
Effect of pretreatment temperature on the photocatalytic activity of microwave irradiated porous nanocrystalline ZnO†
Tarek T.
Ali
*ab,
Katabathini
Narasimharao
*a,
Ivan P.
Parkin
c,
Claire J.
Carmalt
c,
Sanjayan
Sathasivam
c,
Sulaiman N.
Basahel
a,
Salem M.
Bawaked
a and
Shaeel A.
Al-Thabaiti
a
aDepartment of Chemistry, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Kingdom of Saudi Arabia. E-mail: ttali@kau.edu.sa; nkatabathini@kau.edu.sa; Fax: +966-26952292; Tel: +966-500558045
bDepartment of Chemistry, Faculty of Science, Sohag University, P.O. Box 82524, Egypt
cMaterials Chemistry Centre, Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
First published on 8th October 2014
Abstract
Porous nanocrystalline ZnO photocatalysts were successfully synthesized by microwave irradiation and then thermally treated at different temperatures (150 °C, 200 °C, 250 °C and 300 °C). The physico-chemical properties of synthesized samples were determined by using different characterization techniques. The characterization results indicated that the as-synthesized sample was comprised of both ZnO and Zn(OH)2 phases with a particle size of approximately 50 nm. Thermal treatment of the as-synthesized sample at 150 °C resulted in a pure ZnO phase with a particle size of 40 nm. The results also demonstrated that the surface area, pore diameter and bandgap energy reach a maximum value for the ZnO sample treated at 200 °C. The ZnO nanoparticles pretreated at 200 °C showed the highest photocatalytic activity (99% of degradation) in a short reaction time (90 min), which can be attributed to the combined effects of several factors including low crystallite size, relatively high surface area, pore diameter, pore volume and bandgap energy. Reusability results show that the catalysts can be readily separated from the reaction mixture by filtration after the photocatalytic reaction and reused at least five times without any loss of activity.
1. Introduction
Azo dyes are widely used in textile, cosmetic, printing and rubber industries. They often produce environmental pollutants in the form of colored wastewater and these pollutants have been recognized to be toxic, mutagenic and carcinogenic.1 The photocatalytic degradation of organic pollutants in water using photocatalysts has attracted more attention due to their superior ability in environmental detoxification.2,3 Among the different photocatalysts, ZnO nanoparticles have a special role due to their highly semiconducting nature with a wide band gap of 3.37 eV.4 Subsequently, ZnO is used in a wide variety of applications5,6 in addition to photocatalysis. The design and synthesis of ZnO nanoscale structures has recently attracted a great deal of interest.7 Many studies involving photo-degradation of organic pollutants have been investigated using ZnO nanostructures.8 These studies have shown that the morphology of ZnO has a significant impact on its photocatalytic performance.Various synthesis methods have been reported to modify the crystal size and band gap of ZnO.9 Nanocrystalline ZnO with various morphologies (particles, rods, sponges, flowers, etc.) have been effectively prepared by different methods such as ultrasonic synthesis, microwave assisted synthesis, hydrothermal synthesis, sol–gel synthesis, precipitation, thermal decomposition, electrochemical deposition and mechanochemical methods.10 Traditional approaches require high temperatures, which are economically unattractive, and furthermore, many of the reported unconventional methods require more complex devices, expensive raw materials, complex process control, multiple-steps, a long synthesis time and a careful execution. Therefore, it is challenging to adopt a simple and convenient method for the preparation of highly active ZnO nanomaterials for photocatalytic applications.11
The application of microwave heating for chemical synthesis of new materials has grown rapidly.12 Microwave irradiation has been accepted as a promising method to enable rapid heating, high reaction rate and short reaction time as compared to the conventional heating.13
Several studies have been reported on the application of microwave irradiation for synthesis of ZnO nanomaterials with a variety of morphologies, such as nanowires,14 nanorods,15 nanotubes,16 nanoneedles17 and hollow structures.18 Thongtem et al.19 synthesized nanocrystalline ZnO powders through microwave heating. The authors claimed that the process eliminates the complicated synthetic procedures and shortens the reaction time, resulting in a desirable nanocrystalline ZnO material without using any template.
In this work, we report the successful synthesis of porous hexagonal ZnO nanoparticles in an aqueous solution via a microwave irradiation method. The as-synthesized product was thermally treated at various temperatures (150 °C, 200 °C, 250 °C and 300 °C) to study the effect of pretreatment temperature on the morphology and structure of ZnO. The physico-chemical properties of the porous nanocrystalline ZnO were systematically studied by various techniques. The porous ZnO nanoparticles were also tested for photocatalytic degradation of aqueous methyl orange. A schematic representation of the research work is presented in Scheme 1.
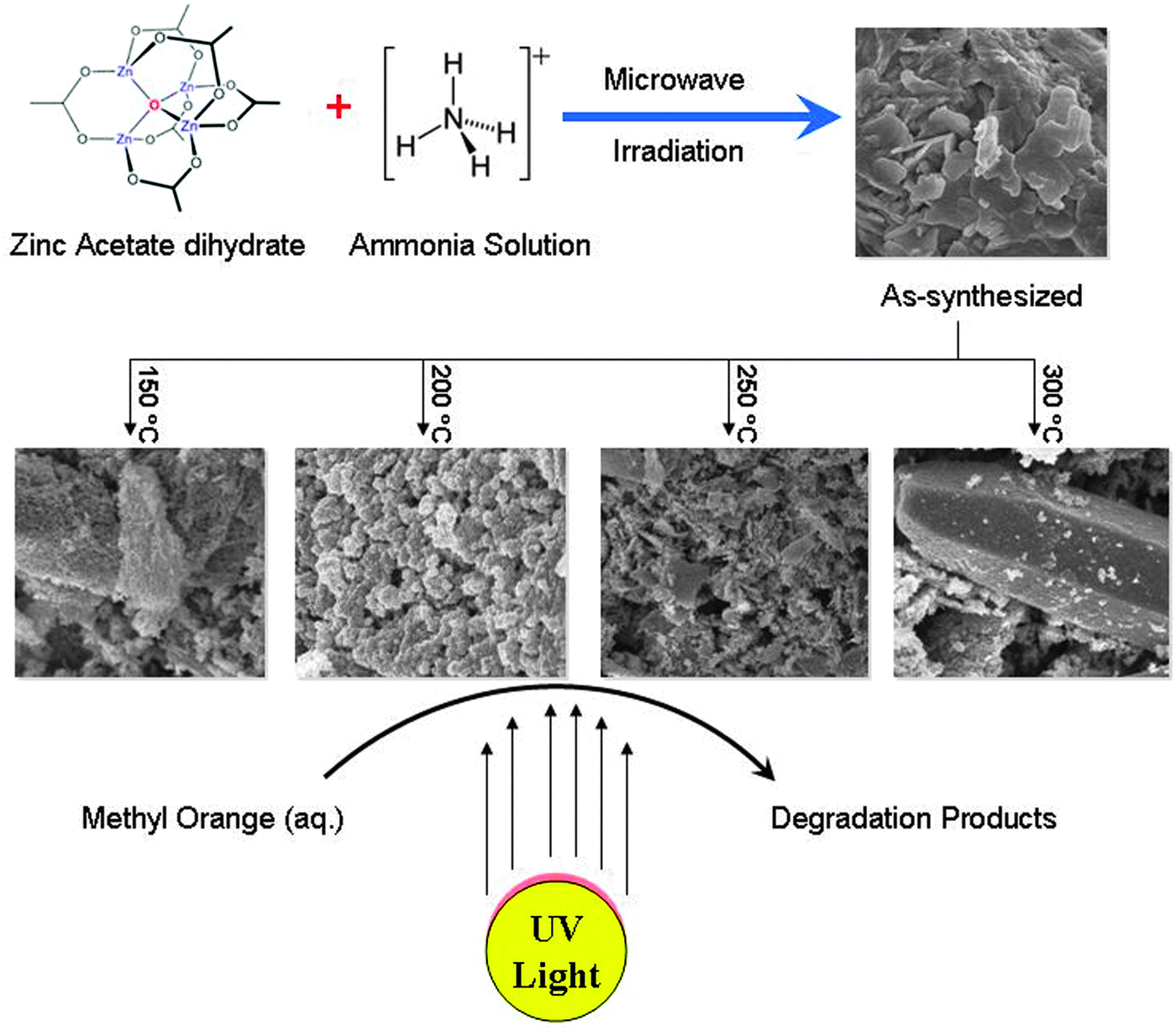 | ||
| Scheme 1 Schematic representation of the research work. | ||
2. Experimental
2.1 Materials
Zinc acetate dihydrate [Zn(CH3COO)2·2H2O], ammonia solution (NH4OH), methyl orange (C14H14N3NaO3S), hydrochloric acid (HCl) and sodium hydroxide (NaOH) were purchased from Aldrich, UK. All chemicals used in this study were of analytical grade and used directly without further purification. Deionized water was used for the preparation of the methyl orange standard solution as well as the respective dilutions.2.2 Method of preparation
Synthesis of porous nanocrystalline ZnO material was carried out using an advanced microwave system (MicroSYNT, Milestone) that possesses the capability of controlling time, temperature and pressure. The ATC–FO (automatic temperature control by fiber optic) system allows for direct continuous monitoring and control of the internal temperature of the reaction vessel (±1 °C precision). A fiber-optic temperature probe, which is housed in a thermowell and protected from chemical exposure by a multiple layer of inert PTFE and ceramic to ensure trouble-free operation, is inserted in the reactor to monitor the reaction temperature. Accurate temperature monitoring and precise feedback control are achieved using the software by continuously adjusting the applied power, resulting in reproducible temperature conditions. A primary solution was prepared by dissolving 0.02 moles of Zn(CH3COO)2·2H2O [5 g] in 30 mL of deionized water and 10 mL of 0.02 molar solution of NH4OH was added drop wise under magnetic stirring at room temperature. The initial pH of the solution was about 12. The solution was vigorously stirred at room temperature resulting in a colorless precursor having a final pH of 9. The total solution was poured into a PTFE sealed vessel and heated in a microwave oven (2.45 GHz) at a power of 700 W for 20 min. The resultant milky solution was centrifuged to separate the precipitate. The precipitate was washed with acetone and deionized water several times and dried in an oven at 60 °C for 12 h and then the powder was calcined at different temperatures (150 °C, 200 °C, 250 °C and 300 °C) under atmospheric conditions in an oven for 3 h (1 °C min−1, isothermal).2.3 Characterization
Thermogravimetric analysis (TGA) was carried out on computerized Shimadzu Thermal Analyzer TA60 Apparatus (Japan). A ceramic sample boat was used for TG analysis. A sample weighing 10 ± 0.1 mg was heated up to 800 °C at 10 °C min−1 in a flow of 40 mL min−1 O2 gas.X-ray powder diffraction (XRD) studies were performed for all of the prepared solid samples using a Bruker diffractometer (Bruker D8 advance target). The patterns were run with copper Kα1 and a monochromator (λ = 1.5405 Å) at 40 kV and 40 mA. The crystallite size of the ZnO was calculated using Scherrer's equation:
D = Bλ/β1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ | (1) |
UV-visible absorption data of the sample were collected using a Perkin-Elmer Lambda-900 spectrophotometer equipped with an integrating sphere in the wavelength range 300–800 nm to measure the reflectance spectra of each sample. A small amount (∼5–10 mg) of the sample in powder form was dispersed and pressed firmly between two microscope glass plates to create a thin absorbing film. The band gap of all the ZnO samples was determined using the Kubelka–Munk method. The Kubelka–Munk transformation (K) was estimated using the equation
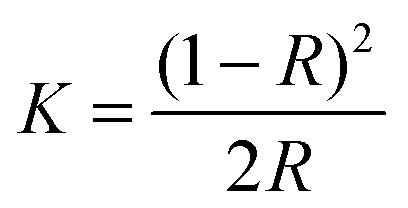 | (2) |
2.4 Photocatalytic degradation of methyl orange
The photocatalytic degradation of methyl orange was performed in a Pyrex glass beaker using synthesized ZnO nanoparticles as photocatalyst under UV illumination for various time intervals. Methyl orange dye (10 ppm) solution was prepared in 100 mL of deionized water and 100 mg of catalyst was added. The resulting suspension was equilibrated by stirring for 45 min to stabilize the absorption of methyl orange dye over the surface of the catalyst, before exposing to UV light. The photocatalytic degradation of methyl orange was examined by measuring the absorbance at regular time intervals by measuring the absorption using a UV-vis spectrophotometer at 465 nm wavelength. Analytical samples were taken from the reaction suspension at regular time intervals and were then analyzed for their absorption using a UV-visible spectrophotometer (Thermo-Fisher-Scientific-Evolution 160). The degradation percentage was calculated using the expression| η = (1 − C/C0) × 100 | (3) |
3. Results and discussion
The microwave assisted as-synthesized ZnO nanoparticles were analyzed by thermogravimetric analysis (TGA) to investigate the influence of the heat treatment of the sample (Fig. 1). The sample exhibited a marked weight loss at a temperature of 140 °C. The major weight loss is primarily caused by the vaporization of water, leaving zinc oxide and hydroxide behind. Furthermore, a minor weight loss was observed at 350 °C, indicating the complete removal of hydroxyl groups on ZnO surface. No further weight loss and thermal effect were observed at temperatures in the range of 400 °C to 800 °C indicating that pyrolysis did not occur in this temperature range, and that a higher temperature is required to infer a sintering process.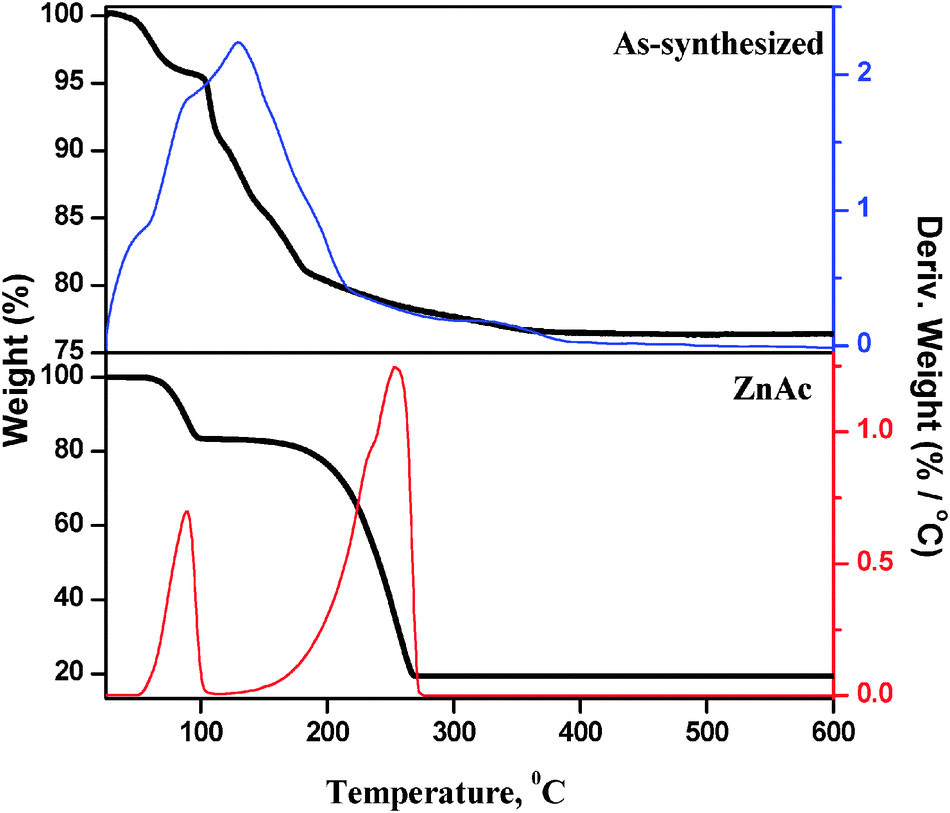 | ||
| Fig. 1 TG pattern of ZnAc precursor and as-synthesized ZnO nanoparticles. | ||
TG experiment was performed with Zn precursor (ZnAc) and the results were compared with those of the microwave treated (as-synthesized) sample. The TG pattern of the ZnAc sample shows weight loss at two stages: the first one is at 89 °C (≈11.0% weight loss) due to dehydration and the second one is at 254 °C (≈65.0% weight loss) due to the decomposition of acetate ions. In comparison, the as-synthesized sample does not show any weight loss at 254 °C, which corresponds to decomposition of acetate ions. This observation clearly shows that the microwave treated sample does not contain zinc acetate precursor. The 25% weight loss in the as-synthesized sample is not only due to the loss of physically adsorbed water, but also due to the loss of hydroxyl groups from zinc hydroxide.
Fig. 2 shows the powder X-ray diffraction patterns of as-synthesized ZnO nanoparticles and thermally treated samples. The as-synthesized samples exhibited diffraction peaks corresponding to both Zn(OH)2 and ZnO phases. It is interesting to note that the as-synthesized sample does not show any diffraction peaks corresponding to zinc acetate. Due to the molecular level heating of microwave irradiation, the zinc acetate within the solution undergoes partial hydrolysis forming acetate ions and zinc ions. ZnO nanoparticles were formed by the following chemical reactions:
| Zn(CH3COO)2 + 2(NH4)OH → Zn(OH)2 + 2CH3COONH4 | (4) |
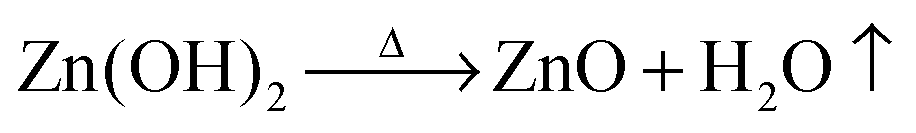 | (5) |
The thermally treated samples all have comparable diffraction patterns indicating that they consist of only the ZnO phase with particles of a hexagonal wurtzite structure [JCPDS file No. 80-0075]. Further, the sharp diffraction peaks show that the synthesized samples are highly crystalline.
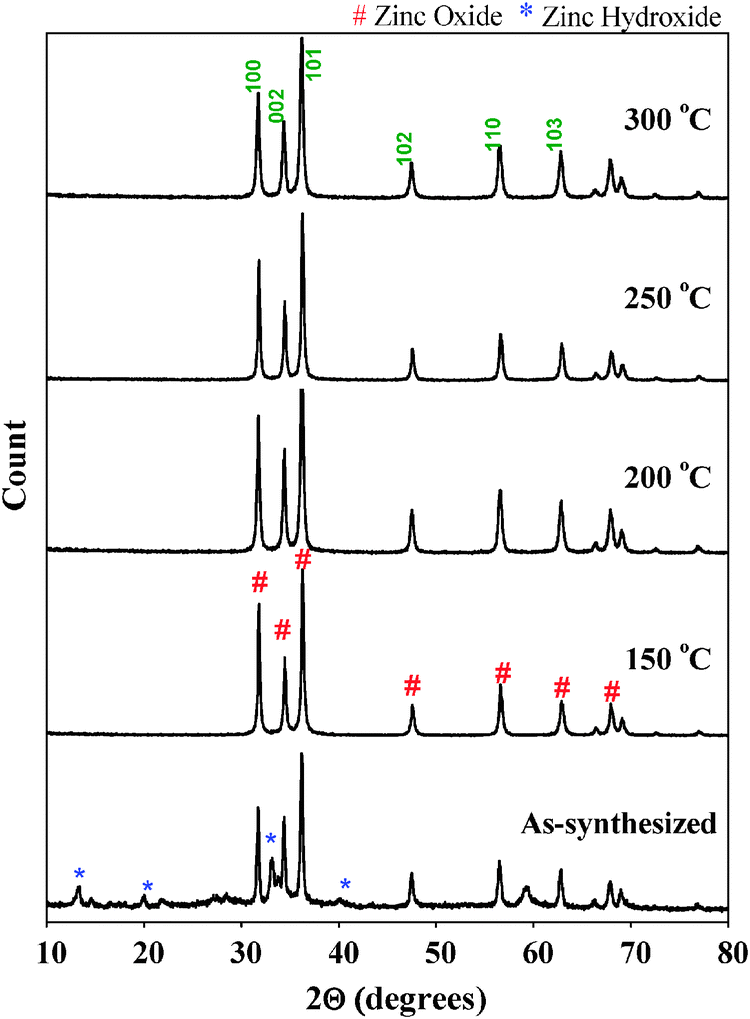 | ||
| Fig. 2 Powder X-ray diffraction patterns of all the samples. | ||
The remarkable crystallinity of the ZnO nanoparticles even at low treatment temperature could be due to the fact that microwave heating enhances crystallinity of the materials at much lower temperatures within shorter times because of uniform and internal heating compared to conventional heating. Parida et al.20 prepared ZnO nanoparticles by microwave irradiation using zinc acetate and sodium carbonate as the zinc precursor and precipitating agent respectively. Based on XRD results, the authors reported the presence of Zn5(CO3)2(OH)6 phase in the sample even after thermal treatment at 300 °C. The authors concluded that as-synthesized ZnO nanoparticles prepared using microwave irradiation needed thermal treatment of up to 400 °C to obtain a pure ZnO phase. It was also reported that the as-synthesized sample prepared under conventional methods required temperatures higher than 150 °C to transform into pure ZnO nanoparticles.10 As shown in Fig. 2, the sample prepared using zinc acetate and ammonium hydroxide and treated with microwave irradiation requires a lower temperature (150 °C) to be transformed into pure ZnO nanoparticles, thus demonstrating that by using zinc acetate and ammonium hydroxide in a microwave irradiated synthesis, it is possible to generate crystalline ZnO nanoparticles at a relatively low temperature.
The particle size of ZnO samples was determined using Scherrer's equation. The full width at half maximum (FWHM) of the (101) index plane of 0.256 nm was used for crystallite size calculation. The crystallite sizes of all the samples are given in Table S1 (ESI†). The crystallite size of the as-synthesized ZnO is approximately 45.4 nm, and it was noted to decrease to 35.0 nm after pretreatment at 150 °C. Upon increasing the pre-treatment temperature to 200 °C, a further decrease of crystallite size to 26 nm was observed. However, a further increase of temperature to 250 °C and 300 °C caused an increase of crystallite size to 37.4 nm and 42.4 nm respectively. From these observations, it appears that there is an optimum pre-treatment temperature (between 150 and 200 °C) to obtain fine ZnO nanoparticles (around 26 nm). In order to study the surface morphology of the ZnO nanoparticles, SEM was used. SEM images of the as-synthesized and thermally treated samples are shown in Fig. 3. The as-synthesized ZnO nanoparticles showed large flake type morphology due to the presence of Zn(OH)2 that was formed because of the partial hydrolysis of zinc acetate precursor according to eqn (1); at this stage crystallization of ZnO nanoparticles was not complete.
 | ||
| Fig. 3 SEM images of as-synthesized (a) and thermally treated samples ((b) 150 °C, (c) 200 °C, (d) 250 °C, (e) 300 °C). | ||
It appears that the microwave method has a problem with higher concentrations since heat conductivity is not homogeneous and this can be observed in SEM images of the samples showing many different sizes and morphology. After the thermal treatment at 150 °C, the morphology of ZnO nanoparticles was transformed into sponge type composed of small spherical particles. The ZnO sample exhibited a good distribution of nanoparticles after being pretreated at 200 °C. The ZnO nanoparticles were agglomerated upon increasing the temperature to 250 °C and 300 °C. The appearance of needle type and hexagonal cylindrical shaped ZnO nanoparticles can be clearly observed in the samples treated at 250 °C and 300 °C.
Fig. 4 shows the TEM images of the as-synthesized and thermally treated samples. The TEM image of the as-synthesized sample shows tightly packed irregular agglomerated particles; however the sample treated at 150 °C shows ZnO nanoparticles consisting of many spherical shaped crystalline particles. Particles with a hexagonal and near-spherical shape can be seen in the TEM images of the ZnO samples treated at 200 °C and 250 °C. The ZnO nanoparticles treated at 200 °C are uniform with compact interconnected particles. The translucent region in the TEM images represents the straight channels and the gray region represents the curved channels. ZnO treated at 200 °C has much more translucent region than other samples which suggests that this sample has many straight channels.
 | ||
| Fig. 4 TEM images of as-synthesized (a) and thermally treated samples ((b) 150 °C, (c) 200 °C, (d) 250 °C, (e) 300 °C). | ||
TEM images of the rest of the ZnO samples show many shadow regions indicating that the nanoparticles have agglomerated in these samples, particularly the ZnO nanoparticles treated at 300 °C. Overall, the thermal treatment results in the particle shape being modified and also the size of the particles increases from 50 to 150 nm, with increasing temperature. This increase in particle size at higher temperatures is likely to be caused by agglomeration of the particles to produce larger particles. Although the particles increased in size, the dispersion of smaller particles can still be observed at 200 °C and 250 °C (Fig. 4). Also the pretreated samples appear to have more inter-particle spaces than as-synthesized ZnO nanoparticles indicating that the sample became less dense with an increase of pre-treatment temperature.
Fig. 5 shows the room temperature UV-vis absorption spectra of the as-synthesized and thermally treated samples. The as-synthesized ZnO nanoparticles show a single broad band at 370 nm. This prominent exciton band at 370 nm corresponds to the ZnO nanostructures; however there are two absorption bands in the spectra of all the thermally treated samples: a strong and narrow near-UV absorption band at 360 nm and a broad but weak spectral band ranging from 450 nm to 600 nm. The absorption maxima values were varied between 360 and 370 nm. The prominent absorption peak is red shifted as compared to the bulk exciton absorption of ZnO (370 nm) which could be due to the size effect of the nanostructures. The absorption peak at 360 nm also indicates good crystallinity of the ZnO nanostructures, with minimal impurity.21 It is clear from Fig. 5 that the absorption of the ZnO nanoparticles treated at different temperatures is stronger than that of the as-synthesized ZnO sample. The absorption in the visible range of wavelength implies that there exist more defect energy levels in the synthesized ZnO nanostructures that are due to the specific experimental synthesis conditions.22
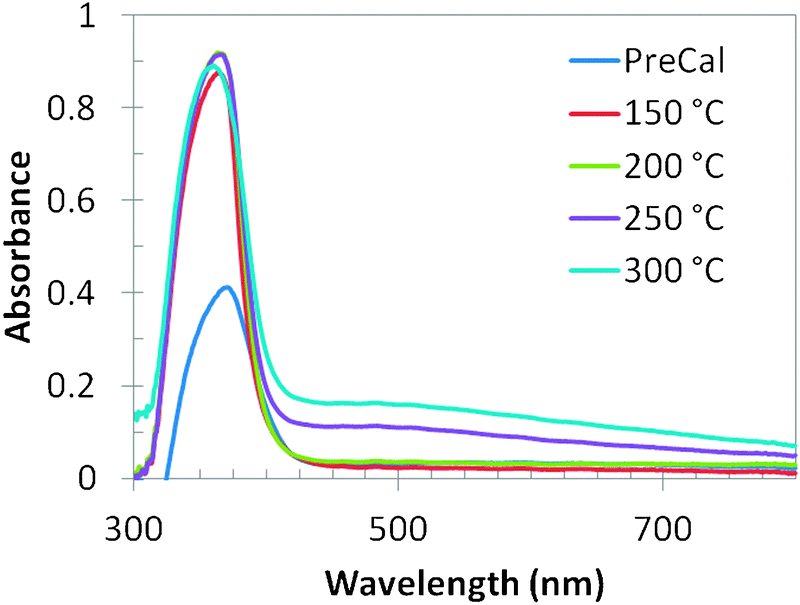 | ||
| Fig. 5 UV-vis absorption spectra of all the samples. | ||
The surface composition and chemical state of the ZnO nanoparticles have been investigated by XPS analysis. The deconvoluted Zn 2p3/2 spectra of all the samples are presented in Fig. 6(A). It was reported that ZnO nanoparticles show a sharp and symmetric XPS peak maximum at 1021.7 eV;23 however three different deconvoluted Zn 2p3/2 peaks were observed in all the ZnO nanoparticles: a major peak at 1022.2 eV and two small peaks at 1020.2 eV and 1023.7 eV. The peak at 1022.2 eV can be ascribed to zinc atoms in the ZnO lattice24 and the small peaks at 1023.7 eV and 1020.2 eV can be assigned to Zn(OH)2 species and Zn adsorbed to H2O molecules respectively.25
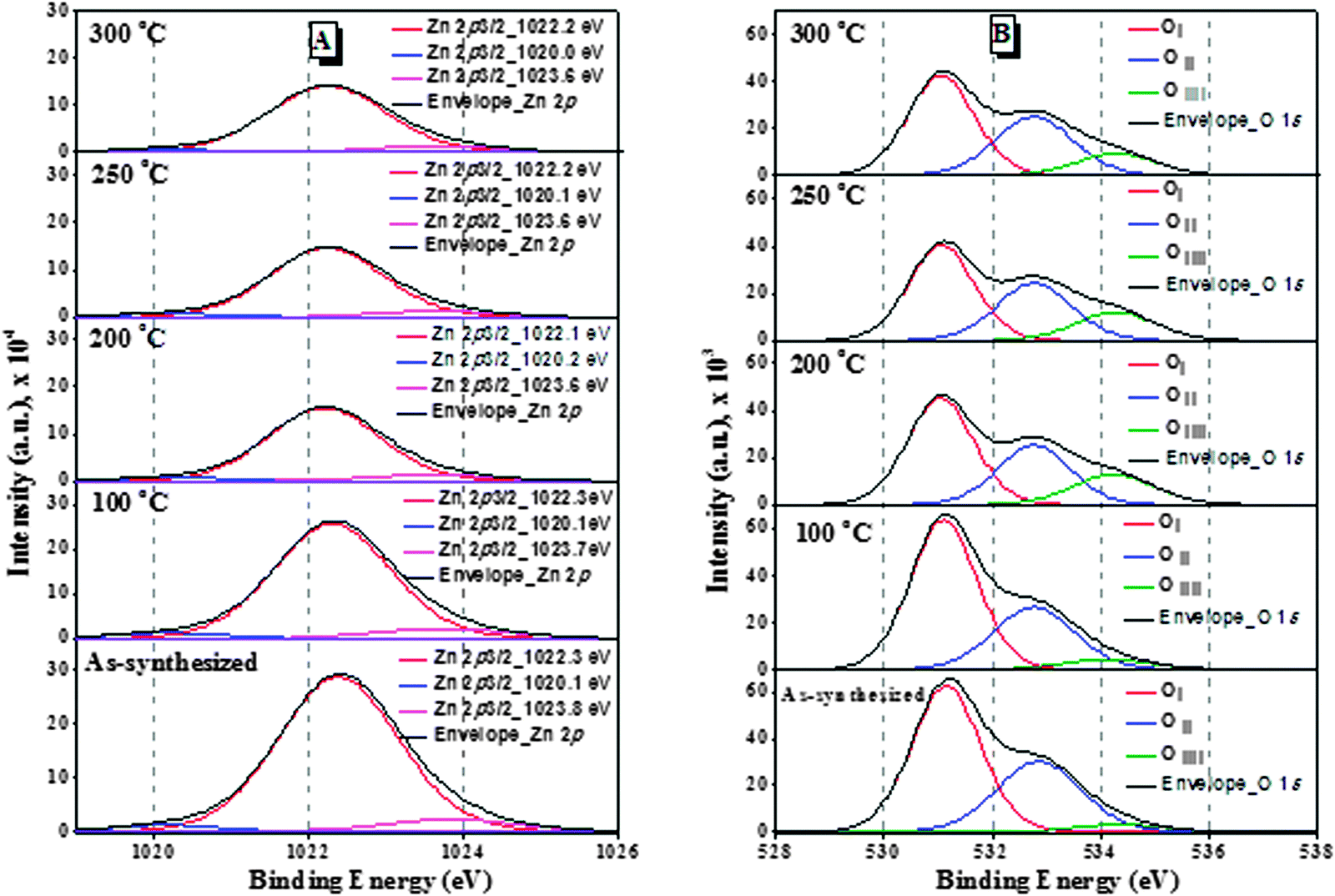 | ||
| Fig. 6 XPS deconvolution spectra of the ZnO samples: (A) Zn 2p3/2 and (B) O 1s. | ||
The binding energies of the peaks are almost the same with the increase of treatment temperature. The deconvolution of the O 1s photoelectron spectra of the ZnO nanoparticles [Fig. 6(B)] showed that the O 1s peaks are wide and asymmetric and different oxygen states are to be distinguished. They are deconvoluted by Lorentzian–Gaussian curve fitting to three components. Ayochi et al. reported that ZnO films exhibited three O 1s peaks.26 The Oi species at the low binding energy of 531.1 eV belong to O2− ions in the ZnO structure.27 The Oii species at 532.5 eV are attributed to O− and O2− ions in the oxygen deficient regions mainly caused by oxygen vacancies.28 The Oiii species at 534.2 eV belong to the absorbed or dissociated oxygen or OH species on the surface of ZnO nanoparticles.29 As shown in Table S1 (ESI†), the percentage of Oii species in ZnO nanoparticles treated at 200 °C is 10.6%, which is higher compared to any other samples of our study. Therefore, the ZnO nanoparticles treated at 200 °C have more oxygen vacancies, which are associated with active sites for photocatalytic activity.
The bandgap energy (eV) was estimated for all the samples and the as-synthesized sample possessed a bandgap energy of 2.93 eV. The bandgap energy was increased to 3.08 eV after the ZnO nanoparticles were treated at 150 °C. Upon increasing the treatment temperature to 200 °C the bandgap energy was further increased to 3.24 eV. Increasing the treatment temperature further (250 °C and 300 °C) resulted in no further increase of the bandgap, rather a decrease to 3.15 eV was observed. The indirect bandgap energy decreased with the increase of treatment temperature beyond 200 °C due to the decrease of the gap between the conduction band edge and the valance band edge of ZnO nanoparticles. These results indicate that the sample treated at 200 °C has the highest bandgap energy among all the samples.
The functional or composition quality of the prepared ZnO nanoparticles was determined by FTIR spectroscopy. Fig. 7 shows the FTIR spectra of the ZnAc precursor, as-synthesized ZnO nanoparticles and thermally treated ZnO nanoparticles. ZnAc precursor shows a broad band centered at 3105 cm−1 due to OH stretching vibration. For the as-synthesized sample, a broad band centered at 3375 cm−1 was observed which also corresponds to the –OH stretching mode. It is known that the OH-stretching band envelope generally appears between 3600 and 3100 cm−1.30 The presence of two carbonyl absorptions due to asymmetric and symmetric stretching modes near 1555 cm−1 and 1390 cm−1 can be seen in both the ZnAc precursor and the as-synthesized sample. The former band is more intense than the latter one. The O–H bending vibration at 1600 cm−1 which is a characteristic peak of bonding water was not clearly observed in the synthesized ZnO sample, which is probably due to the presence of a strong intense band at 1555 cm−1 due to carbonyl absorption. After the microwave and thermal treatment, a new band appeared at 435 cm−1, which can be assigned to Zn–O stretching vibration mode.31 In the spectra of ZnO samples treated at different temperatures, the bands due to the –OH deformation mode and stretching mode from water on the surface are still present, but the intensity of bands decreased with the treatment temperature. However, the intensity of the band due to Zn–O stretching mode at 435 cm−1 appeared to be constant.
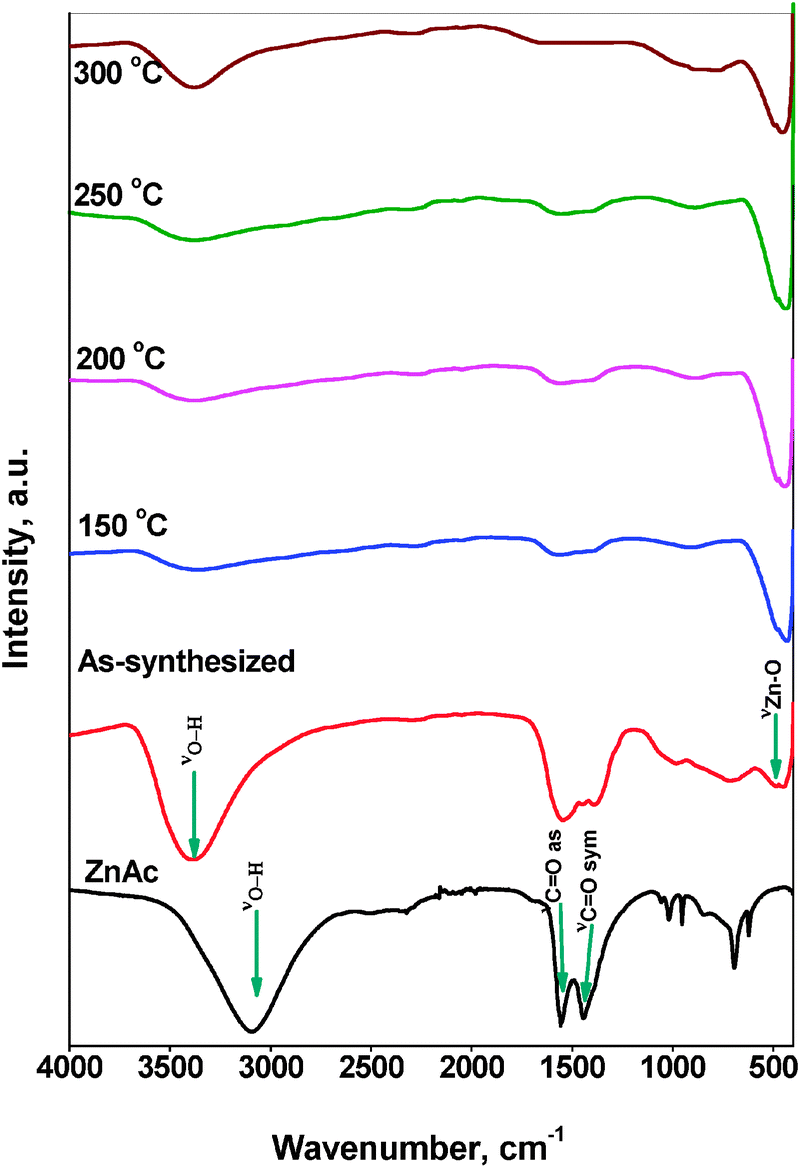 | ||
| Fig. 7 FTIR spectra of ZnAc precursor and of all ZnO samples. | ||
The nitrogen adsorption–desorption isotherms of as-synthesized ZnO and ZnO nanoparticles treated at different temperatures are shown in Fig. 8. All the isotherms can be classified as Type IV according to IUPAC classification and their hysteresis loop could be interpreted as Type H3 representing aggregates of particles forming slit-shaped pores.32 The results from Fig. 8 also indicate that the amount of N2 adsorbed increases gradually with the relative pressure, which also means that the adsorption on these samples proceeds via a multilayer formation. However, at higher pressures the amount adsorbed rises steeply due to the capillary condensation into the pores.
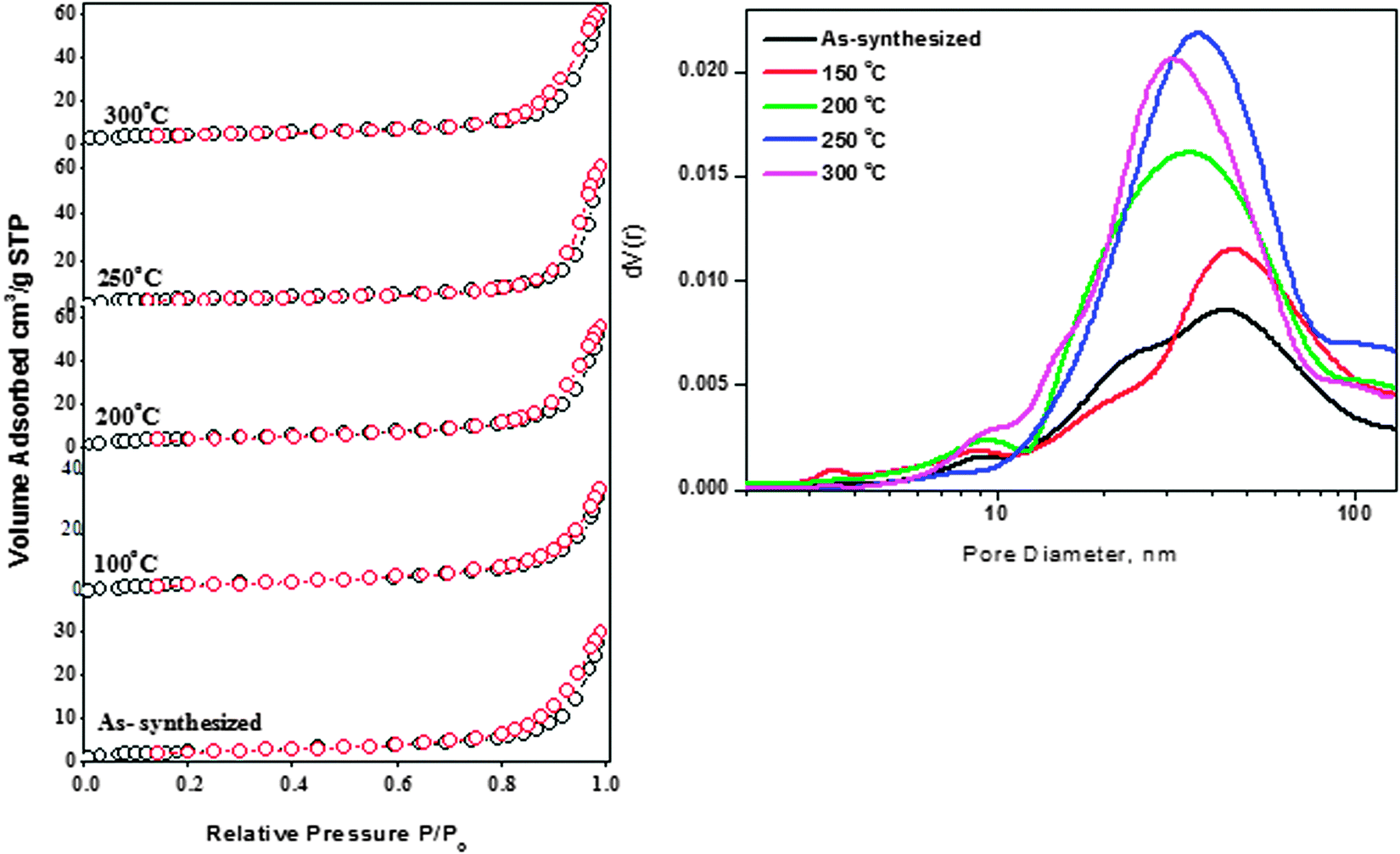 | ||
| Fig. 8 (A) N2 adsorption–desorption isotherms and (B) pore size distribution of ZnO samples. | ||
The N2 volume absorbed for as-synthesized ZnO nanoparticles and those treated at 150 °C is almost similar (30 cc g−1). The increasing trend for N2 volume absorbed for the thermally treated samples can be clearly observed from Fig. 8(A). It was observed that the maximum volume absorbed is for the sample pretreated at 200 °C with a value of 65 cc g−1. It is also interesting to note that the pore volume of the samples (Table S2, ESI†) also increased when the treatment temperature was raised to 200 °C. The pore volume of the ZnO sample treated at 200 °C is twice (0.096 cm3 g−1) that of the as-synthesized sample (0.046 cm3 g−1).
Increasing the temperature further resulted in a slight decrease of pore volume. The changes in the textural properties of thermally treated samples can be attributed to ZnO nanoparticles' surface modifications and subsequent morphology changes during the thermal treatment.33Fig. 8(B) shows the Barrett–Joyner–Halenda (BJH) pore size distribution for as-synthesized ZnO nanoparticles and samples pretreated at different temperatures. As clearly seen from Fig. 8, the pore size distribution peak is located mainly between 15 and 100 nm, indicating that the samples possessed a mesoporous structure, which is in good agreement with the results of Type IV adsorption–desorption isotherms. Pore size distribution of as-synthesized ZnO nanoparticles showed pores of bimodal distribution of around 24 nm and 45 nm (diameter). Disappearance of small-sized pores started upon thermal treatment of the ZnO nanoparticles at 150 °C. An increase of treatment temperature to 200 °C resulted in the complete disappearance of pores at 24 nm and the broad pore size distribution peak appeared at 35 nm. The samples treated at 250 °C and 300 °C also showed a similar behavior as the sample treated at 200 °C and these two samples possessed pores of size 30 nm and 38 nm respectively. It is known that ZnO nanoparticles generally possess a non-porous structure;34 the pores observed were due to inter- and intra-particle agglomeration.
The photocatalytic activity of all the samples was determined by monitoring the degradation of the organic dye, methyl orange. A blank experiment was carried out to confirm that the photo-degradation reaction did not proceed without the presence of either catalyst or the UV radiation. This was in agreement with literature report.35 The as-synthesized ZnO nanoparticles did not show any photocatalytic activity. FTIR analysis clearly demonstrated the presence of zinc acetate in this catalyst; it is possible that zinc acetate might be blocking the active sites on the surface of the catalyst, thus interfering with the photocatalytic activity of the catalyst. Fig. 9 shows the change in the UV-vis absorbance spectra of methyl orange solution (100 ppm) with different irradiation intervals over the ZnO nanoparticles treated at different temperatures. The decrease in the absorbance of the solution was due to the destruction of the homo- and hetero-polyaromatic rings present in the dye molecules. ZnO nanoparticles treated at 200 °C were found to be the most effective catalyst in comparison with the samples treated at other temperatures (Fig. 9).
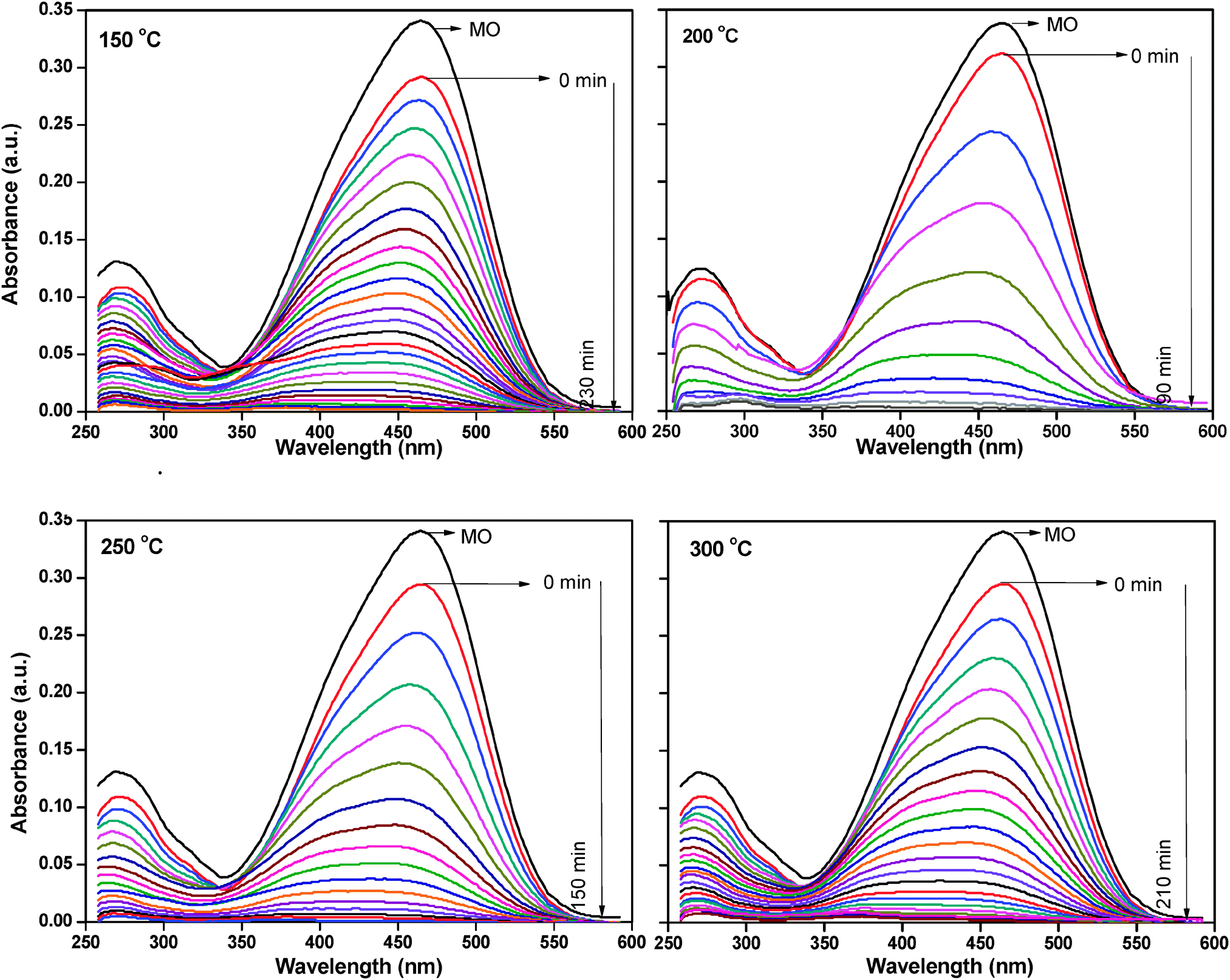 | ||
| Fig. 9 Adsorption changes of methyl orange aqueous solution at 25 °C in the presence of thermally treated ZnO nanoparticles. | ||
Complete degradation of adsorbed dye molecules was observed within 90 min for the ZnO at 200 °C, whereas 240 min, 150 min and 210 min were required for the complete degradation of adsorbed methyl orange molecules for the ZnO nanoparticles treated at 150 °C, 250 °C and 300 °C under similar conditions.
The degradation percentage of ZnO nanoparticles was calculated using eqn (4). Fig. 10 shows percentage changes of methyl orange aqueous solution at room temperature in the presence of thermally treated ZnO nanoparticles. The ZnO photocatalyst treated at 150 °C showed 62% degradation of methyl orange in 90 minutes reaction time, while after treatment at 200 °C, the ZnO nanoparticles showed better photocatalytic activity with 99% degradation in the same reaction time. However, at 250 °C and 300 °C treatment temperature, the degradation decreased to 88% and 72% respectively.
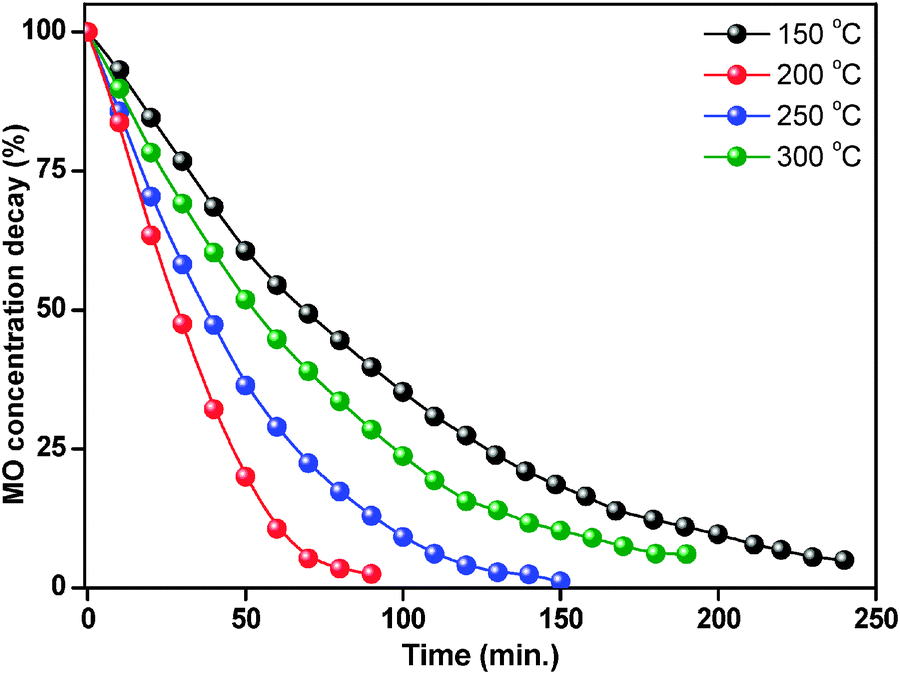 | ||
| Fig. 10 Percentage changes of methyl orange aqueous solution at 25 °C in the presence of thermally treated ZnO nanoparticles. | ||
The role of pH in the photocatalytic degradation of methyl orange was studied in the pH range limited to 2–13 with optimized experimental conditions (100 ppm of methyl orange solution and 50 mg/100 mL of ZnO loading) by considering the solubility of ZnO nanoparticles in acidic as well as in highly basic solutions. The pH of the suspension was adjusted initially and it was not controlled during the course of the reaction.
The rate constant for photocatalytic degradation reaction over ZnO nanoparticles treated at 200 °C, 250 °C and 300 °C was obtained from the slope of the straight line from the plot of ln(C0/Ct) vs. t. The results are presented in Fig. 11. In acidic medium, less photocatalytic degradation of methyl orange was observed. The extent of the photocatalytic degradation of methyl orange was found to increase upon increasing the initial pH of the methyl orange solution, exhibiting maximum photocatalytic degradation at pH 7.0, then decreasing at pH 8.0 and continually decreasing with increasing pH. Below pH 7.0, the active sites on the positively charged catalyst surface are preferentially covered by methyl orange molecules. Thus, the surface concentration of methyl orange is relatively high, while that of OH− and ˙OH is low, and in turn the photocatalytic degradation decreases. On the other hand, above pH 7.0, the catalyst surface is negatively charged by means of metal-bound OH− and consequently the surface concentration of methyl orange is low whilst the hydroxyl radical concentration is high. In addition, methyl orange is not protonated above pH 7.0, and the electrostatic repulsion between the surface charges on the adsorbent and the adsorbate hinders the amount of methyl orange adsorption. Consequently, the surface concentration of methyl orange decreases, resulting in the decrease of photocatalytic degradation at pH 8.0. It appears that pH 7.0 can provide moderate surface concentration of methyl orange and OH−, which react with the holes to form ˙OH, and thereby enhancing the photocatalytic degradation of methyl orange. Yang et al.36 also observed that dumbbell shaped ZnO nanoparticles offered the highest activity for photocatalytic degradation of methyl orange around pH 7.0.
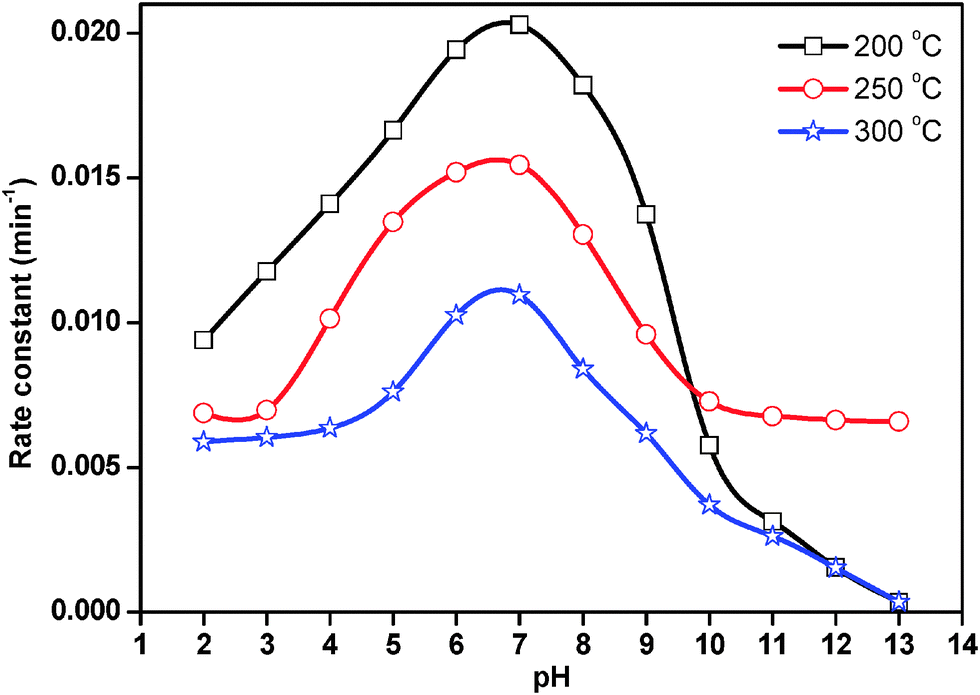 | ||
| Fig. 11 Effect of pH on the rate constant for photocatalytic degradation of methyl orange. | ||
Kaur et al.37 also studied the role of pH in the photocatalytic degradation of methyl orange. The authors carried out the experiment in the pH range of 5–11 at 0.03 mM methyl orange concentration and 1 g L−1 catalyst loading for 40 min. They observed that the degradation rate increases with the increase in pH, exhibiting a maximum at pH 9.
The ZnO nanoparticles with a smaller crystal size, higher oxygen vacancies and a larger specific surface area showed maximum performance. The higher surface area supplies more active sites to adsorb methyl orange, and then facilitates the diffusion and mass transportation of methyl orange molecules and hydroxyl radicals during the photochemical reaction. Moreover, the nanoscale structures favor the movement or transfer of electrons and holes generated inside the crystal to the surface,38 which also enhances the photocatalytic activity. The photocatalytic activity of ZnO nanoparticles appears to be strongly dependent on the surface orientation of the nanocrystals which could result in orientation-dependent charge-transfer processes.39 Thus, as for the hexagonal ZnO nanoparticles, their regular surface orientation with larger emergences of {1010} and {0001} surfaces would be favorable for the separation of photo-induced electrons and holes, leading to relatively high photocatalytic efficiencies. However, in comparison, ZnO nanoparticles pretreated at 200 °C have a smaller size, higher oxygen vacancies and a larger specific surface area, which result in a higher photocatalytic performance.
Although ZnO nanoparticles have previously been investigated in the photocatalytic degradation of methyl orange, the time period required for the degradation of methyl orange appeared to be 2 hours or greater. Wang et al. prepared ZnO nanoparticles with different sizes40 using thermal evaporation and chemical deposition methods. It was observed that 50 nm ZnO nanoparticles degraded 80% methyl orange in 2 h. Zhu et al.41 prepared ZnO nanorod assemblies using a microwave assisted hydrothermal method in the presence of polyethylene glycol. The authors synthesized the sample at 180 °C and observed that the sample required 11 h for complete photocatalytic degradation of methyl orange; the slower rate of reaction reported here is likely due to the size of the nanorods (300 nm diameter and 1 μm length). Saravanan et al.42 synthesized nanosized ZnO by a precipitation method using zinc acetate and sodium hydroxide. The obtained ZnO nanoparticles offered 61% photocatalytic degradation of methyl orange in 2 h. Kumar et al.43 reported the synthesis of ZnO nano-mushrooms by the solution combustion method and their use for the photocatalytic degradation of methyl orange. The authors observed 92% photocatalytic degradation of methyl orange in 210 minutes under optimum conditions. Here, the synthesized ZnO nanoparticles thermally treated at 200 °C offered 99% efficiency in 90 min of reaction time. The observed photocatalytic degradation activity is substantially higher than the activity reported for ZnO nanoparticles in the literature. This is likely to be due to the fact that this sample possessed highly dispersed ZnO nanoparticles of size of about 26 nm.
The surface area of a particle depends mainly on the size of the particle,30 a decrease in the crystallite size tends to increase the surface area. This trend is evident in this work, as shown by the BET surface area results (Table S1, ESI†). The sample thermally treated at 200 °C also possessed relatively high pore volume, pore size and bandgap energy. It is known that the photocatalytic redox reaction mainly takes place on the surface of the photocatalysts and so the surface properties significantly influence the efficiency of the catalyst.44
We also tested the reusability of ZnO nanoparticles treated at 200 °C for subsequent cycles of methyl orange degradation at pH 7. Many of the reported photocatalysts have not been used for the further degradation studies due to the fact that they undergo photocorrosion when subjected to direct illumination with light, and hence their photostability is diminished for further runs. For the reusability study, we collected the white colored catalyst that remained after the reaction, washed, dried, and treated at 100 °C for half an hour and used it for further reactions (0.05 g). Fig. 12 reports the reusability of ZnO nanoparticles. The catalyst was found to be active for five cycles without any major deactivation, and more than 95% degradation was achieved in all experiments within 90 min using 50 mg ZnO. The reusability of ZnO nanoparticles was ascribed to the low photocorrosive effect and high catalytic stability of ZnO nanoparticles.45
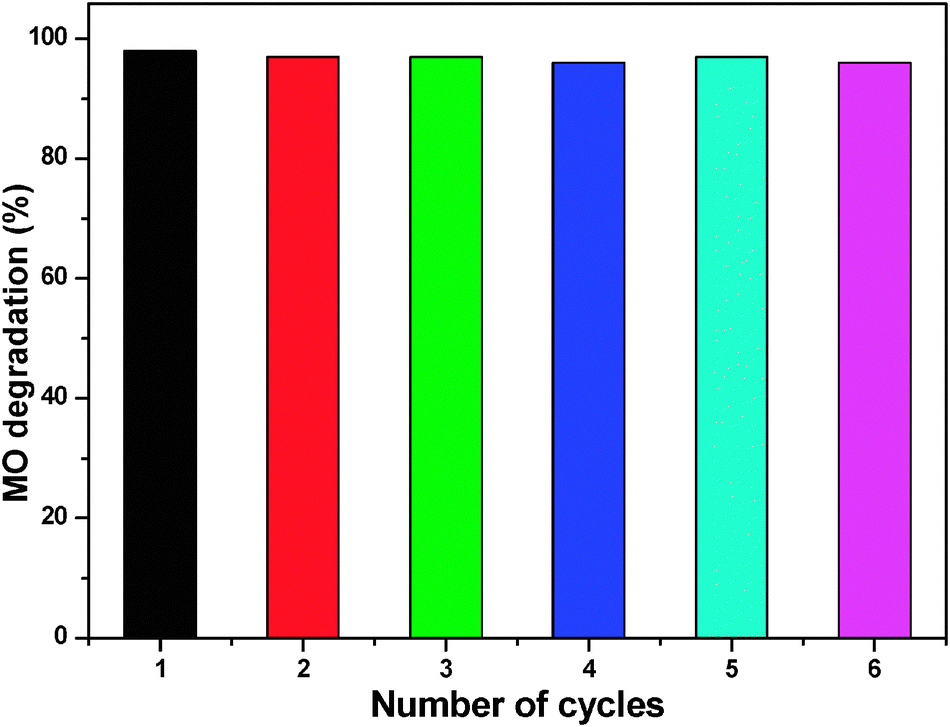 | ||
| Fig. 12 The reusability of ZnO catalyst treated at 200 °C. | ||
4. Conclusions
Porous nanocrystalline ZnO photocatalysts were successfully synthesized by a microwave irradiation method. It is clear that thermal treatment at high temperatures (usually more than 400 °C) is not required to obtain highly active ZnO photocatalysts. The high photocatalytic degradation of methyl orange by the thermally treated ZnO nanoparticles at 200 °C is due to the combined effects of several factors such as small crystallite size, relatively high surface area, pore diameter, pore volume and bandgap energy compared to the same sample treated at 150 °C, 250 °C and 300 °C. The results confirm that there is a positive correlation between the photocatalytic activity and the textural properties of the ZnO nanoparticles, in addition to the purity of the ZnO phase. Superior porosity of ZnO nanoparticles treated at 200 °C could have facilitated the diffusion and mass transportation of methyl orange molecules and hydroxyl radicals during the photochemical reaction. ZnO photocatalyst treated at 200 °C can be reused for five cycles without any loss of activity.Acknowledgements
The Authors thank Felicity Sartain for her support. This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under Grant No. D-5/432. The authors, therefore, acknowledge DSR with thanks for their technical and financial support.References
- B. Swarnalata and Y. Anjaneyulu, J. Mol. Catal. A: Chem., 2004, 223, 161–165 CrossRef PubMed.
- C. C. Chen, J. Mol. Catal. A: Chem., 2007, 264, 82–92 CrossRef CAS PubMed; A. Akyol and M. Bayramoglu, J. Hazard. Mater., 2005, 124, 241–246 CrossRef PubMed.
- M. J. Height, S. E. Pratsinis, O. Mekasuwandumrong and P. Praserthdam, Appl. Catal., B, 2006, 63, 305–312 CrossRef CAS PubMed; S. K. Pardeshi and A. B. Patil, Sol. Energy, 2008, 82, 700–705 CrossRef PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- A. Calzolari, A. Ruini and A. Catellani, J. Am. Chem. Soc., 2011, 133, 5893–5899 CrossRef CAS PubMed.
- T. S. Ko, T. C. Lu, L. F. Zhuo, W. L. Wang, M. H. Liang, H. C. Kuo, S. C. Wang, L. Chang and D. Y. Lin, J. Appl. Phys., 2010, 108, 73504–73505 CrossRef PubMed.
- S. T. Tan, A. A. Umar, A. Balouch, M. Yahaya, C. C. Yap, M. M. Salleh and M. Oyama, Ultrason. Sonochem., 2014, 21, 754–760 CrossRef CAS PubMed.
- M. S. Tokumoto, S. H. Pulcinelli, C. V. Santilli and V. Briois, J. Phys. Chem. B, 2003, 107, 568–574 CrossRef CAS.
- N. Daneshvar, D. G. Salari and A. R. Khataee, J. Photochem. Photobiol., A, 2004, 162, 317–322 CrossRef CAS.
- T. Andelman, Y. Y. Gong, M. Polking, M. Yin, I. Kuskovsky, G. Neumark and S. O'Brien, J. Phys. Chem. B, 2005, 109, 14314–14318 CrossRef CAS PubMed; D. Li and H. Haneda, Chemosphere, 2003, 51, 129–137 CrossRef; L. Yu, F. Qu and X. Wu, J. Alloys Compd., 2010, 504, L1–L4 CrossRef PubMed.
- R. Kumar, G. Kumar and A. Umar, Mater. Lett., 2013, 97, 100–103 CrossRef CAS PubMed.
- Y. He, Powder Technol., 2004, 147, 59–63 CrossRef CAS PubMed.
- R. Gedye, F. Smith, K. Westaway, A. Humera, L. Baldisera, L. Laberge and L. Rousell, Tetrahedron Lett., 1986, 27, 279–282 CrossRef CAS.
- Y. Guo, H. Wang, C. He, L. Qiu and X. Cao, Langmuir, 2009, 25, 4678–4684 CrossRef CAS PubMed.
- S. Manafi, M. R. Rahimipour, B. Yazdani, S. K. Sadrnezhaad and M. H. Amin, Int. J. Eng., Trans. B, 2008, 21, 109–116 Search PubMed.
- K. Ada, M. Gokgoz, M. Onal and Y. Sarıkaya, Powder Technol., 2008, 181, 285–291 CrossRef CAS PubMed.
- H. Yu, Z. Zhang, M. Han, X. Hao and F. Zhu, J. Am. Chem. Soc., 2005, 127, 2378–2379 CrossRef CAS PubMed.
- M. Mazloumi, S. Taghavi, H. Arami, S. Zanganeh, A. Kajbafvala, M. R. Shayegh and S. K. Sadrnezhaad, J. Alloys Compd., 2009, 468, 303–307 CrossRef CAS PubMed.
- T. Thongtema, A. Phuruangratb and S. Thongtem, Ceram. Int., 2010, 36, 257–262 CrossRef PubMed.
- K. M. Parida, S. S. Das and D. P. Das, J. Colloid Interface Sci., 2006, 298, 787–793 CrossRef CAS PubMed.
- M. H. Huang, Y. Wu, H. Feick, N. Tran, E. Weber and P. Yang, Adv. Mater., 2001, 12, 113–116 CrossRef.
- A. Van Dijken, E. A. Meulenkamp, D. Vanmaekelbergh and A. Meijerink, J. Lumin., 2000, 90, 123–128 CrossRef CAS.
- N. Xu, Y. Xu, L. Li, Y. Shen, T. Zhang, J. Wu, J. Sun and Z. Ying, J. Vac. Sci. Technol., A, 2006, 24, 517–520 CAS.
- P. T. Hsieh, Y. C. Chen, K. S. Kao and C. M. Wang, Appl. Phys. A: Mater. Sci. Process., 2008, 90, 317–321 CrossRef CAS.
- S. M. Jejurikar, A. G. Banpurkar, A. V. Limaye, S. K. Date, S. I. Patil, K. P. Adhi, P. Misra, L. M. Kukreja and R. Bathe, J. Appl. Phys., 2006, 99, 14907–14910 CrossRef PubMed.
- R. Ayochi, D. Leinen, F. Martin, M. Gabas, E. Dalchiele and J. R. Ramos-Barrado, Thin Solid Films, 2003, 426, 68–77 CrossRef.
- K. Kotsis and V. Staemmler, Phys. Chem. Chem. Phys., 2006, 8, 1490–1498 RSC.
- C. Liangyuan, L. Zhiyong, B. Shouli, Z. Kewei, L. Dianqing, C. Aifan and C. C. Liu, Sens. Actuators, B, 2010, 143, 620–628 CrossRef PubMed.
- M. Chen, X. Wang, Y. H. Yu, Z. L. Pei, X. D. Bai, C. Sun, R. F. Huang and L. S. Wen, Appl. Surf. Sci., 2000, 158, 134–140 CrossRef CAS.
- A. K. Zak, M. E. Abrishami, W. H. A. Majid, R. Yousefi and S. M. Hosseini, Ceram. Int., 2011, 37(1), 393–398 CrossRef CAS PubMed.
- A. Kajbafvala, S. Zanganeh, E. Kajbafvala, H. R. Zargar, M. R. Bayati and S. K. Sadrnezhaad, J. Alloys Compd., 2010, 497, 325–329 CrossRef CAS PubMed.
- M. Pudukudy and Z. Yaakob, Appl. Surf. Sci., 2014, 292, 520–530 CrossRef CAS PubMed.
- A. Dakhlaoui, M. Jendoubi, L. S. Smiri, A. Kanaev and N. Jouini, J. Cryst. Growth, 2009, 311, 3989–3996 CrossRef CAS PubMed.
- R. Y. Hong, J. H. Li, L. L. Chen, D. Q. Liu, H. Z. Li, Y. Zheng and J. Ding, Powder Technol., 2009, 189, 426–432 CrossRef CAS PubMed.
- S. K. Kansal, M. Singh and D. Sud, J. Hazard. Mater., 2007, 141, 581–590 CrossRef CAS PubMed.
- L.-Y. Yang, S.-Y. Dong, J.-H. Sun, J.-L. Feng, Q.-H. Wu and S.-P. Sun, J. Hazard. Mater., 2010, 179, 438–443 CrossRef CAS PubMed.
- J. Kaur, S. Bansal and S. Singhal, Physica B, 2013, 416, 33–38 CrossRef CAS PubMed.
- W. W. Wang, Y. J. Zhu and L. X. Yang, Adv. Funct. Mater., 2007, 17, 59–64 CrossRef CAS.
- E. S. Jang, J. H. Won, S. J. Hwang and J. H. Choy, Adv. Mater., 2006, 18, 3309–3312 CrossRef CAS; N. Kislov, J. Lahiri, H. Verma, D. Y. Goswami, E. Stefanakos and M. Batzill, Langmuir, 2009, 25, 3310–3315 CrossRef.
- H. Wang, C. Xie, W. Zhang, S. Cai, Z. Yang and Y. Gui, J. Hazard. Mater., 2007, 141, 645–652 CrossRef CAS PubMed.
- Z. Zhu, D. Yang and H. Liu, Adv. Powder Technol., 2011, 22, 493–497 CrossRef PubMed.
- R. Saravanan, V. K. Gupta, V. Narayanan and A. Stephen, J. Mol. Liq., 2013, 181, 133–141 CrossRef CAS PubMed.
- R. Kumar, G. Kumar and A. Umar, Mater. Lett., 2013, 97, 100–103 CrossRef CAS PubMed.
- X. Yang, Y. Wang, L. Xu, X. Yu and Y. Guo, J. Phys. Chem. C, 2008, 112, 11481–11489 CAS.
- H. Fan, X. Zhao, J. Yang, X. Shan, L. Yang, Y. Zhang, X. Li and M. Gao, Catal. Commun., 2012, 29, 29–34 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nj01465k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |