
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic mineralisation of herbicide 2,4,5-trichlorophenoxyacetic acid: enhanced performance by triple junction Cu–TiO2–Cu2O and the underlying reaction mechanism†
Xiaoqiang
An
ab,
Huijuan
Liu
b,
Jiuhui
Qu
b,
Savio J. A.
Moniz
a and
Junwang
Tang
*a
aDepartment of Chemical Engineering, UCL, London, WC1E 7JE, UK. E-mail: Junwang.tang@ucl.ac.uk
bKey Laboratory of Drinking Water Science and Technology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
First published on 17th October 2014
Abstract
A mild and facile photodeposition method was used to fabricate novel Cu–TiO2–Cu2O composite photocatalysts. Due to the in situ rectifying charge carrier separation and enhanced conductivity, the composites present superior photocatalytic activity, leading to more than 90% mineralisation of the toxic 2,4,5-trichlorophenoxyacetic acid herbicide. This result was confirmed by both TOC and UV-vis absorption measurements. The effect of active radicals on the photodegradation of the herbicide was further investigated in order to clarify the underlying mechanism, based on which a hole-dominated photooxidation mechanism was proposed. These results not only offer a green and economical method for constructing triple junction photocatalyst materials, but also shed new insight on the rational design of a low cost and high-efficiency photocatalyst for environmental remediation.
1. Introduction
2,4,5-Trichlorophenoxyacetic acid (2,4,5-T) is one of the most widely used herbicides in agriculture, and is extensively used in developing countries (e.g. Vietnam). As a toxic 2,4,5-trichlorophenol derivative, 2,4,5-T can lead to brain and central nervous system damage in humans if ingested.1,2 Additionally, it is considered to be less readily biodegradable than the analogous phenoxyacetic acid herbicides (e.g. 2,4-dichlorophenoxyacetic acid, 2,4-D) due to the additional chlorine atom on the aromatic ring.3,4Several techniques can be used to degrade 2,4,5-T, including an electrochemical method, peroxi-coagulation and an electro-Fenton method.5,6 Heterogeneous photocatalysis offers a more economically viable route for its decomposition by utilizing widely distributed solar energy. Due to its low toxicity, low-cost, high photostability and high efficiency, the application of TiO2 for the photocatalytic decontamination of water has been intensively investigated over the past few decades.7 One typical proven advantage of TiO2 compared to other photocatalysts is that it can mineralise organic contaminants instead of partially oxidising them, which would result in many intermediates that are more toxic than the original organic substance. Therefore, there is much interest in the modification of TiO2 to improve its efficiency whilst retaining its good stability. However, the fast recombination of photogenerated electron–hole pairs is still a serious challenge in the photocatalytic process, leading to a low reaction efficiency that has to be overcome in order to meet the requirement of practical applications.
Several strategies have been used to improve the photocatalytic activity of TiO2, such as dye sensitization, facet engineering and heterostructured materials, to name but a few.8–11 Modification of the TiO2 photocatalyst with metal nanoparticles has resulted in an improvement in its degradation efficiency. The deposition method and metal species (e.g. Pt, Ag, Au, Cu, Co)12–15 influence the performance of the photocatalyst greatly. Among these, the use of Cu or cuprous oxide (Cu2O) species coupled to TiO2 for the photodegradation of acetonitrile and 2-naphthol has shown some advantages compared to other catalysts due to its lower toxicity, low cost and earth abundance.16,17 However, the oxidation state of the deposited copper is rarely clarified due to the fact that the resulting formation of Cu or Cu2O is highly dependent on the experimental conditions.
It is therefore attractive to synthesise copper incorporated TiO2 photocatalysts for the efficient mineralisation of organic contaminants, e.g. herbicide 2,4,5-T. Furthermore, it is well-known that P25 is much more efficient than any individual phase of TiO2 (rutile, anatase and brookite) for organic contaminant decomposition.18 Modifying the benchmark P25 (composed of 70% anatase and 30% rutile) for a higher reaction efficiency is desirable. In addition, particular attention should be paid to make a fair comparison between the modified samples and fresh P25 because during our studies we found that a pre-treatment process, such as heating P25 at a high temperature (80 °C), dramatically decreases the activity of fresh P25 by decreasing its surface area. In this paper, we report a very mild method to modify P25 to avoid surface area change, and thus a triple junction Cu–TiO2–Cu2O catalyst was successfully fabricated. Consequently, the structure of the photocatalyst was systematically characterized, and the photocatalytic decomposition of a toxic organic pollutant (2,4,5-T) by the triple junction was investigated. Due to the synergic effect of Cu and Cu2O in the composites, our new photocatalyst is superior to pristine P25 for contaminant mineralisation. The enhancement was attributed to the special structure of the heterojunction and the reaction pathway, which is illustrated by a hole-dominated photochemical process, and is also discussed based on a series of control experiments.
2. Experimental section
2.1 Fabrication of the photocatalysts
A mild photodeposition method was used to fabricate the Cu–TiO2–Cu2O photocatalysts.19 In a typical run, 100 mg of commercial P25 (Degussa, surface area = 50 m2 g−1), 6 mg of CuSO4 and 100 mL of DI water was added into a beaker and stirred to form a slurry. 5 mL of methanol was then added, and used as the hole scavenger during the photocatalytic reaction. The solution was irradiated using a 300 W Xe lamp (Newport) for three hours. Finally, the particles were filtered, washed with water and dried in ambient conditions. The exact amount of Cu in the composites was determined using X-ray photoelectron spectroscopic analysis.2.2 Characterization
XRD was performed using a Bruker D4 diffractometer in reflection geometry using Cuα radiation (λ = 1.54 Å, 40 kV, 30 mA). The morphologies of the products were characterized using a transmission electron microscope (HR-TEM, JEOL-2010F, 200 kV) coupled with scanning transmission electron microscopy (STEM). High resolution X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-alpha photoelectron spectrometer using monochromatic Al-Kα radiation. The peak positions were calibrated to carbon (284.5 eV) and plotted using CasaXPS software. The samples were mounted onto conductive tape and attached to a standard sample holder prior to introduction into the instrument. Specific surface area measurements were taken using the BET method (N2 absorption, 77 K, TriStar 3000, Micromeritics) at 77 K. Prior to the analysis, the samples were degassed at 300 °C overnight. The optical properties of the samples were investigated using UV-Vis spectroscopy (UV-Vis, Hitachi 3310). An inductively coupled plasma optical emission spectrometer (ICP-OES) was used to study the stability of Cu–P25 under irradiation. The samples were collected after stirring 1% Cu–P25 solution in the dark for 100 minutes followed by irradiation for 100 minutes.2.3 Electrochemical measurements
5 mg of the photocatalyst and 10 μl of Nafion solution (5 wt%) were dispersed in a 1 ml water/isopropanol mixed solvent (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and sonicated for 30 min to form a homogeneous colloidal suspension. For the measurements, 100 μl of the catalyst colloid was deposited onto FTO conductive glass (ca. 1 cm2) to form a working electrode. In the three electrode photoelectrochemical system, a platinum wire was used as the counter electrode and a Ag/AgCl electrode was used as the reference electrode. The electrolyte was 0.5 M Na2SO4 aqueous solution degassed with Ar. Electrochemical measurements were performed using an Iviumstat potentiostat equipped with Ivium software. Electrochemical impedance spectra (EIS) were recorded under an ac perturbation signal of 10 mV over the frequency range of 1 MHz to 100 mHz.
:1 v/v) and sonicated for 30 min to form a homogeneous colloidal suspension. For the measurements, 100 μl of the catalyst colloid was deposited onto FTO conductive glass (ca. 1 cm2) to form a working electrode. In the three electrode photoelectrochemical system, a platinum wire was used as the counter electrode and a Ag/AgCl electrode was used as the reference electrode. The electrolyte was 0.5 M Na2SO4 aqueous solution degassed with Ar. Electrochemical measurements were performed using an Iviumstat potentiostat equipped with Ivium software. Electrochemical impedance spectra (EIS) were recorded under an ac perturbation signal of 10 mV over the frequency range of 1 MHz to 100 mHz.
2.4 Photocatalytic activity measurements
The photocatalytic activity of the prepared samples was investigated by measuring the photodegradation of 2,4,5-T using UV-Vis spectroscopy and total organic carbon analysis (TOC). In a typical measurement, 10 mg of the photocatalyst was suspended in 100 mL of 50 ppm aqueous 2,4,5-T solution, identical to the concentration in the rivers in Vietnam, as measured by our team very recently. The suspension was stirred in the dark for 2 hours to achieve the equilibrium adsorption. A 300 W Xenon lamp (Newport) was used to provide a full-spectrum emission without any filters, and exhibited broad and strong irradiation from 200 nm to 2400 nm. Upon irradiation, the concentration change of 2,4,5-T was monitored by measuring the UV-Vis spectra of the suspension at regular intervals, using a Shimadzu UV-visible 2550 spectrophotometer. The suspension was filtered to remove any solid material before taking the UV-vis measurements. The apparent rate constant was calculated using the following equation: k = ln(C0/C)/t, where C is the concentration of 2,4,5-T at the irradiation time t, and C0 is the concentration just after the adsorption equilibrium.20 The TOC was measured using a Shimadzu TOC-L analyzer to confirm the degree of mineralisation.3. Results and discussion
To obtain Cu-loaded TiO2 as a photocatalyst for the high-efficiency removal of 2,4,5-T in water, a series of Cu–TiO2–Cu2O triple junctions were synthesized through facile photo-deposition using various copper precursors.Fig. 1a shows the XRD patterns of P25, and of 1% and 1.5% Cu-loaded P25. Typical diffraction peaks corresponding to the anatase (JCPDS 21-1272) and rutile (JCPDS 21-1276) phases are observed in all samples, and the modified samples show similar patterns as pristine P25. This indicates that the mild modification method did not cause a change to the ratio of anatase to rutile in the mixture. However, no characteristic peaks of Cu-related species (Cu, Cu2O, CuO or Cu(OH)2) are observed in the 1% Cu modified P25, which is attributed to the fact that the small amount of copper species is highly dispersed on the surface of P25.21 As shown in Fig. 1b, increasing the loading of Cu to 1.5% results in the appearance of very weak XRD peaks, corresponding to the (200) and (220) planes of Cu2O, and the (111) and (200) planes of Cu respectively.22
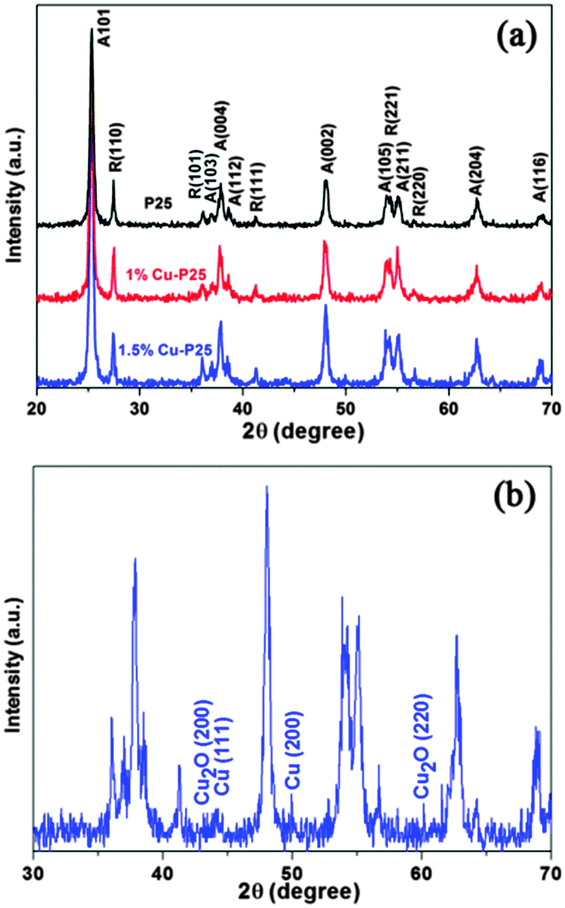 | ||
| Fig. 1 (a) XRD patterns of P25 and P25 loaded with different amounts of copper. (b) The enlarged XRD pattern of 1.5% Cu-loaded P25. | ||
The morphology of the samples was analyzed using TEM. Fig. 2a shows the particle size of the fresh P25, and Fig. 2b shows the 1% Cu-loaded P25. Both consist of nanoparticles and the modification has a negligible effect on the overall size of the products. The inset image clearly shows the existence of small particles with diameters of several nanometers on the surface of larger P25 nanoparticles with diameters in the range of 13–35 nm. Fig. 2c and d and Fig. S1 (ESI†) represent the high-resolution TEM images of 1% Cu-loaded P25 at different regions, and the presence of Cu-related species can be easily distinguished by the differences in the size of nanoparticles. Both figures reveal the characteristic lattice spacing of TiO2. The clear lattice fringes with interplanar spacing of 3.5 Å and 2.4 Å correspond to the (101) plane of anatase and the (001) plane of rutile TiO2 respectively. Besides the lattice fringes of TiO2, the lattice spacings of 2.2 Å, 2.06 Å and 1.8 Å match the (200) plane of Cu2O, and the (111) and (200) planes of Cu respectively.12,23–26 Interestingly, the co-existence of Cu2O and Cu located at different sides of both the anatase and rutile nanoparticles indicate that a triple junction is formed between TiO2, Cu and Cu2O. As reported, Cu2O nanoparticles are usually formed in the initial stage of copper photodeposition. Photoreduction of Cu+ to Cu metal was only observed after relatively long illumination times and using high light intensities.27 Furthermore, the spontaneous oxidation of nano-sized Cu particles was not ruled out, which indicates a much more complicated situation than the other metals.26,28 More recently, Zhang et al. observed that the following photoreduction of Cu2O to Cu was inhibited by the cooperation with TiO2. The photoinduced holes were transferred from TiO2 to Cu, retaining a small amount of Cu which is in turn oxidized to Cu2O. Similarly, it is believed that the final resulting amount of Cu and Cu2O results in the formation of a stable Cu–TiO2–Cu2O triple junction after illumination.29 The formation of a Cu–TiO2 junction was further confirmed by high-angle annular dark field and annular bright field STEM (HAADF-STEM and ABF-STEM) measurements as shown in Fig. S2 (ESI†). The element distributions in the line scan profile provide solid evidence for the formation of Cu nanoparticles on the surface of TiO2.
 | ||
| Fig. 2 TEM images of (a) P25 and (b) 1% Cu-loaded P25; HRTEM images of 1% Cu-loaded P25 at (c) region 1 and (d) region 2. | ||
In order to further confirm the co-existence of different Cu species and identify their chemical states in the junction, 1% Cu-loaded P25 was characterized using XPS and Auger electron spectroscopy. In Fig. 3a, peaks corresponding to the binding energies of 932.5 and 952.4 eV are observed, and are associated with the typical Cu 2p3/2 and Cu 2p1/2 peaks of either Cu0 or Cu2O.30 It is well known that Cu(II) species can be easily identified by the satellite peaks on the higher binding energy of the main peaks, which is related to the open 3d9 shell configuration in the Cu(II) ground state.31 Therefore, the formation of CuO in the 1% Cu-loaded P25 sample can be excluded because of the absence of the shake-up satellite peaks. However, it is difficult to differentiate Cu2O and Cu metal by the Cu 2p XPS peaks as their binding energies are too close.32 In order to further clarify Cu and Cu2O, a Cu L3VV Auger spectrum of 1% Cu-loaded P25 was recorded. The fitted curves in Fig. 3b reveal two peaks centered at 917.2 eV and 919.2 eV, which arise from Cu2O-type oxide and Cu metal respectively.33 In combination with the TEM observations, it is reasonable to conclude that both Cu2O and Cu are present in the modified samples, resulting in a Cu–P25–Cu2O based triple junction.
 | ||
| Fig. 3 (a) High resolution XPS spectrum of Cu 2p; (b) the Cu L3VV Auger spectrum. | ||
The application of the triple junction in the photocatalytic removal of toxic herbicides was evaluated by the photo-mineralisation of 2,4,5-T. As shown in Fig. 4a and Fig. S3 (ESI†), virtually zero degradation of the target pollutant was observed after 100 min full arc illumination in the absence of photocatalysts. When Cu species are loaded on the surface of the P25 nanoparticles, their activity for 2,4,5-T decomposition is enhanced compared with the initial P25, whilst the 1% Cu-loaded P25 sample shows the best photocatalytic performance.34 Furthermore, the kinetic plot for the degradation process follows typical first-order kinetics.35 The apparent rate constant of the 1% Cu-loaded sample was calculated to be 0.0285 min−1, which is 1.6 times higher than that of pristine P25 (k = 0.0183 min−1), indicating that the photocatalytic performance of P25 can be significantly enhanced by the triple junction. This result is even more efficient than the reported photo-induced removal of pollutants over bimetallic Au–Cu and Ag–Cu nanoparticle modified P25.36,37 In order to clarify it as a mineralization process instead of incomplete decomposition, the TOC in the reaction solution was measured and is displayed in Fig. 4b. After 20 minutes of irradiation, the TOC removal efficiency of the 1% Cu modified P25 sample is 41%, which is about 1.6 times larger than that of pristine P25. The junction further results in a 93% TOC removal efficiency compared to 70% for P25 after 100 minutes’ irradiation. The faster TOC reduction rate indicates that the triple junction sample does not only improve the activity of P25, but also enhances the mineralization effect of P25, which is a preferable advantage of TiO2 mentioned above.38
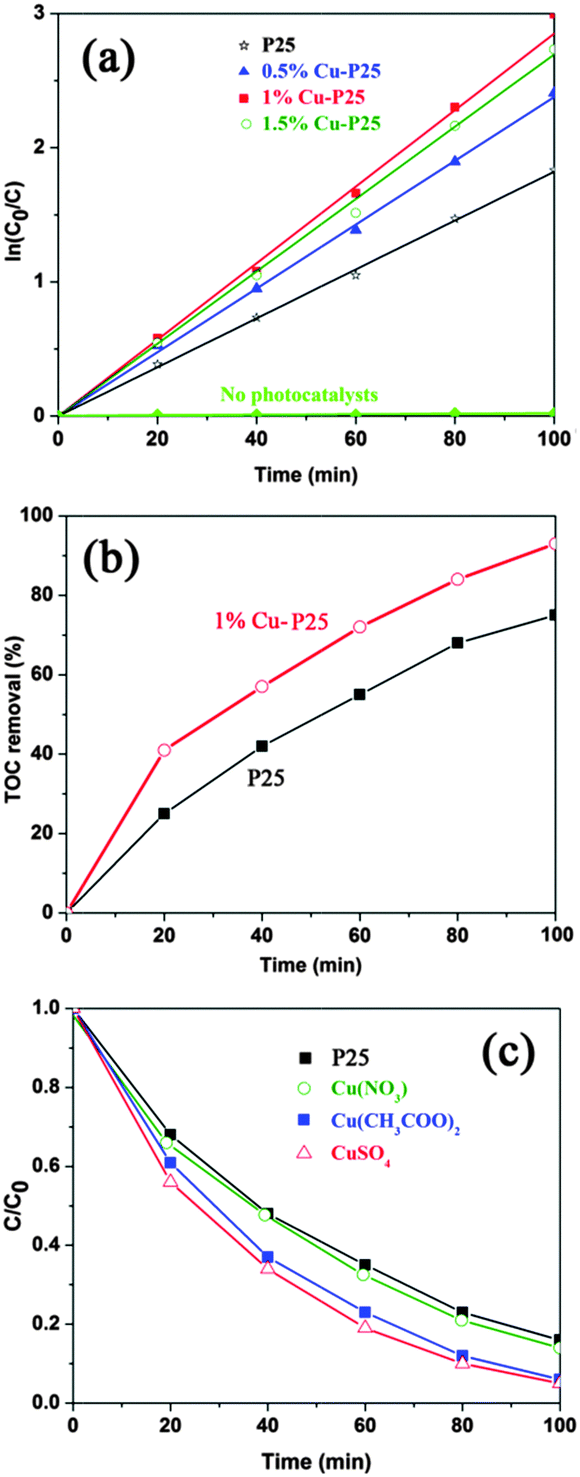 | ||
| Fig. 4 (a) Plots of the pseudo-first-order kinetic model for the products; (b) variation of TOC removal by the photocatalytic degradation of 2,4,5-T; (c) photodegradation of 2,4,5-T over Cu–TiO2–Cu2O fabricated from different copper sources. | ||
Furthermore, the effect of the copper source on the photocatalytic activity was also investigated. As can be seen from Fig. 4c, the junctions fabricated by using CuSO4 and Cu(CH3COO)2 exhibit significantly improved photocatalytic activity. However, only slight enhancement in performance was observed when Cu(NO3)2 was used. The reason for this could be ascribed to the fact that the reduction of Cu2+ was inhibited by the presence of oxidative NO3−, as a much smaller amount of Cu (0.36%) is detected in the XPS measurement (Fig. S4, ESI†). Due to the occurrence of photocatalytic nitrate reduction, it is reasonable to conclude that a certain amount of photo-generated electrons were consumed in this manner.39
To clarify the reasons for the enhanced photoactivity of copper incorporated P25, the samples were further characterized. Based on the BET measurement, the loading of copper has little effect on the surface area of P25, as only a slight increase (from 50 m2 g−1 to 59 m2 g−1) is observed.22 The increased surface area might be ascribed to the more coarse surface resulting from the loading of the metal or metal oxide, which inhibited the overall aggregation of the nanoparticles. Similar results were also observed when coupling Fe2O3 nanoparticles with P25 by an identical photodeposition method.40Fig. 5a represents the photocurrent–time characteristics of bare P25, and the 0.5%, 1% and 1.5% Cu modified samples measured at an applied voltage of 0.1 V (vs. Ag/AgCl). As expected, 1% Cu-P25 exhibits the highest photocurrent response, which is nearly two times higher than the bare P25 electrode and is in good agreement with the measured 1.6 times enhancement in photocatalytic degradation of 2,4,5-T. The improvement is attributed to the enhanced separation of the photogenerated electrons and holes in the triple junction.41 In the electrochemical impedance spectroscopy (EIS) measurements (Fig. 5b), the impedance arc radius of 1% Cu-loaded P25 is much smaller than that of P25, and the 0.5% and 1.5% Cu-loaded P25, which indicates that the conductivity of the sample is also enhanced by the triple junction structure.42
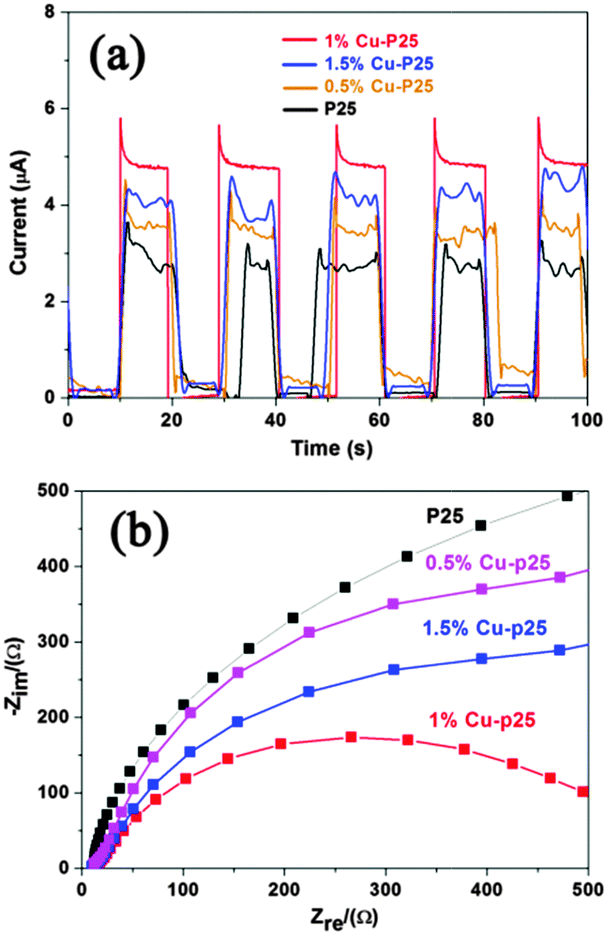 | ||
| Fig. 5 (a) Transient photocurrent of P25 and the Cu–P25 samples; (b) EIS spectra of P25 and the Cu-loaded P25 samples. | ||
It is well known that the photooxidation of organic compounds by inorganic photocatalysts can be ascribed to different reaction pathways dominated by several active species, such as ˙OH, ˙O2− and holes.43 To illustrate the possible mechanism of photodegradation of 2,4,5-T by the triple junction, the role of the active species in the photocatalytic reactions was investigated using different scavengers. Isopropanol was selected as the hydroxyl radical quencher due to its fast reaction with the radical ˙OH.44 As shown in Fig. 6a, the photodegradation of 2,4,5-T is only slightly suppressed in the presence of isopropanol. O2 can capture electrons to generate superoxide radicals, ˙O2−, which have recently been proposed as major photocatalytic oxidants in the photocatalytic oxidation of azo dyes under light irradiation.45 To investigate the effect of ˙O2−, the solution was bubbled with Ar gas to ensure that the reaction was operated in the absence of O2. As shown in Fig. 6b, the decomposition rate of 2,4,5-T is decreased compared to that without a scavenger. It can be inferred that superoxide radicals play an important role in the oxidation reaction. The influence of EDTA was also investigated, as it was used as an efficient hole scavenger.46,47 Surprisingly, no photodegradation of 2,4,5-T is observed, as shown in Fig. 6c. The completely inhibited photocatalytic reaction indicates that the photogenerated holes are the dominant active species, while the ˙O2− radicals are the second most important species, and the ˙OH groups have the smallest effect on photodegradation.
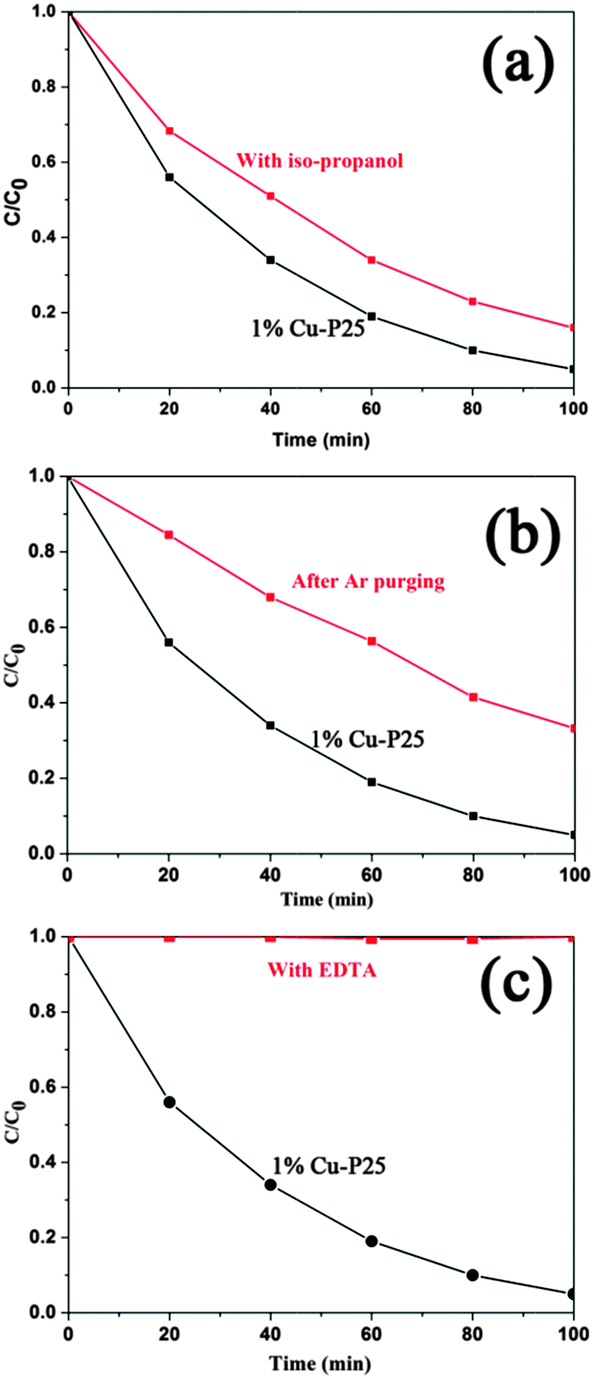 | ||
| Fig. 6 The process of photocatalytic degradation of 2,4,5-T over 1% Cu-loaded P25 in the presence of different scavengers: (a) isopropanol (5 mL), (b) Ar gas bubbling, and (c) EDTA (10 mmol L−1). | ||
Based on the above results, a tentative mechanism was proposed for the photodegradation of 2,4,5-T by the triple junction (Scheme 1). Under illumination, electron–hole pairs are generated in both TiO2 and Cu2O semiconductors. Due to the difference between the conduction bands of TiO2 (conduction band of anatase/rutile: −0.5 V/−0.2 V, valence band of anatase/rutile: +2.7 V/+2.7 V vs. NHE at pH 7) and Cu2O (conduction band: −1.1 V, valence band: +0.9 V vs. NHE at pH 7), electrons can transfer from Cu2O to TiO2 as indicated in Scheme 1.48–50 Subsequently, the electrons are captured by the Cu metal and transformed to superoxide radicals (redox potential +0.89 V vs. SHE).51 Simultaneously, holes in the valence band of TiO2 (including both rutile and anatase) will transfer to Cu2O, which has sufficient over-potential for the oxidation of 2,4,5-T as reported in another study (Fig. S5, ESI†). In addition, the much more negative potential of the holes in the valence band of Cu2O is too high to oxidise water into the ˙OH groups (ENHE(˙OH/H2O) = 2.27 eV) which is not crucial for the expected photocatalytic process.52 It is thus believed that the mitigated recombination of photogenerated charge carriers by the in situ rectifying effect of the junction, the enhanced visible absorption by Cu2O (Fig. S6, ESI†) and the catalytic effect of the Cu metal on the reduction reaction, all contribute to the enhanced mineralisation process of copper loaded P25.53 In the control experiment, 1% Cu2O loaded TiO2 was also fabricated by a simple impregnation method. As shown in Fig. S7 (ESI†), the much higher activity of our triple junction than the binary system indicates its superiority.54
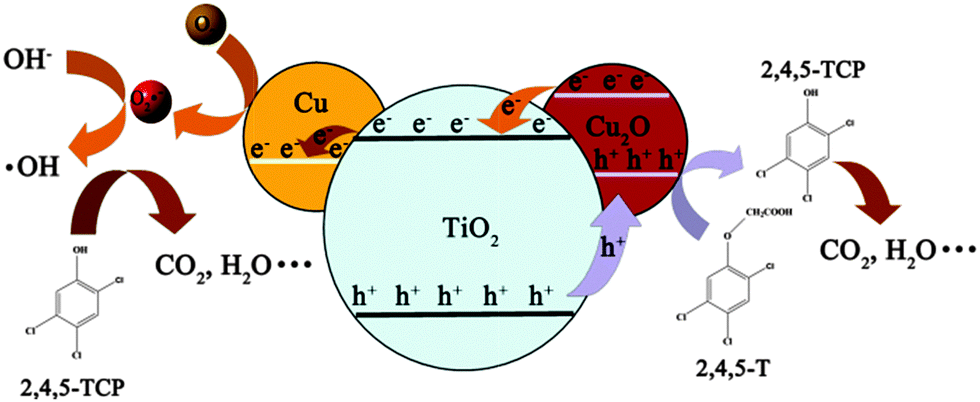 | ||
| Scheme 1 Proposed mechanism of 2,4,5-T photodegradation over Cu–TiO2–Cu2O triple junction. | ||
Generally, the photocatalytic degradation is the result of the oxidation reactions between 2,4,5-T and the active species (photogenerated holes and ˙O2−). The formation of intermediate products during the photodegradation process has been studied by several groups. Barbeni et al. found that 2,4,5-trichlorophenol (2,4,5-TCP) and 2,4,5-trichlorophenyl formate were the two major and longer lived intermediate products from 2,4,5-T degradation.55 Phenol, 2,4,5-trichlorophenol and 2,4-dichlorophenol were detected before the complete mineralisation of 2,4,5-T in another study.56 2,4,5-TCP, 2,4-dichlorophenol and 1,2,4-trichloro-5-methoxy benzene intermediates were also reported by Muneer et al.57 Although various intermediates were detected in these reports, the general consensus is that the removal of the ˙OCH2COOH group to form 2,4,5-TCP is a crucial step for the degradation of 2,4,5-T. Therefore, it is not surprising that 2,4,5-TCP is always detected as the main intermediate in all the above studies, whilst the further degradation of 2,4,5-TCP depends on the active species. Furthermore, another study of the photocatalytic degradation of 2,4-dichlorophenoxyacetic acid suggested that the hole-mediated oxidation results in a high production of 2,4-dichlorophenol while the OH radical-mediated oxidation limits the production of 2,4-dichlorophenol.58,59 Similarly, it is reasonable to suggest that the holes play a dominant role in the formation of 2,4,5-TCP in our research. In the presence of holes, ˙O2− and ˙OH as active species, 2,4,5-TCP can be completely degraded into inorganic products.
The photocatalytic stability of a photocatalyst is crucial to any practical application. Fig. 7 shows the systematic re-use of the triple junction for the photodegradation of 2,4,5-T. The degradation rates are very similar during the three consecutive cycles. After these runs, the samples were characterised by XPS to confirm their chemical stability. The slight increase of Cu2O amount in the Cu L3VV Auger spectrum (Fig. S8, ESI†) indicates that the Cu-loaded P25 exhibits considerable stability after the photocatalytic reaction. The solution of 1% Cu-loaded P25 before irradiation and after 100 minutes’ irradiation was analyzed by ICP-OES. No additional copper ions were detected, which could be ascribed to the low loading amount of Cu and the good stability of Cu–TiO2 under irradiation.
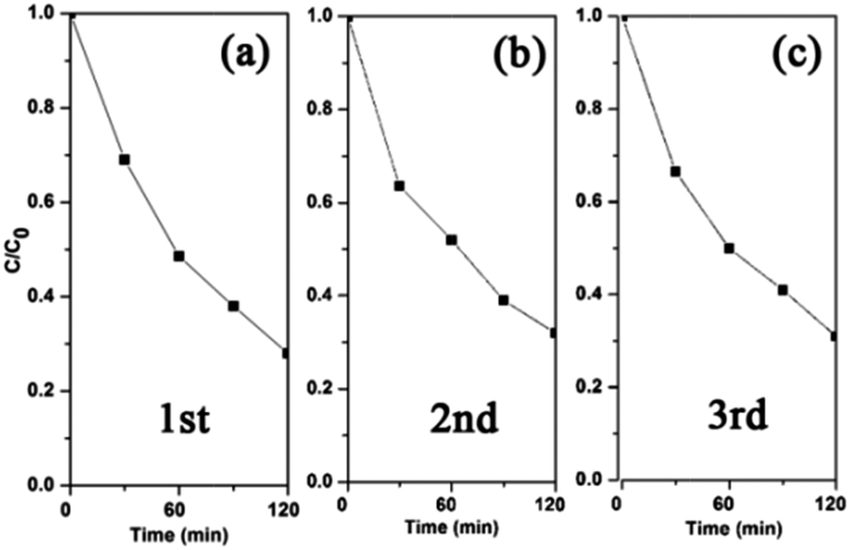 | ||
| Fig. 7 Cycling runs in the photodegradation of 2,4,5-T over 1% Cu-loaded P25. | ||
4. Conclusion
In summary, novel triple junction Cu–TiO2–Cu2O composites were synthesized using an easy, low-cost, green and mild photodeposition method. The composite shows superior photocatalytic activity towards the degradation of a toxic 2,4,5-T herbicide. Compared to pristine P25, the 1% Cu-loaded sample exhibited a 1.6-fold enhancement in apparent rate constant (k = 0.0183 min−1). The influence of the copper precursor and active species on the performance of the photocatalytic reaction was evaluated. A hole-dominant photooxidation mechanism was proposed; ˙O2− radicals also show a promotive effect but ˙OH was found to be less important to the complete degradation of 2,4,5-T into inorganic non-toxic products. The photoresponse and electrochemical impedance studies revealed efficient interfacial separation of the charge carriers and significantly improved the conductivity of the junction materials. As a result, the superior photoactivity of the Cu–Cu2O–TiO2 triple junction can be assigned to the transfer of electrons from Cu2O to TiO2, the enhanced visible absorption by Cu2O and the catalytic effect of the Cu metal on the reduction reaction. Their considerable stability makes Cu–TiO2–Cu2O heterojunctions strong candidates for the application of photocatalytic environmental remediation.Acknowledgements
The authors are grateful to the European Community, Seventh Framework Programme (4G-PHOTOCAT 309636).Notes and references
- M. Yurkova, I. Pozdnyakov, V. Plyusnin, V. Grivin, N. Bazhin, A. Kruppa and T. Maksimova, Photochem. Photobiol. Sci., 2013, 12, 684 CAS.
- Y. Wang and W. Chu, Appl. Catal., B, 2012, 123–124, 151 CrossRef CAS PubMed.
- Y. Wang and W. Chu, Water Res., 2011, 45, 3883 CrossRef CAS PubMed.
- K. Itoh, M. Kinoshita, S. Morishita, M. Chida and K. Suyama, FEMS Microbiol. Ecol., 2013, 84, 124 CrossRef CAS PubMed.
- R. Zona, S. Solar and K. Sehested, Radiat. Phys. Chem., 2012, 81, 152 CrossRef CAS PubMed.
- Y. Wang and W. Chu, Chem. Eng. J., 2013, 215–216, 643 CrossRef CAS PubMed.
- H. Singh, M. Saquib, M. Haque, M. Muneer and D. Bahnemann, J. Mol. Catal. A: Chem., 2007, 264, 66 CrossRef CAS PubMed.
- L. Liu, Z. Liu, A. Liu, X. Gu, C. Ge, F. Gao and L. Dong, ChemSusChem, 2014, 7, 6181 Search PubMed.
- W. Ong, L. Tan, S. Chai, S. Yong and A. Mohamed, Nano Res., 2014, 7, 1528 CrossRef CAS.
- C. Hu, X. Zhang, W. Li, Y. Yan, G. Xi, H. Yang, J. Li and H. Bai, J. Mater. Chem. A, 2014, 2, 2040 CAS.
- O. Lamiel-Garcia, S. Tosoni and F. Illas, J. Phys. Chem. C, 2014, 118, 13667 CAS.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776 CrossRef CAS PubMed.
- S. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390 CAS.
- M. Wang, L. Sun, Z. Lin, J. Cai, K. Xie and C. Lin, Energy Environ. Sci., 2013, 6, 1211 CAS.
- Y. Zou, S. Kang, X. Li, L. Qin and J. Mu, Int. J. Hydrogen Energy, 2014, 39, 15403 CrossRef CAS PubMed.
- S. Inturi, T. Boningari, M. Suidan and P. Smirniotis, Appl. Catal., B, 2014, 144, 333 CrossRef CAS PubMed.
- Q. Jin, M. Fujishima, A. Iwaszuk, M. Nolan and H. Tada, J. Phys. Chem. C, 2013, 117, 23848 CAS.
- D. Scanlon, C. Dunnill, J. Buckeridge, S. Shevlin, A. Logsdail, S. Woodley, C. Catlow, M. Powell, R. Palgrave, I. Parkin, G. Watson, T. Keal, P. Sherwood, A. Walsh and A. Sokol, Nat. Mater., 2013, 12, 798 CrossRef CAS PubMed.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776 CrossRef CAS PubMed.
- X. An, J. C. Yu and J. Tang, J. Mater. Chem. A, 2014, 2, 1000 CAS.
- L. Liu, F. Gao, H. Zhao and Y. Li, Appl. Catal., B, 2013, 134–135, 349 CrossRef CAS PubMed.
- W. Chen, Z. Fan and Z. Lai, J. Mater. Chem. A, 2013, 1, 13862 CAS.
- L. Li, L. Xu, W. Shi and J. Guan, Int. J. Hydrogen Energy, 2013, 38, 816 CrossRef CAS PubMed.
- C. Huang, Z. Long, M. Miyauchi and X. Qiu, CrystEngComm, 2014, 16, 4967 RSC.
- Y. Zou, S. Kang, X. Li, L. Qin and J. Mu, Int. J. Hydrogen Energy, 2014, 39, 15403 CrossRef CAS PubMed.
- W. Foo, C. Zhang and G. Ho, Nanoscale, 2013, 5, 759 RSC.
- J. Jacobs, F. Kampers, J. Rikken, C. Bulle-Lieuwma and O. Koningsberger, J. Electrochem. Soc., 1989, 136, 2914 CrossRef CAS PubMed.
- Y. Wang and C. Wan, J. Photochem. Photobiol., A, 1994, 79, 203 CrossRef CAS.
- Z. Xi, C. Li, L. Zhang, M. Xing and J. Zhang, Int. J. Hydrogen Energy, 2014, 39, 6345 CrossRef CAS PubMed.
- Q. Huang, F. Kang, H. Liu, Q. Li and X. Xiao, J. Mater. Chem. A, 2013, 1, 2418 CAS.
- Z. Zhang, R. Dua, L. Zhang, H. Zhu, H. Zhang and P. Wang, ACS Nano, 2013, 7, 1709 CrossRef CAS PubMed.
- X. Qiu, M. Miyauchi, K. Sunada, M. Minoshima, M. Liu, Y. Lu, D. Li, Y. Shimodaira, Y. Hosogi, Y. Kuroda and K. Hashimoto, ACS Nano, 2012, 6, 1609 CrossRef CAS PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456 CrossRef CAS PubMed.
- S. Oros-Ruiza, R. Zanellaa and B. Prado, J. Hazard. Mater., 2013, 263P, 28 CrossRef PubMed.
- S. Ye, L. Qiu, Y. Yuan, Y. Zhu, J. Xia and J. Zhu, J. Mater. Chem. A, 2013, 1, 3008 CAS.
- Z. Hai, N. Kolli, D. Uribe, P. Beaunier, M. Jose-Yacaman, J. Vigneron, A. Etcheberry, S. Sorgues, C. Colbeau-Justin, J. Chen and H. Remita, J. Mater. Chem. A, 2013, 1, 10829 CAS.
- M. Behnajady and H. Eskandarloo, Chem. Eng. J., 2013, 228, 1207 CrossRef CAS PubMed.
- G. Liao, S. Chen, X. Quan, H. Yu and H. Zhao, J. Mater. Chem., 2012, 22, 2721 RSC.
- K. Doudrick, O. Monzón, A. Mangonon, K. Hristovski and P. Westerhoff, J. Environ. Eng., 2012, 138, 852 CAS.
- S. J. A. Moniz, S. A. Shevlin, X. An, Z-X. Guo and J. Tang, Chem. – Eur. J. DOI:10.1002/chem.201403489.
- X. An, J. Yu, F. Wang, C. Li and Y. Li, Appl. Catal., B, 2013, 129, 80 CrossRef CAS PubMed.
- X. An, X. Yu, J. Yu and G. Zhang, J. Mater. Chem. A, 2013, 1, 5158 CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568 RSC.
- R. Hazime, C. Ferronato, L. Fine, A. Salvador, F. Jaber and J. Chovelon, Appl. Catal., B, 2012, 126, 90 CrossRef CAS PubMed.
- X. Zhang, D. Sun, G. Li and Y. Wang, J. Photochem. Photobiol., A, 2008, 199, 311 CrossRef CAS PubMed.
- P. Ji, J. Zhang, F. Chen and M. Anpo, Appl. Catal., B, 2009, 85, 148 CrossRef CAS PubMed.
- M. Yin, Z. Li, J. Kou and Z. Zou, Environ. Sci. Technol., 2009, 43, 8361 CrossRef CAS PubMed.
- Z. Li, J. Liu, D. Wang, Y. Gao and J. Shen, Int. J. Hydrogen Energy, 2012, 37, 6431 CrossRef CAS PubMed.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171 CrossRef CAS PubMed.
- H. Gerischer, J. Electroanal. Chem., 1977, 82, 133 CrossRef CAS.
- J. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765 RSC.
- M. Hernandez-Alonso, F. Fresno, S. Suareza and J. Coronado, Energy Environ. Sci., 2009, 2, 1231 CAS.
- A. Talebian, M. Entezari and N. Ghows, Chem. Eng. J., 2013, 229, 304 CrossRef CAS PubMed.
- J. Xing, Z. Chen, F. Xiao, X. Ma, C. Wen, Z. Li and H. Yang, Chem. – Asian J., 2013, 8, 1265 CrossRef CAS PubMed.
- M. Barbeni, M. Morello, E. Pramauro, E. Pelizzetti, M. Vincenti, E. Borgarello and N. Serpone, Chemosphere, 1987, 16, 1165 CrossRef CAS.
- A. Chaudhary, M. Hassan and S. Grimes, J. Hazard. Mater., 2009, 165, 825 CrossRef CAS PubMed.
- W. Bahnemann, M. Muneer and M. Haque, Catal. Today, 2007, 124, 133 CrossRef CAS PubMed.
- J. Kim and W. Choi, Appl. Catal., B, 2011, 106, 39 CAS.
- Y. Sun and J. Pignatello, Environ. Sci. Technol., 1995, 29, 2065 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nj01317d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |