
Phosphine oxide-based conjugated microporous polymers with excellent CO2 capture properties†
Shanlin
Qiao‡
a,
Wei
Huang‡
ab,
Zhengkun
Du
a,
Xianghui
Chen
a,
Fa-Kuen
Shieh
c and
Renqiang
Yang
*a
aCAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: yangrq@qibebt.ac.cn; Fax: +86 532 80662778; Tel: +86 532 80662700
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Chemistry, National Central University, 300 Jhong-Da Road, Chung-Li 32001, Taiwan
First published on 26th September 2014
Abstract
Three novel phosphine oxide-based conjugated microporous polymers TEPO-1, TEPO-2 and TEPO-3 are designed and synthesized, which are constructed using the phosphine-based building unit tris(4-ethynylphenyl)phosphine oxide with linkers tris(4-ethynylphenyl)phosphine oxide, triphenylphosphine oxide and tri(thiophen-2-yl)phosphine oxide via Sonogashira–Hagihara homo-coupling or cross-coupling condensation reaction. The adsorption isotherms of N2 reveal that the Brunauer–Emmett–Teller (BET) specific surface areas of TEPO-1, TEPO-2 and TEPO-3 are 485 m2 g−1, 534 m2 g−1 and 592 m2 g−1, respectively. However, these three polymers have strong affinity for CO2, which exhibit relatively high sorption abilities for CO2 (273 K/1.0 bar: 6.52 wt%, 7.62 wt%, and 8.40 wt%). Meanwhile, the TEPOs demonstrate ultrahigh hydrogen uptake and outstanding CO2/CH4 selectivity compared to analogous materials with similar BET surface areas using C, Si and N as the nodes. This work reveals clearly that the gas uptake capacity of material is highly dependent on the length of the rigid skeleton and the modification of functional groups in the monomer structure.
Introduction
Global climate change caused by greenhouse gas emissions has attracted widespread public concern in recent decades. Excessive CO2 emission resulting from the combustion of fossil fuels is considered as a primary factor in the greenhouse effect. Different governments and environmental agencies have encouraged the development of exploring new economic technologies to deal with this issue. Capture of CO2 depending on physical adsorption is an effective way to fight against the global climate warming. In addition, the utilization and storage of new environmentally friendly fuels are also potential methods to meet the environmental demands. Hydrogen, owing to its zero pollution and high calorific value of combustion, is a good candidate as star fuel which has attracted great interest nowadays. However, the shortcomings of its application are the storage and transportation resulting from its lightweight nature and fast diffusion speed. Therefore, finding a good way to store hydrogen and other fuel gases is also one of the effective ways to solve the above problems.Microporous organic polymers (MOPs) have spurred tremendous scientific interest in functional materials research owing to their intrinsic properties of large surface area, narrow pore size distribution, high flexibility in the structure design, excellent physical and chemical stability, and low skeleton density. MOPs have shown great potential in various technological and energy-related applications,1–3 including gas storage,4–7 separation,8–10 heterogeneous catalysis11–15 and as confined environments for reactions.16 MOPs such as covalent organic frameworks (COFs),17–19 polymers of intrinsic microporosity (PIM),20,21 porous aromatic frameworks (PAFs),7,22 hypercrosslinked polymers (HCPs),23,24 crystalline triazine-based organic frameworks (CTFs),25,26 and conjugated microporous polymers (CMPs)9,27 have exhibited high CO2 capture and separation capacities in reported research studies. Normally, aqueous amine–ammonia solution is used to separate CO2 on a large scale relying on the chemical adsorption. The downside of this technology is the severe corrosion problems and high energy consumption during the treating processes. While the CO2 capture and storage relying on physical adsorption on MOPs is a promising method to deal with the problem of excess CO2 emissions, because of the physicochemical stability and lower regeneration energy penalty of MOPs. Meanwhile, MOPs also exhibit great potential in the storage of small gas molecules such as hydrogen, methane and acetylene.
Conjugated microporous polymers (CMPs) as a sub-family of MOPs have attracted extensive attention not only because they can be prepared by different synthetic methods, but also they combine the porosity with the extended π-conjugation skeleton. The adsorption ability of small gases of CMPs relies greatly on the BET surface areas, the pore size and the isosteric enthalpy of materials. In order to enhance the CO2 capture ability of CMPs, several strategies have been employed, for example generating the constricted pores, opening metal-sites,14 post-synthetic modification, and introducing CO2-philic functional groups such as N-rich and O-rich groups into the polymer.28 These novel materials have exhibited a high CO2 uptake capacity, such as APOP-1 (SBET = 1298 m2 g−1, 4.26 mmol g−1 at 273 K/1.31 bar),29 CPOP-1 (SBET = 2220 m2 g−1, 21.2 wt% at 273 K/1.0 bar)30 and Co-CMP (SBET = 965 m2 g−1, 79.3 mg g−1 at 298 K/1.0 bar).14 In the past few years, CMPs were constructed from carbon–carbon bonds or carbon–heteroatoms through transition-metal-mediated cross coupling. Different main group elements of C, N and Si as the nodes of the CMP frameworks have been reported owning their excellent performance in CO2 capture and sequestration. Phosphorus which is a light element with facile chemical modification properties, however, has not received enough attention as the node of CMPs so far.31–33 It is an attractive work to intercalate the phosphorus group into the skeleton and modify it to functionalize the CMPs. In our previous work, we have succeeded in synthesizing two new functional thienyl-phosphine microporous polymers containing the phosphorus group which had exhibited great carbon dioxide capture.33 And the polymer containing P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups show higher CO2 capture capacity than the one with PS groups under the same conditions. Along these lines, here three kinds of CMPs based on the phosphine-oxide unit were designed and synthesized in expectation of excellent performance.
O groups show higher CO2 capture capacity than the one with PS groups under the same conditions. Along these lines, here three kinds of CMPs based on the phosphine-oxide unit were designed and synthesized in expectation of excellent performance.
In order to get different morphologies and a high uptake capacity of carbon dioxide and other gases, two methods were employed to optimize the polymer structures. In this paper, we take TEPO-2 as the reference. Firstly, we design TEPO-1 which has one more triple bond in the monomer structure compared to TEPO-2, which can help us understand the relationship of performance of CMPs with the length of the rigid skeleton. And in attempting to functionalize TEPO-2, TEPO-3 was designed by utilizing the thienyl functional group instead of the benzene ring in TEPO-2. The electron-rich thiophene group is chosen because it is a Lewis base which may have strong interaction with electron-deficient CO2 molecules.34 Finally, the effects of the rigid length and functionalization of the monomer skeleton on the performance of porous materials are investigated.
Experimental section
Materials
All reagents and solvents, unless otherwise specified, were purchased from J&K, Aldrich and Acros Chemical Co. and used as received. Tetrahydrofurane (THF), dimethyl formamide (DMF) and triethylamine (TEA) were distilled prior to use. Tris(4-ethynylphenyl)phosphane oxide (m-1),35,36 tris(4-bromophenyl)phosphine oxide (m-2)37 and tris(5-bromothiophen-2-yl)phosphine oxide (m-3)33 were synthesized according to the literature method. The details of synthesis are shown in the ESI.†Synthesis of TEPO polymers
The TEPO-1 polymer was prepared by palladium-catalyzed homo-coupling of tris(4-ethynylphenyl)phosphine oxide (m-1). And TEPO-2 and TEPO-3 were prepared using Sonogashira–Hagihara coupling condensation reactions between tris(4-ethynylphenyl)phosphine oxide (m-1) and tris(4-bromophenyl)phosphine oxide (m-2) and tris(5-bromothiophen-2-yl)phosphine oxide (m-3), respectively. The molar ratio of ethynyl to bromine functionalities in the monomer feed was set at 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as the reports of CMPs synthesized by the Cooper group and our previous work.33,38 The details of the preparation of TEPOs are as follows.
:1 as the reports of CMPs synthesized by the Cooper group and our previous work.33,38 The details of the preparation of TEPOs are as follows.
Characterization
1H and 13C NMR spectra were recorded on a Bruker AV 600 MHz spectrometer with tetramethylsilane (TMS) as the internal reference. Solid-state 13C CP/MAS NMR measurements were carried out using a Bruker Avance III model 400 MHz NMR spectrometer at a MAS rate of 5 kHz. FT-IR spectra were collected in attenuated total reflection (ATR) mode on a Thermo Nicolet 6700 FT-IR spectrometer. Scanning electron microscopy (SEM) was recorded using a Hitachi S-4800 with acceleration voltage 3 kV and working different distances. Samples were coated by a thin layer of Au before investigation.Gas sorption analysis
The three polymers were degassed at 120 °C for 10 h under vacuum before analysis. BET surface areas and pore size distributions of TEPOs were measured by N2 adsorption–desorption at 77 K. H2 uptake was tested at 77 K up to the pressure of 1.1 bar. CO2 and CH4 isotherms were measured at 273 K and 298 K up to 1.1 bar. All the samples were tested using an Autosorb-iQ-MP-VP volumetric adsorption analyzer after the same degassing procedure.Results and discussion
Structural characterization
The three phosphine-containing microporous polymers were synthesized via Sonogashira–Hagihara coupling condensation reaction, and the structures of TEPOs are shown in Scheme 1. | ||
| Scheme 1 Structures of TEPOs. | ||
The molecular levels of the TEPOs were investigated by 13C solid NMR and 31P solid NMR spectra as shown in Fig. 1. The –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– linkages have been confirmed by the characteristic resonance of triple-bonded carbon with a corresponding peak at around 93 ppm (Fig. 1a). In addition, the signals of the carbon chemical shift in the range of 120–140 ppm are related to the aromatic carbon atoms of building blocks. The resonance at 128 ppm is assigned to the sp2 benzene connected with the alkynylene (CAr–C). The observed appearance of resonance at 133 ppm belongs to the sp2 carbons in the benzene rings (CAr–H). 31P-NMR measurements were carried out to confirm the presence of the –PO bond. It is showed in Fig. 1b that the TEPOs have a signal at 29 ppm corresponding to the –PO stretch. Paying special attention to TEPO-3, there are two obvious peaks which correspond to the different situations of the –PO bond in the structure, and the peak around 4 ppm is assigned to –PO connecting with thiophene groups. The results of 13C NMR and 31P NMR confirm the structure of TEPOs and also indicate that this cross-coupling reaction is effective in the synthesis of conjugated organic polymers.
C– linkages have been confirmed by the characteristic resonance of triple-bonded carbon with a corresponding peak at around 93 ppm (Fig. 1a). In addition, the signals of the carbon chemical shift in the range of 120–140 ppm are related to the aromatic carbon atoms of building blocks. The resonance at 128 ppm is assigned to the sp2 benzene connected with the alkynylene (CAr–C). The observed appearance of resonance at 133 ppm belongs to the sp2 carbons in the benzene rings (CAr–H). 31P-NMR measurements were carried out to confirm the presence of the –PO bond. It is showed in Fig. 1b that the TEPOs have a signal at 29 ppm corresponding to the –PO stretch. Paying special attention to TEPO-3, there are two obvious peaks which correspond to the different situations of the –PO bond in the structure, and the peak around 4 ppm is assigned to –PO connecting with thiophene groups. The results of 13C NMR and 31P NMR confirm the structure of TEPOs and also indicate that this cross-coupling reaction is effective in the synthesis of conjugated organic polymers.
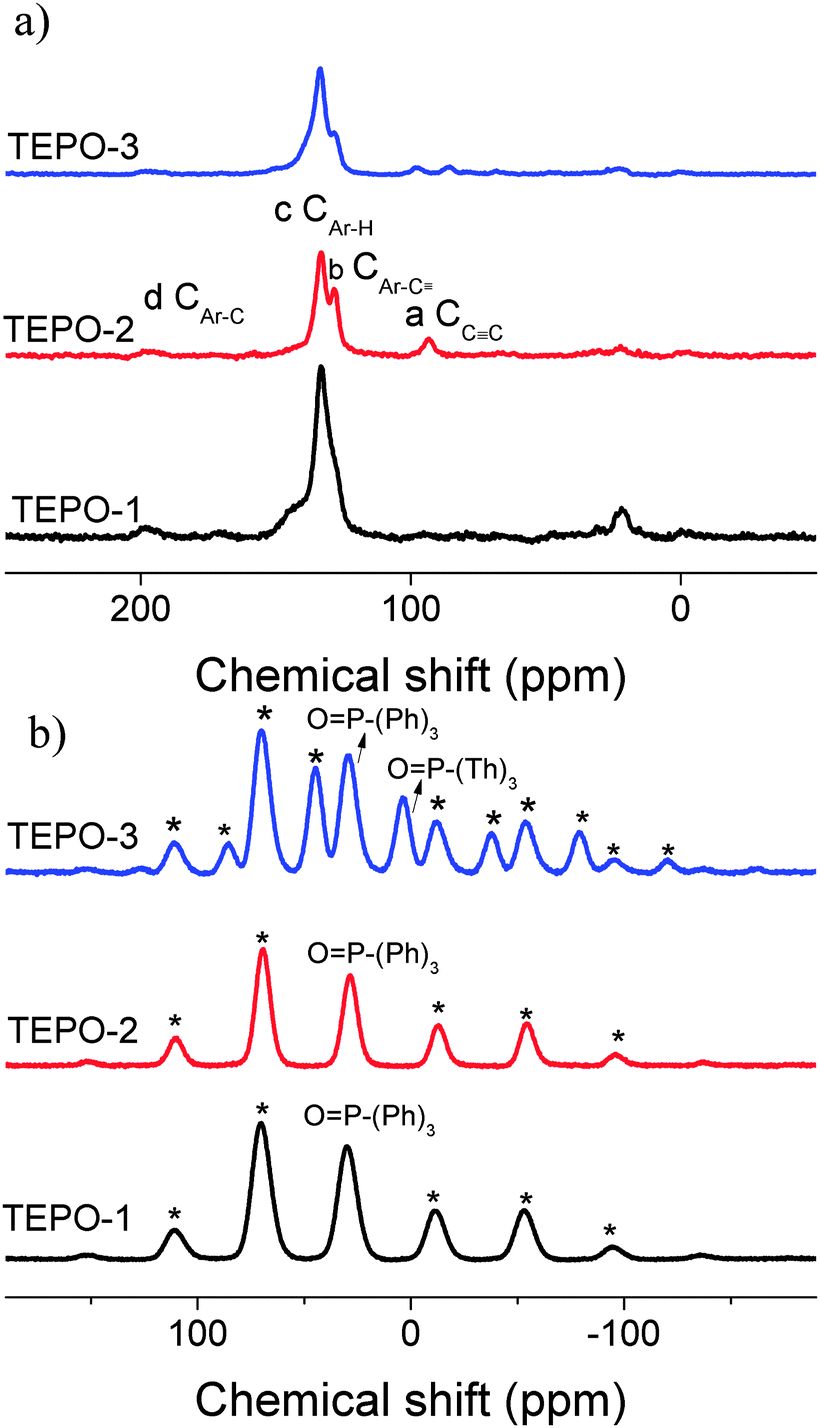 | ||
| Fig. 1 (a) 13C CP-MAS NMR. (b) 31P CP-MAS NMR of the TEPOs. | ||
In the FR-IR spectra (Fig. 2), a strong stretch is observed at 1186 cm−1, which confirms the presence of –PO in the polymers. Meanwhile the band of –CC– stretch at 2202 cm−1 is found in the spectra of TEPOs. SEM images (Fig. 3) for TEPOs show that the polymers have similar amorphous aggregation morphologies as the reported CMPs. The images here are observed from different distances so that one can see the structure clearly. It's easy to recognize that the morphologies of TEPOs are spherular.
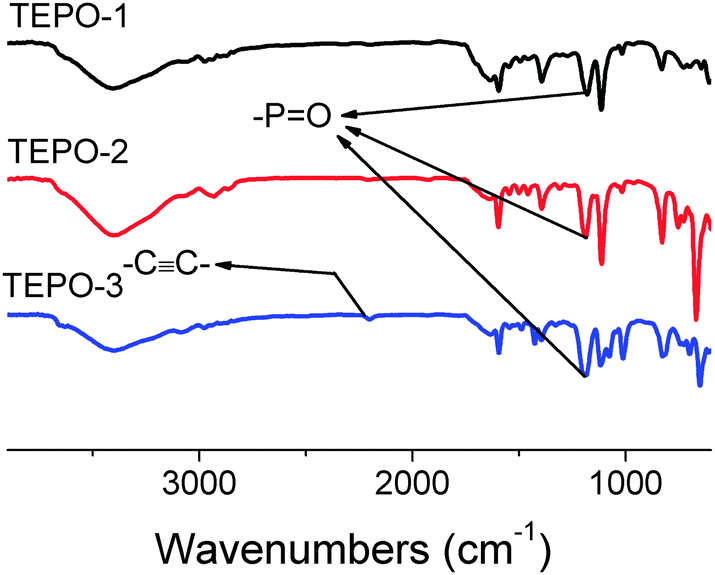 | ||
| Fig. 2 FT-IR spectra of the TEPOs. | ||
 | ||
| Fig. 3 Scanning electron microscopy (SEM) images at different distances for (a) TEPO-1 at 8.2 mm, (b) TEPO-2 at 8.1 mm and (c) TEPO-3 at 6.7 mm with magnification of 30k. | ||
Porosity measurements
The BET surfaces and pore structures of the TEPOs were characterized by nitrogen adsorption–desorption isotherms of samples at 77 K. Samples were degassed at 120 °C for 10 h under vacuum before analysis. The corresponding nitrogen adsorption isotherms and pore size distributions are shown in Fig. 4. According to the IUPAC classification, all of the TEPOs are typical microporous materials, and the full reversible adsorption–desorption of nitrogen with rapid uptake occurred at a low relative pressure (P/P0 = 0–0.1), indicating the permanent microporous nature of all the TEPOs. The BET specific surface areas are 485 m2 g−1, 543 m2 g−1 and 592 m2 g−1 for TEPO-1, TEPO-2 and TEPO-3, respectively. These surfaces are comparable to previous reported phosphine-containing CMPs.33 The upturn in the isotherm of TEPO-1 at relative higher pressures indicates that the TEPO-1 possessed mesopores or macropores, which can result in the condensation of nitrogen in the macropores and interparticle voids.39–42 The difference between TEPO-1 and TEPO-2 can be explained as the general trend, when similar monomers were used, polymers prepared from the larger monomers showed lower surface areas, as the reported CMPs constructed with longer connecting struts have lower BET surface areas.38 As the dynamic effective diameter of the thiophene group is smaller than the benzene ring, TEPO-3 shows a higher BET surface than TEPO-2. This result coincides with our expectation. Analysis on the pore size distribution of the TEPOs (Fig. 4b) shows that all the samples are micropores mainly, as the pore width is less than 2 nm. The pore center for TEPO-1 is located around 1.30 nm, while the pores of TEPO-2 and TEPO-3 are centered at two main sizes 1.08 nm and 1.31 nm. It should be pointed out that there are some mesopores in the TEPO-1 (pore width >2 nm), which is corresponding to its N2 adsorption isotherm (Fig. 4a). The pore size distribution gives us strong proof that the pore structure of materials is highly dependent on the length of the rigid skeleton which can explain why the dominant pore size and pore volume of TEPO-1 are larger than the other two analogues and the TEPO-2 shares a similar pore size distribution as TEPO-3.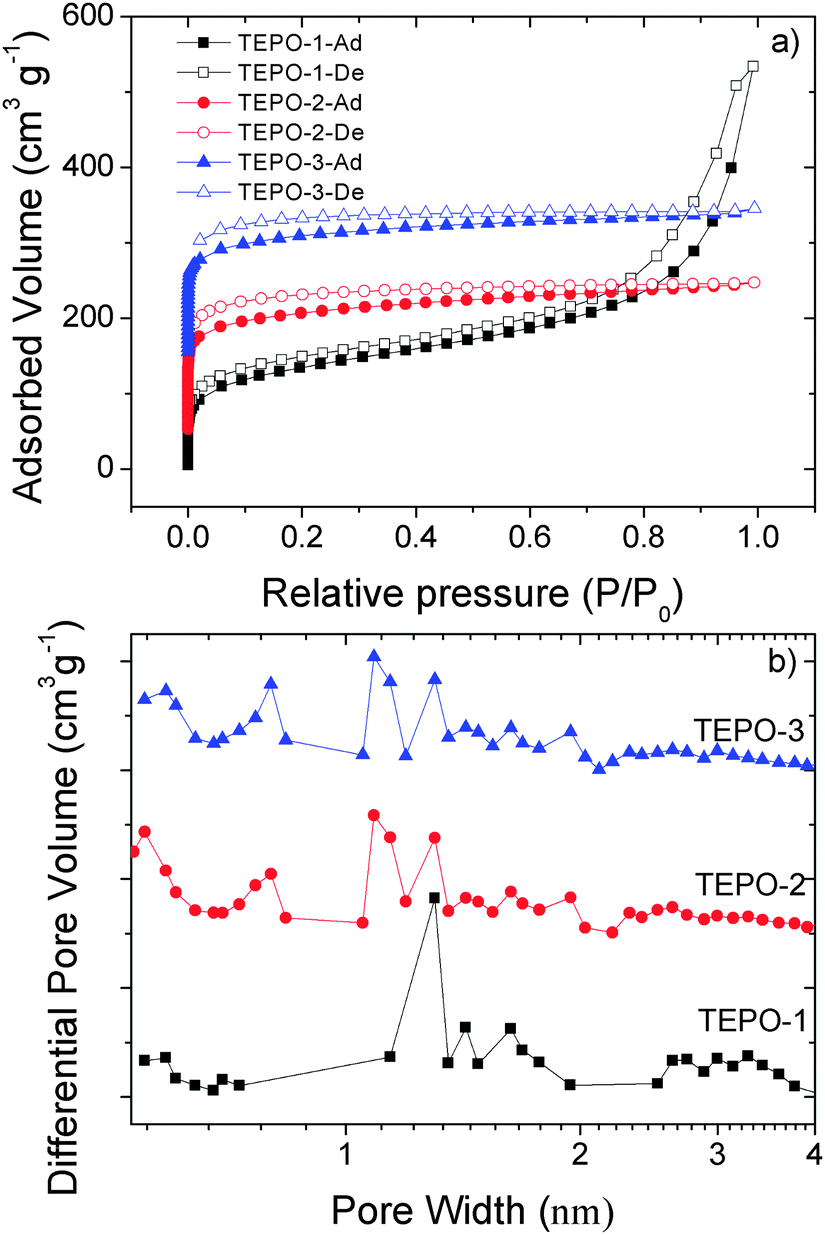 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption isotherms of the TEPOs measured at 77 K. (b) Pore size distribution curves; the curves of TEPO-2 and TEPO-3 were shifted vertically by 2.0 cm3 g−1 and 4.0 cm3 g−1 for clarity. | ||
Gas uptake studies
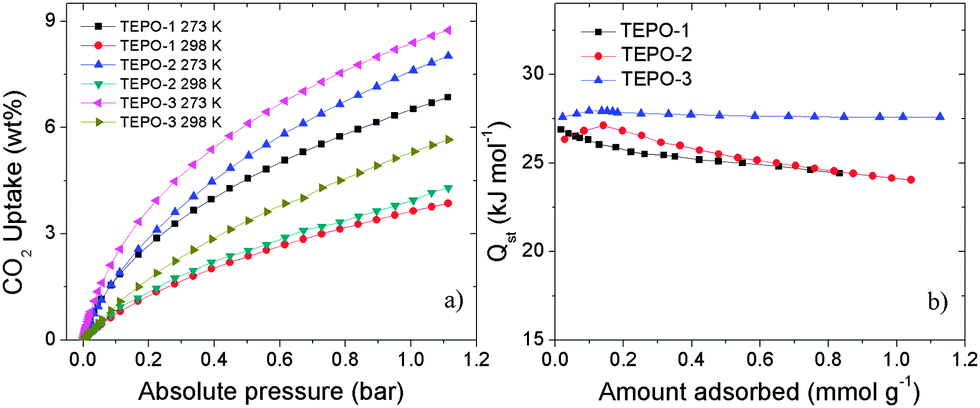 | ||
| Fig. 5 (a) CO2 adsorption properties at 273 K and 298 K. (b) The isosteric heats of CO2 adsorption for the TEPOs. | ||
It is well known that materials with low-density and high-porosity are promising materials for gas storage applications. Conjugated microporous polymers (CMPs) are good candidates for gas adsorption and storage due to their high specific surface area, narrow pore distribution and low framework density. Hydrogen and methane storage capacities of the TEPOs were explored, as both gases have attractive cleaning energy. The hydrogen uptake for TEPOs at 77 K/1.0 bar is 0.89 wt%, 0.97 wt% and 1.02 wt%, respectively (shown in Fig. 6a). Considering that the surface areas of the TEPOs are moderate, the three materials demonstrate ultrahigh hydrogen storage properties under the same condition compared to other microporous polymers whose BET surface areas are less than 600 m2 g−1.
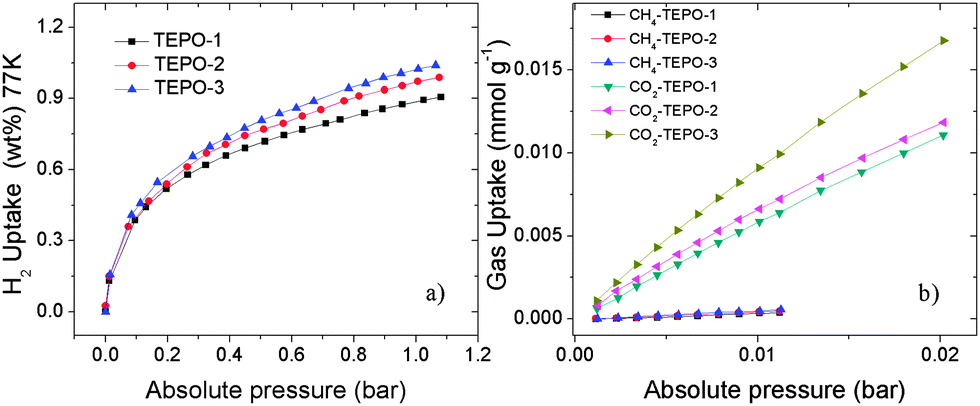 | ||
| Fig. 6 (a) H2 adsorption properties at 77 K. (b) Initial CO2 and CH4 uptake slopes of the TEPOs at 273 K. | ||
The CO2 in the natural gas would reduce the net heat of fuel combustion and corrode pipelines. Therefore, the separation of CO2 over CH4 is important for the industry. The selectivity of CO2/CH4 is calculated from the initial slopes of adsorption isotherms at low pressure (Fig. 6b), and the separation capabilities of these polymers are outstanding. The selectivity of TEPO-1 to TEPO-3 is 12.6, 14.1 and 15.5 at 273 K, which is higher than some reported materials, for example the selectivity of ZIF-78 is 10,43 P-1 is 4,10 and 2-D MOF is 13 which is with open sites.44 Such good selectivity of the TEPOs is expected to arise from the strong interactions between the polymers with polarizable phosphine-oxide units and CO2 molecules. The excellent performance of TEPO-3 may be ascribed to the electron-rich thiophene group which is a Lewis base having strong interaction with electron-deficient CO2 molecules. The detailed porosity structural parameters and gas uptake capacities of TEPOs are shown in Table 1.
| Polymer |
S
BETa [m2 g−1] |
V
Microb [cm3 g−1] |
V
Totalc [cm3 g−1] |
V Micro/VTotal | CO2 uptake [wt%] | H2 uptake [wt%] | CO2/CH4 selectivity | |
|---|---|---|---|---|---|---|---|---|
| 273 K | 298 K | 77 K | 273 K | |||||
| a Surface area calculated from the N2 isotherm. b The micropore volume derived using the t-plot method. c Total pore volume at P/P0 = 0.99. | ||||||||
| TEPO-1 | 485 | 0.11 | 0.82 | 0.13 | 6.52 | 3.65 | 0.89 | 12.6 |
| TEPO-2 | 543 | 0.21 | 0.30 | 0.69 | 7.62 | 3.93 | 0.97 | 14.1 |
| TEPO-3 | 592 | 0.22 | 0.30 | 0.72 | 8.40 | 5.33 | 1.02 | 15.5 |
Conclusions
We have successfully synthesized three tris(4-ethynylphenyl)phosphine oxide (TEPO) based microporous polymers named TEPO-1, TEPO-2 and TEPO-3 through Sonogashira–Hagihara coupling condensation reaction. Although the BET surface area of the polymers is less than 600 m2 g−1, the materials show relatively high sorption abilities for carbon dioxide (6.52 wt% to 8.40 wt% at 273 K/1.0 bar) and hydrogen uptake (0.89 wt% to 1.02 wt% at 77 K/1.0 bar) compared to materials with similar Brunauer–Emmett–Teller (BET) surface areas. Interestingly, the polymers obtained exhibit remarkable performance in separating CO2 over CH4 as the selectivity of CO2/CH4 is as high as 12.6, 14.1 and 15.5 at 273 K, respectively. From the comparison of TEPO-1 and TEPO-2, when monomers with the same chemical component were used, polymers prepared from the larger building blocks showed lower surface areas as they went against to build the stiff porous structure. Compared with TEPO-2, TEPO-3 shows higher gas uptake capacity which can be attributed to the electron-rich thiophene group having a stronger interaction with small gas molecules and a shorter rigid length than the benzene ring. This work clearly reveals that the gas uptake capacity of materials is highly dependent on the length of the rigid skeleton and the modification of functional groups in the monomer structure.Acknowledgements
This work was supported by National Natural Science Foundation of China (21274161, 51173199), the Ministry of Science and Technology of China (2010DFA52310), Shandong Provincial Natural Science Foundation (ZR2011BZ007), and Qingdao Municipal Science and Technology Program (11-2-4-22-hz).Notes and references
- X. Zhu, C. Tian, S. M. Mahurin, S.-H. Chai, C. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478–10484 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- O. K. Farha, A. M. Spokoyny, B. G. Hauser, Y.-S. Bae, S. E. Brown, R. Q. Snurr, C. A. Mirkin and J. T. Hupp, Chem. Mater., 2009, 21, 3033–3035 CrossRef CAS.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng and J. M. Simmons, Angew. Chem., Int. Ed., 2009, 121, 9621–9624 CrossRef.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky and Y. Z. Khimyak, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- S. Qiao, Z. Du and R. Yang, J. Mater. Chem. A, 2014, 2, 1877–1885 CAS.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- M. Rose, A. Notzon, M. Heitbaum, G. Nickerl, S. Paasch, E. Brunner, F. Glorius and S. Kaskel, Chem. Commun., 2011, 47, 4814–4816 RSC.
- J.-X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072–1075 CrossRef CAS PubMed.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 Search PubMed.
- L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- D. M. Vriezema, M. Comellas Aragonès, J. A. Elemans, J. J. Cornelissen, A. E. Rowan and R. J. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- C. Xu and N. Hedin, J. Mater. Chem. A, 2013, 1, 3406–3414 CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- S. Ren, R. Dawson, D. J. Adams and A. I. Cooper, Polym. Chem., 2013, 4, 5585–5590 RSC.
- H. Ren, T. Ben, E. Wang, X. Jing, M. Xue, B. Liu, Y. Cui, S. Qiu and G. Zhu, Chem. Commun., 2010, 46, 291–293 RSC.
- J.-Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670–2672 RSC.
- C. F. Martín, E. Stöckel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475–5483 RSC.
- P. Kuhn, K. Krüger, A. Thomas and M. Antonietti, Chem. Commun., 2008, 5815–5817 RSC.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- W.-C. Song, X.-K. Xu, Q. Chen, Z.-Z. Zhuang and X.-H. Bu, Polym. Chem., 2013, 4, 4690–4696 RSC.
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS.
- S. Qiao, Z. Du, C. Yang, Y. Zhou, D. Zhu, J. Wang, X. Chen and R. Yang, Polymer, 2014, 5, 1177–1182 CrossRef PubMed.
- X. Chen, S. Qiao, Z. Du, Y. Zhou and R. Yang, Macromol. Rapid Commun., 2013, 34, 1181–1185 CrossRef CAS PubMed.
- S. Qiao, Z. Du, W. Huang and R. Yang, J. Solid State Chem., 2014, 69–72 CrossRef CAS PubMed.
- V. H. Gessner and T. D. Tilley, Org. Lett., 2011, 13, 1154–1157 CrossRef CAS PubMed.
- R. Métivier, R. Amengual, I. Leray, V. Michelet and J.-P. Genêt, Org. Lett., 2004, 6, 739–742 CrossRef PubMed.
- C. Liu, Y. Li, Y. Li, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2013, 25, 3320 CrossRef CAS.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS PubMed.
- G. Cheng, T. Hasell, A. Trewin, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 12727–12731 CrossRef CAS PubMed.
- S. Ren, R. Dawson, D. J. Adams and A. I. Cooper, Polym. Chem., 2013, 4, 5585 RSC.
- M. Rose, K. Nicole, B. Winfried, B. Bertram, F. Sven and K. Stefan, Soft Matter, 2010, 6, 3918–3923 RSC.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- B. Mu, F. Li and K. S. Walton, Chem. Commun., 2009, 2493–2495 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis of m-1 to m-3. See DOI: 10.1039/c4nj01477d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |