
Synthesis and optical properties of phenanthromidazole derivatives for organic electroluminescent devices†
Jayaraman
Jayabharathi
*,
Periyasamy
Ramanathan
and
Venugopal
Thanikachalam
Department of Chemistry, Annamalai University, Annamalainagar 608 002, Tamilnadu, India. E-mail: jtchalam2005@yahoo.co.in; Tel: +91 9443940735
First published on 16th October 2014
Abstract
Five phenanthromidazole derivatives, namely 2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (1), 2-(naphthalen-1-yl)-1-phenyl-1H-phenanthro[9,10-d]imidazole (2), 2-(naphthalen-1-yl)-1-p-tolyl-1H-phenanthro[9,10-d]imidazole (3), 1-(4-methoxyphenyl)-2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (4) and 1-(3,5-dimethylphenyl)-2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (5), were synthesised using the TiO2 (R) nanosemiconductor as the catalyst under solvent free conditions and characterised by NMR and single crystal XRD techniques. Their photophysical, electrochemical and electroluminescent properties were carefully analysed. Highly efficient Alq3-based organic light emitting devices have been developed using phenanthromidazoles as functional layers between NPB [4,4-bis(N-(1-naphthyl)-N-phenylamino)biphenyl] and Alq3 [tris(8-hydroxyquinoline)aluminium] layers. Using the device of ITO/NPB/4/Alq3/LiF/Al, a maximum luminous efficiency of 5.99 cd A−1 was obtained with a maximum brightness of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 623 cd m−2 and a power efficiency of 5.25 lm W−1.
623 cd m−2 and a power efficiency of 5.25 lm W−1.
1. Introduction
Arylimidazoles play an important role in materials science and medicinal chemistry due to their optoelectronic properties and high thermal stabilities.1–7 Substituted imidazoles are extensively used as glucagon receptors,8 cannabinoid receptor antagonists,9 modulators of glycoprotein mediated multidrug resistance,10 antibacterial agents,11 anti-allergic agents,12 analgesics,13 antitumor agents14 and pesticides.15 Many of the reported synthetic protocols for imidazoles16–27 suffer from disadvantages such as use of toxic and chlorinated organic solvents, acidic conditions, complex work-up and purification, side reactions, low yield and use of hazardous and expensive reagents. Thus the development of a new catalyst is essential to overcome these shortcomings and to fulfill the criteria of milder reaction conditions, higher yield and reusability of the catalyst.Titanium dioxide finds widespread industrial application28–32 and its utility has been extended to the photodegradation of pesticides33 and carcinogenic dyes.34,35 From a synthetic point of view, titanium dioxide has been used as a green, inexpensive, mild and recyclable heterogeneous Lewis acid potential catalyst in certain organic transformations like Beckmann rearrangement,36 Friedel–Crafts acylation,37 Biginelli condensation,38 in the synthesis of dihydropyrazines,39 piperazines,40 quinoxalines41 and in the photocatalytic oxidation of amines.42
Highly efficient electroluminescence (EL) has been obtained using a bilayer device composed of NPB and Alq3 which are the most widely used hole-transporting and electron-transporting as well as host emitting materials, respectively, in organic light-emitting diodes (OLEDs) due to their high thermal stability.43–45 However, the electron and hole mobilities in Alq3 and NPB are at the level of 10−4–10−5 and 10−3–10−4 cm2 V−1 s−1, respectively, in an electric field of 2 × 106 V cm−1 (ref. 46 and 47) which gives rise to an accumulation of excess holes at the NPB/Alq3 boundary. Insertion of an anodic buffer layer including organic48 and inorganic49 materials between indium-tin oxide (ITO) and a hole-transporting layer (HTL) as a hole injection layer (HIL) could control the hole injection and resulted in enhanced luminous efficiency (LE).50 In this article, we report a simple and straightforward one-pot synthesis of naphthyl phenanthromidazoles51 in good yield using sol–gel synthesised nano-titanium dioxide as an inexpensive heterogeneous and recyclable non-toxic catalyst under solvent free solid-phase conditions. We also present the significant enhancement of the EL performance by inserting the newly synthesised naphthyl phenanthromidazoles52 as a functional layer with the thickness of tens of nanometers between NPB and the Alq3 layer in the device structure, ITO/NPB/naphthyl phenanthromidazoles/Alq3/LiF/Al.
2. Experimental
2.1. Chemicals
1-Naphthaldehyde and phenanthrene-9,10-dione were supplied by Sigma-Aldrich (St. Louis, USA). Aniline, 4-methylaniline, 4-methoxyaniline and 3,5-dimethylaniline were of analytical grade and received from S.D.Fine (Mumbai, India). The solvents used were of spectroscopic grade and supplied by Himedia (Chennai, India).2.2. Synthesis of nanocrystalline TiO2 by the sol–gel method
The TiO2 nanocrystals were prepared by sol–gel hydrolysis of titanium(IV) isopropoxide, followed by calcination. About 1 ml of titanium isopropoxide (Merck, 97%) was dissolved in 20 ml isopropyl alcohol (Merck, 95%) and the solution was dropped slowly into 10 ml of distilled water and the pH was adjusted to 2. After stirring water was added to alkoxide solution, and the formed white sol–gel of hydrous oxide was stirred vigorously for 4 hours at room temperature and then allowed to age overnight. The solid was centrifuged and was redispersed in ethanol to minimise agglomeration. This process was repeated five times and the solid was filtered. The resulting material was then dried and calcinated upto 800 °C for 2 h. The sample was characterised by X-ray diffraction (XRD), scanning electron microscopy (SEM), diffused reflectance spectroscopy (DRS) and solid state photoluminescence spectroscopy. From these results it is found that high acidity [pH 2] favors the formation of rutile phase TiO2.53,542.3. Synthesis of polysubstituted imidazoles
A mixture of naphthaldehyde (1 mmol), phenanthrene-9,10-dione (1 mmol), arylamine (1 mmol) and ammonium acetate (1 mmol) with TiO2 (1 mol%) as the catalyst was stirred continuously at 120 °C with a bar magnet. The progress of the reaction was monitored by TLC (Scheme 1). After completion of the reaction, 10 ml ethyl acetate was added to the reaction mixture and shaken well to dissolve the organic components, the mass was filtered to separate TiO2, and the residue was washed with ethyl acetate. The solid TiO2 residue was further washed with hot acetone and then dried. The product was purified by column chromatography using benzene: ethyl acetate (9:1) as the eluent. The newly synthesised phenanthromidazoles have been characterised by 1H and 13C NMR and mass (MS) spectroscopy.
 | ||
| Scheme 1 Possible mechanism of catalytic synthesis of phenanthromidazoles. | ||
2.4. Measurements
XRD patterns were recorded for the centrifuged and dried samples using an X-ray Rigaku diffractometer with Cu Kα source (30 kV, 100 mA) at a scan speed of 3.0000 deg min−1, a step width of 0.1000 deg, and in a 2θ range of 20–80°. The energy dispersive X-ray (EDS) spectra of the nanosemiconductors were recorded using a JEOL JSM-5610 scanning electron microscope (SEM) equipped with a back electron (BE) detector and EDX. The sample was placed on an adhesive carbon slice supported on copper stubs and coated with a 10 nm thick gold layer using a JEOL JFC-1600 auto fine coater prior to measurement. The 1H and 13C NMR spectra at 400 and 100 MHz, respectively were obtained at room temperature using a Bruker 400 MHz NMR spectrometer (Bruker biospin, California, USA). The mass spectra were obtained using a Thermo Fischer LC-Mass spectrometer in fast atom bombardment (FAB) mode (Thermo, France). The UV-vis and photoluminescence spectra were recorded using a Perkin Elmer Lambda 35 UV-vis spectrophotometer and a PerkinElmer LS55 fluorescence spectrometer, respectively.The lifetime measurements were carried out using a nanosecond time correlated single photon counting (TCSPC) spectrometer Horiba Fluorocube-01-NL lifetime system with Nano LED (pulsed diode excitation source) as the excitation source and TBX-PS as the detector. The quantum yields were measured by comparing the emission intensities of a standard sample and the unknown sample55–57 using the formula 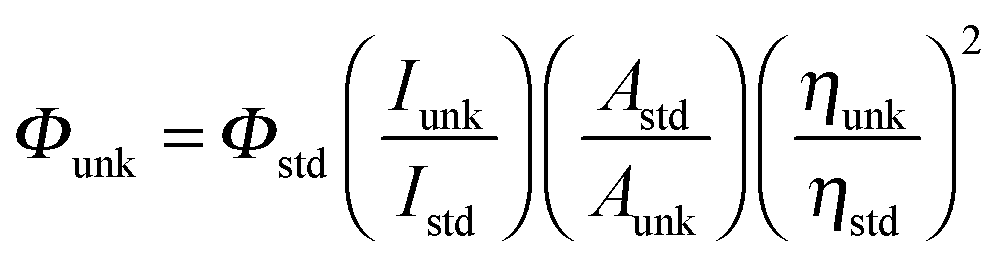 , where Φunk and Φstd are the quantum yields of the sample and the standard, respectively; Iunk and Istd are the integrated emission intensities of the sample and the standard, respectively. Aunk and Astd are the absorbance of the sample and the standard at the excitation wavelength, respectively. ηunk and ηstd are the refractive index of the sample and standard solutions, respectively. Cyclic voltammetry analyses were performed using a CHI electrochemical analyser 604C (CHI electrochemical analyser, USA) at a scan rate of 100 mV s−1 using 0.1 M tetra-(n-butyl)-ammonium hexafluorophosphate as the supporting electrolyte with Ag/Ag+ (0.01 M AgNO3) as the reference electrode and a Pt electrode as the working electrode, standardised for the redox couple ferricinium/ferrocene. All solutions were purged with a nitrogen stream for 10 min before measurement. Thermal analysis of the phenanthromidazoles was conducted using a NETZSCH-Geratebau Gmbh thermal analysis STA 409 PCO. The differential scanning calorimetric (DSC) and thermogravimetric analyses (TGA) were conducted under a nitrogen atmosphere (100 ml min−1). The sensitivity of the instrument was set at 0.01 μg and the sample (10 mg) was heated from 30 to 700 °C at the rate of 10 or 15 or 20 K min−1. DFT calculations were performed using Gaussian-0358 package.
, where Φunk and Φstd are the quantum yields of the sample and the standard, respectively; Iunk and Istd are the integrated emission intensities of the sample and the standard, respectively. Aunk and Astd are the absorbance of the sample and the standard at the excitation wavelength, respectively. ηunk and ηstd are the refractive index of the sample and standard solutions, respectively. Cyclic voltammetry analyses were performed using a CHI electrochemical analyser 604C (CHI electrochemical analyser, USA) at a scan rate of 100 mV s−1 using 0.1 M tetra-(n-butyl)-ammonium hexafluorophosphate as the supporting electrolyte with Ag/Ag+ (0.01 M AgNO3) as the reference electrode and a Pt electrode as the working electrode, standardised for the redox couple ferricinium/ferrocene. All solutions were purged with a nitrogen stream for 10 min before measurement. Thermal analysis of the phenanthromidazoles was conducted using a NETZSCH-Geratebau Gmbh thermal analysis STA 409 PCO. The differential scanning calorimetric (DSC) and thermogravimetric analyses (TGA) were conducted under a nitrogen atmosphere (100 ml min−1). The sensitivity of the instrument was set at 0.01 μg and the sample (10 mg) was heated from 30 to 700 °C at the rate of 10 or 15 or 20 K min−1. DFT calculations were performed using Gaussian-0358 package.
2.5. Device fabrication
The EL devices based on the phenanthromidazoles were fabricated by vacuum deposition of the materials at 5 × 10−6 Torr onto a clean glass precoated with a layer of indium tin oxide as the substrate with sheet resistance of 20 Ω per square. The glass was cleaned by sonication successively in a detergent solution, acetone, methanol and deionised water before use. Organic layers were deposited onto the substrate at a rate of 0.1 nm s−1. LiF and Alq3 were thermally evaporated onto the surface of the organic layer. The thickness of the organic materials and the cathode layers was controlled using a quartz crystal thickness monitor. A series of devices (I, II, III, IV and V) with the multilayer configuration of ITO/NPB (60 nm)/1–5 (20 nm)/Alq3 (30 nm)/LiF (1 nm)/Al (100 nm) were fabricated. Measurements of current, voltage and light intensity were taken simultaneously using a Keithley 2400 sourcemeter (Keithley, Cleveland, Ohio). The EL spectra of the devices were recorded in an ambient atmosphere without further encapsulation.3. Results and discussion
3.1. Characterisation of nano-TiO2 (R)
The nanocrystalline rutile phase of TiO2 was obtained by the sol–gel method, and characterised by X-ray diffraction, high resolution scanning electron microscopy and energy dispersive, UV-visible diffuse reflectance and solid state photoluminescence spectroscopies. Fig. 1 displays the X-ray diffraction pattern (XRD) of the rutile phase of TiO2 nanoparticles and the diffraction pattern of TiO2 (R) matches with the JCPDS pattern of tetragonal primitive (89-4920). The crystal constants a and c are 4 Å and 2.953 Å, respectively. The average crystallite size (L) deduced from the XRD result is 20.44 nm. The Scherrer equation L = 0.9λ/βcosθ (where λ is the wavelength of the X-ray used, θ is the diffraction angle and β is the full width at half maximum of the peak) was used to obtain the mean crystalline size. The specific surface area (S) of the nanocrystal has been deduced by employing the relation, S = 6/ρL (ρ is the material density). The calculated surface area of the rutile phase of TiO2 is 51.72 m2 g−1.
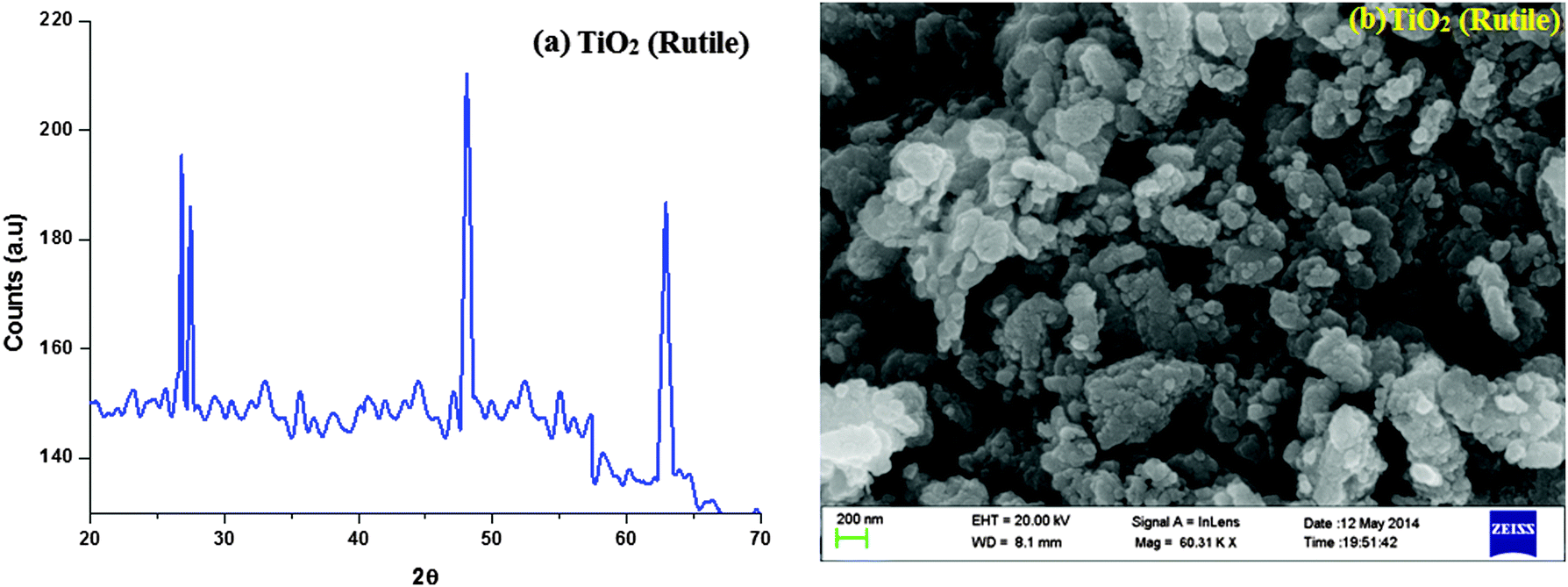 | ||
| Fig. 1 (a) X-ray diffraction pattern (XRD) of TiO2 (rutile); (b) SEM image of TiO2 (rutile). | ||
The scanning electron micrograph (SEM) of the TiO2 (R) nanoparticle is displayed in Fig. 1. The DRS of rutile TiO2 is displayed in Fig. 2 which is presented in terms of F(R), deduced from the recorded reflectance (R) by application of the Kubelka–Munk algorithm [F(R) = (1 − R)2/2R]. The absorption edge of rutileTiO2 is 389 nm and the deduced absorption edge provides the band gap of rutile TiO2 as 3.19 eV. The band gap of the synthesised rutile TiO2 is larger than the literature value. This is because of the smaller size of the synthesised nanoparticles. The quantum confinement effect increases the band gap energy.
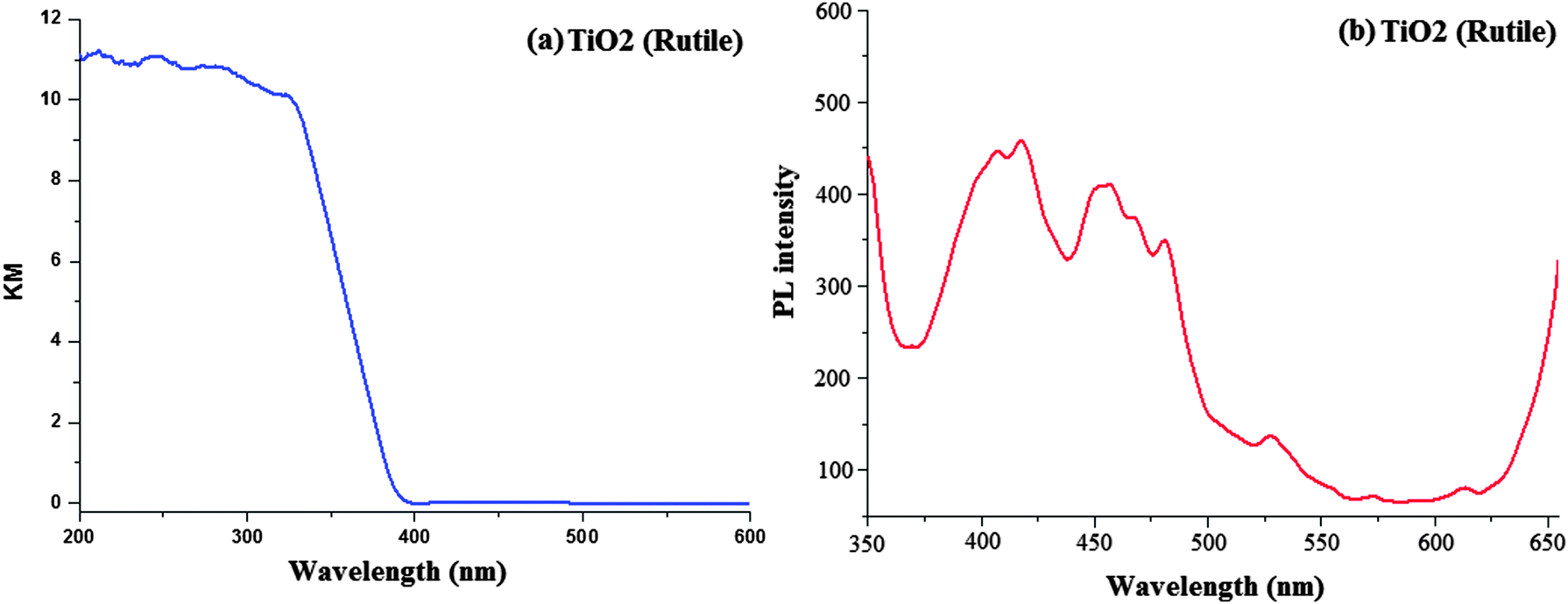 | ||
| Fig. 2 (a) Diffused reflectance spectra of TiO2 (rutile); (b) solid state photoluminescence spectra of TiO2 (rutile). | ||
Fig. 2 displays the solid state photoluminescence spectra of the synthesised rutile TiO2 nanocrystals. Nanocrystals were excited at 340 nm and the solid emission spectrum of rutile TiO2 mainly consists of four emission bands: a strong violet emission at 411 nm (3.01 eV), a blue band at 450 nm (2.75 eV), a blue-green band at 482 nm (2.57 eV) and a weak green band at 530 nm (2.34 eV). The two peaks at 482 and 530 nm are attributed to the transition from the oxygen vacancies with two trapped electrons and one trapped electron to the valence band of TiO2, respectively. The energy levels related to the two kinds of the oxygen vacancies are located at 0.51 and 0.82 eV below the conduction band (CB) of TiO2, respectively. Because of the existence of the energy levels of oxygen vacancies, first the photogenerated electrons in the CB are likely to reach the oxygen vacancies through a non-radiative process and then recombine with the photogenerated holes in the valence band (VB) accompanied by the emission of fluorescence. The broad band in the visible region is also ascribed to the radiative recombination of excitons of the shallow traps identified with oxygen vacancies and Ti4+ adjacent to oxygen vacancies.59
3.2. Catalytic activity of the TiO2 (R) semiconductor
Initially, we have carried out the condensation reaction in the presence of TiO2 (R) (1 mol%), 1-naphthaldehyde (1 mmol), ammonium acetate (1 mmol) and arylamine (1 mmol) in different solvents such as water, ethanol, methanol, chloroform and acetonitrile under refluxing and also under solvent-free conditions at 120 °C (Scheme 1). From these experiments, it was clearly demonstrated that solvent-free conditions are the best for phenanthromidazole synthesis. In the absence of a catalyst under solvent-free conditions at room temperature the yield is very poor even after 24 h. To enhance the yield of the desired product, the temperature of the reaction was increased to 180 °C, but no appreciable increment in the product yield was observed. We found that the presence of a catalytic amount of TiO2 (R) under solvent-free conditions is the best for this synthesis; maximum yield (82%) was obtained at 30 min on loading with 1 mol% of TiO2 (R) at 120 °C (Table 1). Moreover, TiO2 can be recovered and reused several times without significant loss of activity. High product yield, a shorter reaction time, low catalyst loading and easy work-up make this procedure quite simple and more convenient. Our methodology could be a valid contribution to the existing processes of imidazole synthesis.| Entry | Temp. (°C) | Solvent | TiO2 (R) | TiO2 (R) (mol%) | |
|---|---|---|---|---|---|
| Time (min) | Yield (%) | ||||
| a Values in the parentheses correspond to values without the catalyst. | |||||
| 1 | r.t | Solvent-free | 130(380) | 70(trace) | 0.1 |
| 2 | 50 | Solvent-free | 65(250) | 72(22) | 0.1 |
| 3 | 70 | Solvent-free | 30(70) | 76(45) | 0.1 |
| 4 | 90 | Solvent-free | 25(90) | 80(53) | 0.1 |
| 5 | 120 | Solvent-free | 30 | 82 | 1 |
| 6 | 120 | Solvent-free | 25 | 85 | 2 |
| 7 | 120 | Solvent-free | 45 | 88 | 10 |
| 8 | 120 | Water | 100 | 35 | 1 |
| 9 | 120 | Ethanol | 40 | 65 | 1 |
| 10 | 120 | Methanol | 50 | 70 | 1 |
| 11 | 120 | Chloroform | 140 | 42 | 1 |
| 12 | 120 | Acetonitrile | 95 | 62 | 1 |
3.3. XRD characterisation of 1-(4-methoxyphenyl)-2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (4)
The naphthyl phenanthromidazoles have been synthesised using the modified Debus–Radziszewski method, with the use of TiO2 (R) as the catalyst.60 1-(4-Methoxyphenyl)-2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole is a monoclinic crystal and crystallizes in the P21/c space group with cell dimensions of a = 12.3316 (7) Å, b = 21.3475 (11) Å and c = 8.8212 (5) Å and the ORTEP diagram is presented in Fig. 3. The imidazole unit forms dihedral angles of −86.9° (17) [N(2)–C(15)–C(16)–C(17) ] and −102.3° (17) [C(14)–N(2)–C(26)–C(31)] with the adjacent naphthyl and methoxyphenyl ring. In the crystal, molecules are consolidated into a three-dimensional architecture by π–π stacking interactions. The electron donating naphthyl ring and the electron accepting phenanthromidazole moiety would lead to intermolecular electrostatic interaction.61 Optimization of 1-(4-methoxyphenyl)-2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole has been performed by DFT at the B3LYP/6-31G(d,p) level using Gaussian-03. The optimized parameters, namely bond lengths, bond angles and dihedral angles, are slightly higher than those of XRD values (Table 2). The theoretical calculations are of an isolated molecule in the gaseous phase and the XRD results are of the molecule in the solid state. Thermal properties have been investigated by differential scanning calorimetry (DSC) and thermogravimetric analyses (TGA) under a nitrogen atmosphere and the results are displayed in Fig. 3. All phenanthromidazoles exhibit good thermal stability and the decomposition temperature with 5% weight loss (Td5) has been measured as 310, 315, 319, 325 and 334 °C for compounds 1–5, respectively (Table 3). The melting point of phenanthromidazoles 1–5 measured by DSC is 262, 265, 248, 272 and 261 °C, respectively. On the second heating, no melting points were observed, even though it was given enough time to cool in air. Once it became an amorphous solid, it did not revert to the crystalline state at all. After the sample had cooled to room temperature, a second DSC scan performed at 10 °C min−1 revealed a glass transition temperature (Tg) in the range of 65 to 82 °C. The methoxy phenanthromidazole 4 has the highest melting point, which may due to the intermolecular C–H⋯π interactions that can induce more condensed molecular packing. The high Tm and Td5 values indicate that they could form morphologically stable amorphous film upon vacuum thermal evaporation, which is highly important for device fabrication62 since the high Tm and Td5 could improve the lifetime of the devices.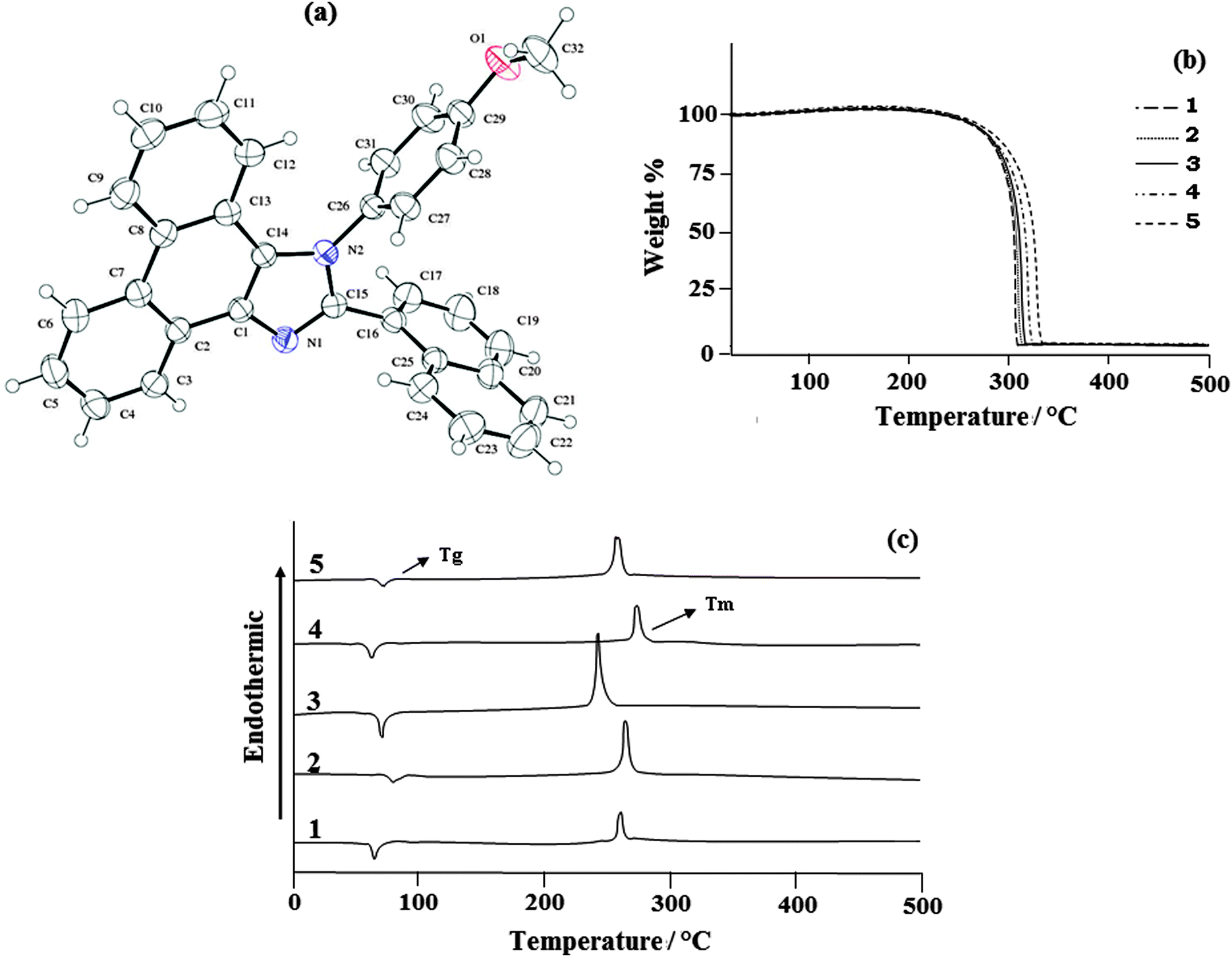 | ||
| Fig. 3 (a) ORTEP diagram of 1-(4-methoxyphenyl)-2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole (4); (b) TG-DTA (upper); and (c) DSC (bottom) curves of phenanthromidazoles at a heating rate of 20 °C min−1. | ||
| Connectivity | Bond lengths XRD | Connectivity | Bond angles XRD | Connectivity | Torsional angles XRD |
|---|---|---|---|---|---|
| a Values in the parentheses correspond to theoretical values. | |||||
| C(1)–C(14) | 1.3744(1.3746) | C(14)–C(1)–C(2) | 121.82(121.84) | N(1)–C(1)–C(14)–N(2) | 0.24(0.26) |
| C(1)–C(2) | 1.4279(1.429) | N(1)–C(1)–C(2) | 126.92(126.94) | N(1)–C(1)–C(2)–C(3) | −3.2(−3.4) |
| C(13)–C(14) | 1.4349(1.4351) | C(1)–C(14)–C(13) | 123.28(123.29) | C(1)–C(2)–C(7)–C(8) | 1.71(1.73) |
| C(1)–N(1) | 1.3809(1.3810) | C(14)–C(1)–N(1) | 111.22(111.24) | N(1)–C(1)–C(14)–N(2) | 0.24(0.26) |
| C(15)–N(1) | 1.3132(1.3135) | N(2)–C(14)–C(13) | 131.68(131.69) | N(1)–C(15)–N(2)–C(14) | 1.15(1.17) |
| C(15)–N(2) | 1.3710(1.3712) | C(14)–N(2)–C(26) | 128.04(128.06) | N(2)–C(15)–C(16)–C(17) | −86.86(−86.88) |
| C(14)–N(2) | 1.3897(1.3899) | C(31)–C(26)–N(2) | 120.32(120.34) | N(2)–C(15)–C(16)–C(25) | 94.40(94.42) |
| C(15)–C(16) | 1.4779(1.4780) | C(27)–C(26)–N(2) | 119.36(119.38) | N(2)–C(26)–C(27)–C(28) | −176.51(−176.53) |
| C(16)–C(17) | 1.363(1.3650) | C(27)–C(26)–C(31) | 120.31(120.33) | C(13)–C(14)–N(2)–C(26) | 1.6(1.8) |
| C(16)–C(25) | 1.4214(1.423) | C(26)–C(27)–C(28) | 120.55(120.57) | C(31)–C(26)–N(2)–C(14) | −102.27(17) |
| C(17)–C(18) | 1.395(1.3970) | C(29)–C(28)–C(27) | 119.24(119.25) | C(27)–C(26)–N(2)–C(14) | 76.45(76.47) |
| C(26)–N(2) | 1.4375(1.4377) | C(28)–C(29)–C(30) | 120.08(120.10) | N(2)–C(26)–C(31)–C(30) | 177.13(177.15) |
| C(26)–C(27) | 1.3681(1.3681) | O(1)–C(32)–H(32A) | 109.5 | N(2)–C(26)–C(27)–C(28) | −176.51(−176.53) |
| C(26)–C(31) | 1.380(1.3840) | O(1)–C(32)–H(32B) | 109.5 | C(26)–C(27)–C(28)–C(29) | −0.8(−0.9) |
| C(27)–C(28) | 1.3799(1.3810) | O(1)–C(32)–H(32C) | 109.5 | C(27)–C(28)–C(29)–C(30) | −1.2(−1.4) |
| C(28)–C(29) | 1.377(1.3790) | C(29)–O(1)–C(32) | 117.59(117.61) | C(28)–C(29)–C(30)–C(31) | 1.9(1.11) |
| C(30)–C(31) | 1.372(1.3730) | O(1)–C(29)–C(28) | 123.97(123.99) | C(29)–C(30)–C(31)–C(26) | −0.5(−0.7) |
| C(29)–C(30) | 1.382(1.3840) | O(1)–C(29)–C(30) | 115.94(115.95) | C(30)–C(29)–O(1)–C(32) | 178.73(178.75) |
| C(29)–O(1) | 1.3696(1.3698) | C(30)–C(31)–C(26) | 119.39(119.41) | C(28)–C(29)–O(1)–C(32) | −0.2(−0.4) |
| C(32)–O(1) | 1.423(1.4250) | C(31)–C(30)–C(29) | 120.38(120.40) | C(18)–C(19)–C(20)–C(25) | 1.1(1.3) |
| C(17)–C(16)–C(15) | 120.72(120.73) | C(16)–C(17)–C(18)–C(19) | −1.6(−1.8) | ||
| C(25)–C(16)–C(15) | 119.17(119.19) | C(23)–C(24)–C(25)–C(16) | −178.90(−178.91) | ||
| Compd. |
λ
absa (nm) |
λ
emia (nm) |
λ
absb (nm) |
T g/Tm/Td5 (°C) | HOMOc (eV) | LUMOd (eV) |
|---|---|---|---|---|---|---|
| a Measured in the CH2Cl2 solvent at room temperature. b Solid state. c Calculated by comparing with ferrocene (Fc) (4.8 eV) and calibrated using E1/2 (Fc/Fc+) = 0.22 V. d HOMO − 1239/λ. | ||||||
| 1 | 250, 286, 339 | 422, 367 | 241 | 65/262/310 | −5.40 | −2.32 |
| 2 | 260, 283, 228 | 423 | 268 | 82/265/315 | −5.36 | −2.37 |
| 3 | 260, 284, 229 | 435, 363 | 272 | 74/248/319 | −5.32 | −2.36 |
| 4 | 251, 285, 308 | 414 | 272 | 64/272/325 | −5.30 | −2.38 |
| 5 | 261, 283, 229 | 409, 365 | 268 | 74/261/334 | −5.38 | −2.35 |
The electronic spectral studies of naphthyl phenanthromidazoles were performed in dichloromethane and the absorption and emission spectra are displayed in Fig. 4. The absorption maxima around 250 nm may originate from the aryl ring and the absorption band between 280–339 nm is assigned to π → π* electronic transition of the phenanthromidazole ring.63 phenanthromidazole derivatives show emission at 422, 423, 435, 414 and 409 nm, respectively, in CH2Cl2.
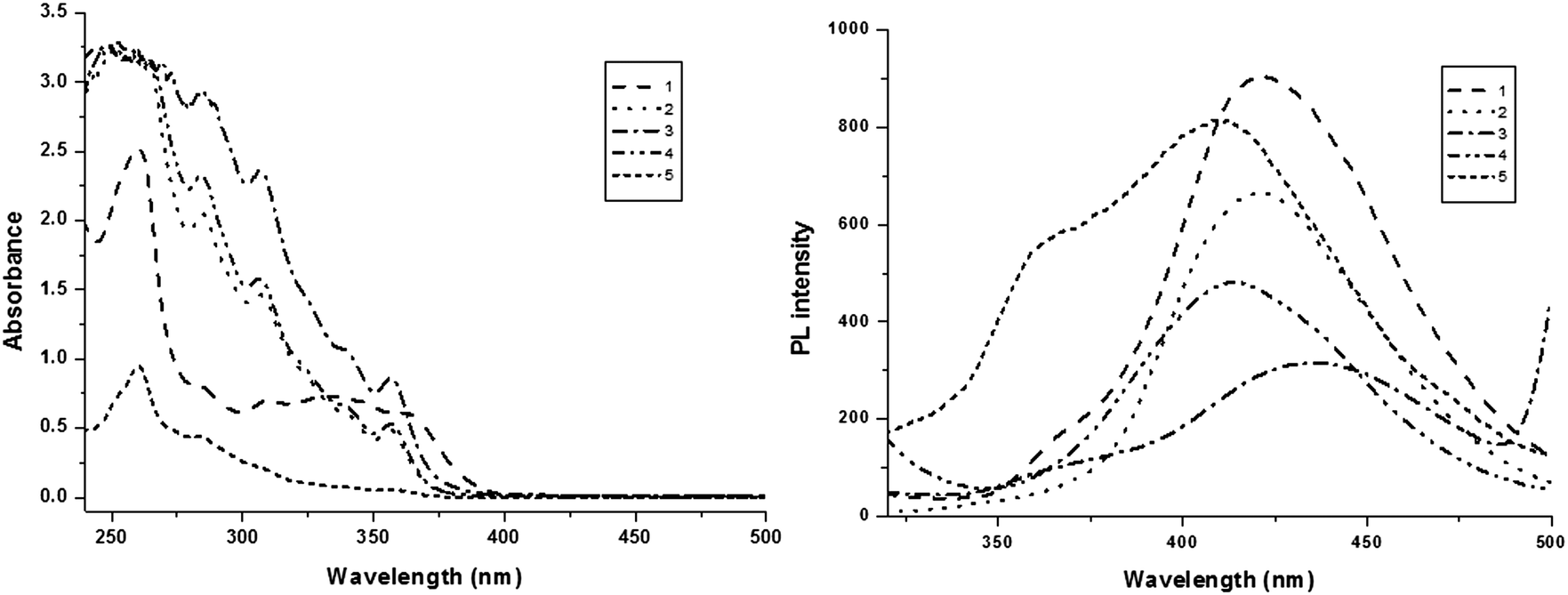 | ||
| Fig. 4 Absorption and emission spectra of phenanthromidazoles in CH2Cl2. | ||
The electronic spectra of the phenanthromidazole materials show remarkable differences in solution and in the solid state (Fig. 5). This may be due to restrained intermolecular aggregation of the bulky and non-coplanar part of these compounds. It can also be correlated with the dihedral parameters of the molecules as shown in Table 4 and Fig. 5. The red shift can be explained by the stronger π–π stacking interactions due to a smaller dihedral angle and the larger dihedral angle results in a smaller peak shift. The quantum yield (Φ) measured in the CH2Cl2 solution is 0.56 (1), 0.55 (2), 0.53 (3), 0.62 (4) and 0.59 (5). The radiative (kr) and non-radiative (knr) decays of the excited state of these compounds have been obtained using the quantum yield and lifetime (τ) and are listed in Table 4. The lifetime decay curve is shown in Fig. 6. The radiative lifetime of these compounds falls in the range of 2.09–2.53 ns. Radiative emission of naphthyl phenanthromidazole derivatives is predominant over non-radiative emission.
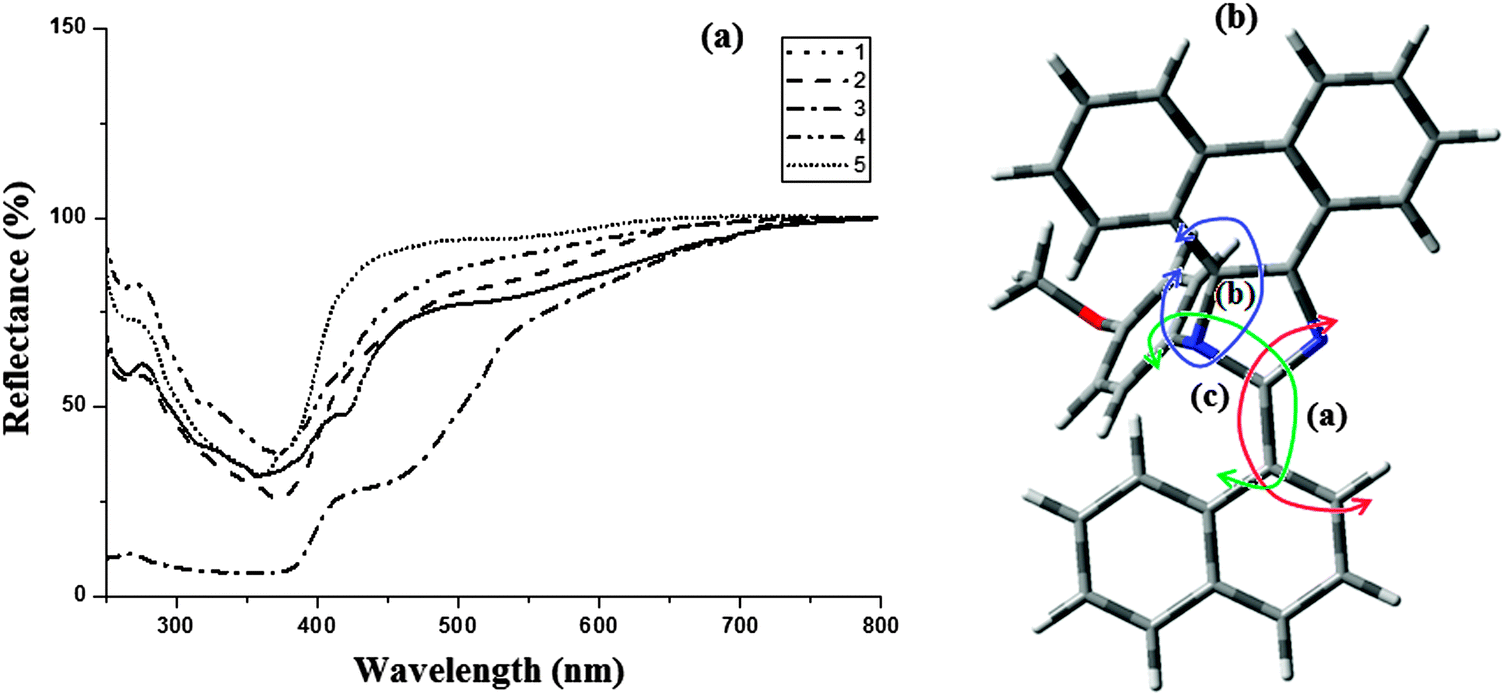 | ||
| Fig. 5 (a) Absorption spectra of phenanthromidazoles in the solid state; (b) molecular modeling of phenanthromidazole using Gaussian-03. | ||
| Compd. | Anglea | Angleb | Anglec | Φ | k r | k nr |
|---|---|---|---|---|---|---|
| a Dihedral angle between the imidazole ring and aryl ring. b Dihedral angle between the imidazole ring and naphthyl ring. c Dihedral angle between the aryl ring and naphthyl ring. | ||||||
| 1 | 179 | 85 | 74 | 0.56 | 0.24 | 0.19 |
| 2 | 182 | 154 | 75 | 0.55 | 0.23 | 0.19 |
| 3 | 174 | 85 | 79 | 0.53 | 0.21 | 0.19 |
| 4 | 185 | 154 | 118 | 0.62 | 0.27 | 0.16 |
| 5 | 180 | 86 | 116 | 0.59 | 0.28 | 0.20 |
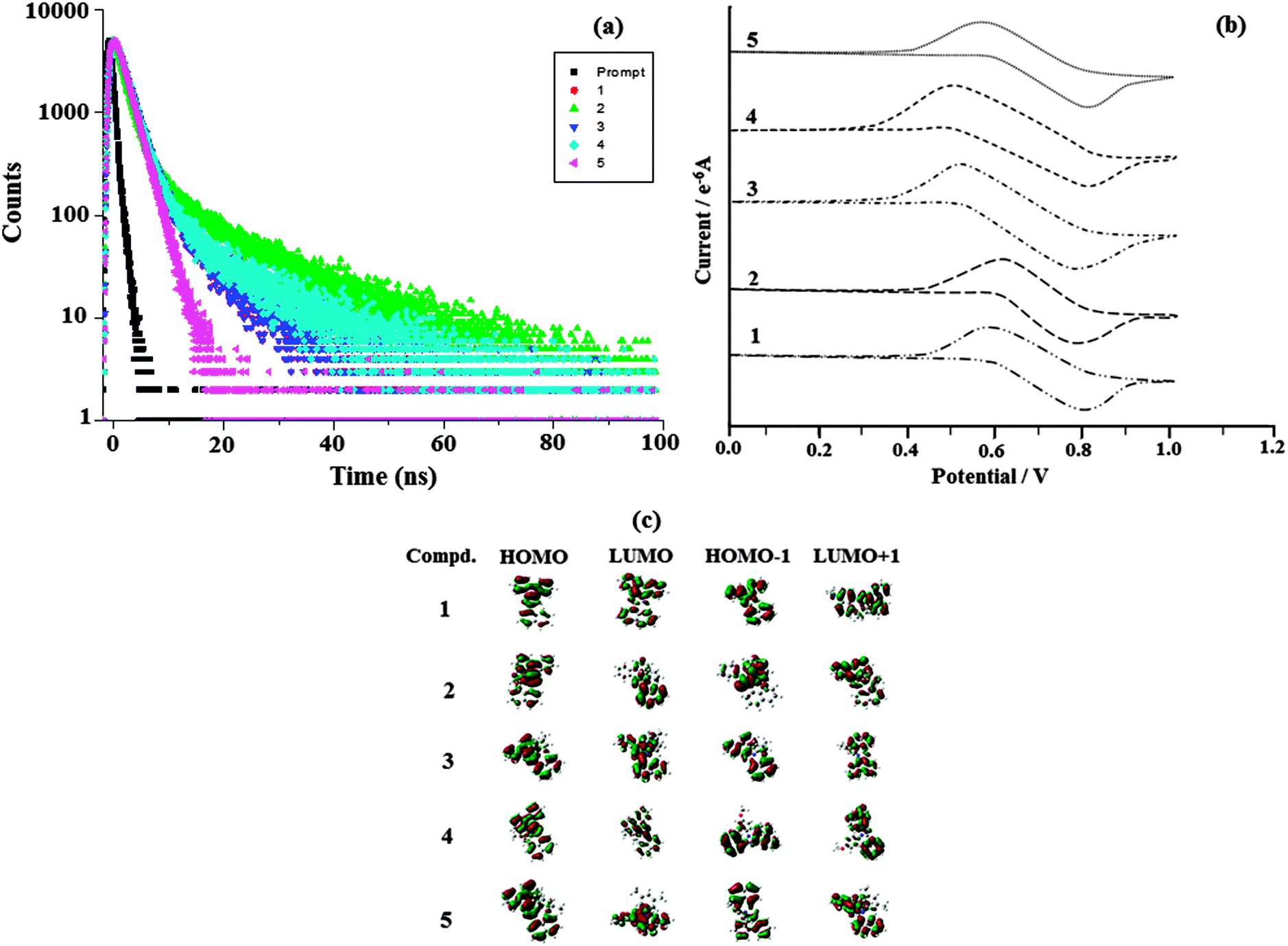 | ||
| Fig. 6 (a) Lifetime spectra of naphthyl phenanthromidazoles 1–5; (b) cyclic voltammogram of phenanthromidazoles 1–5; and (c) frontier molecular orbitals of phenanthromidazoles. | ||
DFT calculations show that the methoxy phenanthromidazole possesses a larger dihedral angle with the imidazole and naphthyl ring. In addition the large dihedral angle between them suppresses the intermolecular π–π stacking in the solid state and prevents self-quenching of emission intensity. Examination of the quantum yield and radiative (kr) and non-radiative (knr) rate constants of the synthesised methoxy phenanthromidazole exhibits better photophysical properties. The quantum yield and radiative (kr) rate constant are larger for the methoxy phenanthromidazole than others and knr is the least among the studied molecules.
The electrochemical properties of phenanthromidazoles have been examined by cyclic voltammetry and the redox potentials have been measured from the plot potential versus current, which is shown in Fig. 6. These compounds exhibit one reversible oxidation wave with an oxidation onset potential of 0.60 V (1), 0.62 V (2), 0.56 V (3), 0.50 V (4) and 0.58 V (5), which gives the HOMO energies of −5.40 eV (1), −5.36 eV (2), −5.32 eV (3), −5.30 eV (4) and −5.38 eV (5) by comparison to ferrocene (EHOMO = Eox + 4.8 eV).64 The much smaller HOMO energies support the hole injection ability of these phenanthromidazoles and the calculated values are displayed in Table 3. The LUMO energies have been deduced from the HOMO energies (Fig. 6) and the lowest-energy absorption edges of the UV-vis absorption spectra.64 The LUMO energies of 2.32 eV (1), −2.37 eV (2), −2.36 eV (3), 2.38 eV (4) and −2.35 eV (5) are close to that of 1,3,5-tris(N-phenylimidazol-2-yl)benzene (−2.40 eV) revealing that the electron injection abilities of phenanthromidazoles are similar to 1,3,5-tris(N-phenylimidazol-2-yl)benzene. This reflects the more balanced carrier injection properties existing in these materials. This result gives us a new direction to introduce new building block emitters by using different substituents to tune the HOMO and LUMO energies.65,66 To investigate the compound relationship between structure and properties, the ground-state structure and electronic properties were predicated using the B3LYP/6-31G(d,p) level as shown in Fig. 6. The electron density of HOMO orbitals is mainly populated on the electron-donating arylamine moieties while the LUMO orbitals are mainly located on the electron-accepting phenanthromidazole moiety. The electron density of HOMO and LUMO orbitals exhibits almost complete separation, which is preferable for efficient charge transporting properties.67
Wang et al.63 fabricated OLEDs with the phenyl substituted phenanthromidazoles and their results could be compared with the present work in which naphthyl substituted phenanthromidazoles are used as the functional layer between NPB and ALq3. The naphthyl substituted phenanthromidazoles have a much shallower HOMO energy than phenyl substituted phenanthromidazoles (∼−5.58 eV). The shallow HOMO effectively prevents the leakage of holes into the electron transport layer. Attaching the naphthyl moiety instead of phenyl to the phenanthromidazoles plays a key role in controlling the HOMO energy levels of the naphthyl phenanthromidazoles. With the shallow HOMO, it can effectively lower the hole injection barrier from the NPB layer into the phenanthromidazoles resulting in higher efficiency. Furthermore, the radiative emission of the synthesised naphthyl phenanthromidazoles is predominant over non-radiative emission. To investigate the EL properties of phenanthromidazoles, a series of devices (I, II, III, IV and V) with the device configuration of ITO/NPB (10 nm)/1–5 (30 nm)/Alq3 (60 nm)/LiF (1 nm)/Al have been fabricated (Fig. 7).
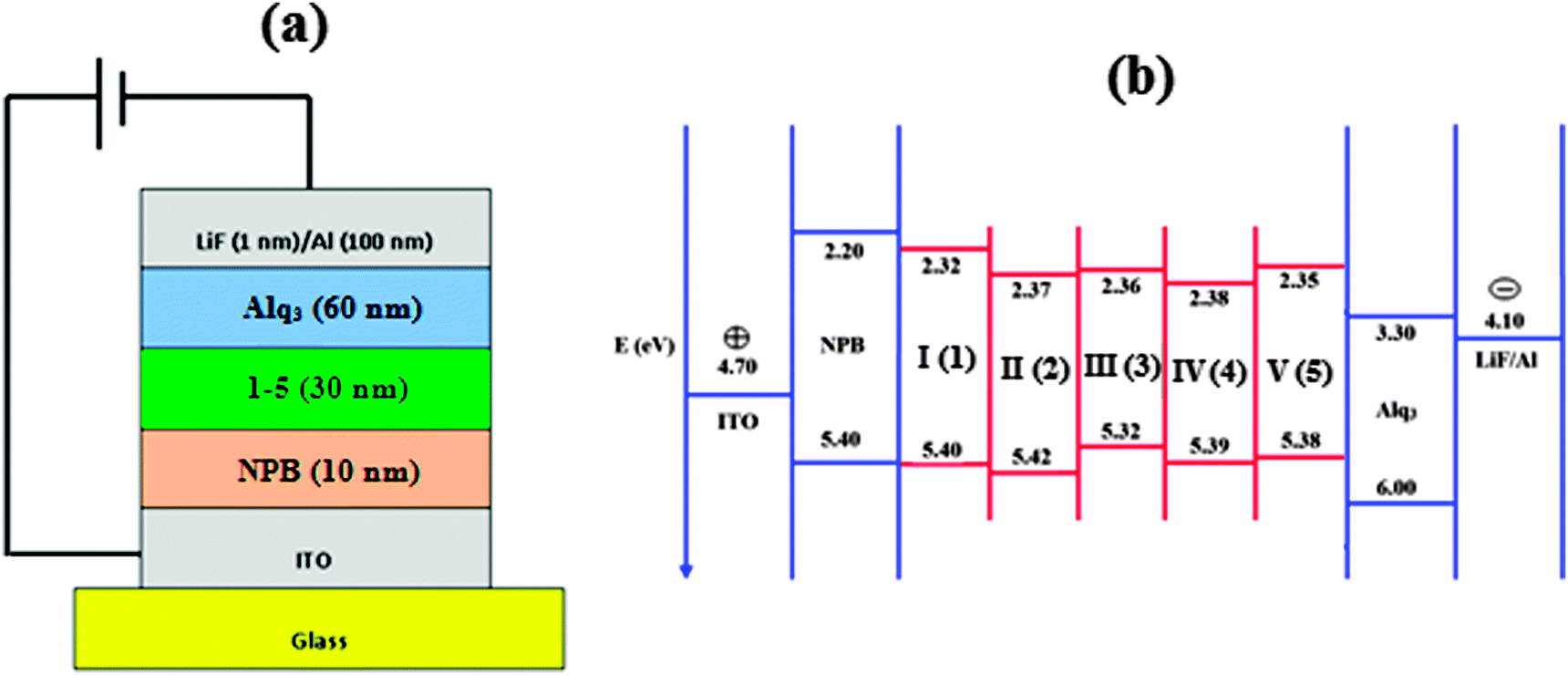 | ||
| Fig. 7 (a) General structure of a multilayer OLED device; (b) schematic energy level diagram of LUMO–HOMO for devices I–V. | ||
The devices exhibit emission between 393–431 nm and the current density–brightness–voltage and the luminous efficiencies are listed in Table 5. Fig. 8 and 9a and b show the brightness–voltage, power, current efficiencies and the external quantum yield–current density characteristics of the devices, respectively. Devices showed very high brightness and colour stability under different driving voltages. The improved efficiency of devices I–V is due to the decrease of the leakage of holes through the phenanthromidazole layer, which results in balancing the electrons and holes in the emitting layer and thus eliminating the non-productive hole current.
| Device | EL | V (V) | L (cd m−2) | η ex (%) | η c (cd A−1) | η p (lm W−1) |
|---|---|---|---|---|---|---|
| a Voltage (V) required for 1 cd m−12. b The brightness (L), current efficiency (ηc), external quantum yield (ηex%), and power efficiency (ηp). | ||||||
| I | 431 | 4.3 | 31982 |
2.90 | 4.01 | 3.92 |
| II | 393 | 4.0 | 34129 |
2.21 | 4.21 | 3.81 |
| III | 419 | 2.8 | 37562 |
3.56 | 4.08 | 3.97 |
| IV | 422 | 2.5 | 40623 |
3.92 | 5.99 | 5.25 |
| V | 409 | 3.5 | 38143 |
2.85 | 5.41 | 5.02 |
| VI63 | — | 2.7 | 37360 |
— | 3.22 | — |
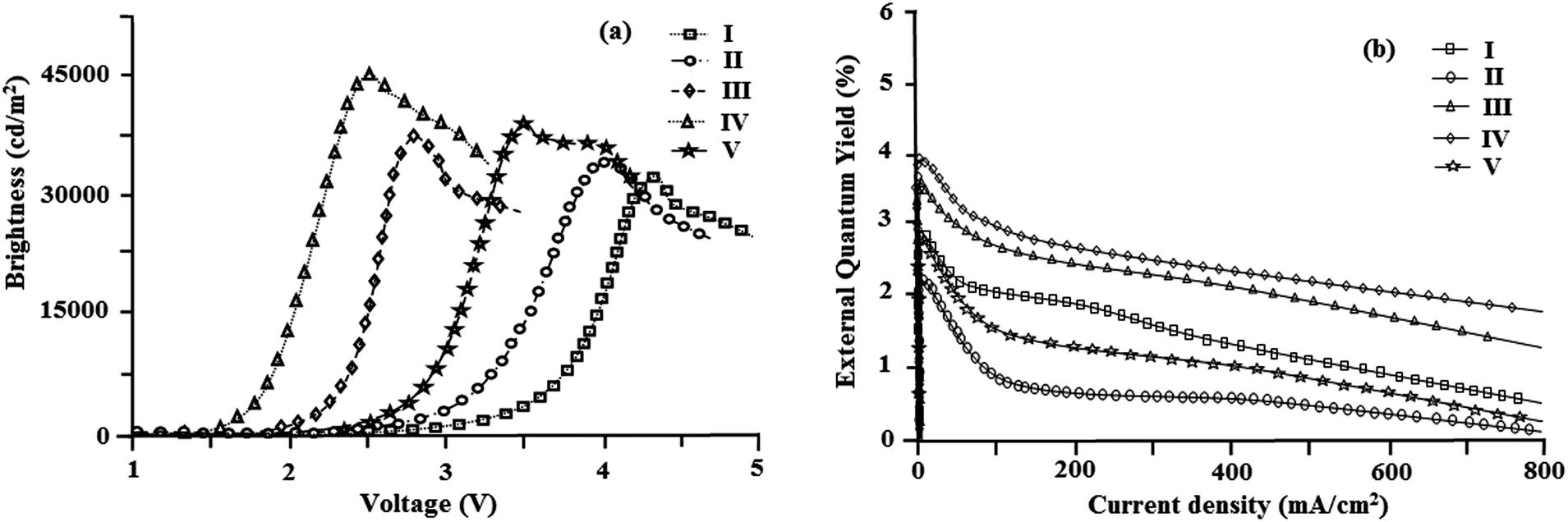 | ||
| Fig. 8 (a) Plot of brightness vs. voltage; (b) plot of external quantum yield vs. current density. | ||
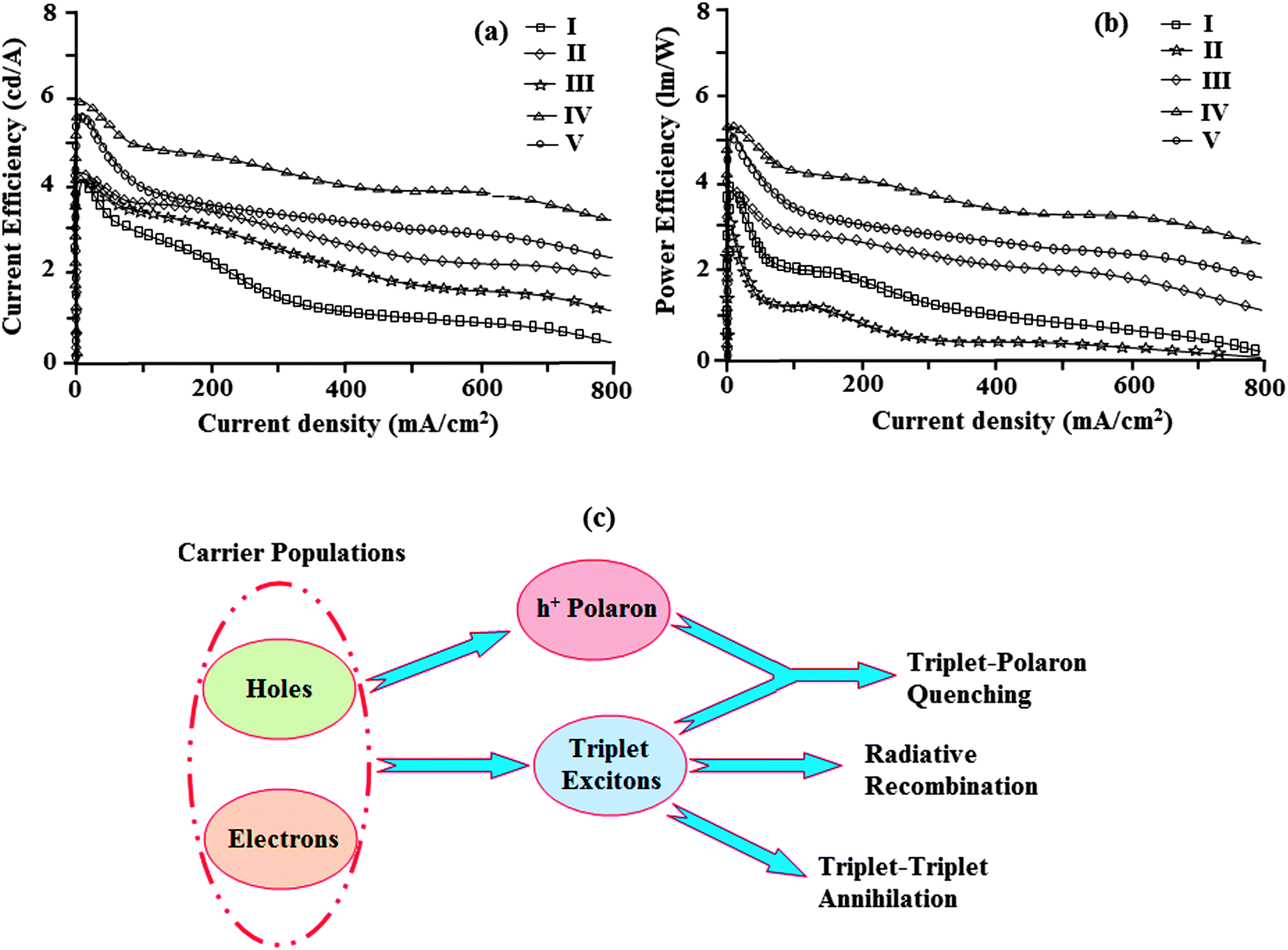 | ||
| Fig. 9 (a) Plot of current efficiency vs. current density; (b) plot of power efficiency vs. current density; and (c) a schematic illustration of TTA and TPA processes. | ||
All devices show quite appreciable efficiencies and brightness. Of the five devices, device II shows poor efficiency followed by devices I and V. The external quantum efficiency of device II is low; even at a driven voltage of 4.0, the external quantum efficiency is 2.2%. The efficiency roll-off may be due to triplet–triplet annihilation [TTA − 3M* + 3N* → 1M + 3N*] and triplet–polaron annihilation [TPA − 3M* + N− → 1M + N−*] as reported in the literature.68 Baldo et al.,69 reported that triplet–triplet annihilation (TTA) is the dominant factor for the external quantum efficiency roll-off. Furthermore, Reineke et al.,70 and Aziz et al.,71 advocated that triplet–polaron annihilation (TPA) could be the only source of efficiency roll-off at high current densities. A simple illustration of these processes is shown in Fig. 9c. Though all devices show high efficiencies and brightness, device IV with methoxy phenanthromidazole shows emission at 422 nm with a maximum current efficiency of 5.99 cd A−1, an external quantum efficiency of 3.92% and a maximum luminance of 40623 cd m−2 at a low turn-on voltage of 2.5 V. From the electroluminescent studies it was concluded that the device made from methoxy phenanthromidazole 4 exhibits a higher device performance, which provides the possibility of application in low cost devices. The performance of this device resulted from its high thermal stability and balanced charge injection properties.
Zhang et al.72 reported that incorporation of thiophene at the C-2 position of the phenanthromidazole, namely 1-(4-tert-butylphenyl)-2-(5-(pyren-1-yl)thiophen-2-yl)-1H-phenanthro[9,10-d]imidazole, shows a maximum brightness of 15960 cd m−2 at 4.4 V, a current efficiency of 2.93 cd A−1, a power efficiency of 3.02 lm W−1 and an external quantum efficiency of 0.88%. In the present study the reported naphthyl phenanthromidazole derivatives show appreciable brightness, external quantum yield, power and current efficiencies when compared with the thiophene substituted phenanthromidazole derivatives. A comparison of the reference device (VI) with the configuration of ITO/NPB (40 nm)/Alq3 (60 nm)/LiF (1 nm)/Al reported by Wang et al.63 shows that the current efficiency of the synthesised naphthyl phenanthromidazoles is higher than the standard one.
4. Conclusion
Nanocrystalline rutile TiO2 catalyses the synthesis of naphthyl phenanthromidazole derivatives under solvent free conditions. The high Tm and Td5 values indicate that these compounds are thermally stable and can be used to fabricate devices. The naphthyl phenanthromidazole based devices exhibit higher efficiencies compared with the thiaphene phenanthromidazole based devices. phenanthromidazoles with shallow HOMO energy effectively prevent the leakage of holes into the electron transport layer and thus improve the efficiency.The external quantum efficiency of device II based on 2-(naphthalen-1-yl)-1-phenyl-1H-phenanthro[9,10-d]imidazole is low when compared with other devices; even at a high driven voltage of 4.0, the external quantum efficiency is 2.2%. The efficiency roll-off may be due to triplet–triplet annihilation [TTA − 3M* + 3N* → 1M + 3N*] and triplet–polaron annihilation [TPA − 3M* + N− → 1M + N−*]. The device based on 1-(4-methoxyphenyl)-2-(naphthalen-1-yl)-1H-phenanthro[9,10-d]imidazole shows a brightness of 40623 cd m−2, a current efficiency of 5.99 cd A−1, a power efficiency of 5.25 lm W−1 and an external quantum efficiency of 3.92%.
Acknowledgements
One of the authors, Prof. J. Jayabharathi, is thankful to DST [No. SR/S1/IC-73/2010], DRDO (NRB-213/MAT/10-11), UGC (F. No. 36-21/2008 (SR)) and CSIR (NO 3732/NS-EMRII) for providing funds to this research study.References
- N. S. Hush, Coord. Chem. Rev., 1985, 64, 135–157 CrossRef CAS.
- R. A. Marcus, J. Phys. Chem., 1989, 93, 3078–3086 CrossRef CAS.
- I. R. Gould, R. H. Young, R. E. Moody and S. Farid, J. Phys. Chem., 1991, 95, 2068–2080 CrossRef CAS.
- I. R. Gould, D. Noukakis, L. Gomez-Jahn, R. H. Young, J. L. Goodman and S. Farid, J. Chem. Phys., 1993, 176, 439–456 CAS.
- J. Cortes, H. Heitele and J. Jortner, J. Phys. Chem., 1994, 98, 2527–2536 CrossRef CAS.
- R. S. Mulliken and W. B. Person, Molecular Complexes: A Lecture and Reprint Volume, VCH, Weinheim, 1969 Search PubMed.
- J. Nelson, S. A. Haque, D. R. Klug and J. R. Durrant, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 205321 CrossRef.
- S. E. de Laszlo, C. Hacker, B. Li, D. Kim, M. Maccoss, N. Mantle, J. V. Pivnichny, L. Colwell, G. E. Koch, M. A. Cascieri and W. K. Hagmann, Bioorg. Med. Chem. Lett., 1999, 9, 641–646 CrossRef CAS.
- P. A. Eyers, M. Craxton, N. Morrice, P. Cohen and M. Goedert, Chem. Biol., 1998, 5, 321–328 CrossRef CAS.
- M. J. Newman, J. C. Rodarte, K. D. Benbatoul, S. J. Romano, C. Zhang, S. Krane, E. J. Moran, R. T. Uyeda, R. Dixon, E. S. Guns and L. D. Mayer, Cancer Res., 2000, 60, 2964–2972 CAS.
- M. Antolini, A. Bozzoli, C. Ghiron, G. Kennedy, T. Rossi and A. Ursini, Bioorg. Med. Chem. Lett., 1999, 9, 1023–1028 CrossRef CAS.
- J. W. Black, G. J. Durant, J. C. Emmett and C. R. Ganellin, Nature, 1974, 248, 65–67 CrossRef CAS.
- Ü. Uçucu, N. G. Karaburun and İ. Işikdağ, Farmaco, 2001, 56, 285–290 CrossRef.
- L. Wang, K. W. Woods, Q. Li, K. J. Barr, R. W. McCroskey, S. M. Hannick, L. Gherke, R. B. Credo, Y. H. Hui, K. Marsh, R. Warner, J. Y. Lee, N. Zielinski-Mozng, D. Frost, S. H. Rosenberg and H. L. Sham, J. Med. Chem., 2002, 45, 1697–1711 CrossRef CAS PubMed.
- T. Maier, R. Schmierer, K. Bauer, H. Bieringer, H. Buerstell and B. Sachse, US Pat., 4820335, 1989; Chem. Abstr. 1989, 111, 1949w Search PubMed.
- (a) S. A. Siddiqui, U. C. Narkhede, S. S. Palimkar, T. Daniel, R. J. Lahoti and K. V. Srinivasan, Tetrahedron, 2005, 61, 3539–3546 CrossRef CAS PubMed; (b) M. M. Heravi, M. Zakeri, N. Karimi, M. Saeedi, H. A. Oskooie and N. T. Hosieni, Synth. Commun., 2010, 40, 1998–2006 CrossRef CAS.
- (a) J. Wang, R. Mason, D. VanDerveer, K. Feng and X. R. Bu, J. Org. Chem., 2003, 68, 5415–5418 CrossRef CAS PubMed; (b) S. Sarshar, D. Siev and M. M. Mjalli, Tetrahedron Lett., 1996, 37, 835–838 CrossRef CAS; (c) T. F. Gallagher, G. L. Seibel, S. Kassis, J. T. Laydon, M. J. Blumenthal, J. C. Lee, D. Lee, J. C. Boehm, S. M. Fier-Thompson, J. W. Abt, M. E. Soreson, J. M. Smietana, R. F. Hall, R. S. Garigipati, P. E. Bender, K. F. Erhard, A. J. Krog, G. A. Hofmann, P. L. Sheldrake, P. C. McDonnell, S. Kumar, P. R. Young and J. L. Adams, Bioorg. Med. Chem., 1997, 5, 49–64 CrossRef CAS.
- A. Shaabani and A. Rahmati, J. Mol. Catal. A: Chem., 2006, 249, 246–248 CrossRef CAS PubMed.
- S. Kantevari, S. V. N. Vuppalapati, D. O. Biradar and L. Nagarapu, J. Mol. Catal. A: Chem., 2007, 266, 109–113 CrossRef CAS PubMed.
- M. Kidwai, P. Mothsra, V. Babsal and R. Goyal, Monatsh. Chem., 2006, 137, 1189–1194 CrossRef CAS.
- L. M. Wang, Y. H. Wang, H. Tian, Y. F. Yao, J. H. Shao and B. Liu, J. Fluorine Chem., 2006, 127, 1570–1573 CrossRef CAS PubMed.
- G. V. M. Sharma, Y. Jyothi and P. S. Lakshmi, Synth. Commun., 2006, 36, 2991–3000 CrossRef CAS.
- S. Balalaie and A. Arabanian, Green Chem., 2000, 2, 274–276 RSC.
- M. M. Heravi, K. Bakhtiari, H. A. Oskooie and S. Taheri, J. Mol. Catal. A: Chem., 2007, 263, 279–281 CrossRef CAS PubMed.
- K. Sivakumar, A. Kathirvel and A. Lalitha, Tetrahedron Lett., 2010, 51, 3018–3021 CrossRef CAS PubMed.
- (a) J. F. Hayes, M. B. Mitchell and C. Wicks, Heterocycles, 1994, 38, 575–585 CrossRef CAS; (b) L. Revesz, F. Bonne and P. Makavou, Tetrahedron Lett., 1998, 39, 5171–5174 CrossRef CAS.
- N. J. Liverton, J. W. Butcher, C. F. Claiborne, D. A. Claremon, B. E. Libby, K. T. Nguyen, S. M. Pitzenberger, H. G. Selnick, G. R. Smith, A. Tebben, J. P. Vacca, S. L. Varga, L. Agarwal, K. Dancheck, A. J. Forsyth, D. S. Fletcher, B. Frantz, W. A. Hanlon, C. F. Harper, S. J. Hofsess, M. Kostura, J. Lin, S. Luell, E. A. O'Neill, C. J. Orevillo, M. Pang, J. Parsons, A. Rolando, Y. Sahly, D. M. Visco and S. J. O'Keefe, J. Med. Chem., 1999, 42, 2180–2190 CrossRef CAS PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- D. A. Tryk, A. Fujishima and K. Honda, Electrochim. Acta, 2000, 45, 2363–2376 CrossRef CAS.
- L. G. Phillips and D. M. Barbano, J. Dairy Sci., 1997, 80, 2726–2731 CrossRef CAS.
- J. P. Hewitt, Cosmet. Toiletries, 1999, 114, 59–63 Search PubMed.
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC.
- M. Mahalakshmi, B. Arabindoo, M. Palanichamy and V. Murugesan, J. Hazard. Mater., 2007, 143, 240–245 CrossRef CAS PubMed.
- M. Abu Tariq, M. Faisal and M. Muneer, J. Hazard. Mater., 2005, 127, 172–179 CrossRef CAS PubMed.
- O. S. Mohamed, A. E-A. M. Gaber and A. A. Abdel-Wahab, J. Photochem. Photobiol., A, 2002, 148, 205–210 CrossRef CAS.
- H. Sharghi and M. Hosseini Sarvari, J. Chem. Res. (S), 2003, 176 CrossRef CAS.
- M. A. Pasha, K. Manjula and V. P. Jayashankara, Synth. React. Inorg. Met.-Org. Chem., 2006, 36, 321–324 CrossRef CAS.
- M. Z. Kassaee, H. Masrouri, F. Movahedi and R. Mohammadi, Helv. Chim. Acta, 2010, 93, 261–264 CrossRef CAS.
- K. V. Subba Rao, B. Srinivas, A. R. Prasad and M. Subrahmanyam, Chem. Commun., 2000, 1533–1534 RSC.
- K. V. Subba Rao and M. Subrahmanyam, Photochem. Photobiol. Sci., 2002, 1, 597–599 CAS.
- V. Jeena and R. S. Robinson, Beilstein J. Org. Chem., 2009, 5, 1–4 Search PubMed.
- X. J. Lang, H. W. Ji, C. C. Chen, W. H. Ma and J. C. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934–3937 CrossRef CAS PubMed.
- (a) C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS PubMed; (b) C. W. Tang, S. A. Vanskyke and C. H. Chen, J. Appl. Phys., 1989, 65, 3610–3616 CrossRef CAS PubMed.
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS PubMed; (b) X. H. Zhang, B. J. Chen, X. Q. Lin, O. Y. Wong, C. S. Lee, H. L. Kwong, S. T. Lee and S. K. Wu, Chem. Mater., 2001, 13, 1565–1569 CrossRef CAS; (c) K. Q. Ye, J. Wang, H. Sun, Y. Liu, Z. C. Mu, F. Li, S. M. Jiang, J. Y. Zhang, H. X. Zhang, Y. Wang and C. M. Che, J. Phys. Chem. B, 2005, 109, 8008–8016 CrossRef CAS PubMed; (d) H. Bi, K. Q. Ye, Y. F. Zhao, Y. Yang, Y. Liu and Y. Wang, Org. Electron., 2010, 11, 1180–1184 CrossRef CAS PubMed.
- L. S. Hung and C. H. Chen, Mater. Sci. Eng., R, 2002, 39, 143–222 CrossRef.
- (a) G. G. Malliaras, Y. Shen, D. H. Dunlap, H. Murata and Z. H. Kafafi, Appl. Phys. Lett., 2001, 79, 2582–2584 CrossRef CAS PubMed; (b) S. C. Tse, H. H. Fong and S. K. So, J. Appl. Phys., 2003, 94, 2033–2037 CrossRef CAS PubMed; (c) S. C. Tse, S. K. So, M. Y. Yeung, C. F. Lo, S. W. Wen and C. H. Chen, Jpn. J. Appl. Phys., 2006, 45, 555 CrossRef CAS.
- (a) S. C. Tse, K. C. Kwok and S. K. So, Appl. Phys. Lett., 2006, 89, 262102 CrossRef PubMed; (b) K. K. Tsung and S. K. So, Appl. Phys. Lett., 2008, 92, 103315 CrossRef PubMed.
- (a) Y. Shirota, Y. Kuwabara, H. Inada, T. Wakimoto, H. Nakada, Y. Yonemoto, S. Kawami and K. Imai, Appl. Phys. Lett., 1994, 65, 807 CrossRef CAS PubMed; (b) S. A. VanSlyke, C. H. Chen and C. W. Tang, Appl. Phys. Lett., 1996, 69, 2160–2162 CrossRef CAS PubMed; (c) S. A. Carter, M. Angelopoulos, S. Karg, P. J. Brock and J. C. Scott, Appl. Phys. Lett., 1997, 70, 2067–2069 CrossRef CAS PubMed.
- (a) Z. B. Deng, X. M. Ding, S. T. Lee and W. A. Gambling, Appl. Phys. Lett., 1999, 74, 2227–2229 CrossRef CAS PubMed; (b) H. J. Jiang, Y. Zhou, B. S. Ooi, Y. W. Chen, T. Wee, Y. L. Lam, J. S. Huang and S. Y. Liu, Thin Solid Films, 2000, 363, 25–28 CrossRef CAS; (c) C. F. Qiu, H. Y. Chen, Z. L. Xie, M. Wong and H. S. Kwok, Appl. Phys. Lett., 2002, 80, 3485–3487 CrossRef CAS PubMed; (d) Y. Kurosaka, N. Tada, Y. Ohmori and K. Yoshino, Synth. Met., 1999, 102, 1101–1102 CrossRef CAS; (e) I. M. Chan, T. Y. Hsu and E. C. Hong, Appl. Phys. Lett., 2002, 81, 1899–1901 CrossRef CAS PubMed; (f) W. P. Hu, M. Matsumura, K. Furukawa and K. Torimitsu, J. Phys. Chem. B, 2004, 108, 13116–13118 CrossRef CAS.
- (a) J. Kwon, T. H. Kwon, H. S. Cho, M. K. Kim, I. S. Shin, D. Y. Shin, S. J. Park and J. I. Hong, New J. Chem., 2008, 32, 1368–1372 RSC; (b) L. Aubouy, N. Huby, L. Hirsch, A. van der Lee and P. Gerbier, New J. Chem., 2009, 33, 1290–1300 RSC.
- (a) J. Jayabharathi, V. Thanikachalam, V. Kalaiarasi and K. Jayamoorthy, J. Photochem. Photobiol., A, 2014, 275, 114–126 CrossRef CAS PubMed; (b) J. Jayabharathi, V. Thanikachalam, V. Kalaiarasi and K. Jayamoorthy, Spectrochim. Acta, Part A, 2014, 120, 389–394 CrossRef CAS PubMed; (c) C. Karunakaran, J. Jayabharathi, V. Kalaiarasi and K. Jayamoorthy, Spectrochim. Acta, Part A, 2014, 118, 182–186 CrossRef CAS PubMed; (d) J. Jayabharathi, V. Kalaiarasi, V. Thanikachalam and K. Jayamoorthy, J. Fluoresc., 2014, 24, 625–637 CrossRef CAS PubMed; (e) J. Jayabharathi, V. Thanikachalam, P. Ramanathan and A. Arunpandiyan, Spectrochim. Acta, Part A, 2014, 121, 551–558 CrossRef CAS PubMed; (f) J. Jayabharathi, P. Ramanathan, V. Thanikachalam and A. Arunpandiyan, Spectrochim. Acta, Part A, 2014, 133, 201–206 CrossRef CAS PubMed; (g) J. Jayabharathi, C. Karunakaran, V. Thanikachalam and P. Ramanathan, New J. Chem., 2014, 38, 4321–4335 RSC; (h) V. Thanikachalam, A. Arunpandiyan, J. Jayabharathi and p. Ramanathan, RSC Adv., 2014, 4, 6790–6806 RSC; (i) V. Thanikachalam, J. Jayabharathi, A. Arunpandiyan and P. Ramanathan, J. Fluoresc., 2014, 24, 377–387 CrossRef CAS PubMed.
- I. Yoshikatsu and M. Teruo, J. Org. Chem., 1979, 44, 41–49 CrossRef.
- J. Nikkanen, T. Kanerva and T. Mäntyla, J. Cryst. Growth, 2007, 304, 179–183 CrossRef CAS PubMed.
- H. Cheng, J. Ma, Z. Zhao and L. Qi, Chem. Mater., 1995, 7, 663–671 CrossRef CAS.
- J. Jayabharathi, V. Thanikachalam and K. Saravanan, J. Photochem. Photobiol., A, 2009, 208, 13–20 CrossRef CAS PubMed.
- J. Jayabharathi, V. Thanikachalam, M. Venkatesh Perumal and N. Srinivasan, Spectrochim. Acta, Part A, 2011, 79, 236–244 CrossRef CAS PubMed.
- S. Okada, K. Okinaka, H. Iwawaki, M. Furugori, M. Hashimoto, T. Mukaide, J. Kamatani, S. Igawa, A. Tsuboyama, T. Takiguchi and K. Ueno, Dalton Trans., 2005, 1583–1590 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03 (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- C. Karunakaran and P. Gomathisankar, ACS Sustainable Chem. Eng., 2003, 1, 1549–1555 Search PubMed.
- M. Veeranarayana Reddy and Y. T. Jeong, J. Fluorine Chem., 2012, 142, 45–51 CrossRef CAS PubMed.
- A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., 2006, 118, 3242–3245 CrossRef.
- C. Fan, Y. H. Chen, Z. Q. Jiang, C. L. Yang, C. Zhong, J. G. Qin and D. G. Ma, J. Mater. Chem., 2010, 20, 3232–3237 RSC.
- Y. Yuan, D. Li, X. Zhang, X. Zhao, Y. Liu, J. Zhang and Y. Wang, New J. Chem., 2011, 35, 1534–1540 RSC.
- (a) S. Ranjan, S. Y. Lin, K. C. Hwang, Y. Chi, W. L. Ching, C. S. Liu, Y. T. Tao, C. H. Chien, S. M. Peng and G. H. Lee, Inorg. Chem., 2003, 42, 1248–1255 CrossRef CAS PubMed; (b) S. L. Lin, L. H. Chan, R. H. Lee, M. Y. Yen, W. J. Kuo, C. T. Chen and R. J. Jeng, Adv. Mater., 2008, 20, 3947–3952 CrossRef CAS.
- (a) A. P. Kulkarni, A. P. Gifford, C. J. Tonzola and S. A. Jenekhe, Appl. Phys. Lett., 2005, 86, 061106 CrossRef PubMed; (b) W. L. Yu, J. Pei, Y. Cao and W. Huang, J. Appl. Phys., 2001, 89, 2343–2350 CrossRef CAS PubMed.
- (a) J. Staudigel, M. Stossel, F. Steuber and J. Simmerer, J. Appl. Phys., 1999, 86, 3895–3910 CrossRef CAS PubMed; (b) Y. Kawabe and J. Abe, Appl. Phys. Lett., 2002, 81, 493–495 CrossRef CAS PubMed; (c) M. T. Lee, H. H. Chen, C. H. Liao, C. H. Tsai and C. H. Chen, Appl. Phys. Lett., 2004, 85, 3301–3303 CrossRef CAS PubMed; (d) Q. X. Tong, S. L. Lai, M. Y. Chan, Y. C. Zhou, H. L. Kwong, C. S. Lee and S. T. Lee, J. Phys. Chem. C, 2009, 113, 6227–6230 CrossRef CAS.
- Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Qin and D. Ma, Adv. Funct. Mater., 2010, 20, 304–311 CrossRef CAS.
- (a) Q. Wang, I. W. H. Oswald, M. R. Perez, H. Jia, A. A. Shahub, Q. Qiao, B. E. Gnade and M. A. Omary, Adv. Funct. Mater., 2014, 24, 4746–4752 CrossRef CAS; (b) Q. Wang, I. W. H. Oswald, M. R. Perez, H. P. Jia, B. E. Gnade and M. A. Omary, Adv. Funct. Mater., 2013, 23, 5420–5428 CrossRef CAS; (c) C. Fan and C. Yang, Chem. Soc. Rev., 2010, 39, 2387–2398 RSC; (d) Q. Wang, J. Ding, D. Ma, Y. Cheng, L. Wang, X. Jing and F. Wang, Adv. Funct. Mater., 2009, 19, 84–95 CrossRef CAS; (e) B. Q. Wang, J. Ding, D. Ma, Y. Cheng, L. Wang and F. Wang, Adv. Mater., 2009, 21, 2397–2401 CrossRef.
- M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10967 CrossRef CAS.
- S. Reineke, K. Walzer and K. Leo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 125328 CrossRef.
- D. Song, S. Zhao, Y. Luo and H. Aziz, Appl. Phys. Lett., 2010, 97, 243304 CrossRef PubMed.
- Y. Zhang, S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, Z. C. Wen, G. Q. Zhang, S. T. Lee, H. L. Kwonge and C. S. Lee, J. Mater. Chem., 2011, 21, 8206–8214 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 972157. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01515k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |