | Ti-MMM-2/H2O2 | 1:9:7:6.5 |
1:6:113 |
0.9 | 0.9 | 12v |
| 29 | [Mn2L22O3]2+/MeCO2H/H2O2 | 1:42:37:34 |
1:26:200 |
0.35 | 4.1 | 12w,x |
| 30 | “Mn”/oxalic acid/H2O2 | 1:91:99:68 |
0.3 | 13 | 12y | |
| 31 | [Mn2L22O3]2+/oxalic acid/oxone | 1:30:28:30 |
1:12:150 |
0.5 | 0.2 | 12z |
| 32 | Cu(MeCN)4+/TBHP | 1:14:9:13 |
12aa | |||
| 33 | “Cu4”/TBHP | 1:34:23:21 |
1:16:130 |
0.4 | 0.1 | 12ab |
| 34 | Cu(H3L3)(NCS)/TBHP | 1:13:8:7 |
1:15:150 |
0.6 | 0.1 | 12ac |
| 35 | FeCl3/L4/m-CPBA | 1:29:30:27 |
1:21:211 |
0.25 | 3.0 | 12ad |
| 36 | TS-1/H2O2 | 1:80:193:100 |
No products | No products | No products | 12ae |
The oxidation of saturated hydrocarbons with TBHP catalyzed by complexes 1a and 2 proceeds more selectively in comparison with the oxidation using H2O2. Thus, parameters collected in entries 3 and 4 of Table 2 testify that the oxidation with TBHP involves the interaction of the alkane with the tert-butoxy radical tert-BuO˙ (comparison with other oxidations where this radical takes part is collected in entries 32–34 of Table 2). Remarkable peculiarity was found in the case of the oxidation of n-octane (Fig. S10, ESI‡). The regioselectivity parameter for the oxidation with TBHP catalyzed by 1a is similar to that found previously in the reaction with the systems containing a tetracopper(II) triethanolaminate complex [O⊂Cu4{N(CH2CH2O)3}4(BOH)4][BF4]2/TBHP12a,b and a dinuclear manganese complex [Mn2(R-LMe2R)2(μ-O)2]3+(PF6)3 (where LMe2R is 1-(2-hydroxypropyl)-4,7-dimethyl-1,4,7-triazacyclononane)/oxalic acid/TBHP12y containing a strongly hindered reaction center. These systems were believed to operate without the participation of free hydroxyl radicals. The regioselectivity in the oxidation of linear alkanes catalyzed by multicopper complexes resembles the selectivity observed for the case of cytochrome P450.1h,s,u
In the oxidation of methylcyclohexane (MCH) the position 2 relative to the methyl group of the substrate (isomeric products P6 and P7 in Fig. S11 and S12, ESI‡) is much less reactive than the corresponding positions 3 and 4, respectively (products P8 + P10 and P9 + P11), regarding the formation of alcohol products. This behaviour indicates a noticeable steric hindrance which is apparently due to the involvement of a reactive Cu-center surrounded by bulky substituents. Similar isomer distribution has been found by us previously for the oxidation of methylcyclohexane with the “Cu4”–TBHP system where “Cu4” is the tetracopper(II) triethanolaminate complex [O⊂Cu4{N(CH2CH2O)3}4(BOH)4][BF4]2.12a,b Mizuno and coworkers used the bulky divanadium-substituted phosphotungstate, [γ-H2PV2W10O40]3−, as a catalyst for the methylcyclohexane oxidation and obtained the following distribution of isomers (%): P5 (19), P6 + P7 (6), P8 + P10 (44), P9 + P11 (24).13a
The oxidation of cis- and trans-isomers of 1,2-dimethylcyclohexane (DMCH) with TBHP catalyzed by complexes 1a and 2 proceeds stereoselectively. Moreover, the inversion of configuration has been noticed in the case of trans-1,2-DMCH: the trans/cis ratios of 0.40 and 0.53 (Table 2, entries 3 and 4) have been measured for 1a and 2, respectively. Similarly, it has been found earlier that the oxidation of trans-1,2-DMCH by the “Cu4”–TBHP system proceeds with a substantial inversion of configuration, as attested by the respective trans/cis product molar ratio of 0.1.12ab In all cases (complexes 1a, 2 and “Cu4”) the oxygenation reaction of 1,2-DMCH occurs in a narrow cleft between ligand shells1h,13b due to bulky ligands which surround copper centers, thus resulting in the inversion of configuration.
2.4. Kinetic analysis of the cyclohexane (RH) oxidation with H2O2
In our kinetic analysis we will operate with the initial rate of the cyclohexyl hydroperoxide formation, W0 = (d[ROOH]/dt)0, which is equal to the initial rate of the oxygenate formation. This initial rate W0 was determined from the slope of a dotted straight line which is tangent to the kinetic curve (an example is presented by the dotted line 1a in Fig. 4).Assuming that the mode of the initial rate W0 dependence on the initial concentration of cyclohexane (Fig. 8A) reflects a concurrence between the alkane and acetonitrile for the oxidizing species OS generated in the H2O2 decomposition process, we can propose the following kinetic scheme which describes the rate of ROOH accumulation:
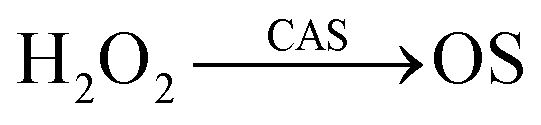 | (i) |
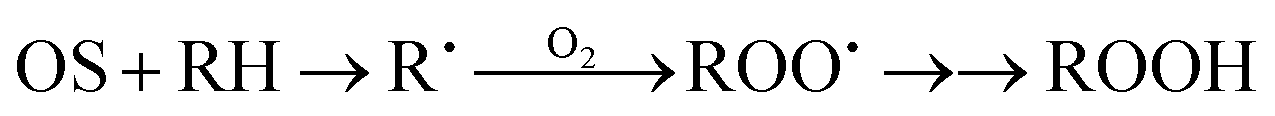 | (1) |
| OS + CH3CN → products | (2) |
Here (i) is a stage of generation of oxidizing species OS with the rate Wi defining the interaction of a catalytically active species CAS with H2O2; stage (1) is the sequence of transformations of RH into ROOH with the rate-limiting step in the interaction between alkane RH and OS (characterized by the rate constant k1); stage (2) is the rate-limiting step of the acetonitrile transformation into products during the interaction between OS and CH3CN (rate constant k2).
The analysis of the proposed kinetic scheme in a quasi-stationary approximation relative to OS allows us to obtain the following expression for the initial rate of ROOH accumulation:
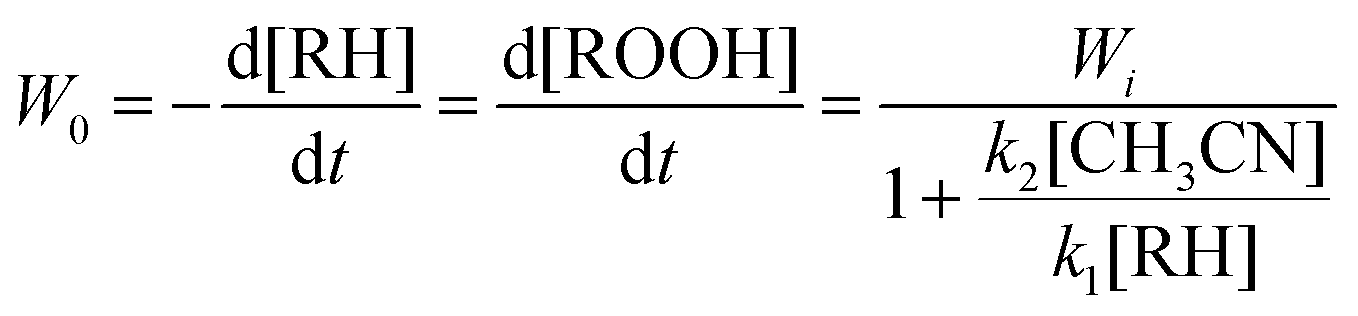 | (3) |
The experimental data demonstrated in Fig. 8A are in agreement with eqn (3). Indeed, there is a linear dependence of (d[ROOH]dt)−1 on 1/[RH]0 as shown in Fig. 8B. The analysis of this dependence led to the parameters k2[CH3CN]/k1 = 0.25 M and Wi = 1.6 × 10−5 M s−1 for the conditions described in the legend to Fig. 8. In addition, the data on regio- and bond-selectivity of alkane oxidation with H2O2 (see above, Section 2.3, Table 2) indicate that the oxidizing species OS in the system under investigation is the hydroxyl radical. However, it should be noted that the values of parameters k2[CH3CN]/k1 = 0.25 M and k2/k1 = 0.015 measured using the kinetic data presented in Fig. 8 are slightly larger than the values expected for free hydroxyl radicals. Indeed, these values for the catalytic systems which generate hydroxyl radicals have been reported to vary in the intervals k2[CH3CN]/k1 = 0.10–0.20 M and k2/k1 = 0.006–0.012 (Table 3). The enhanced value of the k2[CH3CN]/k1 parameter in the case of catalysis with complex 1a indicates that the interaction of the hydroxyl radical with acetonitrile is more efficient than with cyclohexane. In all cases the value k2[CH3CN]/k1 was measured on the basis of analysis of dependence W0 on [RH]0. Here the volume concentrations of CH3CN and cyclohexane have been assumed to be constant in all volumes of the reaction solution. The enhanced efficiency of the reaction of the hydroxyl radical with acetonitrile can be explained if we assume that the oxidizing species (hydroxyl radicals) are generated in the interaction of H2O2 with copper ions in close proximity between Cu and H2O2. Copper ions in compound 1a (at least in the initial period of the oxidation reaction when the globule is not disintegrated) are surrounded with bulky ligands which obstruct the approach of the reactants to the reaction centers from the main bulk of the solvent. As nitric acid in low concentration is a necessary component of the reaction mixture we suppose that the acid promotes (in the initial period partial) decoordination of some ligands around copper ions and, as a result, easing the approach of the components of the reaction mixture to the metal center.1h,s,u,13b One can compare the role of acid in this experiment with the role of oyster knife which opens the valves of the mollusk shell before eating. Small and hydrophilic acetonitrile molecules more easily penetrate into the catalyst shell than voluminous hydrophobic cyclohexane molecules. It means that the local concentration of acetonitrile inside the shell will be higher than its concentration in the bulk of the solution. In the frames of this model we can conclude that the enhanced k2[CH3CN]/k1 value is obtained as a result.
| Entry | System | k 2[CH3CN]/k1 (M) | k 2/k1 | Ref. |
|---|---|---|---|---|
| a The concentration [CH3CN]0 was assumed to be 17 M. Abbreviations. PCA is pyrazine-2-carboxylic acid. Cu4 is tetracopper(II) triethanolaminate complex [O⊂Cu4{N(CH2CH2O)3}4(BOH)4][BF4]2. Complex [Co4Fe2OSae8]·4DMF·H2O, where H2Sae = salicylidene-2-ethanolamine. Cp2*Os is decamethylosmocene. Cp2Fe is ferrocene. Fe2(TACN) is an iron(III) complex with 1,4,7-triazacyclononane. | ||||
| 1 | H2O2/O2/1a/HNO3 | 0.25 | 0.015 | This work |
| 4 | H2O2/O2/(n-Bu4N)VO3/PCA | 0.14 | 0.008 | 12a |
| 5 | H2O2/O2/“Cu4”/CF3COOH | 0.20 | 0.012 | 12t |
| 6 | H2O2/O2/“Cu4”/HCl | 0.10 | 0.006 | 12t |
| 7 | H2O2/O2/[Co4Fe2OSae8]/HNO3 | 0.14 | 0.008 | 12s |
| 8 | H2O2/O2/Cp2*Os/py | 0.09–0.19 | 0.0055–0.011 | 12l |
| 10 | H2O2/O2/Cp2Fe/Py/PCA | 0.19 | 0.011 | 12h |
| 11 | H2O2/O2/“Fe2(TACN)”/PCA | 0.19 | 0.011 | 12f |
2.5. Oxidation of cyclohexane with H216O2 under an 18O2 atmosphere
The experiments with isotopically labeled 18O2 are a useful mechanistic probe to test the involvement and the mode of involvement of molecular oxygen in the radical reactions. It has previously been shown14a that the cyclodecane oxidation under Gif conditions (FeIII/pyridine/CH3COOH/H2O2) in an 18O2 atmosphere results in a ca. 50% degree of 18O incorporation in the cyclodecanone where the yield of ketone was ca. 15%. However, the yield of the corresponding alcohol (as well as some important reaction conditions) was not reported. The results of that work clearly pointed to an involvement (reduction) of air oxygen in open-air reactions and they inspired us to investigate the process of such a type in more detail using the catalytic system based on copper complex 1a.The accumulation of labeled oxygenated products with time was studied for conditions [1a]0 = 4.1 × 10−4 M and 60 °C under the atmosphere of 18O2. The yields and isotopic abundances were measured after reduction of the reaction samples with PPh3. The highest degree (50%) of 18O incorporation into the cyclohexanol was observed at the beginning of the reaction (Fig. 9A). The percentage of labeled alcohol decreases with reaction time reaching 28% of Cy–18OH after 5 h. This effect can be explained by a weak catalase activity of the catalytic system which produces unlabeled oxygen 16O2 from the hydrogen peroxide H216O2. One may expect the incorporation of the 18O isotope into the formed triphenylphosphine oxide (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3) via the following reaction scheme where the alkyl hydroperoxide is reduced to alcohol by phosphine:
PPh3) via the following reaction scheme where the alkyl hydroperoxide is reduced to alcohol by phosphine:
| Cy–18O18OH + PPh3 → Cy–18OH + 18OPPh3 |
Simultaneously the reduction of remaining non-labeled hydrogen peroxide gives 16OPPh3:
| H16O16OH + PPh3 → H16OH + 16OPPh3 |
The catalytic system based on complex 1a demonstrates nearly linear growth of the 18O incorporation degree into OPPh3, and the fitted line does not pass through the zero point.
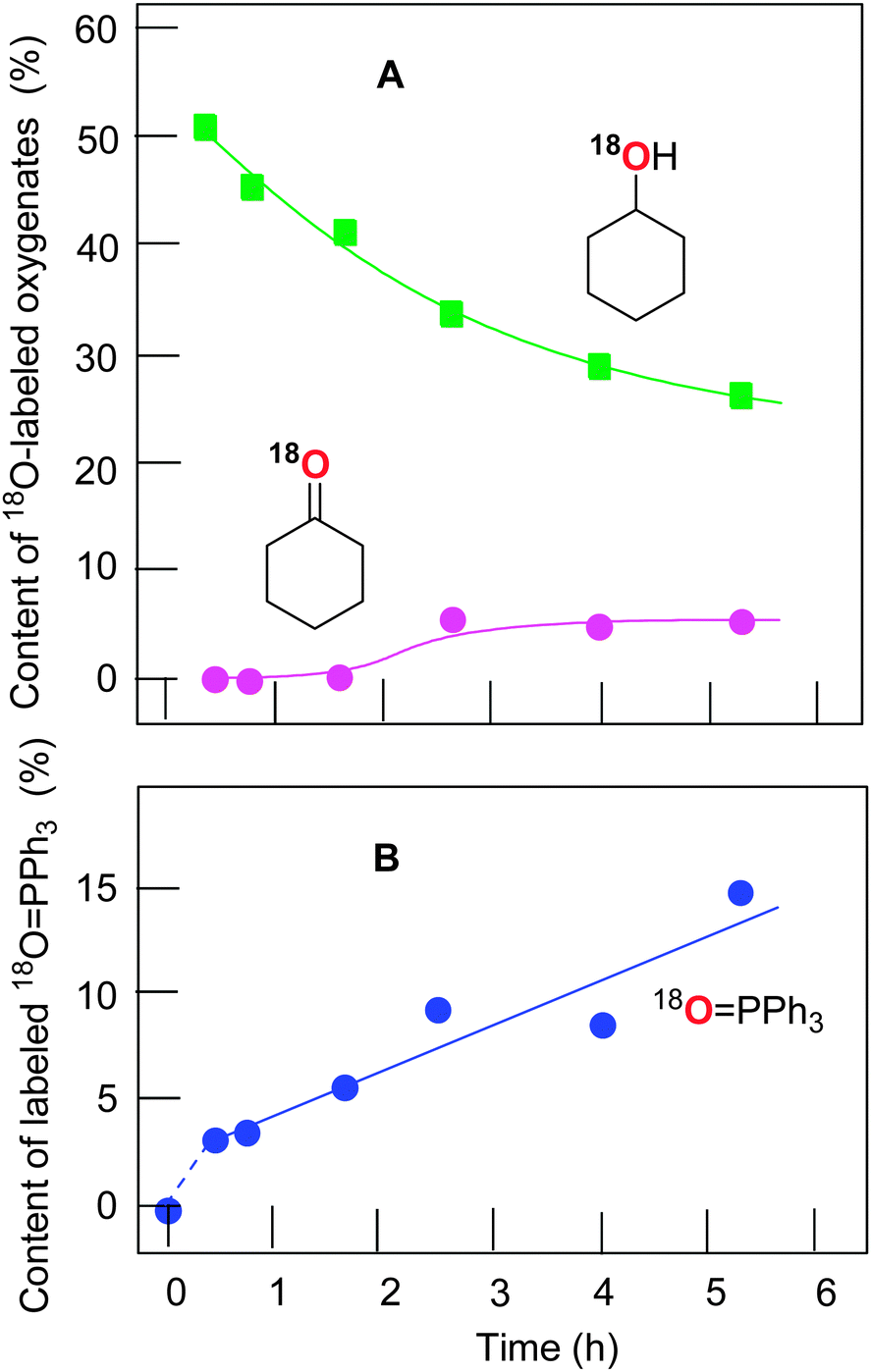 | ||
| Fig. 9 Incorporation of the labeled oxygen into the cyclohexanol and cyclohexanone (Graph A) and triphenylphosphine oxide (Graph B; circles are experimental data, the solid line is an exponential fit) in the course of the cyclohexane oxidations with subsequent reduction of the reaction sample with PPh3. Conditions: [1a]0 = 4.1 × 10−5 M; [HNO3]0 = 0.4 M; [H2O2]0 = 1 M; [H2O]total = 2.6 M; [cyclohexane]0 = 0.46 M; 60 °C; 18O2, 1 bar. The yields and isotopic abundances were measured after reduction of the reaction samples with PPh3. | ||
The dependence of the 18O incorporation into the cyclohexanone differs from that obtained for the alcohol (compare Graphs A and B in Fig. 10). The maximum concentration of the 18O-labeled ketone does not exceed 1 × 10−4 M (yield is 0.02% based on cyclohexane). We can clearly see in Fig. 10 (Graph B) that the yield of cyclohexanone–16O is much higher than that of labeled cyclohexanone. It is necessary to emphasize that the ketone yield after reduction with PPh3 is equal to the real yield of this compound in the reaction. Unlabeled ketone cannot be formed from the unlabeled peroxide Cy–16O–16OH. Indeed, the labeled hydroperoxide Cy–18O–18OH is present in the reaction solution and its decomposition would lead to the formation of cyclohexanone–18O in addition to cyclohexanol–18O (Fig. 11). Thus, some (small) amount of cyclohexanone (which is really present in the reaction mixture) does not contain 18O and we can conclude that this amount is formed not from CyOOH. It is reasonable to assume that this unlabeled cyclohexanone is produced in an alternative pathway which apparently does not involve hydroxyl radicals and ROOH. Molecular oxygen 16O2 from the atmosphere is not incorporated into the ketone in this route.
 | ||
| Fig. 10 Kinetic curves of accumulation with time of cyclohexanol (Graph A) and cyclohexanone (Graph B) containing both partially labeled (the sum 16O + 18O) and completely 18O-labeled oxygenates. The yields and isotopic abundances were measured after reduction of the reaction samples with PPh3. | ||
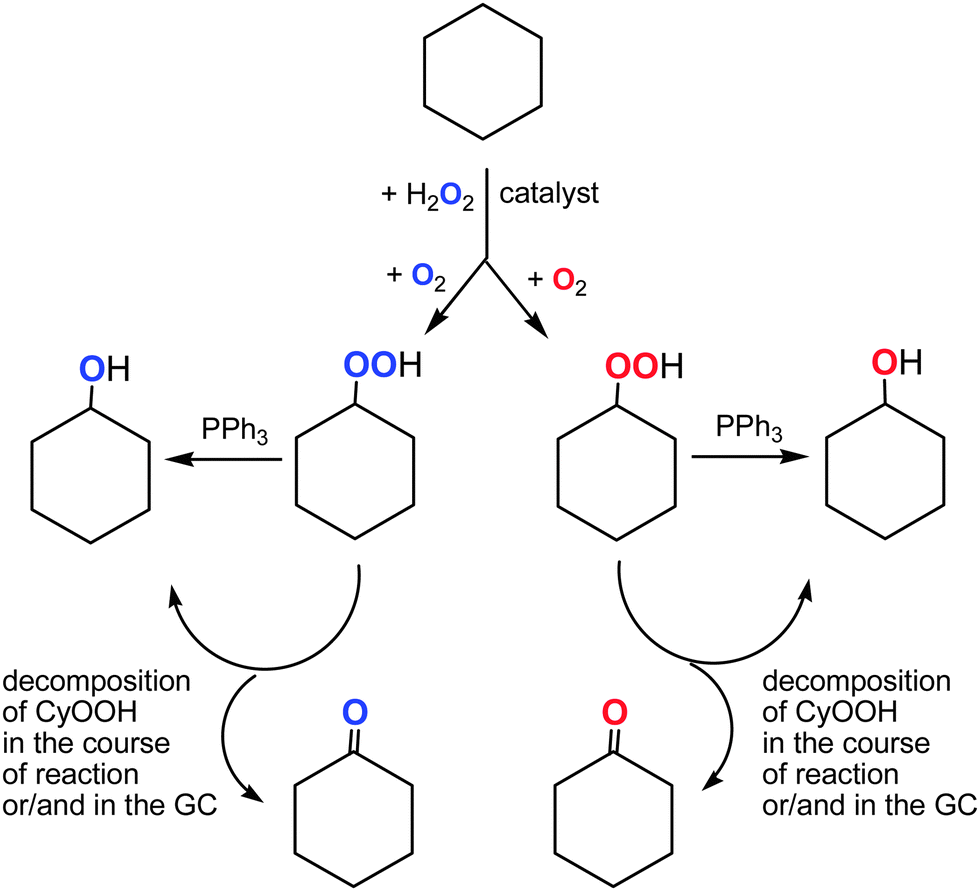 | ||
| Fig. 11 Transformations of cyclohexane in the presence of labeled dioxygen 18O2. | ||
These results are in conformity with our recent data obtained14b in the oxidation of cyclohexane with hydrogen peroxide in the presence of an osmium complex under the atmosphere of 18O2. In that case, a relatively high 18O incorporation degree into the cyclohexanone was observed after 2 h reaction time (50% of 18O, maximum concentration of labeled cyclohexanone was 5 × 10−4 M), with the subsequent decay until 2% level at 4 h reaction time due to production of unlabeled cyclohexanone–16O (see Fig. 10A in ref. 14b). In contrast, the reaction catalyzed by complex 1a shows five times smaller amount of labeled cyclohexanone (1 × 10−4 M) under similar reaction conditions. Increased amounts of labeled cyclohexanone–18O in the osmium-catalyzed process14b can be explained by operating some minor mechanisms that produce negligible amounts of labeled cyclohexanone–18O at the beginning of the reaction.
With the cyclohexyl hydroperoxide (cyclo-C6H11OOH) as the main reaction product one may expect the formation of isomeric cyclohexane dihydroperoxides, cyclo-C6H10(OOH)2, as the main over-oxidation products which produce 1,2-, 1,3-, and 1,4-cyclohexanediols upon reduction with PPh3. However, the analysis of the chromatographic patterns (Fig. 12) revealed a large number of byproducts, apart from expected cyclohexanediols and hydroxycyclohexanones (Fig. 12). The total yield of by-products does not exceed 5% based on the cyclohexane.

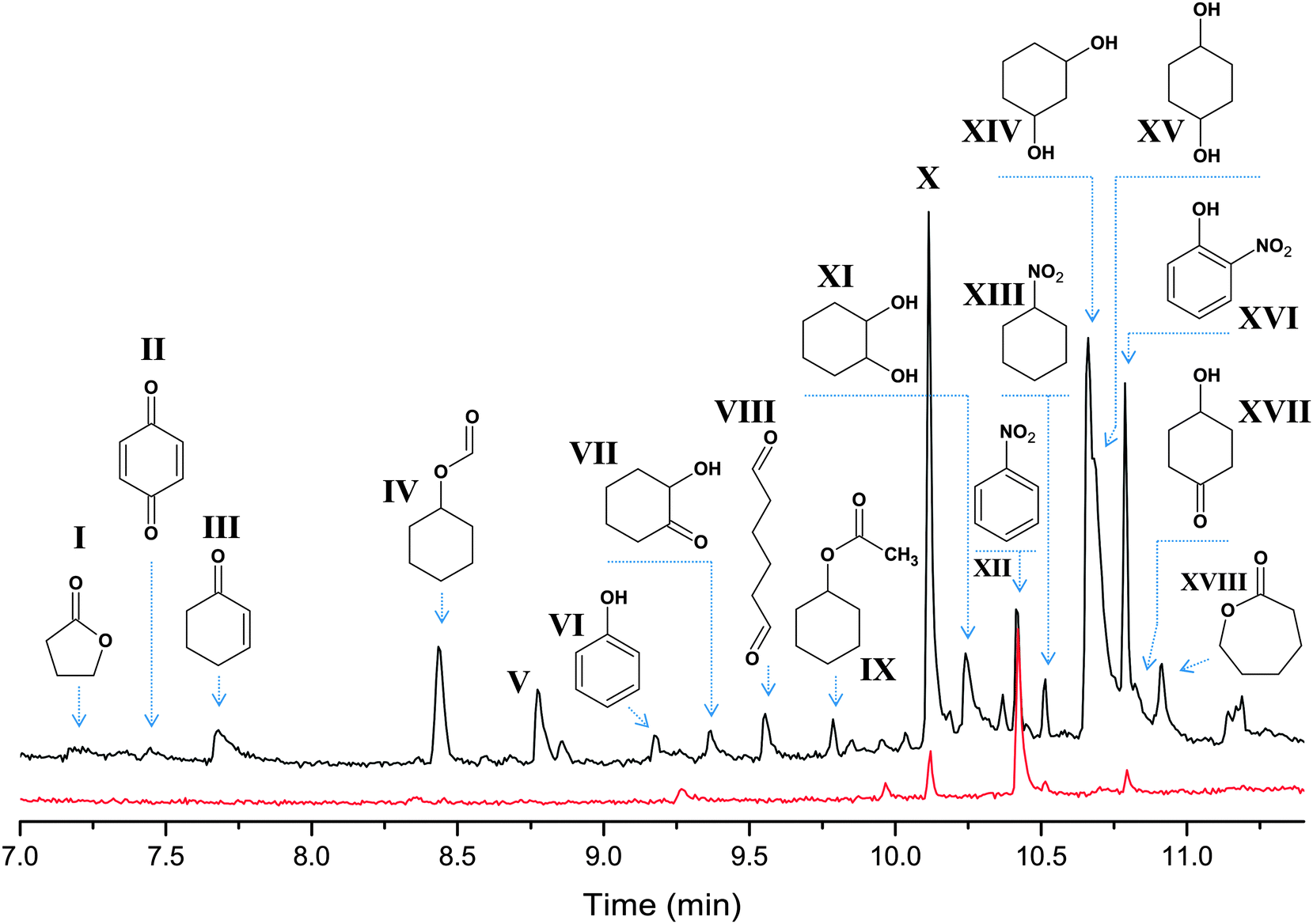 | ||
| Fig. 12 The oxidation of cyclohexane by the 1a–HNO3–H2O2 system in the 18O2 atmosphere. Chromatograms of the reaction samples taken after 26 min (bottom, red line) and 240 min (top, black line) time intervals and reduced with PPh3 show the accumulation of over-oxidation products. The peaks of main products (cyclohexanol and cyclohexanone) appear at 5.8 and 6.1 min, respectively, and are omitted for clarity. The full mass-spectra of the selected products are presented in the ESI.‡ | ||
Taking into account our observation that the major oxidation mechanism results in ca. 28% of 18O incorporation (at the end of the reaction) one may expect the following distribution of labeled diols: 52:40:8% for 16O–16O, 16O–18O and 18O–18O combinations, respectively. The peaks of 1,2- and 1,4-cyclohexanediols (XI and XV, respectively) became discernible after a reaction time of 2.5 h. The analysis of molecular ion peaks of the respective mass spectra (Fig. S13, ESI‡) revealed a constant level of non-labeled and single labeled diol (55 and 45%, respectively), with no any peaks at 120 m/z, attributable to doubly labeled 1,2-diol XI. The peaks of 1,4-cyclohexanediol XV are overlapped with those of 1,3-diol (XIV), and careful analysis allowed us to only evaluate the mass spectrum of XV from the chromatogram taken at 310 min reaction time (Fig. S13, ESI‡). The 62:32:6 ratio was found for 16O–16O, 16O–18O and 18O–18O combinations, respectively.
Furthermore, the comparison of the mass spectra of 1,3-diol (XIV), which has a very weak molecular ion peak (Fig. S13, ESI‡), with the reference spectra from the NIST database14c definitely shows the presence of 18O labeling. A strong peak at 98 m/z (Fig. S13, ESI‡), which can be attributed to the [M–H2O]+ ion, shows 29% of 18O incorporation (98 → 100 m/z shift) in the respective ion (samples taken at 4 and 5.2 h). Although the 98:100 m/z intensity ratio shows the 18O labeling of only one hydroxyl group and does not allow us to evaluate double labeled species, it should be dependent on the overall percentage of 18O in the cyclohexanediol molecule. The mass spectra of 1,2-diols (XI) demonstrate 20 to 32% of 18O incorporation into the [M–H2O]+ ion (98 → 100 m/z), and the mass spectrum of 1,4-diol (XV) exhibits 29% of 18O in the [M–H2O]+ ion. These values are comparable with that found for 1,3-diol (XIV) and, therefore, one can conclude that the amount of doubly labeled 1,3-diols (if they are formed at all) should be low (less than 10%), with the amount of single labeled species (16O–18O) comparable with that for 1,2- and 1,4-cyclohexanediols. These results are in agreement with the expected 18O incorporation level into cyclohexanediols.
The study of distribution of 18O in hydroxycyclohexanones is complicated by their low amounts and interfering of the respective peaks with those of other products. Nevertheless, we were able to detect the isotopic composition of 2- and 4-hydroxycyclohexanones (VII and XVII, respectively) at 240 and 310 min and 3-hydroxycyclohexanone (not shown in Fig. S13, ESI‡) at 310 min. Comparing the intensities of 114, 116 and 118 m/z peaks, one can see that doubly labeled species are almost absent, while the incorporation of 18O into 3- and 4-hydroxycyclohexanones is at the 33% level. Surprisingly, 1,2-hydroxycyclohexanone showed a much lower level of 18O incorporation, 14% (samples taken at 240 and 310 min) and ca. 12% (at 160 min). One can assume two general ways (processes) towards the hydroxycyclohexanones, particularly 2-isomer VII, depicted in Fig. 13.
Process 1 means the radical mechanism through attack of the HO˙ with the subsequent reaction with O2, while process 2 introduces a ketone group in a way similar to that of cyclohexanone formation (Fig. 13). From the fact that [Cy–18OH] reaches a plateau at 2 h reaction time, while [Cy–16OH] continues to increase until 240 min (Fig. 10), one can suppose that after 2 h the primary radical mechanism (process 1) does not lead to 18O incorporation due to depletion of the 18O2 amount. Hence, reactions a, b and c should lead to pure 16O products. The peaks for hydroxycyclohexanones appear after 160 min of reaction time (concentration of cyclohexanone of ca. 3 mM) and, from above considerations, radical process 1 cannot bring the labeled oxygen into hydroxycyclohexanones.
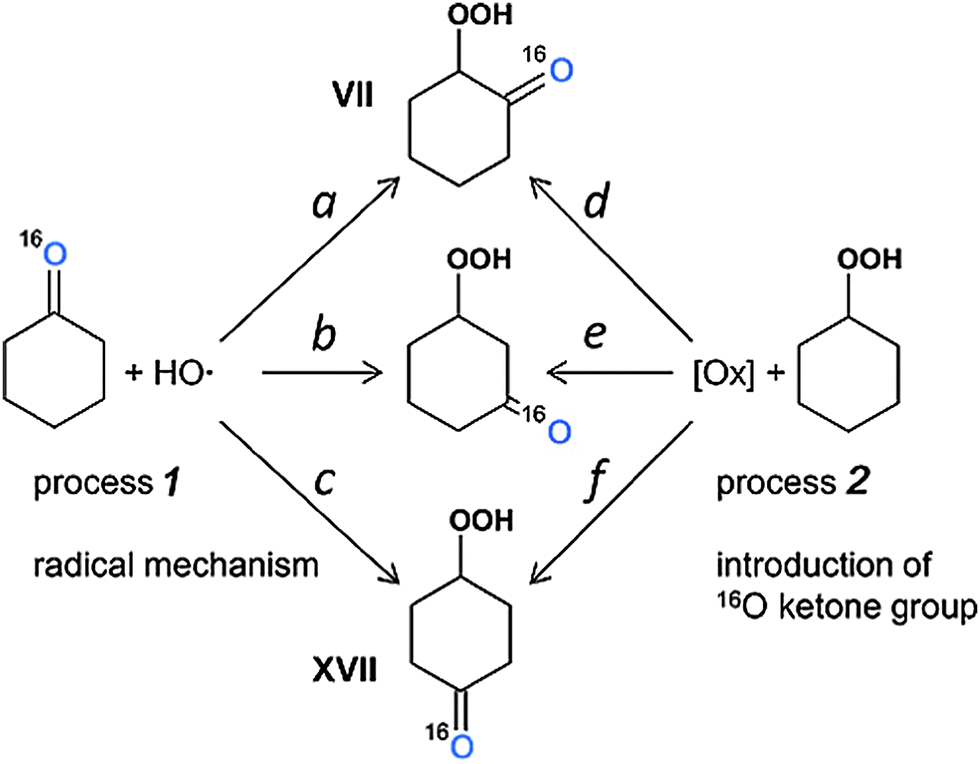 | ||
| Fig. 13 Two main processes (1 and 2) which could lead to different isomers of hydroxycyclohexanones (shown as the respective hydroperoxides; pathways a–f). [Ox] means oxidation to form a ketone in a way similar to oxidation of cyclohexane to cyclohexanone, which does not involve O2 participation. Route d is suggested to be unfavourable, compared to other pathways. | ||
Process 2 is assumed to proceed through the same mechanism, as for the cyclohexanone formation, giving pure 16O ketone (Fig. 9 and 10). Since process 2 starts from the mixture of unlabeled and labeled Cy–OOH, the 16O:18O ratio of Cy–OOH is maintained in the respective hydroxycyclohexanones. Therefore, the ratios of the reaction rates a/d, b/e and c/f define the 18O incorporation level. From this point of view, the only way to get a reduced amount of the 18O labeled 2-hydroxycyclohexanone (VII) is the reduced reaction rate of process d, compared to e and f. One may explain this effect due to the presence of the bulky –OOH group, which either sterically hinders the 2-positions of the C6 ring, or reacts with attacking species, preventing formation of product VII.
A number of by-products, such as I–IV, are commonly observed in the oxidation of cyclohexane via the attack of the hydroxyl radical. The 18O amounts in I–IV vary from 10 to 30%, according to the mass-spectra (see Fig. S13, ESI‡). The mass spectra of hexanedial (VIII) and cyclohexyl acetate (IV) suggest that these products are 18O free. The peak appearing at 8.7 min (V) was not recognized, although the respective mass spectrum (Fig. S13, ESI‡) suggests the molecular weight of 114 (as for hydroxycyclohexanones) and the presence of single and doubly labeled species with 55:40:5 for 16O–16O, 16O–18O and 18O–18O combinations, respectively.
All the chromatograms reveal noticeable peaks of nitrobenzene, nitrocyclohexane and o-nitrophenol (XII, XIII and XVI, respectively), all containing only 16O. Speculatively, the formation of these products could be due to reactions involving a nitric acid co-catalyst, present in a large concentration (0.4 M). Notably, the peaks of these byproducts are clearly seen at the chromatogram taken at 26 min reaction time, and their intensity increases with time (pointing that these are not just admixtures in the starting reagents).
The most intensive peak at 10.1 min (X) reveals a mass spectrum, which could not be assigned to any compound from the NIST database. The spectral pattern suggests the presence of a C3H7+ fragment (group of peaks in the 27–43 m/z range).14d The respective mass spectrum observed in the oxidation of C6D12 (Fig. S14, ESI‡) suggests that compound X contains at least 11 H atoms (83 → 94 m/z shift) and the weak peak at 98 m/z, showing +10 shift (98 → 108 m/z), does not represent a molecular ion of X. Also, it is clear that X has an 18O labeled part (+2 m/z peaks at 59 and 100 m/z) and, therefore, contains at least one O atom. The shape of the peak points to the absence of a carboxylic group or more than one hydroxyl group, which would result in an elongated tail of the peak. In spite of revealing different peak intensity ratios, the mass spectrum taken at low ionization energy (10 eV) does not show peaks with an m/z higher than 98 (Fig. S14, ESI‡). From the above considerations and from the comparison with the mass spectra of known compounds, we assume that X has CnH12O composition (n = 6 or 7), probably containing an alkene fragment.
3. Conclusions
In the current study, we have found that local-structure parameters around copper atoms in complexes 1a, 1b and 2 revealed by Cu K-edge extended X-ray absorption fine structure (EXAFS) are fully consistent with those established by X-ray crystallographic studies.Complex 1a is a good pre-catalyst for the alkane hydroperoxidation with hydrogen peroxide in air in an acetonitrile solution in the presence of nitric acid. The kinetic analysis as well as selectivity parameters measured in the oxidation of linear and branched alkanes indicated that the oxidizing species in the reaction is the hydroxyl radical. The oxidations of saturated hydrocarbons with tert-butyl hydroperoxide catalyzed by complexes 1a and 2 exhibited unusual selectivity parameters which are apparently due to the steric hindrance created by bulky siloxane ligands surrounding reactive copper centers. The regioselectivity in the oxidation of linear alkanes catalyzed by multicopper complexes resembles the selectivity observed for the case of cytochrome P450. The oxidation of trans-1,2-dimethylcyclohexane with tert-butyl hydroperoxide catalyzed by complexes 1a and 2 proceeds stereoselectively with the inversion of configuration. We have observed very different reactivities of our complexes and simple copper salts relative to different hydrocarbons. In cases of some hydrocarbons the complexes are effective catalysts whereas simple salts are almost inactive. In other cases the reactivity of the complexes and simple salts can be comparable. Such a selectivity will be a subject of our further studies.
The oxidation of cyclohexane with H216O2, catalyzed by complex 1a, in an atmosphere of 18O2 gave cyclohexyl hydroperoxide, CyOOH, containing 50% of 18O at the beginning of the reaction and 30% after 5 h reaction time. All the cyclohexanone formed was found to be 100% 16O. We assume that unlabeled cyclohexanone is formed not from CyOOH but is produced in an alternative pathway which apparently does not involve hydroxyl radicals and ROOH. These observations were confirmed by studying the incorporation of 18O into the main by-products (cyclohexanediols and hydroxycyclohexanones), where hydroxycyclohexanones were suggested to not contain doubly 18O labeled species. Furthermore, the incorporation degree of 18O into the hydroxycyclohexanones was found to be dependent on their isomer structure: while the 3- and 4-isomers reveal expected ca. 30% of 18O, the 2-isomer shows twice lower amount of 15% only. This could point to the presence of steric effect during the formation of hydroxycyclohexanones, which could be formed, in part, through the process which does not involve hydroxyl radicals. This assumption is in accordance with the absence of 18O-labeled cyclohexanone (main product) in the catalytic system.
4. Experimental
4.1. EXAFS study
Cu K-edge EXAFS spectra were measured at the Structural Materials Science beamline of the Kurchatov Synchrotron Radiation Source (National Research Center “Kurchatov Institute”, Moscow). The spectra were measured in the transmission mode using two ionization chambers filled with appropriate N2–Ar gas mixtures. The energy scale was calibrated against a Cu foil spectrum (E0 = 8979 eV). Data reduction and analysis was performed using the IFEFFIT software suite.154.2. Catalytic alkane oxidation
Hydrogen peroxide and TBHP were used as 50% and 70% solutions in H2O, respectively. The reactions of alkanes were typically carried out in air in thermostated Pyrex cylindrical vessels with vigorous stirring and using MeCN as the solvent (the total volume of the reaction solution was typically 5 mL). Typically, precatalyst 1a or 2 and the cocatalyst (nitric acid) were introduced into the reaction mixture in the form of stock solutions in acetonitrile. The substrate was then added and the reaction started when hydrogen peroxide was introduced in one portion. (CAUTION: the combination of air or molecular oxygen and H2O2 with organic compounds at elevated temperatures may be explosive!). The reactions were stopped by cooling and after addition of nitromethane as a standard compound analyzed by GC (instrument ‘HP 5890 – Serie-II’; fused silica capillary columns column Hewlett-Packard; the stationary phase was polyethyleneglycol: INNOWAX with parameters 25 m × 0.2 mm × 0.4 μm; carrier gas was helium with a column pressure of 15 psi). Attribution of peaks was made by comparison with chromatograms of authentic samples. The quantification of alkyl hydroperoxides and ketones (aldehydes) and alcohols present in the reaction solution was performed using a simple GC method developed previously by Shul'pin,10 based on comparison of chromatograms of the reaction samples before and after reduction with triphenylphosphine.4.3. Experiments with 18O2
A Perkin-Elmer Clarus 600 gas chromatograph, equipped with two capillary columns (SGE BPX5; 30 m × 0.32 mm × 25 μm), one having EI-MS (electron impact) and the other one with FID detectors, was used for analyses of the reaction mixtures. Helium was used as the carrier gas. All EI mass spectra were recorded with 70 eV energy, unless stated otherwise.Labeled dioxygen (99% of 18O) was purchased from CortecNet. Freshly prepared catalytic reaction mixtures were frozen with liquid nitrogen, pumped and filled with N2 a few times in order to remove air. Then mixtures were pumped again, vacuum pump turned off, Schlenk flasks with vacuum inside were heated up to 20 °C and immediately filled with 18O2 gas using a syringe through a septa. The mixtures were then heated up to 60 °C with a possibility of gas flow to compensate excessive pressure. The 16O and 18O compositions of the oxygenated products were determined by the relative abundances of mass peaks at m/z = 57/59 (for cyclohexanol) and 98/100 (for cyclohexanone), unless stated otherwise.
Acknowledgements
The authors thank the Russian Foundation for Basic Research (grants 12-03-00084-a, 14-03-00713, 14-03-31772 and 14-03-31970 mol_a), the FCT (projects PTDC/QUI-QUI/119561/2010, PTDC/QUI-QUI/121526/2010 and PEst-OE/QUI/UI0100/2013; fellowship SFRH/BPD/42000/2007) (Portugal) and the “Science without Borders Program, Brazil–Russia”, CAPES (grant A017-2013) for support. M.M.V., L.S.S. and G.B.S. express their gratitude to the FCT and Group V of Centro de Química Estrutural for making it possible for them to stay at the Instituto Superior Técnico, University of Lisbon, as invited scientists and to perform a part of the present work (all the funding for the invited scientist fellowship comes entirely from this Group).References
- (a) R. H. Crabtree, Chem. Rev., 1985, 85, 245–269 CrossRef CAS; (b) A. E. Shilov and G. B. Shul'pin, Usp. Khim., 1990, 59, 1468–1491 CrossRef CAS PubMed; A. E. Shilov and G. B. Shul'pin, Russ. Chem. Rev., 1990, 59, 853–867 Search PubMed; (c) in Catalytic Oxidations with Hydrogen Peroxide as Oxidant, ed. G. Strukul, Kluwer Academic: Dordrecht, The Netherlands, 1992 Search PubMed; (d) A. Sen, Acc. Chem. Res., 1998, 31, 550–551 CrossRef CAS; (e) G. B. Maravin, M. V. Avdeev and E. I. Bagrii, Neftekhimiya, 2000, 40, 3–21 CAS; (f) V. V. Vasil'eva, A. I. Nekhaev, I. Y. Shchapin and E. I. Bagrii, Kinet. Catal., 2006, 47, 610–623 CrossRef; (g) A. A. Shteinman, Usp. Khim., 2008, 77, 1013–1035 CrossRef PubMed; (h) G. B. Shul'pin, Org. Biomol. Chem., 2010, 8, 4217–4228 RSC; (i) E. G. Chepaikin, Russ. Chem. Rev., 2011, 80, 363–396 CrossRef CAS PubMed; (j) K. Schröder, K. Junge, B. Bitterlich and M. Beller, Top. Organomet. Chem., 2011, 33, 83–109 CrossRef; (k) B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885–898 CrossRef CAS PubMed; (l) B. M. Prince and T. R. Cundari, Organometallics, 2012, 31, 1042–1048 CrossRef CAS; (m) A. Sivaramakrishna, P. Suman, E. V. Goud, S. Janardan, C. Sravani, T. Sandep, K. Vijayakrishna and H. S. Clayton, J. Coord. Chem., 2013, 66, 2091–2109 CrossRef CAS; (n) A. M. Kirillov and G. B. Shul'pin, Coord. Chem. Rev., 2013, 257, 732–754 CrossRef CAS PubMed; (o) D. Munz, D. Meyer and T. Strassner, Organometallics, 2013, 32, 3469–3480 CrossRef CAS; (p) O. A. Mironov, S. M. Bischof, M. M. Konnick, B. G. Hashiguchi, W. A. Goddard, M. Ahlquist and R. A. Periana, J. Am. Chem. Soc., 2013, 135, 14644–14658 CrossRef CAS PubMed; (q) A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed; (r) A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710–15713 CrossRef CAS PubMed; (s) G. B. Shul'pin, Dalton Trans., 2013, 42, 12794–12818 RSC; (t) D. Munz and T. Strassner, Angew. Chem., Int. Ed., 2014, 53, 2485–2488 CrossRef CAS PubMed; (u) G. B. Shul'pin, Selectivity in C–H functionalizations, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk, K. Poeppelmeier and L. Casella, Elsevier, 2nd edn, 2013, ch. 6.04, vol. 6, pp. 79–104 Search PubMed.
- Recent reviews: (a) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed; (b) T. Punniyamurthy and L. Rout, Coord. Chem. Rev., 2008, 252, 134–154 CrossRef CAS PubMed; (c) in Copper-Oxygen Chemistry, ed. K. Karlin and S. Itoh, J. Wiley & Sons, Inc., 2011 Search PubMed; (d) A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, Adv. Inorg. Chem., 2013, 65, 1–31 CrossRef CAS; (e) A. M. Kirillov, M. V. Kirillova and A. J. L. Pombeiro, Coord. Chem. Rev., 2012, 256, 2741–2759 CrossRef CAS PubMed; (f) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- Recent selected original publications: (a) J. Le Bras and J. Muzart, J. Mol. Catal. A: Chem., 2002, 185, 113–117 CrossRef CAS; (b) S. Velusamy and T. Punniyamurthy, Tetrahedron Lett., 2003, 44, 8955–8957 CrossRef CAS PubMed; (c) M. Zhu, X. Wei, B. Li and Y. Yuan, Tetrahedron Lett., 2007, 48, 9108–9111 CrossRef CAS PubMed; (d) L. S. Shul'pina, K. Takaki, T. V. Strelkova and G. B. Shul'pin, Pet. Chem., 2008, 48, 219–222 CrossRef; (e) C. Di Nicola, F. Garau, Y. Y. Karabach, L. M. D. R. S. Martins, M. Monari, L. Pandolfo, C. Pettinari and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2009, 666–676 CrossRef CAS; (f) E. G. Chepaikin, A. P. Bezruchenko, G. N. Menchikova, N. I. Moiseeva and A. E. Gekhman, Kinet. Catal., 2010, 51, 666–671 CrossRef CAS; (g) P. Roy and M. Manassero, Dalton Trans., 2010, 39, 1539–1545 RSC; (h) C. Wang, Y. Zhang, B. Yuan and J. Zhao, J. Mol. Catal. A: Chem., 2010, 333, 173–179 CrossRef CAS PubMed; (i) L. R. Martins, E. T. Souza, T. L. B. de Souza, S. Rachinski, C. B. Pinheiro, R. B. Faria, A. Casellato, S. P. Machado, A. S. Mangrich and M. Scarpellini, J. Braz. Chem. Soc., 2010, 21, 1218–1229 CrossRef CAS PubMed; (j) M. N. Kopylovich, K. T. Mahmudov, M. F. C. G. da Silva, P. J. Figiel, Y. Y. Karabach, M. L. Kuznetsov, K. V. Luzyanin and J. L. Pombeiro, Inorg. Chem., 2011, 50, 918–931 CrossRef CAS PubMed; (k) A. Rahman, S. M. Al Zahrani and A. A. Nait Ajjou, Chin. Chem. Lett., 2011, 22, 691–693 CrossRef CAS PubMed; (l) R. R. Fernandes, J. Lasri, M. F. C. G. da Silva, J. A. L. da Silva, J. J. R. Fraústo da Silva and J. L. Pombeiro, Appl. Catal., A, 2011, 402, 110–120 CrossRef CAS PubMed; (m) M. N. Kopylovich, A. C. C. Nunes, K. T. Mahmudov, M. Haukka, T. C. O. Mac Leod, L. M. D. R. S. Martins, M. Kuznetsov and J. L. Pombeiro, Dalton Trans., 2011, 40, 2822–2836 RSC; (n) S. Goberna-Ferrón, V. Lillo and J. R. Galan-Mascarós, Catal. Commun., 2012, 23, 30–33 CrossRef PubMed; (o) A. L. Maksimov, Y. S. Kardasheva, V. V. Predeina, M. V. Kluev, D. N. Ramazanov, M. Y. Talanova and E. A. Karakhanov, Pet. Chem., 2012, 52, 318–326 CrossRef CAS; (p) P. Nagababu, S. Maji, M. P. Kumar, P. P.-Y. Chen, S. S.-F. Yu and S. I. Chan, Adv. Synth. Catal., 2012, 354, 3275–3282 CrossRef CAS; (q) M. N. Kopylovich, M. J. Gajewska, K. T. Mahmudov, M. V. Kirillova, P. J. Figiel, M. F. C. G. da Silva, B. Gil-Hernández, J. Sanchiz and J. L. Pombeiro, New J. Chem., 2012, 36, 1646–1654 RSC; (r) S. Biswas, A. Dutta, M. Debnath, M. Dolai, K. K. Das and M. Ali, Dalton Trans., 2013, 42, 13210–13219 RSC; (s) H. Hosseini-Monfared, N. Asghari-Lalami, A. Pazio, K. Wozniak and C. Janiak, Inorg. Chim. Acta, 2013, 406, 241–250 CrossRef CAS PubMed; (t) M. Nandi and P. Roy, Indian J. Chem., 2013, 52B, 1263–1268 Search PubMed; (u) H. Hosseini-Monfared, S. Alavi and M. Siczek, Chin. J. Catal., 2013, 34, 1456–1461 CrossRef CAS; (v) O. Perraud, A. B. Sorokin, J. P. Dutasta and A. Martinez, Chem. Commun., 2013, 49, 1288–1290 RSC; (w) P. Nagababu, S. S.-F. Yu, S. Maji, R. Ramu and S. I. Chan, Catal.: Sci. Technol., 2014, 4, 930–935 RSC.
- (a) R. N. Austin and J. T. Groves, Metallomics, 2011, 3, 775–787 RSC; (b) E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef CAS PubMed.
- (a) R. L. Lieberman, K. C. Kondapalli, D. B. Shrestha, A. S. Hakamian, S. M. Smith, J. Telser, J. Kuzelka, R. Gupta, A. S. Borovik, S. J. Lippard, B. M. Hoffman, A. C. Rozenzweig and T. L. Stemmler, Inorg. Chem., 2006, 45, 8372–8381 CrossRef CAS PubMed; (b) S. I. Chan, V. C.-C. Wang, J. C.-H. Lai, S. S.-F. Yu, P. P.-Y. Chen, K. H.-C. Chen, C. C.-L. Chen and M. K. Chan, Angew. Chem., Int. Ed., 2007, 46, 1992–1994 CrossRef CAS PubMed; (c) S. I. Chan and S. S.-F. Yu, Acc. Chem. Res., 2008, 41, 969–979 CrossRef CAS PubMed; (d) A. C. Rosenzweig, Biochem. Soc. Trans., 2008, 36, 1134–1137 CrossRef CAS PubMed; (e) A. Miyaji, M. Suzuki, T. Baba, T. Kamachi and I. Okura, J. Mol. Catal. B: Enzym., 2008, 57, 211–215 CrossRef PubMed; (f) A. S. Hakemian, K. C. Kondapalli, J. Telser, B. M. Hoffman, T. L. Stemmler and A. C. Rozenzweig, Biochemistry, 2008, 47, 6793–6801 CrossRef CAS PubMed; (g) Y. Shiota and K. Yoshizawa, Inorg. Chem., 2009, 48, 838–845 CrossRef CAS PubMed; (h) R. Balasubramanian, S. M. Smith, S. Rawat, L. A. Yatsunyk, T. L. Stemmler and A. C. Rosenzweig, Nature, 2010, 465, U115–U131 CrossRef PubMed No. 7294; (i) A. Miyaji, Methods Enzymol., 2011, 495, 211–225 CAS; (j) Y. Shiota, G. Juhász and K. Yoshizawa, Inorg. Chem., 2013, 52, 7907–7917 CrossRef CAS PubMed.
- (a) Q. Zhu, Y. Lian, S. Thyagarajan, S. E. Rokita, K. D. Karlin and N. V. Blogh, J. Am. Chem. Soc., 2008, 230, 6304–6305 CrossRef PubMed; (b) P. J. Donoghue, J. Tehranchi, C. J. Cramer, R. Sarangi, E. I. Solomon and W. B. Tolman, J. Am. Chem. Soc., 2011, 133, 17602–17605 CrossRef CAS PubMed; (c) A. N. Pham, G. Xing, C. J. Miller and T. D. Waite, J. Catal., 2013, 301, 54–64 CrossRef CAS PubMed.
- (a) A. N. Bilyachenko, M. S. Dronova, A. I. Yalymov, A. A. Korlyukov, L. S. Shul'pina, D. E. Arkhipov, E. S. Shubina, M. M. Levitsky, A. D. Kirilin and G. B. Shul'pin, Eur. J. Inorg. Chem., 2013, 5240–5246 CrossRef CAS; (b) M. S. Dronova, A. N. Bilyachenko, A. I. Yalymov, Y. N. Kozlov, L. S. Shul'pina, A. A. Korlyukov, D. E. Arkhipov, M. M. Levitsky, E. S. Shubina and G. B. Shul'pin, Dalton Trans., 2014, 43, 872–882 RSC.
- (a) R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Roesky, Chem. Rev., 1996, 96, 2205–2236 CrossRef CAS PubMed; (b) V. Lorenz, A. Fischer, S. Gießmann, J. W. Gilje, Y. Gun'ko, K. Jacob and F. T. Edelmann, Coord. Chem. Rev., 2000, 206–207, 321–368 CrossRef CAS; (c) R. W. J. M. Hanssen, R. A. van Santen and H. C. L. Abbenhuis, Eur. J. Inorg. Chem., 2004, 675–683 CrossRef CAS; (d) H. W. Roesky, G. Anantharaman, V. Chandrasekhar, V. Jancik and S. Singh, Chem. – Eur. J., 2004, 10, 4106–4114 CrossRef CAS PubMed; (e) V. Lorenz and F. T. Edelmann, Adv. Organomet. Chem., 2005, 53, 101–153 CrossRef CAS; (f) M. M. Levitsky, B. G. Zavin and A. N. Bilyachenko, Russ. Chem. Rev., 2007, 76, 847–866 CrossRef CAS PubMed; (g) P. Jutzi, H. M. Lindemann, J.-O. Nolte and M. Schneider, Synthesis, Structure, and Reactivity of Novel Oligomeric Titanasiloxanes, in Silicon Chemistry: From the Atom to Extended Systems, ed. P. Jutzi and U. Schubert, Wiley, 2007, pp. 372–382 Search PubMed; (h) F. T. Edelmann, Metallasilsesquioxanes. Synthetic and Structural Studies, in Silicon Chemistry: From the Atom to Extended Systems, ed. P. Jutzi and U. Schubert, Wiley, 2007, pp. 383–394 Search PubMed; (i) A. J. Ward, A. F. Masters and T. Maschmeyer, Adv. Silicon Sci., 2011, 135–166 CrossRef CAS.
- (a) V. S. Kulikova, M. M. Levitsky and A. L. Buchachenko, Russ. Chem. Bull., 1996, 45, 2870–2872 CrossRef; (b) V. S. Kulikova, M. M. Levitsky, A. F. Shestakov and A. E. Shilov, Russ. Chem. Bull., 1998, 47, 435–437 CrossRef CAS.
- (a) G. B. Shul'pin, J. Mol. Catal. A: Chem., 2002, 189, 39–66 CrossRef; (b) G. B. Shul'pin, C. R. Chim., 2003, 6, 163–178 CrossRef; (c) G. B. Shul'pin, Mini-Rev. Org. Chem., 2009, 6, 95–104 CrossRef; (d) G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina, A. R. Kudinov and D. Mandelli, Inorg. Chem., 2009, 48, 10480–10482 CrossRef PubMed; (e) G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina and P. V. Petrovskiy, Appl. Organomet. Chem., 2010, 24, 464–472 Search PubMed.
- (a) G. B. Shul'pin, D. Attanasio and L. Suber, Russ. Chem. Bull., 1993, 42, 55–59 CrossRef; (b) G. B. Shul'pin, R. S. Drago and M. Gonzalez, Russ. Chem. Bull., 1996, 45, 2386–2388 CrossRef; (c) G. B. Shul'pin, Y. Ishii, S. Sakaguchi and T. Iwahama, Russ. Chem. Bull., 1999, 48, 887–890 CrossRef; (d) G. B. Shul'pin, G. S. Mishra, L. S. Shul'pina, T. V. Strelkova and A. J. L. Pombeiro, Catal. Commun., 2007, 8, 1516–1520 CrossRef PubMed.
- (a) G. B. Shul'pin, Y. N. Kozlov, G. V. Nizova, G. Süss-Fink, S. Stanislas, A. Kitaygorodskiy and V. S. Kulikova, J. Chem. Soc., Perkin Trans. 2, 2001, 1351–1371 RSC; (b) Y. N. Kozlov, V. B. Romakh, A. Kitaygorodskiy, P. Buglyó, G. Süss-Fink and G. B. Shul'pin, J. Phys. Chem. A, 2007, 111, 7736–7752 CrossRef CAS PubMed; (c) M. V. Kirillova, M. L. Kuznetsov, V. B. Romakh, L. S. Shul'pina, J. J. R. Fraústo da Silva, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2009, 267, 140–157 CrossRef CAS PubMed; (d) M. Sutradhar, N. V. Shvydkiy, M. F. C. G. da Silva, M. V. Kirillova, Y. N. Kozlov, A. J. L. Pombeiro and G. B. Shul'pin, Dalton Trans., 2013, 42, 11791–11803 RSC; (e) L. S. Shul'pina, M. V. Kirillova, A. J. L. Pombeiro and G. B. Shul'pin, Tetrahedron, 2009, 65, 2424–2429 CrossRef PubMed; (f) G. B. Shul'pin, G. V. Nizova, Y. N. Kozlov, L. Gonzalez Cuervo and G. Süss-Fink, Adv. Synth. Catal., 2004, 346, 317–332 CrossRef; (g) G. V. Nizova, B. Krebs, G. Süss-Fink, S. Schindler, L. Westerheide, L. Gonzalez Cuervo and G. B. Shul'pin, Tetrahedron, 2002, 58, 9231–9237 CrossRef CAS; (h) G. B. Shul'pin, M. V. Kirillova, L. S. Shul'pina, A. J. L. Pombeiro, E. E. Karslyan and Y. N. Kozlov, Catal. Commun., 2013, 31, 32–36 CrossRef PubMed; (i) L. S. Shul'pina, E. L. Durova, Y. N. Kozlov, A. R. Kudinov, T. V. Strelkova and G. B. Shul'pin, Russ. J. Phys. Chem. A, 2013, 87, 1996–2000 CrossRef; (j) V. B. Romakh, B. Therrien, G. Süss-Fink and G. B. Shul'pin, Inorg. Chem., 2007, 46, 3166–3175 CrossRef CAS PubMed; (k) E. E. Karslyan, L. S. Shul'pina, Y. N. Kozlov, A. J. L. Pombeiro and G. B. Shul'pin, Catal. Today, 2013, 218–219, 93–98 CrossRef CAS PubMed; (l) G. B. Shul'pin, M. V. Kirillova, Y. N. Kozlov, L. S. Shul'pina, A. R. Kudinov and A. J. L. Pombeiro, J. Catal., 2011, 277, 164–172 CrossRef PubMed; (m) G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina, W. Carvalho and D. Mandelli, RSC Adv., 2013, 3, 15065–15074 RSC; (n) G. B. Shul'pin, A. R. Kudinov, L. S. Shul'pina and E. A. Petrovskaya, J. Organomet. Chem., 2006, 691, 837–845 CrossRef PubMed; (o) G. B. Shul'pin, G. Süss-Fink and L. S. Shul'pina, Chem. Commun., 2000, 1131–1132 RSC; (p) G. B. Shul'pin, J. Chem. Res., Synop., 2002, 351–353 CrossRef; (q) D. Mandelli, K. C. Chiacchio, Y. N. Kozlov and G. B. Shul'pin, Tetrahedron Lett., 2008, 49, 6693–6697 CrossRef CAS PubMed; (r) I. Gryca, B. Machura, J. G. Malecki, L. S. Shul'pina, A. J. L. Pombeiro and G. B. Shul'pin, Dalton Trans., 2014, 43, 5759–5776 RSC; (s) D. S. Nesterov, E. N. Chygorin, V. N. Kokozay, V. V. Bon, R. Boča, Y. N. Kozlov, L. S. Shul'pina, J. Jezierska, A. Ozarowski, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2012, 51, 9110–9122 CrossRef CAS PubMed; (t) M. V. Kirillova, Y. N. Kozlov, L. S. Shul'pina, O. Y. Lyakin, A. M. Kirillov, E. P. Talsi, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2009, 268, 26–38 CrossRef CAS PubMed; (u) G. B. Shul'pin, M. V. Kirillova, T. Sooknoi and A. J. L. Pombeiro, Catal. Lett., 2008, 128, 135–141 CrossRef PubMed; (v) A. J. Bonon, D. Mandelli, O. A. Kholdeeva, M. V. Barmatova, Y. N. Kozlov and G. B. Shul'pin, Appl. Catal., A, 2009, 365, 96–104 CrossRef CAS PubMed; (w) G. B. Shul'pin and J. R. Lindsay Smith, Russ. Chem. Bull., 1998, 47, 2379–2386 CrossRef; (x) G. B. Shul'pin, M. G. Matthes, V. B. Romakh, M. I. F. Barbosa, J. L. T. Aoyagi and D. Mandelli, Tetrahedron, 2008, 64, 2143–2152 CrossRef PubMed; (y) V. B. Romakh, B. Therrien, G. Süss-Fink and G. B. Shul'pin, Inorg. Chem., 2007, 46, 1315–1331 CrossRef CAS PubMed; (z) G. B. Shul'pin, Y. N. Kozlov, L. S. Shul'pina and A. J. L. Pombeiro, Tetrahedron, 2012, 68, 8589–8599 CrossRef PubMed; (a a) G. B. Shul'pin, J. Gradinaru and Y. N. Kozlov, Org. Biomol. Chem., 2003, 1, 3611–3617 RSC; (a b) M. V. Kirillova, A. M. Kirillov, D. Mandelli, W. A. Carvalho, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2010, 272, 9–17 CrossRef CAS PubMed; (a c) A. M. Kirillov, M. V. Kirillova, L. S. Shul'pina, P. J. Figiel, K. R. Gruenwald, M. F. C. G. da Silva, M. Haukka, A. J. L. Pombeiro and G. B. Shul'pin, J. Mol. Catal. A: Chem., 2011, 350, 26–34 CrossRef CAS PubMed; (a d) G. B. Shul'pin, H. Stoeckli-Evans, D. Mandelli, Y. N. Kozlov, A. Tesouro Vallina, C. B. Woitiski, R. S. Jimenez and W. A. Carvalho, J. Mol. Catal. A: Chem., 2004, 219, 255–264 CrossRef PubMed; (a e) G. B. Shul'pin, T. Sooknoi, V. B. Romakh, G. Süss-Fink and L. S. Shul'pina, Tetrahedron Lett., 2006, 47, 3071–3075 CrossRef PubMed.
- (a) K. Kamata, K. Yonehara, Y. Nakagawa, K. Uehara and N. Mizuno, Nat. Chem., 2010, 2, 478–483 CrossRef CAS PubMed; (b) G. B. Shul'pin, Selectivity in C–H functionalizations, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk, K. Poeppelmeier and L. Casella, Elsevier, 2nd edn, 2013, ch. 6.04, vol. 6, pp. 79–104 Search PubMed.
- (a) C. Knight and M. J. Perkins, J. Chem. Soc., Chem. Commun., 1991, 925–927 RSC; (b) M. M. Vinogradov, Y. N. Kozlov, D. S. Nesterov, L. S. Shul'pina, A. J. L. Pombeiro and G. B. Shul'pin, Catal.: Sci. Technol., 2014, 4, 3214–3226 RSC; (c) US National Institute of Standards and Technology (NIST) Mass Spectral Library, ver. 2.0f, build Apr. 1, 2009; (d) J. H. Gross, Mass Spectrometry, A Textbook, Springer-Verlag, Berlin Heidelberg, 2nd edn, 2011 Search PubMed.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS.
Footnotes |
| † This paper is dedicated in memory of Aleksandr Evgenievich Shilov (1930–2014). |
| ‡ Electronic supplementary information (ESI) available: Fig. S1–S14. See DOI: 10.1039/c4nj01163e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |