
Effect of stacking mode on the mechanofluorochromic properties of 3-aryl-2-cyano acrylamide derivatives†
Qingbao
Song
a,
Yongsheng
Wang
a,
Chenchen
Hu
b,
Yujian
Zhang
*b,
Jingwei
Sun
a,
Kunyan
Wang
b and
Cheng
Zhang
*a
aState Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou, P. R. China. E-mail: czhang@zjut.edu.cn
bDepartment of Materials Chemistry, Huzhou University, Xueshi Road 1#, Huzhou, P. R. China. E-mail: sciencezyj@foxmail.com
First published on 30th October 2014
Abstract
Three structurally simple 3-aryl-2-cyano acrylamide derivatives, 2-cyano-3-(2-methoxyphenyl)-2-propenamide (1), 2-cyano-3-(3-methoxyphenyl)-2-propenamide (2) and 2-cyano-3-(4-methoxyphenyl)-2-propenamide (3) were synthesized. They exhibited different optical properties due to their distinct face-to-face stacking mode. The as-prepared crystals of 1 exhibited green luminescence and the emission peak did not change after grinding treatment. However, the emission peaks of 2 (Φf = 12%) and 3 (Φf = 16%) exhibited an obvious red-shift upon grinding, and their corresponding quantum yields decreased to 8% and 10%, respectively. Differential scanning calorimetry and powder X-ray diffractometry data indicated that the optical properties of 2 and 3 could be attributed to the transformation from the crystalline phase to the amorphous phase. X-ray crystal structures, infrared spectroscopy and data from fluorescence lifetime experiments further validated the relationship between fluorescence switching, stacking mode and molecular interactions.
Introduction
Mechanochromic fluorescent (MCF) materials revealing reversible fluorescent changes under external force stimuli1 could be applied to sensors, memory chips and security inks.2 Recently, rapid progress has been achieved in these material systems and in understanding the formation mechanism of MCF behaviour. For instance, luminophores with a highly twisted conformation were reported recently,3–5 whose molecular structure adopted a more planar conformation after grinding that induced a longer wavelength. In addition, the excitonic interaction was substantially altered in accord with various stacking modes, where excimer formation resulted in a red-shift of fluorescence.6 To date, the overwhelming majority of MCF materials exhibited a phase transformation from the crystalline state to the (partial) amorphous state.7 However, there is some doubt about whether organic powders with a transition will reveal MCF behaviour. Thus, it is still essential to further understand the relationship between molecular packing characteristics and the resulting MCF properties at the molecular level.Recently, Yang's group reported that 9,10-bis(alkoxystyryl)-anthracene isomers8 indicated MCF behaviour which was both chain length-dependent9 and position-dependent. Further, short alkyl-containing oOC3 and mOC3 exhibited more remarkable MCF activity than pOC3, which was related to the molecular conformation and stacking modes. In addition, our group prepared new isomers containing arylamines, in which changing the position of the cyano group had a great effect on the MCF properties and packing modes.10 Clearly, employing structural isomers to investigate the formation mechanism of MCF properties is of significant interest.
We have synthesized three isomers, 2-cyano-3-(2-methoxyphenyl)-2-propenamide (o-MPCPA), 2-cyano-3-(3-methoxyphenyl)-2-propenamide (m-MPCPA) and 2-cyano-3-(4-methoxyphenyl)-2-propenamide (p-MPCPA) (Fig. 1), by a simple Knoevenagel reaction. Among them, the m-MPCPA and p-MPCPA samples with a blue fluorescence exhibited remarkable MCF properties. Their fluorescence changed to green after grinding treatment. For the o-MPCPA, the phase transition also underwent an obvious change upon grinding, but MCF behaviour was not observed. X-ray crystal structures and data from fluorescence lifetime experiments further validated the phase transition of MPCPA (o-, m-, p-). However, the intermolecular interactions remained unchanged for luminophore o-MPCPA, which might be the main factor causing its abnormal behaviour.
 | ||
| Fig. 1 Molecular structures of 3-aryl-2-cyano acrylamide derivatives o-MPCPA, m-MPCPA and p-MPCPA. | ||
Results and discussion
Single crystals of MPCPA (o-, m- and p-) were obtained by slow evaporation of ethanol–n-hexane mixtures. Fig. 2 exhibits the intermolecular interactions and packing modes of the neighbouring molecules. It was found that the aromatic ring and cyano group together with the acrylamide for the three MPCPA isomers were located on the same plane. For o-MPCPA, the crystal structure contained eight molecules in the unit cell and was monoclinic with space group I2/a. As depicted in Fig. S2C (ESI†), there were two types of hydrogen bonds between adjacent molecules with distances of 2.379 Å and 2.113 Å. The luminogens adopted an antiparallel H-type packing mode in order to fit into the crystalline lattice. Viewed from the top, the acrylamide was placed directly above the aromatic ring of the neighbouring molecule with an interplanar distance of 3.5329 Å (Fig. 2a). Importantly, the aromatic rings did not overlap between the adjacent molecules. Such an antiparallel H-type packing mode did not result in π–π interactions, which contributed to an enhancement of the quantum yields of the crystals. By comparison, the luminogens m-MPCPA and p-MPCPA contained four molecules in their unit cells. As depicted in Fig. S3C and S4C (ESI†), multiple C–H/N and C–H/O hydrogen bonds as well as H-type packing were also observed, which induced a molecular column. Interestingly, both of the luminogens m-MPCPA and p-MPCPA adopted head-to-head or parallel arrangements. The aromatic ring was stacked directly above the benzene ring of the neighbouring molecule, with interplanar distances of 3.5931 Å and 3.4903 Å for m-MPCPA and p-MPCPA respectively (Fig. 2b and c). The interplanar distance was less than the range of the effective intermolecular interaction, but π–π overlap was slightly reduced due to the slip-stacking along the long axis of the m-/p-MPCPA molecule (Fig. 2b and c).11 Thus, there was a weak π–π interaction between aromatic rings, which caused fluorescence quenching. | ||
| Fig. 2 The packing modes and distances between adjacent molecules in the single-crystal structures of MPCPA (o-, m- and p-). | ||
In the crystalline state, the luminophore o-MPCPA exhibited a relatively high luminescence with a quantum yield (Φf) of 23%. In sharp contrast, the quantum yields of m-MPCPA and p-MPCPA were decreased to 12% and 16%, respectively. As depicted in Fig. S4 and Table S1 (ESI†), the radiative rate constant (kF = Φf/τf) of o-MPCPA was 3.7 × 107 s−1, which was similar to that of m-/p-MPCPA (approximately 4.0 × 107 s−1). However, the crystalline powder of o-MPCPA yielded a low non-radiative rate constant [knr = (1 − Φf)/τ] of 1.2 × 108 s−1, which increased to 3.1 × 108 s−1 for m-/p-MPCPA. The results indicated the non-radiative deactivation pathways were obviously unblocked in m-/p-MPCPA, which led to a lower Φf as a result of the weak π–π interactions. This explanation was in accordance with the head-to-head arrangements (Fig. 2b and c). Fig. 3 shows the fluorescence images of o-MPCPA, m-MPCPA and p-MPCPA samples after a cycle of grinding, heating or solvent fuming. Of the crystalline powders, o-MPCPA, with green luminescence, did not exhibit MCF behaviour as there was not an obvious spectral shift upon grinding (Fig. 4A). However, the m-MPCPA and p-MPCPA samples exhibited more remarkable MCF properties (Fig. 4B). The white crystals of m-MPCPA emitted blue-purple light (λmax = 452 nm) under UV light, which changed to green (λmax = 470 nm) after the grinding treatment. Upon fuming with solvent vapours such as CH2Cl2 and ethyl acetate, the luminescence recovered its original state (see Fig. S6, ESI†). For p-MPCPA, the sample emitted blue-purple light (λmax = 450 nm) under UV light, which became green (λmax = 478 nm) after grinding treatment. Clearly, the MCF behaviour of MPCPA depended on the change in the methoxy position, and followed the order p-MPCPA > m-MPCPA > o-MPCPA.
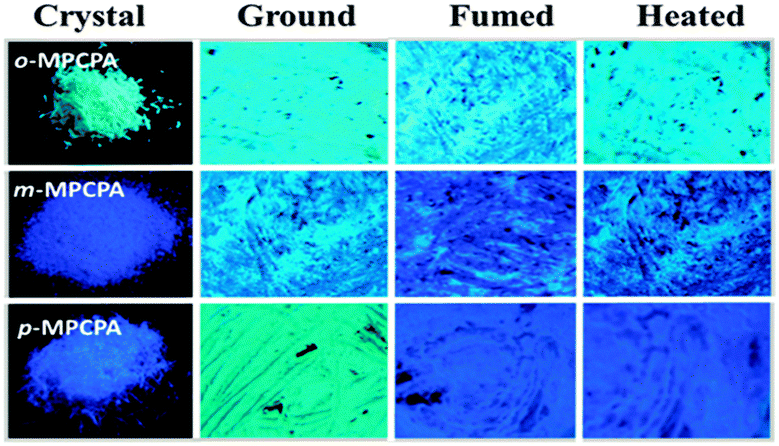 | ||
| Fig. 3 Fluorescence images of o-MPCPA, m-MPCPA and p-MPCPA under a 365 nm UV light: as-prepared, ground, fumed and heated. | ||
 | ||
| Fig. 4 Fluorescence spectra of o-MPCPA, m-MPCPA and p-MPCPA in crystalline form (●) and after grinding treatment (○). | ||
To further investigate the MCF behaviour, differential scanning calorimetry (DSC) measurements and powder wide-angle X-ray diffraction patterns (PXRD) of MPCPA (o-, m-, p-) powders in various states were obtained. As depicted in Fig. S6 (ESI†), the ground powders of the three isomers indicated an obvious cold-crystallization peak before the isotropic melting transition, which did not exist in the crystal. These were observed at approximately 109 °C, 85 °C and 132 °C for the ground samples of o-MPCPA, m-MPCPA and p-MPCPA, respectively. In previous reports, the existence of a cold-crystallization peak indicated that a ground powder was a metastable state transforming into a more stable packing structure via annealing, which was related to the MCF behaviour. However, in our case, the o-MPCPA samples did not exhibit MCF properties even though the ground o-MPCPA powders showed an evident cold-crystallization peak (Fig. S7A, ESI†). Powder X-ray diffraction patterns showed that the pristine powders of MPCPA clearly exhibited numerous sharp and intense reflection peaks, which indicated a well-defined microcrystalline structure. These peaks of the MPCPA samples almost disappeared and became weak, broad and diffused peaks upon grinding. The results showed that the crystal lattice was significantly disrupted and a new amorphous phase was formed (Fig. 5). The ground samples could be recovered to the original states after solvent or heating treatment. Thus, the MCF properties of m-/p-MPCPA could be attributed to the transformation from the crystalline phase to the amorphous phase, which coincided with previous results.7 Conspicuously, an evident phase transition of the o-MPCPA sample which did not produce MCF behaviour was also observed. What were the reasons for this abnormal phenomenon?
 | ||
| Fig. 5 PXRD profiles of o-MPCPA, m-MPCPA and p-MPCPA powders in the different states. | ||
IR spectra of o-MPCPA powders were identical before and after grinding (Fig. 6A). In contrast, two IR peaks at 3163 and 3366 cm−1 (νN–H) existed for the luminogen m-MPCPA, which disappeared upon grinding (Fig. 6B). Further, an IR peak at 3163 cm−1 (νN–H) existed for the luminogen p-MPCPA, which drifted upon grinding (Fig. 6C). Previously, researchers usually chose a strongly twisted conjugated backbone12,13 as the research object of MCF fluorophores. The disappearance and drifting of some IR peaks implied that the compounds probably had a more planar conformation and stronger molecular interactions. However, since we synthesized MPCPA luminogens with planar conformations, the disappearance and drifting of some IR peaks of m-MPCPA and p-MPCPA could only be caused by stronger molecular interactions. Thus, the disappearance and drifting of some IR peaks implied that the luminogens m-MPCPA and p-MPCPA probably had stronger molecular interactions than those of o-MPCPA in the ground state.
 | ||
| Fig. 6 IR spectra of o-MPCPA, m-MPCPA and p-MPCPA in crystalline form (black curve) and after grinding treatment (blue curve). | ||
Time-resolved fluorescence experiments further verified this hypothesis and the weighted mean lifetimes (τ) are illustrated in Fig. S5 and Table S1 (ESI†). The τ values of o-MPCPA before and after grinding were similar. By contrast, the τ values of the m-MPCPA and p-MPCPA samples in different states revealed significant differences: the respective values were 2.99 and 2.62 ns for the original samples, and 4.61 and 5.22 ns for the ground samples. The increase in lifetime revealed that the excitonic coupling was obviously enhanced. The as-prepared crystals of o-MPCPA exhibited a quantum yield (Φf) of 23%, which was scarcely changed upon grinding (Φf = 24%). However, for the luminogens m-MPCPA (Φf = 12%) and p-MPCPA (Φf = 16%), the quantum yields were decreased to 8% and 10% upon grinding, respectively. Furthermore, the radiative rate constants (kF) for m-MPCPA and p-MPCPA were decreased from 4.0 × 107 s−1 and 6.1 × 107 s−1 to 1.7 × 107 s−1 and 1.9 × 107 s−1, respectively. The results indicated that the π–π interaction was further enhanced, which blocked the radiative deactivation pathways. However, this process did not exist for the o-MPCPA sample. What were the reasons that mechanical grinding did not alter the excited state of the o-MPCPA sample? As shown in Fig. 2a, the acrylamide was placed above the aromatic ring of the neighbouring molecule. The interplanar distance was effectively reduced upon grinding, which has been demonstrated in other MCF molecules. However, the decrease in distance did not lead to the enhancement of π–π interactions. In sharp contrast, for m-MPCPA and p-MPCPA with head-to-head packing, the intermolecular interaction between the adjacent aromatic rings was enhanced as a result of the excitonic coupling. Such strong π–π interactions between the neighbouring molecules blocked the decay of the excited species through radiative pathways, resulting in the altering of the excited state as well as the weak Φf of m-MPCPA (8%) and p-MPCPA (10%). In summary, luminogen o-MPCPA revealed an obvious phase transition but this did not cause any variation in π–π interactions between adjacent molecules, owing to the antiparallel H-type arrangement. Hence, the luminogen o-MPCPA did not show MCF properties despite the change in phase state.
Conclusions
In summary, we have synthesized novel luminogenic 3-aryl-2-cyano acrylamide derivatives (o-MPCPA, m-MPCPA and p-MPCPA), with differences in the methoxy position. It was found that the optical properties and stacking mode depended on the change in the methoxy position. The luminogen o-MPCPA adopted an antiparallel H-type packing mode. However, both of the luminogens m-MPCPA and p-MPCPA adopted head-to-head or parallel arrangements. Thus, the luminogen o-MPCPA exhibited a relatively higher quantum yield than the luminogens m-MPCPA and p-MPCPA due to the distinct packing mode and molecular interactions. The molecular conformation and stacking modes of the luminogens MPCPA (o-, m-, p-) were altered under the stimulation of mechanical force. However, the luminogen o-MPCPA did not exhibit MCF behaviour. As far as we know, this is the first example of a luminogen showing an evident phase transition, which did not induce MCF behaviour. The interplanar distance was generally reduced upon grinding. However, the decrease in distance did not lead to an increase in π–π interactions, which was the reason for the abnormal behaviour of o-MPCPA. Thus, MCF behaviour is not only dependent on molecular conformation and stacking modes, but is also strongly related to molecular interaction.Experimental
2-Cyano-3-(2-methoxyphenyl)-2-propenamide (o-MPCPA), 2-cyano-3-(3-methoxyphenyl)-2-propenamide (m-MPCPA) and 2-cyano-3-(4-methoxyphenyl)-2-propenamide (p-MPCPA) were synthesized (Fig. 1) by a simple Knoevenagel reaction under mild conditions in good yield.Procedure for o-MPCPA: A mixture of 2-(methoxy)benzaldehyde (4.08 g, 30 mmol), 2-cyanoacetamide (2.61 g, 31 mmol) and L-proline (0.69 g, 6 mmol) in ethanol (20 ml) was heated under reflux for 2 h; the solid product was filtered, washed with ethanol, dried (91% yield) and upon crystallization from ethanol solution gave rise to the pure product. Finally, the desired luminophore 2-cyano-3-(2-methoxyphenyl)-2-propenamide (o-MPCPA) was fully characterized by 1H NMR, 13C NMR and HRMS. 1H NMR (500 MHz, CDCl3) δ 8.81 (s, 1H), 8.19 (dd, J = 7.8, 1.2 Hz, 1H), 7.56–7.48 (dd, J = 7.8, 1.6 Hz, 1H), 7.07 (t, J = 7.6 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.35 (d, 2H), 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.5, 159.3, 148.8, 134.7, 129.1, 120.9, 120.9, 117.4, 111.2, 102.7, 55.7. HRMS (ESI) calcd for C11H10N2O2 ([M + H]+): 203.0821. Found: 203.0807. Crystallographic data for o-MPCPA: C11H10N2O2, M = 202.21 g mol−1, monoclinic, a = 13.7381(9) Å, b = 8.8770(4) Å, c = 17.5640(9) Å, β = 103.368(6)°, V = 2083.9(2) Å3, T = 293(2) K, R(int) = 0.0214, space group I2/a, Dcalc = 1.289 Mg m−3, Z = 8; the final R indices were R1 = 0.0448, wR2 = 0.1153 [I > 2σ(I)], CCDC 999360.
Procedure for m-MPCPA: A mixture of 3-(methoxy)benzaldehyde (4.08 g, 30 mmol), 2-cyanoacetamide (2.61 g, 31 mmol) and L-proline (0.69 g, 6 mmol) in ethanol (20 ml) was heated under reflux for 2 h; the solid product was filtered, washed with ethanol, dried (92% yield) and upon crystallization from ethanol solution gave rise to the pure product. Finally, the desired luminophore 2-cyano-3-(3-methoxyphenyl)-2-propenamide (m-MPCPA) was fully characterized by 1H NMR, 13C NMR and HRMS. 1H NMR (500 MHz, CDCl3) δ 8.31 (s, 1H), 7.54–7.48 (m, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.11–7.09 (m, 1H), 6.43 (s, 1H), 6.19 (s, 1H), 3.87 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.1, 160.0, 154.1, 132.8, 130.3, 123.8, 119.7, 117.1, 114.6, 103.3, 55.4. HRMS (ESI) calcd for C11H10N2O2 ([M + H]+): 203.0821. Found: 203.0822. Crystallographic data for m-MPCPA: C11H10N2O2, M = 202.21 g mol−1, monoclinic, a = 15.6778(10) Å, b = 3.9598(3) Å, c = 17.4076(14) Å, β = 108.928(8)°, V = 1022.26(13) Å3, T = 293(2) K, R(int) = 0.0235, space group P2(1)/c, Dcalc = 1.314 Mg m−3, Z = 4; the final R indices were R1 = 0.0430, wR2 = 0.1138 [I > 2σ(I)], CCDC 999359.
Procedure for p-MPCPA: A mixture of 4-(methoxy)benzaldehyde (4.08 g, 30 mmol), 2-cyanoacetamide (2.61 g, 31 mmol) and L-proline (0.69 g, 6 mmol) in ethanol (20 ml) was heated under reflux for 2 h; the solid product was filtered, washed with ethanol, dried (94% yield) and upon crystallization from ethanol solution gave rise to the pure product. Finally, the desired luminophore 2-cyano-3-(4-methoxyphenyl)-2-propenamide (p-MPCPA) was fully characterized by 1H NMR, 13C NMR and HRMS. 1H NMR (500 MHz, DMSO) δ 8.11 (s, 1H), 7.97 (d, J = 8.8 Hz, 2H), 7.79 (s, 1H), 7.72–7.59 (m, 1H), 7.14 (t, J = 5.8 Hz, 2H), 3.86 (s, 3H). 13C NMR (126 MHz, DMSO) δ 163.1, 162.6, 150.1, 132.4, 124.4, 117.0, 114.8, 102.9, 55.6, 54.7. HRMS (ESI) calcd for C11H10N2O2 ([M + H]+): 203.0821. Found: 203.0824. Crystallographic data for p-MPCPA: C11H10N2O2, M = 202.21 g mol−1, monoclinic, a = 3.920(2) Å, b = 10.792(6) Å, c = 23.124(12) Å, β = 93.463 (8)°, V = 976.5 (9) Å3, T = 293(2) K, R(int) = 0.1143, space group P2(1)/c, Dcalc = 1.375 Mg m−3, Z = 4; the final R indices were R1 = 0.0824, wR2 = 0.2367 [I > 2σ(I)], CCDC 999361.
Acknowledgements
The authors gratefully acknowledge the support of National Natural Science Foundation of China (51403060, 51273179), National Basic Research Program of China (2011CBA00700), International S&T Cooperation Program, China (2012DFA-51210) and Natural Science Foundation of Zhejiang Province (LQ14B040003).Notes and references
- (a) Y. Q. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Z. Sun, H. H.-Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40–42 RSC; (b) B.-K. An, S. H. Gihm, J. W. Chung, C. R. Park, S.-K. Kwon and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 3950–3957 CrossRef CAS PubMed; (c) H. Y. Zhang, Z. L. Zhang, K. Ye, J. Y. Zhang and Y. Wang, Adv. Mater., 2006, 18, 2369–2372 CrossRef CAS; (d) X. Fan, J. L. Sun, F. Z. Wang, Z. Z. Chu, P. Wang, Y. Q. Dong, R. R. Hu, B. Z. Tang and D. C. Zou, Chem. Commun., 2008, 2989–2991 RSC.
- (a) Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed; (b) C. Weder, J. Mater. Chem., 2011, 21, 8235–8236 RSC; (c) Z. G. Chi, X. Q. Zhang, B. J. Xu, X. Zhou, C. P. Ma, Y. Zhang, S. W. Liu and J. R. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC; (d) S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS; (e) S. J. Lim, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef CAS PubMed.
- (a) X. Q. Zhang, Z. G. Chi, H. Y. Li, B. J. Xu, X. F. Li, W. Zhou, S. W. Liu, Y. Zhang and J. R. Xu, Chem. – Asian J., 2011, 6, 808–811 CrossRef CAS PubMed; (b) X. Q. Zhang, Z. G. Chi, J. Y. Zhang, H. Y. Li, B. J. Xu, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef CAS PubMed.
- (a) W. Z. Yuan, Y. Q. Tan, Y. Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. F. Feng, H. H. Y. Sung, Y. W. Lu, I. D. Williams, J. Z. Sun, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed; (b) Y. Q. Dong, J. W. Y. Lam, A. J. Qin, J. X. Sun, J. Z. Liu, Z. Li, J. Z. Sun, H. H. Y. Sung, I. D. Williams, H. S. Kwokc and B. Z. Tang, Chem. Commun., 2007, 3255–3257 RSC.
- (a) Y. L. Wang, W. Liu, L. Y. Bu, J. F. Li, M. Zheng, D. T. Zhang, M. X. Sun, Y. Tao, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2013, 1, 856–862 RSC; (b) L. Y. Bu, M. X. Sun, D. T. Zhang, W. Liu, Y. L. Wang, M. Zheng, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2013, 1, 2028–2035 RSC; (c) W. Liu, Y. L. Wang, L. Y. Bu, J. F. Li, M. Sun, D. T. Zhang, M. Zheng, C. Yang, S. F. Xue and W. J. Yang, J. Lumin., 2013, 143, 50–55 CrossRef CAS.
- (a) M. J. Teng, X. R. Jia, S. Yang, X. F. Chen and Y. Wei, Adv. Mater., 2012, 24, 1255–1261 CrossRef CAS PubMed; (b) J. Luo, L. Y. Li, Y. L. Song and J. Pei, Chem. – Eur. J., 2011, 17, 10515–10519 CrossRef CAS PubMed; (c) X. Q. Zhang, Z. G. Chi, Y. Zhang, S. W. Liu and J. R. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC.
- (a) S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed; (b) C. Kitamura, T. Ohara, N. Kawatsuki, A. Yoneda, T. Kobayashi, H. Naito, T. Komatsuc and T. Kitamura, CrystEngComm, 2007, 9, 644–647 RSC; (c) H. Y. Zhang, Z. L. Zhang, K. Q. Ye, J. Y. Zhang and Y. Wang, Adv. Mater., 2006, 18, 2369–2372 CrossRef CAS; (d) R. R. Wei, P. S. Song and A. J. Tong, J. Phys. Chem. C, 2013, 117, 3467–3474 CrossRef CAS; (e) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed; (f) S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS; (g) X. Q. Zhang, Z. G. Chi, B. J. Xu, C. J. Chen, X. Zhou, Y. Zhang, S. W. Liu and J. R. Xu, J. Mater. Chem., 2012, 22, 18505–18513 RSC.
- W. Liu, Y. L. Wang, M. X. Sun, D. T. Zhang, M. Zheng and W. J. Yang, Chem. Commun., 2013, 49, 6042–6044 RSC.
- (a) X. Q. Zhang, Z. G. Chi, B. R. Xu, L. Jiang, X. Zhou, Y. Zhang, S. W. Liu and J. R. Xu, Chem. Commun., 2012, 48, 10895–10897 RSC; (b) X. Q. Zhang, Z. G. Chi, X. Zhou, S. W. Liu, Y. Zhang and J. R. Xu, J. Phys. Chem. C, 2012, 116, 23629–23638 CrossRef CAS.
- Y. J. Zhang, G. L. Zhuang, M. Ouyang, B. Hu, Q. B. Song, J. W. Sun, C. Zhang, C. Gu, Y. X. Xu and Y. G. Ma, Dyes Pigm., 2013, 98, 486–492 CrossRef CAS.
- (a) S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed; (b) X. Q. Zhang, Z. Y. Ma, Y. Yang, X. Y. Zhang, X. R. Jia and Y. Wei, J. Mater. Chem. C, 2014, 2, 8932–8938 RSC.
- H. Y. Li, X. Q. Zhang, Z. G. Chi, B. J. Xu, W. Zhou, S. W. Liu, Y. Zhang and J. R. Xu, Org. Lett., 2011, 13, 556–559 CrossRef CAS PubMed.
- Y. J. Zhang, J. W. Sun, G. L. Zhuang, M. Ouyang, Z. W. Yu, F. Cao, G. X. Pan, P. S. Tang, C. Zhang and Y. G. Ma, J. Mater. Chem. C, 2014, 2, 195–200 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 999359–999361. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01492h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |