DOI:
10.1039/C4NJ01377H
(Paper)
New J. Chem., 2015,
39, 664-675
DNA-interacting and biological properties of copper(II) complexes from amidino-O-methylurea
Received
(in Montpellier, France)
14th August 2014
, Accepted 4th November 2014
First published on 14th November 2014
Abstract
The interaction of two copper(II) complexes, namely [Cu(L1)Cl2]2 (1) and [Cu(L2)Cl2]2 (2), where L1 = 1-amidino-O-methylurea and L2 = N-(benzyl)-amidino-O-methylurea, with DNA has been thoroughly investigated using different characterization techniques, including electronic absorption spectroscopy, viscosity measurements, fluorescence spectroscopy, circular dichroism spectroscopy, thermal denaturation, stoichiometric determination, gel electrophoresis and atomic-force microscopy. The coordination compounds exhibit DNA binding potential by non-intercalation and DNA-cleaving ability through the oxidative pathway. Indeed, both complexes display antibacterial properties (against three bacteria involved in human-food poisoning, i.e. Salmonella, E. coli and Campylobacter). Furthermore, their cytotoxicity has been tested against three cancer cell lines, which are the small cell lung carcinoma (NCI-H187), the oral cavity carcinoma (KB) and the breast adenocarcinoma (MCF-7) and it was revealed that they are more cytotoxic than cisplatin against the NCI-H187 cancer cell line.
1. Introduction
The application of metal coordination compounds in biology has received a great deal of attention from the scientific community, because the use of chemical approaches and small molecules to control or restore (altered) biological processes has shown great potential. For instance, inorganic small molecules that can bind to DNA via two possible binding modes, covalent bonds and non-covalent interactions (i.e. intercalation and non-intercalation) are of paramount importance to medicinal chemistry. DNA has become a privileged target for metal-based therapeutic agents,1 after the great impact of the square-planar anticancer agent [Pt(NH3)2Cl2], viz. cisplatin,2 in the treatment of various cancers, such as testicular, ovarian, head and neck as well as small cell lung cancers.3 The molecular mechanism of cisplatin is believed to involve in the coordination with the N7 guanine, leading to apoptosis.4 However, treatment with cisplatin causes severe side effects and (acquired) cell resistance to the drug representing a significant problem; therefore, the design and development of new metal complexes exhibiting higher efficiencies, lower toxicities and target-specific properties is required. Hence, numerous platinum and some non-platinum (ruthenium, gold, cobalt, iron, zirconium and copper) compounds have been described in the literature together with their cytotoxicity behavior.5–12 In particular, complexes based on copper, which is an important trace element in the human body involving essential enzymatic functions due to its Cu(II)/Cu(I) redox process,13 have attracted the attention of researchers worldwide.14–17 Moreover, copper is cheaper and gives the complexes with diverse geometries. Therefore, copper is suitable to be an alternative element providing the benefits in both design and various applications. A number of copper(II) coordination compounds have thus shown some interesting DNA-binding and cleavage properties, as well as antibacterial activities.12,18–22
Over the past few years, we have been focusing on the development of copper(II) complexes from amidino-O-alkylurea ligands to increase their biological properties. This ligand system shows high potential to form hydrogen-bonding interactions,23 which are of paramount importance in the biological system and significantly useful in designs of DNA-interacting agents. There are several copper(II) coordination compounds prepared from amidino-O-alkylurea derivatives including 1-n-butylamidino-O-alkylurea, 1-phenylamidino-O-methylurea, 1-phenylamidino-O-i-butylurea and 1-amidino-O-2-alkoxyethylurea as described in the literature, which display interesting biological properties such as DNA-binding abilities, or activities against fungal and bacterial pathogens.24–26 These evidences inform the great potential of such compounds for medicinal applications. Herein, two copper(II) complexes of 1-amidino-O-methylurea (L1) and N-(benzyl)-amidino-O-methylurea (L2) have been prepared which formed the dimeric copper(II) complexes [Cu(L1)Cl2]2 (1) and [Cu(L2)Cl2]2 (2) (Fig. 1) and adopted an approximate square-pyramidal geometry.27 Their binding ability to nucleobases has been previously investigated and found that they are indeed capable of binding to the N3 cytosine by the replacement of the chloride ligands.28 In addition, the related monomeric copper(II) complexes [Cu(L1)2Cl2] and [Cu(L2)2Cl2] with a square-planar geometry29 have also been reported to exhibit potential DNA-interacting properties and antimicrobial activities against Campylobacter.30 To continue our research, the complexes 1 and 2 have been intensively studied in terms of the DNA interacting and biological properties. Furthermore, a difference in the N-substituted sidearm, –NH2 and –NHCH2Ph of the L1 and L2 ligands, respectively, has been examined the effect on both properties.
 |
| | Fig. 1 Proposed structures of the copper(II) complexes 1 (R = H) and 2 (R = CH2C6H5). | |
In the present study, the potential DNA interactions and antiproliferative behavior of the dimeric complexes 1 and 2 have been examined. Several techniques including electronic absorption titration, viscosity measurements, fluorescence spectroscopy, circular dichroism spectroscopy, thermal analysis and stoichiometric determination have been employed to monitor their prospective interaction with calf thymus DNA (CT-DNA). Their potential DNA-cleaving abilities have been investigated by gel electrophoresis and atomic-force microscopy (AFM), using pBR322 plasmid DNA. Their antimicrobial properties against three Gram-negative bacteria (namely Salmonella, E. coli and Campylobacter) have been determined applying the agar-well diffusion method. In addition, their cytotoxicity towards three human cancer cell lines, i.e. small cell lung carcinoma (NCI-H187), epidermoid carcinoma of the oral cavity (KB) and breast adenocarcinoma (MCF-7) has been evaluated using the resazurin microplate assay (REMA).
2. Experimental
2.1. Materials and instrumentation
The sodium salt of calf thymus DNA (CT-DNA, Type I fibrous) was purchased from Sigma-Aldrich. Plasmid pBR322 DNA (4361 bp, 0.25 μg μL−1) was obtained from Bio Basic INC and Roche Farma, S.A. Ethidium bromide (EB) solution (10 mg mL−1) and tris(hydroxymethyl)aminomethane (Tris base) were purchased from Promega. Agarose (D-1, Low EEO) was purchased from Pronadisa. N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) was obtained from Sigma-Aldrich. All reagents were of molecular biology grade and used without further purification. The coordination compounds [Cu(L1)Cl2]2 (1) and [Cu(L2)Cl2]2 (2) were prepared as reported earlier.28
Electronic absorption spectra were recorded using an Agilent 8453 UV-Vis spectrophotometer. Fluorescence determination was performed on a Shimadzu RF–5301PC spectrofluorophotometer. Circular dichroism (CD) spectra were recorded on a Jasco J-810 spectropolarimeter. The amount of copper for each stoichiometric determination was determined using a Perkin Elmer AAnalyst 100 atomic absorption spectrometer. The electrophoretic band intensities were visualized with a Bio-Rad Gel Doc 2000 system using the LABWORK software. The AFM images were obtained by using a Nanoscope V Multimode 8 AFM (Bruker AXS) operating in the PEAK FORCE tapping mode. Commercial Si-tip on Nitride lever cantilevers (SNL, Bruker) with a force constant of 0.4 N m−1 was used.
2.2. DNA-binding studies
The DNA stock solution, prepared in Tris-buffer (containing 5 mM Tris-HCl and 50 mM NaCl at pH 7.1), gave a UV-absorbance ratio A260/A280 of about 1.8–1.9 (where A260 and A280 are the absorbances of a DNA sample at 260 and 280 nm, respectively), indicating that DNA was sufficiently free of protein contamination.31 The DNA stock solution was kept at 4 °C and used within 4 days. A 10-fold dilution of the DNA concentration was determined spectrophotometrically at 260 nm, by using the molar extinction coefficient value of 6600 M−1 cm−1.32 The stock solutions of 1 and 2 prepared in Tris-buffer were highly soluble and showed the similar blue color to their solid phase, thus, suggesting the stability of both compounds under these conditions. The stock solution was freshly prepared before utilization. All experiments were carried out in Tris-buffer at pH 7.1, except for the thermal denaturation study in HEPES-buffer (containing 40 mM HEPES and 10 mM MgCl2 at pH 7.1).
2.2.1. Electronic absorption titrations.
Absorption titration experiments were carried out with a constant concentration of the copper(II) complexes, viz. 50 μM, and varying the concentration of CT-DNA (0–200 μM) in Tris-buffer. The complex and DNA solutions were incubated at 37 °C for 24 h. Subsequently, the spectra were recorded using a UV-Vis spectrophotometer at ambient temperature. To subtract the absorption due to the DNA itself (in each sample), solutions of free CT-DNA (namely in the absence of copper compound) at the corresponding concentrations (0–200 μM) were used as blanks before recording the absorption band of each sample. To compare the DNA-binding strength of the two complexes, their intrinsic binding constant (Kb) was calculated from the plot of [DNA]/(εa − εf) vs. [DNA] using eqn (1); Kb is given by the ratio of the slope to the y intercept.33| | 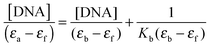 | (1) |
In eqn (1), [DNA] is the concentration of DNA, εa is given by Aobsd/[Cu], εf is the extinction coefficient of the free metal complex and εb is the extinction coefficient of the metal complex in the fully bound form.
2.2.2. Viscosity measurements.
Viscosity experiments were carried out using an Ubbelodhe viscometer, immersed in a water bath at 37 ± 0.1 °C. The viscosity of a 100 μM solution of CT-DNA was determined in the presence of the complexes, using different [Complex]/[DNA] ratios, in the range of 0.00–1.00 with 0.10 intervals. The flow time for each sample was recorded in triplicate with a digital stopwatch, and an average flow time was calculated. The relative viscosity values (η) were determined from the flow time of the DNA-containing solutions (t) corrected by the flow time of the free buffer (t0): η = (t − t0)/t0.34 The data are presented as the plot of the relative viscosity, i.e. (η/η0)1/3vs. [Complex]/[DNA]. η0 and η are the viscosity of the free DNA solution and the DNA–complex solution, respectively.
2.2.3. Fluorescence spectroscopy.
Emission intensity measurements of ethidium bromide (EB) in the presence of free CT-DNA and in the presence of complexes 1 and 2 were performed in Tris-buffer. The CT-DNA solution (50 μM) was pretreated with EB (30 μM) for 30 min at room temperature and stored in the dark. Solutions of the complexes were then prepared (from the previous stock solution of EB–DNA) with the [Complex]/[DNA] ratios in the range of 0.0–2.0 with 0.2 intervals; these solutions were kept in dark for 30 min before measurements. The emission intensities (between 550 and 700 nm) were obtained through excitation at 500 nm. The association binding constant (Ka) and the number of binding site (n) can be calculated according to eqn (2).35| |  | (2) |
Where F0 and F are the fluorescence intensities of the DNA–EB complex in the absence and the presence of copper(II) compounds, respectively. [Q] is the concentration of copper(II) complexes and n is the number of binding site. The linear plot of log(F0 − F)/F vs. log[Q] gives Ka and n as, respectively, the value of the antilogarithm of the intercept and the slope.
2.2.4. Circular dichroism (CD) spectroscopy.
Solutions of CT-DNA (100 μM) with the [Complex]/[DNA] ratios of 0.0, 0.5 and 1.0 were incubated at 37 °C for 24 h. The CD spectra of these solutions were recorded from 200 to 400 nm using a quartz cuvette with an optical path length of 5 mm and a scanning rate of 50 nm min−1. The data were collected in triplicate with a time constant of 1 s and a spectral bandwidth of 1.0 nm. The background signal due to the buffer was subtracted.
2.2.5. Thermal denaturation.
The DNA denaturation experiments were performed in HEPES-buffer in the presence of the copper(II) complexes, applying the [Complex]/[DNA] ratios of 0.5, 1.0, 1.5 and 2.0, by monitoring the variation of the absorption intensity of DNA (100 μM) at 259 nm when the temperature was increased from 25.0 to 100.0 °C with interval temperature of 5 °C. The data are presented as a curve of the relative absorbance intensity (A/A0, A0 = absorbance value at 25 °C) at 259 nm vs. the temperature. The melting temperature (Tm) was determined from the maximum of the first derivative curve or tangentially from the graph at the midpoint of the transition curve. ΔTm was defined as the difference between Tm of the free DNA and Tm of the bound DNA.
2.2.6. Stoichiometric determination.
The copper(II)–DNA complex stoichiometry was determined applying a procedure described in the literature.36 The complex solution (3 mM, 1 mL) was added to the CT-DNA solution (ca. 3 mM, 1 mL), and the resulting mixture was incubated at 37 °C for 24 h. Precipitation of the DNA–copper(II) complex was achieved by adding absolute ethanol (4 mL) and aqueous NaCl (2 mM, 0.2 mL). The solution containing the precipitate was stored at −70 °C for 1 h, and the precipitate was subsequently isolated by centrifugation at 4 °C (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 30 min). The supernatant was separated by pouring it out slowly. Next, deionized water (25 mL) was added to dissolve the DNA–copper(II) precipitate, and the DNA concentration was calculated (from triplicate experiments) by the absorption intensity at 259 nm using ε = 6600 M−1 cm−1.32 The amount of copper(II) was determined by atomic absorption spectroscopy, hence providing the Cu (mmol)/DNA (mol base) ratio.
000 rpm, 30 min). The supernatant was separated by pouring it out slowly. Next, deionized water (25 mL) was added to dissolve the DNA–copper(II) precipitate, and the DNA concentration was calculated (from triplicate experiments) by the absorption intensity at 259 nm using ε = 6600 M−1 cm−1.32 The amount of copper(II) was determined by atomic absorption spectroscopy, hence providing the Cu (mmol)/DNA (mol base) ratio.
2.3. Nuclease activity
2.3.1. Gel electrophoresis.
The DNA-cleaving properties of the complexes in the absence and presence of ascorbic acid (H2ASC) were investigated using agarose gel electrophoresis. Plasmid pBR322 DNA (0.2 μg, ∼30 μM base pairs) was treated with the copper(II) complex (in the range of 200–1000 μM). All samples (10 μL in Tris-buffer) were incubated at 37 °C for 24 h. Then, a loading buffer (2 μL) consisting of 0.25% bromophenol blue, 0.25% xylene cyanol and 30% glycerol was added. The mixture was subjected to gel electrophoresis on 2.5% agarose in a 1× TAE buffer (40 mM Tris-acetate and 1 mM EDTA) at 50 V for 4 h. After electrophoresis, the gel was stained with ethidium bromide for 5 min and then photographed under UV light. The intensities of the supercoiled DNA (Form I) were corrected by multiplying with the factor value of 1.22 (for the plasmid pBR322 DNA) because the intercalation between EB and Form I DNA is relatively weak compared to that of nicked (Form II) and linear (Form III) DNA.37 The efficiency of DNA cleavage was obtained according to the modified eqn (3).38 The percentage of DNA cleavage activity derived from the volume (band intensity × area) of supercoiled DNA (Form I) was calculated by the following eqn (3).| | | %DNA cleavage activity = {[(volume of SC-DNA)control − (volume of SC-DNA)sample]/(volume of SC-DNA)control} × 100 | (3) |
In the presence of ascorbic acid (100 μM), the complexes (10–600 μM) were incubated for 1 h and electrophoresed in 0.8% agarose gel in a 1× TAE buffer at 50 V for 1.5 h.
2.3.2. Atomic-force microscopy (AFM).
Plasmid pBR322 DNA (0.2 μg), heated at 60 °C for 15 min to obtain an open circular form, was incubated with the copper(II) complexes (200 μM) containing ascorbic acid (100 μM) in 20 μL HEPES-buffer at 37 °C for 1 h. Milli-Q water and all solutions for the AFM studies were filtered through 0.2 μm FP030/3 filters (Scheicher and Schuell GmbH, Germany) to obtain clear AFM images. After incubation, a drop (8 μL) of each sample was placed onto peeled mica disks (PELCO Mica Discs, 9.9 mm diameter; Ted Pella, Inc. California, USA) and allowed to adsorb for 2 min at room temperature. The samples were rinsed for 5 s with a stream of Milli-Q water directed onto the surface, which was subsequently blown dried with compressed argon before imaging.
2.4.
In vitro antibacterial activity
The screening of the in vitro antibacterial activity of the copper(II) complexes (50 mg mL−1) against three Gram-negative bacteria (Salmonella, E. coli and Campylobacter) was performed by the agar-well diffusion method.39 Active culture of the bacteria was transferred into 0.75% (w/w) semisolid brucella agar (10 mL) at 50 °C. Subsequently, the incubated medium was swirled to distribute the cell culture of bacteria and held at room temperature for 30 min. A well of 6 mm diameter was made aseptically. The plates were placed at 37 °C for 48 h under the appropriate conditions to allow the cell culture growth. Each complex was dissolved in water to give the final concentration of 0.50% and transferred into a well aseptically. The inhibitory activity of the complex on bacteria was obtained from a clearing zone around the disc. The minimum inhibitory concentration (MIC) values were also determined by the two-fold serial dilution method in liquid media containing the tested complexes.40 The experiments were carried out in duplicate.
2.5. Anticancer activity assay
Cancer-cell growth inhibition tests were carried out against three human cancer cell lines, namely the KB (epidermoid carcinoma of oral cavity, ATCC CCL-17), MCF-7 (breast adenocarcinoma, ATCC HTB-22) and NCI-H187 (small cell lung carcinoma, ATCC CRL-5804) cell lines, using the resazurin microplate assay (REMA), as described in the literature.41 Ellipticine, doxorubicin and tamoxifen were used as positive controls, and 0.5% DMSO was used as a negative control. To investigate the potential cytotoxic behaviors of the copper(II) complexes, cells at a logarithmic growth phase were harvested and diluted in fresh medium to 7 × 104 cells mL−1 for KB, and 9 × 104 cells mL−1 for MCF-7 and NCI-H187. Subsequently, 5 μL of the test sample diluted in 5% DMSO, and 45 μL of cell suspension were added to 384-well plates and then incubated at 37 °C in 5% CO2. After the incubation period (3 days for KB and MCF-7, and 5 days for NCI-H187), 12.5 μL of 62.5 μg mL−1 resazurin solution were added to each well, and the plates were then incubated at 37 °C for 4 h. The fluorescence signal was measured using a SpectraMax M5 multi-detection microplate reader (Molecular devices, USA) at the excitation and emission wavelengths of 530 nm and 590 nm, respectively. The inhibition percentage of the cell growth was calculated with eqn (4).| |  | (4) |
where FUT and FUC are the mean fluorescence units from, respectively, the cells treated and untreated with the copper(II) complexes. The anticancer activity of the complexes was expressed as the 50% inhibitory concentration (IC50), which is determined from the dose–response curves using the SOFTMax Pro software (Molecular devices, USA). The plotted data were obtained from six different concentrations of two-fold serially diluted test samples. The complexes with IC50 > 50 μg mL−1 were considered to be inactive.42
3. Results and discussion
3.1. DNA-binding studies
DNA is the classical pharmacological target of metal-based anticancer agents; therefore, the study of the potential interaction of coordination compounds with DNA is of paramount importance for the development of molecules with potential medical applications. There are four possible ways in which small molecules can bind to double-stranded DNA; (i) intercalation between two adjacent base pairs and perpendicular to the helical axis; (ii) outside-edge binding to the sugar-phosphate backbone of the helix through electrostatic interactions; (iii) groove binding with functional groups into either the major or minor groove43 and (iv) the covalent interaction between DNA and metal complexes at the nitrogen atoms of nucleobases.4 Therefore, several characterization techniques have been employed to study the interactions of copper(II) complexes 1 and 2 with CT-DNA.
3.1.1. Electronic absorption titrations.
Intensity changes and wavelength shifts are observed in the electronic absorption spectra of 1 and 2 when the concentration of DNA is increased, thus indicating the interaction of the compounds with the biomolecular helix (Fig. 2). The intercalation of a small molecule into double-stranded DNA generally results in hypochromism, viz. a decrease in absorption intensity associated with a red shift, which arises from strong π → π* stacking interactions between the planar aromatic ligand and DNA base pairs.44 In contrast, non-intercalative interactions will enhance the absorption intensity (hyperchromism) that is ascribed to DNA conformation changes upon complex binding, which lead to the alteration of the structure of the DNA double-helix.45
 |
| | Fig. 2 Absorption titration spectra of 50 μM solutions of 1 (a) and 2 (b), in the absence (⋯) and presence (—) of increasing amounts of CT-DNA (0–200 μM). The arrows show the change upon DNA concentration increase in Tris-buffer (pH = 7.1), after incubation at 37 °C for 24 h. | |
The electronic spectra of 1 and 2 (Fig. 2) actually display intense absorption bands at λmax = 226 and 209 nm, respectively, which are assigned to π–π* intraligand transitions.30,46–48 Upon increasing the DNA concentration, both 1 and 2 show hyperchromism with a blue shift of 2 and 5 nm, respectively. Such a behavior suggests that both compounds most likely interact through a non-intercalative binding mode with DNA, probably via (i) hydrogen bonds between base pairs and the ligands; (ii) electrostatic attractions between the positively charged metal center and the negatively charged phosphate backbone and (iii) the coordination bonding of the copper(II) centers with cytosine nucleobases of DNA as previously reported.28 Furthermore, the comportments of 1 and 2 (Table 1) are comparable to those of related copper(II) compounds described in the literature.25,26,30
Table 1 Effect of the addition of CT-DNA on the UV absorption bands of a series of compounds and corresponding intrinsic binding constants
| Compound |
λ
max (nm) |
Effect on intensity |
Shift (nm) |
K
b
(M−1) |
Ref. |
|
L3 = 1-phenylamidino-O-methylurea.
L4 = 1-phenylamidino-O-i-butylurea, tmen = N,N,N′,N′-tetramethylethylenediamine.
L5 = 1-amidino-O-2-methoxyethylurea.
L6 = 1-amidino-O-2-ethoxyethylurea.
L7 = 1-amidino-O-2-buthoxyethylurea.
|
| Ethidium bromide (EB) |
285 |
Hypochromism |
+1 |
5.35 × 106 |
This work |
| [Cu(L1)Cl2]2, (1) |
226 |
Hyperchromism |
−2 |
5.63 × 104 |
This work |
| [Cu(L2)Cl2]2, (2) |
209 |
Hyperchromism |
−5 |
1.07 × 105 |
This work |
| [Cu(L1)2]Cl2 |
228 |
Hyperchromism |
0 |
5.67 × 104 |
30
|
| [Cu(L2)2]Cl2 |
229 |
Hyperchromism |
−1 |
1.16 × 105 |
30
|
| [Cu(L3)2(H2O)]2(Cl2)2a |
232 |
Hyperchromism |
0 |
1.30 × 105 |
25
|
| [Cu(L4)(tmen)]2(Cl2)2·2H2Ob |
232 |
Hyperchromism |
0 |
1.50 × 106 |
25
|
| [Cu(L5)2](ClO4)2·H2Oc |
224 |
Hyperchromism |
−2 |
4.08 × 104 |
26
|
| [Cu(L6)2](ClO4)2·2/3H2Od |
224 |
Hyperchromism |
−2 |
1.39 × 104 |
26
|
| [Cu(L7)2](ClO4)2·H2Oe |
225 |
Hyperchromism |
−3 |
3.06 × 104 |
26
|
The intrinsic binding constants (Kb) are 5.63 × 104 M−1 for 1 and 1.07 × 105 M−1 for 2, hence indicating that 2 interacts stronger than 1 with DNA. This difference is probably due to the presence of phenyl rings in the ligand L2, which may provide additional π-stacking interactions with DNA bases. The DNA-binding affinities of 1 and 2 are apparently lower than that of the intercalating agent EB (Table 1); however, it should be mentioned that the copper(II) compounds may interact with a different binding mode (not as an intercalator). Although, the electronic absorption studies have confirmed that the complexes 1 and 2 can bind to DNA, it is necessary to carry out further experiments to prove the binding mode.
3.1.2. Viscosity measurements.
Viscosity measurements were carried out to clarify further the interaction mode of the two compounds with DNA. For instance, partial and/or non-intercalation of the complexes may result in DNA bending or kinking, which gives rise to a reduction of the effective DNA length associated with a change of viscosity of the DNA solution.49 The intercalating agent EB produces an increase of the relative viscosity for a ratio [EB]/[DNA] within the range of 0.0–0.3 (Fig. 3), as a result of the separation of DNA base pairs to accommodate the binding molecule. When [EB]/[DNA] ≥ 0.4, the relative viscosity does not change anymore, most likely because the DNA binding sites have reached saturation. For 1 and 2, their effect on the viscosity of a CT-DNA solution is reversed (Fig. 3). Indeed, when the complex concentration is increased, the relative viscosity of the DNA solution steadily decreases, again indicating that partial and/or non-intercalative modes of interaction take place with 1 and 2. This behavior is similar to those observed copper(II) complexes described in the literature.26,30,48 The viscosity decrease caused by 2 is lower than that induced by 1, thus suggesting that the bending or the kinking effect of 2 on the DNA helix is inferior. This difference is probably due to the phenyl ring of the ligand L2 which can also interact with DNA through partial intercalation, therefore enhancing the binding of 2, as observed as well by UV-Vis spectroscopy.
 |
| | Fig. 3 Effect of the addition of increasing amounts of ethidium bromide (♦), 1 (■) and 2 (▲) on the relative viscosity of CT-DNA (100 μM), in Tris-buffer (pH = 7.1) at 37 °C. | |
3.1.3. Effect of the complexes on the emission spectra of the DNA–EB complex.
EB fluoresces when bound to DNA (λem = 592 nm). Hence, the binding of a second molecule may lead to fluorescence quenching through displacement of EB and/or by accepting the excited state electron of the EB through a photo-electron transfer mechanism.50,51 The emission spectra of the EB–DNA complex with increasing concentrations of 1 and 2 are shown in Fig. 4. A moderate decrease of the intensity is noticed, hence suggesting that the compounds slightly interact with DNA. However, it should be mentioned here that the mode(s) of interaction of 1 and 2 most likely differ from that of EB;19,52 two potential types of binding, i.e. pathways (i) and (ii), are shown below.| | | CuL + DNA–EB ⇌ DNA–CuL + EB K | (i) |
and/or| | | CuL + DNA–EB ⇌ EB–DNA–CuL K′ | (ii) |
The UV-Vis studies and viscosity measurements suggest that both 1 and 2 most likely interact with DNA by non-intercalative modes. Hence, the main quenching process of the DNA–EB complex by the copper(II) compounds probably involves the formation of a non-fluorescent [DNA–EB–copper complex] species, i.e. pathway (ii), through a so-called fluorescence static quenching process. The association binding constant (Ka) and the number of equivalent binding sites (n) on the biomolecule can be calculated using eqn (2).35 The binding parameters for 1 and 2 are Ka = 3.73 × 103 M−1; n = 1.02 and Ka = 1.64 × 104 M−1; n = 1.16, respectively. These values indicate that the two complexes have distinct binding affinities towards DNA, as also suggested by the UV-Vis data (see Kb values above). Accordingly, 2 exhibits greater DNA-binding potential than 1.
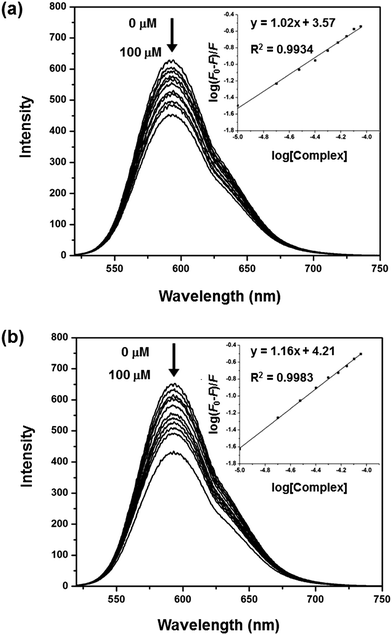 |
| | Fig. 4 Fluorescence emission spectra of the EB–DNA complex in Tris-buffer (pH = 7.1) in the absence and presence of 1 (a) and 2 (b). [EB] = 30 μM, [DNA] = 50 μM, [Complex] = 0–100 μM, λex = 500 and λem = 592 nm. | |
3.1.4. Circular dichroism (CD) spectroscopy.
To examine the potential DNA conformational changes induced by the metal-based compounds, CD spectra of CT-DNA in the absence and presence of the complexes have been recorded. The CD spectrum of B-DNA displays a positive band at 277 nm attributable to base stacking and a negative band at 246 nm that arises from the right-handed helicity of DNA.53 Any modification(s) of the base-stacking pattern or helicity of the strands will produce either a change in the band position or their intensity, or even both. It is known that simple electrostatic or groove-binding interactions of small molecules with DNA do not cause any (significant) alteration of the intensity of the two bands. Conversely, intercalators augment the intensity of these bands.54 The CD spectra of CT-DNA in the presence of 1 and 2, using the [Complex]/[DNA] ratios of 0.0, 0.5 and 1.0, are depicted in Fig. 5. Upon increasing the complex concentration, the intensity and the position of the negative band are modified for 1 while they remain unchanged for 2. For the positive band, a slight red shift is observed, from 277 to 280 nm for 1, and from 277 to 279 nm for 2, with a small decrease in ellipticity. This red shift ascribed to π–π* transitions,55 associated with an ellipticity diminution, indicates that the complexes somewhat modify the DNA-base stacking, without provoking significant changes in the supramolecule helicity. These results give further evidence that both complexes interact with DNA via a non-intercalative binding mode.
 |
| | Fig. 5 CD spectra of CT-DNA (100 μM) in the absence and presence of 1 (a) and 2 (b), at the [Complex]/[DNA] ratios of 0.0 (—), 0.5 (---) and 1.0 (⋯). | |
3.1.5. DNA-melting analysis.
The thermal behavior of DNA in the presence of small molecules can give valuable information about their interaction with the double helix. Double-stranded DNA dissociates when the temperature is raised, as the result of the breaking of hydrogen bonds between base pairs. This DNA denaturation is associated with a hyperchromic effect in the absorbance spectrum of the biomolecule. The temperature at which 50% of the DNA denatured is defined as the melting temperature, i.e. Tm. Generally, the intercalation of small molecules between base pairs gives rise to a stabilization of the DNA structure, which is denoted by an augmentation of Tm; actually, ΔTm values in the range of 10–14 °C can be observed, depending on the DNA-binding affinity of the interacting molecule.56 ΔTm < 10 °C would be indicative of weak DNA-binding abilities of the complexes that would most likely interact with DNA through a non-intercalative pathway.57 Conversely, a decrease in Tm of DNA will arise from DNA distortions induced by the interacting molecules.58 The thermal denaturation profiles of CT-DNA obtained in the absence and presence of 1 and 2 are shown in Fig. 6, and the consequent melting temperatures and melting temperature variations are listed in Table 2.
 |
| | Fig. 6 DNA melting curves of CT-DNA (100 μM) (■) in the absence and presence of 1 (a) and 2 (b), at the [Complex]/[DNA] ratios of 0.5 (●), 1.0 (▲), 1.5 (▼) and 2.0 (♦). | |
Table 2 Melting temperatures (Tm) of CT-DNA in the presence of 1 and 2, at different [Complex]/[DNA] ratios
| [Complex]/[DNA] |
T
m (°C) |
ΔTma (°C) |
|
1
|
2
|
1
|
2
|
|
T
m of CT-DNA = 82.1 °C.
|
| 0.5 |
82.6 |
82.4 |
+0.5 |
+0.3 |
| 1.0 |
82.7 |
82.6 |
+0.6 |
+0.5 |
| 1.5 |
82.8 |
82.9 |
+0.7 |
+0.8 |
| 2.0 |
83.3 |
83.4 |
+1.2 |
+1.3 |
The Tm value of free CT-DNA is 82.1 °C. Upon increase of the complex concentration, a slight increase of Tm is observed. The corresponding ΔTm values are in the range of 0.5–1.2 °C for 1, and 0.3–1.3 °C for 2. The low ΔTm values (close to zero) suggest that the DNA-binding mode of the complexes is non-intercalative.
3.1.6. Stoichiometry of the DNA-binding agents.
Determination of the DNA-binding stoichiometry of the copper(II) complexes, expressed as Cu (mmol)/DNA (mol base), is an additional method to obtain more information regarding their interaction with the double helix. The stoichiometric ratio can be determined from atomic absorption and UV-Vis spectroscopic measurements. Hence, 1 and 2 exhibit Cu (mmol)/DNA (mol base) ratios of 54 and 46, respectively. These values can be analyzed using the Cu (mmol)/DNA (mol base) ratios of known copper(II) compounds, namely [Cu(H2O)6]2+ (ratio > 150) and Cu(II)-dipeptide complexes (ratio < 42).36 The large ratio observed for [Cu(H2O)6]2+ indicates a poorly selective DNA binding, most likely owing to the fact that these cationic species interact with the negatively-charged phosphate groups on the DNA backbone. In contrast, the small value for Cu(II)-dipeptide complexes suggests more efficient DNA interactions. In the present case, complex 1 appears to interact electrostatically with DNA (as [Cu(H2O)6]2+), whereas 2, which contains aromatic moieties, may partially intercalate DNA, which is reflected by the Cu (mmol)/DNA (mol base) ratio lower than 50.
3.2. Nuclease activity of the complexes towards plasmid pBR322 DNA
3.2.1. Gel electrophoresis.
Agarose gel electrophoresis is a common and simple technique that is used to visualize (damaging) interactions of the complexes with DNA. For instance, different (typical) shapes of plasmid DNA can be observed by electrophoresis, namely the normal supercoiled form (Form I), the circular nicked form (Form II that results from a single-strand cut), and the linear form (Form III, which is obtained when both strands are broken). Hence, Form I will migrate more rapidly than Form III, and Form II, which is the bulkiest, will be the slowest in the gel (as the separation is not only by charge but also by size).
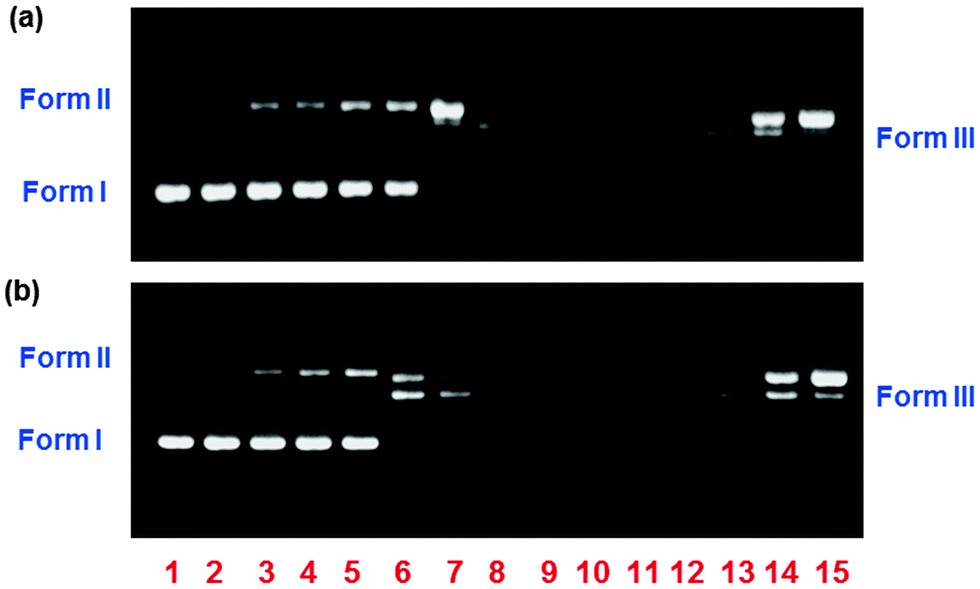
The potential interacting/cleaving properties of 1 and 2 were assessed by electrophoretic mobility measurements with plasmid pBR322 DNA. Fig. 7 shows the results of electrophoretic separations of the plasmid DNA induced by the different concentrations of the complexes 1 (Fig. 7a) and 2 (Fig. 7b) after 24 h for incubation and their behaviors are also comparable. Band intensities of Form II are significantly enhanced when treated by 1 and 2 from 200 μM to 400 μM (lanes 2–3), indicating that both complexes can cleave the supercoiled form (Form I) to nicked form (Form II). In the concentration from 600 μM to 1000 μM (lanes 4–6), both Forms I and II gradually decrease, suggesting the degradation of plasmid DNA into undetectable single-stranded pieces. Their cleaving behaviors at a large amount differ from the previous reports in which square planar copper(II) complexes ([Cu(L1)2]Cl2 and [Cu(L2)2]Cl2) show DNA cleavage from Form I to Form II.30 Band intensities were measured to determine the amount of each DNA form. The percentage of DNA cleavage activity (Fig. 8) was calculated by eqn (3). It is found that complex 2 shows considerably higher DNA cleavage efficiency than 1, demonstrating that 2 displays better cleaving activity than 1.
 |
| | Fig. 7 Agarose gel electrophoresis images of pBR322 plasmid DNA incubated for 24 h at 37 °C in Tris-buffer (5 mM Tris-HCl/50 mM NaCl, pH = 7.1) with increasing concentrations of 1 (a) and 2 (b), without adding reducing agents. Lane 1: DNA control; lanes 2–6: DNA + [complex] (200, 400, 600, 800 and 1000 μM, respectively). | |
 |
| | Fig. 8 Percentage of DNA cleavage induced by complexes 1 and 2. | |
It is known that the DNA cleaving potential of copper(II) complexes can be enhanced by adding the exogenous agents such as hydrogen peroxide and ascorbic acid (H2ASC), which act as reducing agents. Herein, H2ASC is used to mimic the reducing environment that can be found inside the cells. Hence, H2ASC will induce the formation of copper(I) species (from 1 and 2), which would allow the potential formation of reactive oxygen species (ROS) that are able to cleave DNA. When H2ASC is added, completely different circumstances are achieved (Fig. 9). The corresponding H2ASC (100 μM) does not affect free DNA (lane 2). In the presence of H2ASC, Form II can be observed at the low concentration (10 μM) for both complexes. Moreover, Form III is also found at 50 μM for 1 (lane 7, Fig. 9a) and 40–50 μM for 2 (lanes 6, 7, Fig. 9b). At 60–100 μM (lanes 8–12), both 1 and 2 are capable of inducing the DNA degradation of plasmid DNA into very small single-stranded pieces which are observed as smear on gel. Upon enhancing the concentration of both from 200 μM to 600 μM (lanes 13–15), very strange features can be observed; indeed, the bands ascribed to Forms II and III reappear and increase.
 |
| | Fig. 9 Agarose gel electrophoresis images of pBR322 plasmid DNA incubated for 1 h at 37 °C in Tris-buffer (5 mM Tris-HCl/ 50 mM NaCl, pH = 7.1) with increasing concentrations of 1 (a) and 2 (b), in the presence of a reducing agent, i.e. ascorbic acid (H2ASC; 100 μM). Lane 1: DNA control; lane 2: DNA + H2ASC; lanes 3–15: DNA + H2ASC + [complex] (10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 400 and 600 μM, respectively). | |
As a result, it is preliminarily suggested that both 1 and 2 at concentrations ≤100 μM with H2ASC (100 μM) are capable of promoting DNA cleavage through an oxidative DNA damage pathway which gives the reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), hydroxyl radical (OH˙), singlet oxygen (1O2) or superoxide (O2−) that cleave DNA.58 The roles of copper(II) complexes showing their enzymatic behaviors can be explained by the proposed mechanism of in situ generation of H2O2 by oxidation of the cuprous complex under aerobic conditions in the presence of ascorbic acid acting as a reducing agent as shown below.59
| | | H2ASC + H2O ⇌ HASC− + H3O+ | (iii) |
| | | HASC− + 2Cu(II) → ASC + 2Cu(I) + H+ | (iv) |
| | | 2Cu(I) + O2 + 2H+ → 2Cu(II) + H2O2 | (v) |
| | | HASC− + O2 + H+ → ASC + H2O2 | (iv) + (v) |
The first step (iii) is the hydrolysis of ascorbic acid (H2ASC) generating the ascorbate form (HASC−), the second step (iv) is the reduction of the Cu(II) complex to the Cu(I) complex by HASC−. Then, the reaction of the cuprous state with dioxygen (v) leads to the generation of H2O2. Overall reactions (iv) + (v) correspond to the autooxidation of ascorbate which may involve a single two-electron oxidation step.60 Hydrogen peroxide then reacts with another equivalent of the Cu(I) through the Fenton reaction (vi) which gives the reactive species, hydroxyl radicals, responsible for DNA strand scission.59
| | | Cu(I) + H2O2 → Cu(II) + OH− + OH˙ | (vi) |
Hence, an excess amount of the copper(II) complexes (200–600 μM) cannot be reduced by the limited amount of ascorbic acid and may induce several undesirable secondary reactions. One of them is the formation of Cu(OH)2, which may precipitate, resulting in the reduction of DNA cleavage efficiency. The unusual phenomenon that is the reverse DNA cleavage altering from the Form III to Form II most likely arised from the fact that the concentration of copper(II) complexes (200–600 μM) is huge when compared with H2ASC (100 μM). This situation has also been found in the Fenton reaction catalyzed by ferrous ions which produced Fe(OH)3 precipitates due to ferrous ion excess.61,62 In summary, complexes 1 and 2 are both capable of cleaving plasmid DNA through the oxidative pathway and 2 also shows better activity than 1 in both with and without reducing agents corresponding to their DNA binding capabilities. The reason for this may arise from the presence of phenyl rings on ligand L2 which possibly gives additional binding positions on DNA.
3.2.2. AFM analysis of the morphological changes of pBR322 DNA induced by the complexes.
The consequence of the interaction of 1 and 2 on the DNA structure has been analyzed further using atomic-force microscopy (AFM). Thus, pBR322 plasmid DNA (0.2 μg) was incubated for 1 h at 37 °C with 1 and 2, at a complex concentration of 200 μM in the presence of ascorbic acid (H2ASC, 100 μM) in HEPES-buffer (pH = 7.1). After the incubation time, the samples were subsequently investigated by AFM and the corresponding images are depicted in Fig. 10. It shows that the original open circular structures of DNA (Fig. 10a) are not affected by H2ASC (Fig. 10b). In contrast, both compounds 1 and 2 with H2ASC induce significant alterations of the DNA morphology; in fact, the biomolecule is completely broken into small pieces (Fig. 10c and d), corroborating the data achieved by electrophoresis (see lane 13 in Fig. 9), and therefore confirming that the complexes act as efficient DNA cleavers.
 |
| | Fig. 10 AFM images of (a) pure pBR322 plasmid DNA; (b) plasmid DNA + H2ASC; (c) plasmid DNA + H2ASC + 1; (d) plasmid DNA + H2ASC + 2. Conditions: 0.2 μg DNA per sample; [H2ASC] = 100 μM; [Complex] = 200 μM. Incubation for 1 h at 37 °C in HEPES-buffer (pH = 7.1). | |
3.3.
In vitro antibacterial activity
According to the evidence showing DNA binding and cleaving behaviors of the copper(II) complexes, it guided us that both 1 and 2 may inhibit the growth of bacteria. Some copper(II) complexes containing amidino-O-alkylurea derivatives show antibacterial activities towards Bacillus,24E. coli, K. pneumonia and P. mirabilis,25 and Campylobacter.30 Therefore, the potential antibacterial activities of complexes 1 and 2 against Salmonella, E. coli and Campylobacter (which are known as human-food poisoning bacteria) have been examined. The corresponding results are presented as inhibition zone diameters (IZDs) and minimum inhibitory concentrations (MICs) (Table 3). Complexes 1 and 2 are capable of inhibiting the growth of all tested bacteria. Hence, 1 exhibits MIC values of 25.00 mg mL−1 for Salmonella and E. coli. Its inhibition activity is significantly higher towards Campylobacter, with a MIC value of 3.12 mg mL−1. These data perfectly correlate with the corresponding IZDs; indeed, the larger inhibition zone is achieved with Campylobacter, which indicates that 1 is more active against this bacterium. For complex 2, the MIC values obtained suggest that its antibacterial properties follow the order Campylobacter > Salmonella > E. coli. Interestingly, the antibacterial activity of 2 towards Salmonella is greater than that of 1, which is in agreement with the better DNA-binding and cleavage properties of 2. Finally, it should be mentioned that dimeric [Cu(L1)Cl2]2 (1) and [Cu(L2)Cl2]2 (2) are less efficient against Campylobacter than the corresponding monomeric complexes [Cu(L1)2Cl2] and [Cu(L2)2Cl2] which have been previously reported,30 it is probably because the latter display slightly better DNA-interacting properties. Enrofloxacin antibacterial drug was also tested as a reference. However, the results indicate that both 1 and 2 may be applied for the growth inhibition of a series of bacteria involved in human-food poisoning. Although, their obtained antibacterial activities expressed by MIC values are in the mg mL−1 (mM) range and much too large to practical uses when compared with enrofloxacin, it is the preliminary data leading us to the development of the metal complexes to be the antibacterial agents.
Table 3 Antibacterial activities of 1 and 2 and the related compounds against three Gram-negative bacteria, determined using the agar-well diffusion method
| Compound |
Inhibition-zone diameter (mm) |
MIC (mg mL−1) |
|
Salmonella
|
E. coli
|
Campylobacter
|
Salmonella
|
E.coli
|
Campylobacter
|
|
Data from ref. 30.
N.D. = not determined.
|
| [Cu(L1)Cl2]2, (1) |
11.0 |
12.0 |
22.5 |
25.00 |
25.00 |
3.12 |
| [Cu(L2)Cl2]2, (2) |
15.0 |
11.0 |
18.0 |
12.50 |
25.00 |
3.12 |
| [Cu(L1)2]Cl2a |
N.D.b |
N.D.b |
9.0 |
— |
— |
1.52 |
| [Cu(L2)2]Cl2a |
N.D.b |
N.D.b |
14.5 |
— |
— |
0.78 |
| Enrofloxacin |
10.0 |
10.0 |
15.0 |
0.002 |
0.008 |
0.015 |
3.4. Cytotoxicity assays
The positive results obtained from the DNA-interactions of the copper(II) complexes encouraged us to investigate their in vitro cytotoxicity against three human cancer cell lines, namely the small cell lung carcinoma (NCI-H187), the oral cavity carcinoma (KB) and the breast adenocarcinoma (MCF-7) by resazurin microplate assay. The corresponding IC50 values are summarized in Table 4.
Table 4 Cancer-cell growth inhibition of 1 and 2 and other compounds on three human cancer cell lines, assessed through the resazurin microplate assay
| Compound |
IC50a (μg mL−1) |
| NCI-H187 |
KB |
MCF-7 |
|
50% inhibitory concentrations in the REMA method.
|
| [Cu(L1)Cl2]2, (1) |
49.42 |
Inactive |
Inactive |
| [Cu(L2)Cl2]2, (2) |
47.63 |
22.51 |
Inactive |
| [Cu(L1)2]Cl2 |
Inactive |
Inactive |
Inactive |
| [Cu(L2)2]Cl2 |
Inactive |
Inactive |
Inactive |
| Cisplatin |
Inactive |
27.01 |
Inactive |
Both 1 and 2 can inhibit the growth of NCI-H187 cells with similar IC50 values (Table 4), but are inactive towards MCF-7 cells. For the KB cell line, only complex 2 shows some activity, with an IC50 value of 22.51 μg mL−1. The better cytotoxicity of complex 2 corroborates the stronger DNA binding ability compared to complex 1. The existence of phenyl rings on the ligand L2 may serve additional interactions providing better reactivity for the complex 2. Moreover, their anticancer activities have also been compared with cisplatin and the related monomeric compounds [Cu(L1)2]Cl2 and [Cu(L2)2]Cl2. Under this condition, cisplatin is capable of inhibiting only the growth of KB cells with less cytotoxicity than 2. On the other hand, though the compounds [Cu(L1)2]Cl2 and [Cu(L2)2]Cl2 appear to have slightly higher DNA binding potential than 1 and 2, respectively, (see Table 1), they are not active against all tested cell lines. Reasons for the different anticancer behaviors of the monomeric and dimeric copper(II) compounds can be explained by the structures and compared with cisplatin. For the monomeric compounds, after losing chloride anions by hydrolysis, the hydrolyzed forms, [Cu(L1)2]2+ and [Cu(L2)2]2+, which show the square-planar structures like cisplatin,29 are inactive, probably due to the CuN4 chromophore in which the copper(II) center is fixed by coordination with the two N,N-bidentate ligands, L1 and L2, thus it cannot further bind with DNA nucleobases via coordinative bonds as cisplatin. In the case of 1 and 2 which are dimeric compounds containing square-pyramidal geometry around the copper(II) center (Fig. 1), there are two terminal chloride ligands available to be replaced and then interact with DNA like cisplatin.4 These assays thus indicate that the copper(II) compounds of the type 1 and 2 may find applications as potential cytotoxic agents for specific cancer cell lines (such as the KB cell line).
4. Conclusions
Two copper(II) complexes [Cu(L1)Cl2]2 (1) and [Cu(L2)Cl2]2 (2) from hydrophilic ligands, namely amidino-O-methylurea derivatives, have been prepared and their DNA-interacting properties have been assessed. Both complexes have DNA binding potential through non-intercalation such as hydrogen bonds, electrostatic interactions and partial intercalation. In addition, these water-soluble complexes are efficient DNA cleavers and act as antibacterial agents, being lethal for a series of prokaryotic microorganisms involved in human-food poisoning, viz. Salmonella, E. coli and Campylobacter. Moreover, these metal-based molecules exhibit cytotoxic properties towards NCI-H187 cancer cells (small cell lung carcinoma), which are not affected by the well-known cisplatin. Complex 2 containing aromatic rings on sidechains of the ligands L2 shows the greater DNA binding and cleaving efficiencies, thus revealing higher antibacterial activity against Campylobacter as well as they are more effective than complex 1 to inhibit the growth of cancer cell types such as the small cell lung carcinoma (NCI-H187) and the oral cavity carcinoma (KB). Such a result has pointed out that the functional groups on the sidechain of the ligands have a significant effect on the DNA-interacting and biological properties of metal complexes.
The present study therefore shows high potential of (amidino-O-methylurea)-based complexes for biological applications. Indeed, functional groups can be easily introduced into amidino-O-methylurea derivatives (see R group in Fig. 1), thus providing an easy access to numerous water-soluble ligands (which is of paramount importance for biological applications) that can bind biometals like copper and generate potentially bioactive metal agents.
Acknowledgements
This work was financially supported by Center of Excellence for Innovation in Chemistry (PERCH-CIC) and the Development and Promotion of Science and Technology Talents Projects (DPST) (to A.M.). We also thank the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Communication, through the Advanced Functional Materials Cluster of Khon Kaen University. P.G. acknowledges ICREA (Institució Catalana de Recerca i Estudis Avançats), the Ministerio de Economía y Competitividad of Spain (Project CTQ2011-27929-C02-01), and the support of COST Actions CM1003 and CM1105.
References
-
Metal-DNA Chemistry, ACS Symposium Series, ed. T. D. Tullius, American Chemical Society 402, Washington DC, 1989, pp.1–23 Search PubMed.
- B. Rosenberg, L. van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
- P. J. Loehrer and L. H. Einhorn, Ann. Intern. Med., 1984, 100, 704–713 CrossRef CAS PubMed.
-
A. Eastman, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, VHCA & Wiley-VCH, Zurich & Germany, 1999, pp. 111–134 Search PubMed.
- Y.-Y. Xie, H.-L. Huang, J.-H. Yao, G.-J. Lin, G.-B. Jiang and Y.-J. Liu, Eur. J. Med. Chem., 2013, 63, 603–610 CrossRef CAS PubMed.
- S. David, R. S. Perkins, F. R. Fronczek, S. Kasiri, S. S. Mandal and R. S. Srivastava, J. Inorg. Biochem., 2012, 111, 33–39 CrossRef CAS PubMed.
- M. N. Patel, B. S. Bhatt and P. A. Dosi, Inorg. Chem. Commun., 2013, 29, 190–193 CrossRef CAS PubMed.
- S. D. Schimler, D. J. Hall and S. L. Debbert, J. Inorg. Biochem., 2013, 119, 28–37 CrossRef CAS PubMed.
- D. Wallis, J. Claffey, B. Gleeson, M. Hogan, H. Müller-Bunz and M. Tacke, J. Organomet. Chem., 2009, 694, 828–833 CrossRef CAS PubMed.
- A. Esparza-Ruiz, C. Herrmann, J. Chen, B. O. Patrick, E. Polishchuk and C. Orvig, Inorg. Chim. Acta, 2012, 393, 276–283 CrossRef CAS PubMed.
- F. A. Beckford, J. Thessing, A. Stott, A. A. Holder, O. G. Poluektov, L. Li and N. P. Seeram, Inorg. Chem. Commun., 2012, 15, 225–229 CrossRef CAS PubMed.
- I. Ali, W. A. Wani, K. Saleem and M.-F. Hseih, Polyhedron, 2013, 56, 134–143 CrossRef CAS PubMed.
- B. R. Stern, J. Toxicol. Environ. Health, Part A, 2010, 73, 114–127 CrossRef CAS PubMed.
-
S. J. Lippard and J. M. Berg, Principles of bioinorganic chemistry, University Science Books, Mill Valley, California, 1994 Search PubMed.
- M. Jagadeesh, S. K. Kalangi, L. S. Krishna and A. V. Reddy, Spectrochim. Acta, Part A, 2014, 118, 552–556 CrossRef CAS PubMed.
- S. Sayen, A. Carlier, M. Tarpin and E. Guillon, J. Inorg. Biochem., 2013, 120, 39–43 CrossRef CAS PubMed.
- B. Duff, V. R. Thangella, B. S. Creaven, M. Walsh and D. A. Egan, Eur. J. Pharmacol., 2012, 689, 45–55 CrossRef CAS PubMed.
- G.-Y. Li, K.-J. Du, J.-Q. Wang, J.-W. Liang, J.-F. Kou, X.-J. Hou, L.-N. Ji and H. Chao, J. Inorg. Biochem., 2013, 119, 43–53 CrossRef CAS PubMed.
- V. C. Silveira, H. Benezra, J. S. Luz, R. C. Georg, C. C. Oliveira and A. M. C. Ferreira, J. Inorg. Biochem., 2011, 105, 1692–1703 CrossRef PubMed.
- M. N. Patel, P. A. Dosi, B. S. Bhatt and V. R. Thakkar, Spectrochim. Acta, Part A, 2011, 78, 763–770 CrossRef PubMed.
- A. Kellett, O. Howe, M. O′Connor, M. McCann, B. S. Creaven, S. McClean, A. F.-A. Kia, A. Casey and M. Devereux, Free Radical Biol. Med., 2012, 53, 564–576 CrossRef CAS PubMed.
- S. Kashanian, M. M. Khodaei, H. Roshanfekr, N. Shahabadi and G. Mansouri, Spectrochim. Acta, Part A, 2012, 86, 351–359 CrossRef CAS PubMed.
- P. Hubberstey, U. Suksangpanya and C. Wilson, CrystEngComm, 2000, 26, 141–145 RSC.
- O. I. Singh, M. Damayanti, N. R. Singh, R. K. H. Singh, M. Mohapatra and R. M. Kadam, Polyhedron, 2005, 24, 909–916 CrossRef CAS PubMed.
- S. P. Devi, R. K. B. Devi, M. Damayanti, N. R. Singh, R. K. H. Singh and R. M. Kadam, J. Coord. Chem., 2011, 64, 1586–1601 CrossRef CAS.
- S. P. Devi, R. K. B. Devi, N. S. Devi, L. J. Singh and R. K. H. Singh, Polyhedron, 2012, 47, 1–8 CrossRef PubMed.
- M. J. Begley, P. Hubberstey and C. H. M. Moore, J. Chem. Res., Synop., 1986, 172–173 CAS.
- A. Meenongwa, U. Chaveerach and K. Siriwong, Inorg. Chim. Acta, 2011, 366, 357–365 CrossRef CAS PubMed.
- U. Suksangpanya, A. J. Blake, P. Hubberstey, D. J. Parker, S. J. Teat and C. L. Wilson, CrystEngComm, 2003, 5, 10–22 RSC.
- U. Chaveerach, A. Meenongwa, Y. Trongpanich, C. Soikum and P. Chaveerach, Polyhedron, 2010, 29, 731–738 CrossRef CAS PubMed.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- M. F. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- A. Wolfe, G. H. J. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS.
- G. Cohen and H. Eisenberg, Biopolymers, 1969, 8, 45–55 CrossRef CAS.
- X.-Z. Feng, Z. Lin, L.-J. Yang, C. Wang and C. C.-L. Bai, Talanta, 1998, 47, 1223–1229 CrossRef CAS.
- G. Facchin, E. Kremer, D. A. Barrio, S. B. Etcheverry, A. J. Costa-Filho and M. H. Torre, Polyhedron, 2009, 28, 2329–2334 CrossRef CAS PubMed.
- M. F. Shubsda, J. Goodisman and J. C. Dabrowiak, J. Biochem. Biophys. Methods, 1997, 34, 73–79 CrossRef CAS.
- M. N. Patel, P. A. Dosi and B. S. Bhatt, Polyhedron, 2010, 29, 3238–3245 CrossRef CAS PubMed.
-
C. H. Collins and P. M. Lyne, Microbiological Methods, University Park Press, Baltimore, 1970, p. 422 Search PubMed.
- E. J. L. Lana, F. Carazza and J. A. Takahashi, J. Agric. Food Chem., 2006, 54, 2053–2056 CrossRef CAS PubMed.
- J. O′Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef.
- M. J. O'Neill, D. H. Bray, P. Boardman, J. D. Phillipson and D. C. Warhurst, Planta Med., 1985, 51, 394–398 CrossRef PubMed.
-
G. M. Blackburn, M. J. Gait and D. Loakes, in Nucleic Acid in Chemistry and Biology, ed. D. M. Williams, RSC Publishing, Cambridge, 3rd edn, 2006, pp. 342–382 Search PubMed.
- J. M. Kelly, A. B. Tossi, D. J. McConnell and C. OhUigin, Nucleic Acids Res., 1985, 13, 6017–6034 CrossRef CAS PubMed.
- Q. Li, P. Yang, H. Wang and M. Guo, J. Inorg. Biochem., 1996, 64, 181–195 CrossRef CAS.
- Y.-J. Liu, X. Y. Wei, F.-H. Wu, W.-J. Mei and L.-X. He, Spectrochim. Acta, Part A, 2008, 70, 171–176 CrossRef PubMed.
- S. Tabassum, R. A. Khan, F. Arjmand, M. Aziz, A. S. Juvekar and S. M. Zingde, Carbohydr. Res., 2011, 346, 2886–2895 CrossRef CAS PubMed.
- F. Arjmand, S. Parveen, M. Afzal and M. Shahid, J. Photochem. Photobiol., B, 2012, 114, 15–26 CrossRef CAS PubMed.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS.
- B. C. Baguley and M. LeBret, Biochemistry, 1984, 23, 937–943 CrossRef CAS.
- R. F. Pasternack, M. Cacca, B. Keogh, T. A. Stephenson, A. P. Williams and F. J. Gibbs, J. Am. Chem. Soc., 1991, 113, 6835–6840 CrossRef CAS.
- E. Nyarko, N. Hanada, A. Habib and M. Tabata, Inorg. Chim. Acta, 2004, 357, 739–745 CrossRef CAS PubMed.
- V. I. Ivanov, L. E. Minchenkova, A. K. Schyolkina and A. I. Poletayev, Biopolymers, 1973, 12, 89–110 CrossRef CAS PubMed.
- B. Norden and F. Tjerneld, Biopolymers, 1982, 21, 1713–1734 CrossRef CAS PubMed.
- E. K. Efthimiadou, Y. Sanakis, M. Katsarou, C. P. Raptopoulou, A. Karaliota, N. Katsarosa and G. Psomas, J. Inorg. Biochem., 2006, 100, 1378–1388 CrossRef CAS PubMed.
- G. A. Neyhart, N. Grover, S. R. Smith, W. Kalsbeck, T. A. Fairley, M. Cory and H. H. Thorp, J. Am. Chem. Soc., 1993, 115, 4423–4428 CrossRef CAS.
- G. L. Eichhorn and Y. A. Shin, J. Am. Chem. Soc., 1968, 90, 7323–7328 CrossRef CAS.
-
B. Halliwell and J. M. C. Gutteridge, Free Radicals Biol. Med., Oxford Science Publication, Oxford, 3rd edn, 1999, pp. 42–43 Search PubMed.
- D. S. Sigman and C.-H. B. Chen, Acc. Chem. Res., 1986, 19, 180–186 CrossRef CAS.
- I. Yamazaki and L. H. Piette, Biochim. Biophys. Acta, 1961, 50, 62–69 CrossRef CAS.
-
O. G. Rojas, C. G. Quintero, M. R. Bolívar, A. Romero and A. Rodríguez, CIMMACS '10 Proceedings of the 9th WSEAS international conference on computational intelligence, man-machine systems and cybernetics, Wisconssin, 2010, 251–258.
-
J. Blanco, PhD thesis, Universidad Politécnica de Cataluña, Barcelona, España, 2009.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.