
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTitanocene(III) pseudohalides: an ESR and structural study†
Martin
Křižan
ab,
Jan
Honzíček
a,
Jaromír
Vinklárek
b,
Zdeňka
Růžičková
b and
Milan
Erben
*b
aInstitute of Chemistry and Technology of Macromolecular Materials, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
bDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic. E-mail: milan.erben@upce.cz; Fax: +420466037068; Tel: +420466037163
First published on 30th October 2014
Abstract
A series of complexes of the type [Ti(′Cp)2Y] {′Cp = η5-C5H5 (Cp), η5-C5H4SiMe3 (SiCp), η5-1,3-C5H3(Me3Si)2 (Si2Cp); Y = Cl−, CN−, NCS−, NCSe−, dicyanamide (dca), tricyanomethanide (tcm) and 1,2,3,4-thiatriazol-5-thiolate (ttt)} were prepared and studied by ESR spectroscopy. Ring-substituted compounds [Ti(SiCp)2Cl], [Ti(Si2Cp)2Cl], [Ti(SiCp)2CN] and [Ti(Si2Cp)2CN] dissolved in coordinating solvents exist in an equilibrium with solvated species. The thiatriazolthiolate ligand in complexes [Ti(′Cp)2(ttt)] behaves as a S,N-chelator to yield a thermally unstable 4-membered chelate ring having the ESR signal with significant splitting due to coupling with one 14N nucleus. Solutions of [Ti(′Cp)2Y] (Y = NCS−, NCSe−, dca and tcm) readily dissolve the additional pseudohalide salt giving anionic species [Ti(′Cp)2Y2]− with a characteristic ESR pattern caused by super-hyperfine splitting by two 14N nuclei. Cyanide complexes dissolve additional KCN as well, but no coupling to ligand nuclei (neither 14N nor 13C) was observed. The frozen-solution ESR spectra of [Ti(Si2Cp)2Cl], [Ti(Si2Cp)2CN] and [Ti(′Cp)2Y] (Y = NCS−, NCSe− and ttt) are typical for monomeric species of rhombic symmetry. The remaining complexes form dimeric {[Ti(Cp)2Cl]2, [Ti(SiCp)2Cl]2, [Ti(′Cp)2dca]2, and [Ti(′Cp)2tcm]2} or trimeric {[Ti(SiCp)2CN]3} adducts under these conditions. The nuclearity of [Ti(Cp)2CN] cannot be accurately determined by ESR spectroscopy due to its poor solubility but the presence of species with a higher spin ground state was confirmed (similar to the case of the above-mentioned dimers and trimer) by the observation of formally forbidden half-field transition. The reported results of the crystal structure analysis of [Ti(SiCp)2Cl], [Ti(Cp)2tcm], [Ti(SiCp)2tcm], [Ti(SiCp)2dca], [Ti(SiCp)2CN], and [TiCp2(NCS)]2O corroborate the findings based on spectroscopic measurements.
Introduction
Coordination behaviour of pseudohalides toward mid- and late transition metals has been extensively studied with the view to elucidate the bonding pattern of the ligand in these compounds. It was proved that pseudohalides could behave as bridging ligands connecting transition metals to give complexes with various topological structures, including isolated molecular clusters, 1D chains and 2D or 3D networks. In the past few decades, there has been continuous interest in these multimetallic complexes because of their fascinating structural features and interesting magnetic properties.1 The very common spacers used for the connection of transition metals to oligomeric and polymeric structures are cyanides, azides, cyanates, thiocyanates, selenocyanates, dicyanamides, tricyanomethanides together with related anionic ligands such as tetracyanoethanide or tetracyanoquinodimethanide and many others.2 It has been shown that these species can effectively transmit magnetic coupling between the metal ions through the π-bond system of the bridging unit.3 By contrast, paramagnetic pseudohalide complexes of early transition metals are relatively sparse and these are generally limited to mononuclear vanadium(IV)4,5 compounds of the type [VCp2Y2] (Cp = η5-bonded cyclopentadienyl, Y− is a linear or planar pseudohalide anion) and few examples of inorganic niobium(IV) complexes.6 Only three Ti(III) derivatives of the type [TiCp2Y] with a linear pseudohalide ligand Y− = CN−, NCS− and NCO− have been described previously.7 Based on spectroscopic measurements, the oligomeric nature of these compounds was supposed and lastly approved by the X-ray diffraction analysis of polymorphic [TiCp2(CN)].8,9Titanium(III) compounds are investigated for their catalytic properties in both polymerization and hydrogenation of various olefins. For the study of the origin, structure and nuclearity of catalytically active species, the spectroscopic techniques are routinely used. The electron spin resonance (ESR) spectroscopy represents an excellent choice for this purpose, as it allows selective and very sensitive identification of paramagnetic species in the reaction mixture without additional signals of diamagnetic compounds. For the study of titanium(III) complexes, various ESR methods including electron spin echo envelope modulation (ESEEM), electron nuclear double resonance (ENDOR) and hyperfine sublevel correlation spectroscopy (HYSCORE) techniques in both the X- and Q-band region were used recently.10
In this paper, we have studied the properties of titanocene(III) complexes bearing both linear and nonlinear pseudohalide ligands in the solution and in the solid state. As all studied complexes are paramagnetic d1-systems, the ESR spectroscopy was advantageously utilized. Gathered information was used for the examination of the bonding pattern of the pseudohalide ligand toward the titanium(III) central atom.
Results and discussion
Synthesis and characterization of pseudohalide complexes
The syntheses of bis(cyclopentadienyl)titanium(III) pseudohalides were straightforward using the corresponding organometallic monochlorides and alkali metal pseudohalide salts (Scheme 1). Unsubstituted derivatives of the [TiCp2Y] type were precipitated from aqueous solutions of [TiCp2Cl] (1a) by addition of KY, where Y is CN−, SCN−, SeCN−, dca or tcm. Ring-substituted compounds [Ti(η5-C5H4SiMe3)2Y] and [Ti{η5-1,3-C5H3(SiMe3)2}Y] were prepared similarly from corresponding monochlorides using deoxygenated THF as a solvent. Unsubstituted compounds 2a–CN, 2a–dca and 2a–tcm are sparingly soluble in THF or toluene, whereas all the remaining titanium(III) complexes are largely soluble even in less polar solvents (pentane, cyclohexane, etc.). Selenocyanate complexes 2–NCSe showed only limited stability in the solution decomposing to elemental selenium and respective titanocene(III) cyanides 2–CN. | ||
| Scheme 1 Synthesis of the studied complexes and their notation. | ||
We have also performed reactions of titanocene(III) monochlorides with cyclic pseudohalide, 1,2,3,4-thiatriazol-5-thiolate (ttt). From the aqueous solutions, green [TiCp2(ttt)] (2a–ttt) instantly precipitates but it apparently evolves nitrogen bubbles and its colour gradually turns to orange. The analysis of the resulting orange solid showed its diamagnetic nature and spectroscopic data are identical to those for the known complex [TiCp2(NCS)]2O (3a).11 The most characteristic features are very strong IR bands at ∼2050 cm−1 (NCS stretching) and ∼720 cm−1 (deformation of the Cp2Ti–O–TiCp2 moiety). The structure of this product has been also confirmed by its single-crystal X-ray diffraction analysis, vide infra. The formation of this compound could be explained by the decomposition of the ttt ligand giving elemental nitrogen, sulphur and thiocyanate anion.12 The resulting complex 2a–NCS is oxidized by released sulphur to titanium(IV) species which is further hydrolysed to give 3a and hydrogen sulphide (Scheme 2). The ability of elemental sulphur to oxidize bis(cyclopentadienyl)–titanium(III) complexes has been proven by the reaction of 1a with S8 in toluene. This reaction proceeds readily, fast change from dark brown to blood red colour is observable and the ESR signal of starting 1a vanishes. Subsequent hydrolysis gave a dinuclear oxo-bridged complex [TiCp2(Cl)]2O and hydrogen sulphide. Using sterically hindered, ring-substituted chlorides [Ti(η5-C5H4SiMe3)2Cl] (1b) and [Ti{η5-1,3-C5H3(Me3Si)2}2Cl] (1c) allowed us to prepare thermally more robust thiatriazolate complexes 2b–ttt and 2c–ttt, respectively, and characterize them spectroscopically at lowered temperature. Explosive properties of these complexes together with the sensitivity to the air disabled their characterization in the solid state by infrared or Raman spectroscopy. Solution IR spectra affirmed thermal decomposition of the ttt ligand as its characteristic bands at 1280 and 1196 cm−1 rapidly vanish, whereas strong absorption due to thiocyanate stretch (∼2050 cm−1) grows up.
 | ||
| Scheme 2 The formation of 2a–ttt and suggested decomposition giving complex 3a. | ||
The attempts to synthesize azide, cyanate and nitrosodicyanomethanide derivatives were not successful in our hands. The reaction of 1a or 1b with sodium azide yielded yellow solid and liberation of nitrogen has been observed. In IR and Raman spectra of isolated compounds no bands in the region 2030–2090 cm−1, characteristic for the presence of the azide moiety, were detected and these compounds are ESR silent in both solution and in the solid state. Likewise, in electronic UV-Vis spectra no absorptions corresponding to d–d transitions were found and thus we anticipate that the oxidation of titanium(III) takes place in the reaction. The decomposition of the cyanate complex [TiCp2(NCO)] has been reported elsewhere and hence we did not study this compound.13 Nitrosodicyanomethanide (ndcm) salt reacts with 1a in aqueous solution immediately giving a diamagnetic orange compound containing Ti–O–Ti bonds due to oxidizing behaviour of the ndcm ligand. The identity of the isolated product has been proved by the comparison of its IR, Raman, UV-Vis and 1H-NMR spectra with an authentic sample of [TiCp2(ndcm)]2O.14
Vibrational spectroscopy affirmed the presence of the bent bis(cyclopentadienyl)titanium fragment in studied compounds (characteristic bands at 3100, 1130, 830 and 270 cm−1).15 The infrared spectroscopy has also been used as a sensitive probe of possible oxidation to titanium(IV) complexes of the type [TiCp2(Y)]2O as the presence of a very strong band at 720–740 cm−1 is a typical feature for titanocene compounds containing the Ti–O–Ti bond.14,16 In the case of ring-substituted derivatives a strong and sharp IR band at ∼1250 cm−1, corresponding to symmetric deformation of CH3 groups attached to a silicon atom, was found.17 Compounds 2–NCS and 2–NCSe show two strong bands in the region 2055–2011 cm−1 that is typical for the N-bonded terminal thiocyanate or selenocyanate ligand, respectively, as observed for corresponding titanocene(IV) complexes.13,18 Additionally, the energy of νCN vibration in these compounds is significantly lower than the wavenumber span typical for bridging the thiocyanate or selenocyanate ligand (2180–2150 cm−1).19 In the solution, IR spectra of 2–NCS and 2–NCSe contain only one band corresponding to νCN at ∼2050 cm−1. Therefore, two bands of νCN originate from vibrational interaction of ligands in the solid state. From vibrational spectra it could not be unambiguously deduced whether this interaction is due to weak association of molecules into pseudohalide-bridged oligomers or due to site-symmetry effects in the crystal lattice.
Tricyanomethanides 2a–tcm and 2b–tcm have identical IR and Raman spectra in the νCN stretch region. A distinct medium IR band at 2228 ± 2 cm−1 (Ra: strong band at 2230 ± 4 cm−1) could be assigned to symmetric cyanide stretch and two strong bands at 2185 ± 4 and 2167 ± 3 cm−1 (Ra: very strong and broad band at 2170 cm−1) to antisymmetric vibrational motion of CN groups. The narrow wavenumber span (∼61 cm−1) of νCN bands is similar to that found in a free tricyanomethanide anion (52 cm−1) or in the [TiCp2(tcm)]2O complex (65 cm−1) indicating that all CN bonds in 2–tcm have very similar force constants.14,20
Dicyanamide compounds 2a–dca and 2b–dca showed rather complex IR spectra with six medium-to-strong bands in the region 2300–2100 cm−1. Raman spectra were not obtained because of strong sample fluorescence. Observed IR bands could not be reliably assigned due to the presence of combination modes enhanced by Fermi resonance with cyanide stretching fundamentals.21 Hence, the analysis of this region is not useful for the determination of the bonding mode of the dca ligand in the studied complexes.
Cyanide complex 2a–CN gives a weak, broad band of CN stretching at 2091 cm−1 in both IR and Raman spectra. Since this compound forms trimeric and tetrameric species in the solid state,8,9 we expect that prepared samples of 2a–CN consist from the mixture of both oligomers. Hence, the assignment of observed broad bands to vibrational motions of the molecule is dubious. Very low solubility of 2a–CN also disabled the vibrational study in the solution. Compound 2b–CN shows one weak, broadened IR band centred at 2095 cm−1 accompanied with two medium Raman bands at 2090 and 2128 cm−1. Assuming the trimeric structure of studied 2b–CN compounds (see below), the symmetry of the [Ti(CN)]3 core is of D3h and three νCN stretching vibrations belong to A1′ + E′ species. Symmetric stretch A1′ is Raman only active (assigned to the Raman band at 2128 cm−1) and antisymmetric E′ stretch (the broad band at ∼2090 cm−1) is both IR and Raman active. Very low intensity of the νCN band in IR spectra of both 2a–CN and 2b–CN is somewhat anomalous, but it should be attributed to only small change in dipole moment during the vibrational motion of the cyanide bridge in cyclic oligomers.19 On the contrary, 1,3-disubstituted 2c–CN shows a single, narrow and much stronger absorption band at 2098 cm−1 in both IR and Raman spectra. This is indicative for the isolated, probably terminal, cyano group having no significant vibrational interaction with another one. With regard to high steric crowding due to the presence of Me3Si substituents, we do not suppose forming of 2c–CN oligomers. This presumption was further affirmed by ESR data obtained both from solution and glassy spectra.
Molecular structures
Within the framework of this study, we have prepared green single-crystals of 1b suitable for the X-ray diffraction analysis. Its structure is given in the Fig. 1 and confirms the dimeric arrangement in the solid state. Cyclopentadienyl rings in 1b adopt nearly staggered conformation with trimethylsilyl groups located at the opposite sides of the bent fragment. The Si1 and Si2 atoms are displaced slightly above the cyclopentadienyl ring, with an angle between the Cp plane and the carbon–silicon bond vector of 8.5(3)° and 8.1(2)°, respectively. The observed Ti⋯Ti distance is in the range typical for similar titanium(III) bent metallocene complexes (3.912–4.017 Å) and does not imply any direct metal–metal bond.22 | ||
| Fig. 1 ORTEP drawing of 1b. Thermal ellipsoids are drawn at 30% probability level; hydrogen atoms and the second position of disordered Si2 atom are omitted for clarity. Representative bond distances (Å) and angles (°): Ti1–Cg1 2.073(3), Ti1–Cg2 2.076(2), Ti1–Cl1 2.555(2), Ti1–Cl1a 2.562(1), Cg1–Ti1–Cg2 131.65(11), Cl1–Ti1–Cl1a 77.00(4), Ti1–Cl1–Ti1a 103.00(4), Ti1⋯Ti1a 4.0046(17). | ||
Tricyanomethanide complexes 2a–tcm and 2b–tcm form cyclic dimers in the solid state, see Fig. 2 and 3, respectively. In the crystal of 2a–tcm, two crystallographically independent but essentially identical molecules are present. The structures are centrosymmetric with a 12-membered ring composed of two titanium atoms and two tricyanomethanide ligands. The Ti atoms in both structures are pseudotetrahedrally coordinated with a large Ti⋯Ti separations of 7.64(6) Å. The central ring is almost planar with the deviation from the mean plane defined by all 12 ring atoms not exceeding 0.048(1) Å [molecule B: 0.134(2) Å] and 0.071(1) Å in 2a–tcm and 2b–tcm, respectively. Terminal cyano groups are slightly tilted from the macrocycle, but the angle between the central plane and vector of the respective C–N bond is lower than 12°.
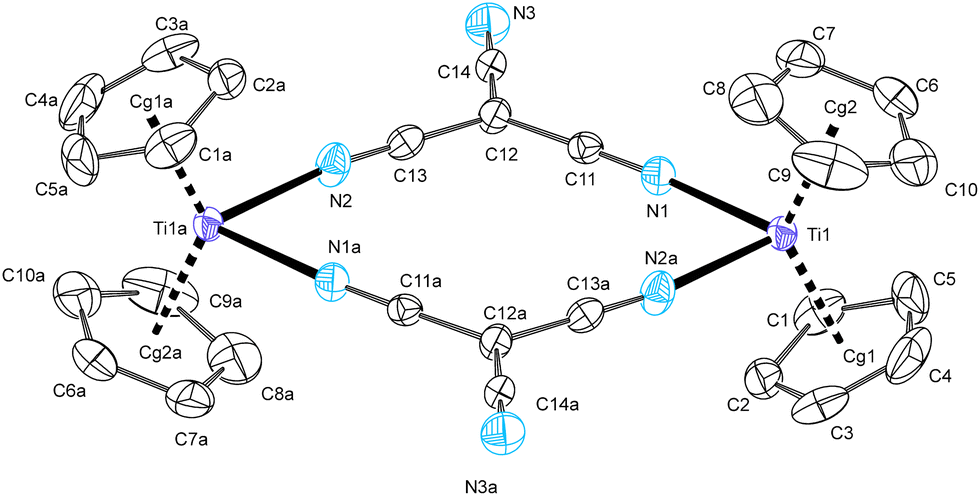 | ||
| Fig. 2 ORTEP drawing of dimeric 2a–tcm, molecule A. Thermal ellipsoids corresponds to 50% probability level; hydrogen atoms as well as the disordered THF molecule are omitted for the clarity. Representative bond distances (Å) and angles (°): Ti1–Cg1 2.046(2), Ti1–Cg2 2.042(2), Ti1–N1 2.165(3), Ti1–N2a 2.147(3), N1–C11 1.150(4), C11–C12 1.402(4), C12–C14 1.418(4), C14–N3 1.150(4), C12–C13 1.401(4), C13–N2 1.145(4), Cg1–Ti1–Cg2 133.85(8), N1–Ti1–N2a 78.91(10), Ti1⋯Ti1a 7.6927(11) [molecule B: 7.5787(12)]. | ||
 | ||
| Fig. 3 ORTEP drawing of the 2b–tcm dimer. Thermal ellipsoids are drawn at 50% probability level; hydrogen atoms and one molecule of co-crystallized toluene are omitted for the clarity. Representative bond distances (Å) and angles (°): Ti1–Cg1 2.0569(11), Ti1–Cg2 2.0586(11), Ti2–Cg3 2.0459(12), Ti2–Cg4 2.0408(12), Ti1–N1 2.152(2), Ti1–N5 2.156(2), N5–C39 1.153(3), C39–C38 1.403(3), C38–C40 1.419(3), C40–N6 1.146(3), C38–C37 1.398(3), C37–N4 1.154(3), Cg1–Ti1–Cg2 132.99(4), Cg3–Ti2–Cg4 134.17(5), N1–Ti1–N5 79.22(7), N2–Ti2–N4 78.33(7), Ti1⋯Ti2 7.6611(8). | ||
Tricyanomethanide ligands in both complexes have all C–N and C–C distances in very narrow ranges 1.145(6)–1.156(2) and 1.398(3)–1.413(6) Å. The Ti–N bonds in 2a–tcm and 2b–tcm are longer (by ca. 0.1 Å) than that observed in the corresponding titanocene(IV) complex [TiCp2(tcm)2].14 This could be due to different ionic radii for Ti3+ (0.81 Å) and Ti4+ (0.75 Å) (ref. 23) and also due to the presence of d-electron in SOMO which is slightly antibonding toward Ti–N interaction. The Cp rings in the crystal of 2a–tcm are near the eclipsed arrangement, whereas Cp rings of each titanocene moiety in 2b–tcm show a different pattern. The Cp ligands bonded to Ti2 are nearly to be eclipsed and SiMe3 substituents are located on the opposite sides of bent fragment [Si3–Cg3⋯Cg4–Si4 dihedral of 169.9(1)°]. The second titanocene fragment in 2b–tcm has a partially staggered conformation with one bulky SiMe3 group placed above the central macrocycle. The observed mutual arrangement of trimethylsilyl functions is not usual in monosubstituted titanocenes and it has been reported only for few titanium(IV) complexes but not yet for titanium(III) ones.24
The dicyanamide complex 2b–dca crystallizes as a centrosymmetric dimer as well. There are two independent molecules with minor structural differences in the unit cell of 2b–dca, one of them being depicted in Fig. 4. In this structure a 12-membered macrocycle consisting of two titanium atoms and two dca ligands is significantly puckered, with N3 deviating by 0.109(2) Å from the mean plane to the largest extent [0.161(2) Å in molecule B]. Titanium atoms are coordinated in a distorted tetrahedral environment with Ti–N distances of 2.166(2) Å [molecule B: 2.156(3) Å]. Analysing internal geometric parameters in dca ligands, it is evident that all cyanide groups, much like as in the studied tcm derivatives, are equivalent within the experimental error. Uniformity of internal bonds and high symmetry of tcm and dca moieties indicate the presence of a fully delocalized π-electron system in these bridging ligands.
 | ||
| Fig. 4 ORTEP drawing of dimeric 2b–dca, molecule A. Thermal ellipsoids are drawn at 50% probability level; hydrogen atoms are omitted for the clarity. Representative bond distances (Å) and angles (°): Ti1–Cg1 2.0558(11), Ti1–Cg2 2.0517(15), Ti1–N1 2.166(2), Ti1–N3a 2.166(2), N1–C17 1.145(4), C17–N2 1.302(4), N2–C18 1.297(4), C18–N3 1.149(4), N1–C17–N2 174.0(3), C17–N2–C18 120.7(2), N2–C18–N3 174.7(3), Cg1–Ti1–Cg2 132.90(6), N1–Ti1–N3a 78.57(9), Ti1⋯Ti1a 7.6897(8) [molecule B: 7.6362(9)]. | ||
The 2b–CN behaves as a cyclic trimer in the solid state, bearing three titanium atoms linked with cyanide bridges, see Fig. 5. Triangular structure has been reported recently for the unsubstituted complex 2a–CN prepared from [TiCp2(η2-Me3SiC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSiMe3)] and cyanuric chloride,8 whereas in an earlier report it has been described as a tetramer with titanium atoms occupying vertices of a heavily distorted tetrahedron.9 The 9-membered ring in 2b–CN is planar with mean deviation from an ideal plane of 0.013 Å. Similarly as in the above-mentioned cyclic structures of 2a–CN, cyanide atoms are disordered over two sites that results in indistinguishableness of each carbon and nitrogen atom in the bridge.8,9 Titanium atoms are pseudotetrahedrally coordinated; Cp rings are partially staggered and positioning of Me3Si groups is constrained by their mutual steric hindrance. The mean observed Ti⋯Ti separation is 5.39 Å.
CSiMe3)] and cyanuric chloride,8 whereas in an earlier report it has been described as a tetramer with titanium atoms occupying vertices of a heavily distorted tetrahedron.9 The 9-membered ring in 2b–CN is planar with mean deviation from an ideal plane of 0.013 Å. Similarly as in the above-mentioned cyclic structures of 2a–CN, cyanide atoms are disordered over two sites that results in indistinguishableness of each carbon and nitrogen atom in the bridge.8,9 Titanium atoms are pseudotetrahedrally coordinated; Cp rings are partially staggered and positioning of Me3Si groups is constrained by their mutual steric hindrance. The mean observed Ti⋯Ti separation is 5.39 Å.
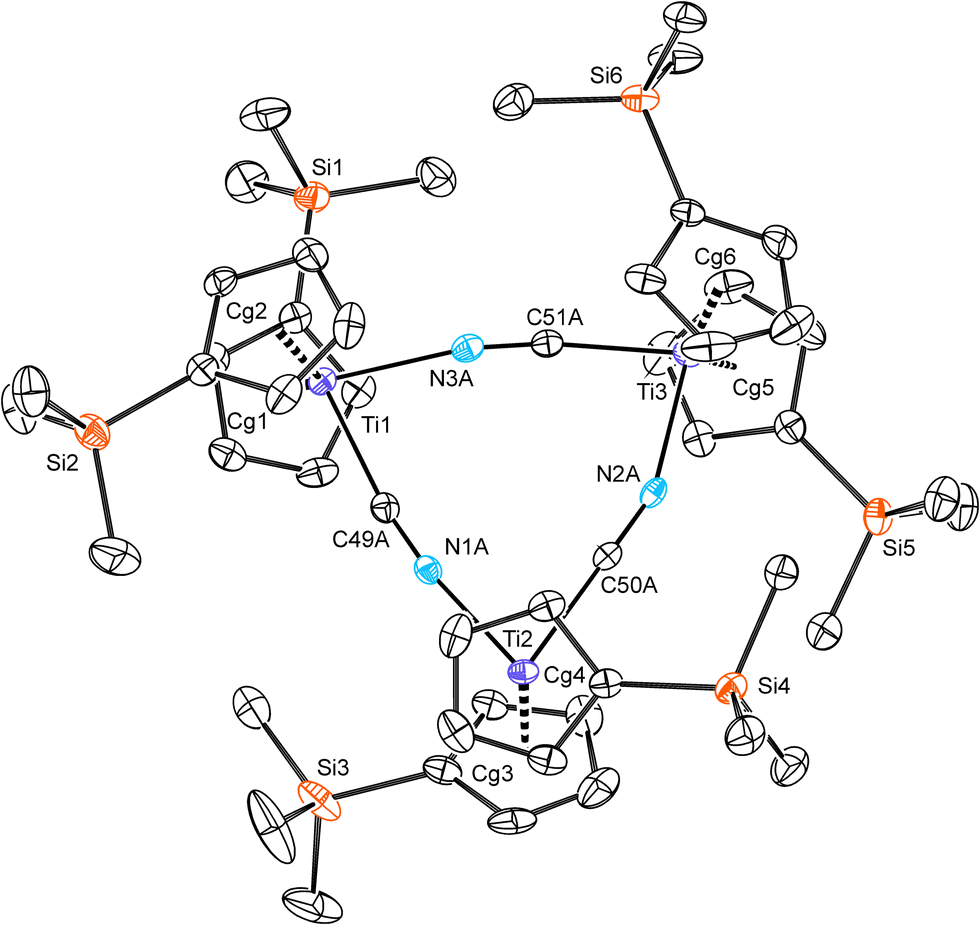 | ||
| Fig. 5 ORTEP drawing of the trinuclear 2b–CN. Thermal ellipsoids are drawn at 50% probability level; only one set of positions of disordered cyano groups is shown. For the clarity, only silicon and core atoms are numbered, hydrogen atoms are omitted. Representative bond distances (Å) and angles (°): Ti1–Cg1 2.0612(11), Ti1–Cg2 2.0572(10), Ti1–N3 2.151(3), N3–C51 1.157(3), Ti3–C51 2.139(2), Cg1–Ti1–Cg2 139.35(6), N3–Ti1–C49 80.73(8), Ti1⋯Ti2 5.3770(8), Ti2⋯Ti3 5.4262(8), Ti3⋯Ti1 5.3787(8). | ||
The molecules of 2b–CN show interesting crystal packing in the solid state. Triangular molecules are stacked in columns with each macrocycle twisted by 60° toward the next one. This arrangement gives rise to tubes with a hexagram-shaped projection along the c-axis (Fig. 6). It should be also noted that identical crystals of 2b–CN were repeatedly isolated from the solutions of the respective selenocyanate compound during the attempts to prepare single-crystals of 2b–SeCN. This observation confirms the instability of studied selenocyanate titanocene(III) complexes.
 | ||
| Fig. 6 Crystal packing diagram of 2b–CN viewed along the c-axis. | ||
Fig. 7 shows the perspective view of dinuclear μ-oxo compound 3a of which single-crystals were grown from chilled THF extracts of freshly precipitated 2a–ttt. Its structure proves the decomposition of the thiatriazole ligand according to Scheme 2. This molecule consists of two pseudotetrahedrally coordinated titanium(IV) units connected with a slightly bent oxo-bridge. The two [TiCp2(NCS)]+ fragments are rotated against each other around their Ti–O bond vectors by the dihedral angle N1–Ti⋯Ti2–N2 of 80.22(10)°. The near to linearity of Ti–O–Ti and the shortening of Ti–O bonds indicate additional π-bonding of the central oxygen atom to two titanium atoms, the phenomenon that is well known in a series of related pseudohalides of the type [TiCp2(Y)]2O (Y = tcm, dca, N3−, CN−, NCSe−).4,14,16,25
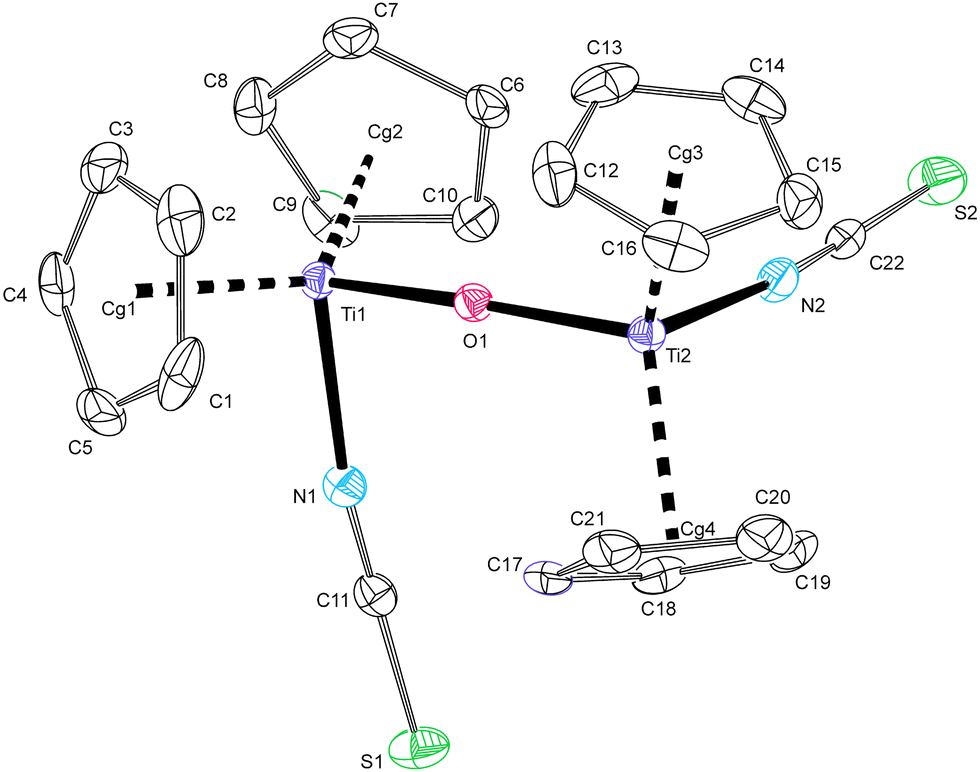 | ||
| Fig. 7 Molecular structure of 3a at 30% probability level, hydrogen atoms and two molecules of co-crystallized THF are omitted. Bond distances (Å) and angles (°): Ti1–Cg1 2.0739(15), Ti1–Cg2 2.0769(15), Ti2–Cg3 2.0785(16), Ti2–Cg4 2.0746(15), Ti1–N1 2.069(3), Ti1–O1 1.833(2), N1–C11 1.158(4), C11–S1 1.626(3), Ti2–N2 2.077(3), Ti2–O1 1.833(2), N2–C22 1.155(4), C22–S2 1.622(3), Cg1–Ti1–Cg2 130.20(7), Cg3–Ti2–Cg4 131.97(7), Ti1–O1–Ti2 170.06(12), N1–Ti1–O1 93.61(10), N2–Ti2–O1 94.67(10), Ti1⋯Ti2 3.6523(8). | ||
Fluid solution behavior
ESR spectra of starting chloride 1a measured in various solvents were identical to those described elsewhere.26,27 In THF or 2-methyltetrahydrofuran (meTHF) a narrow signal of 1a solvate was solely observed whereas in toluene, benzene or hexane, a broad band of dimeric species dominates.28Freshly prepared 1b dissolved in nonpolar solvent (toluene, hexane, cyclohexane) give two ESR bands with giso of 1.977 and 1.961, respectively, the latter being broader (ΔH ∼ 12 G) and very intense. Solutions of 1b in coordinating solvents such as THF, meTHF or diglyme show a narrow signal at 1.977 accompanied with weak and broad high-field resonance. Upon cooling the solution to 220 K, the line at 1.961 vanishes and a narrow single line with well-resolved hyperfine splitting to Ti nuclei is observable. More bulky complex 1c dissolved in meTHF or toluene gives strong broadened line (giso = 1.961, ΔH ∼ 8 G) accompanied with weak, narrow resonance at a lower field. In both complexes, the high-field line has been attributed to non-solvated species, whereas the low-field one is due to solvent adduct. The solvate signal of 1b and 1c in toluene solutions originates from the synthesis carried out in THF and could be only hardly removed by repeated vacuum sublimation. The lowering of temperature shifts the equilibrium toward solvate species. Similar ESR behaviour was described for the series of halide complexes [Ti{η5-C5H(5−n)Men}2X] (n = 0–5), where X is Cl, Br or I. In that paper, the reluctance of highly substituted titanocene(III) halides toward solvation has been attributed to decreased Lewis acidity due to electron-releasing properties of methyl groups bonded in the Cp ring.28 As we know, the electron-releasing impact of the trimethylsilyl function is much lower (approximately half of that for the methyl substituent in titanocene(IV) complexes),29 and thus the bulkiness of the Me3Si group in both 1b and 1c is a more important factor for the solvation process than simple electronic effects. Steric hindrance in the 1c substantially protects the titanium atom and only a weak signal of solvate has been observed even at low temperatures in coordinating solvents.
Thiocyanate complexes 2–NCS give a resonance line flanked with satellites due to coupling with 47Ti and 49Ti nuclei. Complexes 2–NCSe, 2–dca, 2–tcm and 2a–CN have significantly broadened signals from which the interaction with titanium isotopes could not be determined, see Table 1. Line broadening is probably due to equilibrium between monomeric and oligomeric species in the solution. In meTHF trimethylsilyl-substituted derivatives 2b–CN and 2c–CN show two ESR bands with a pattern very similar to parent chlorides 1b and 1c, respectively. The broad line at giso = 1.960 was attributed to the non-solvated complex, whereas the narrow line at 1.978 with noticeable hyperfine splitting to Ti nuclei is due to solvated species. Similarly as in 1b and 1c, the intensity of the lower-field band increases with decreasing temperature.
| Neutral species | Anionic species | |||||
|---|---|---|---|---|---|---|
| Compd | g iso | a iso(Ti) | ΔH | Compd | g iso | a iso(Ti)/aiso(2N) |
| a Coupling constants and signal half-heights (ΔH) are given in Gauss. b Not determined due to broadened or weak resonance line. c In parenthesis is coupling constant aiso(1N). d ESR spectra of the parent neutral complex did not change after the addition of the respective salt. e Two bands were observed, for details see the text. f Value could not be accurately determined due to the broadened line. g Not observed. | ||||||
| 1a | 1.981 | 12.3 | 4.5 | —d | ||
| 1b | 1.977 | 12.5 | 4.8 | —d | ||
| 1.960 | —b | 9.3 | ||||
| 1c | 1.977 | 9.4 | 3.3 | —d | ||
| 1.961 | —b | 8.4 | ||||
| 2a–NCS | 1.979 | 12.8 | 3.5 | 4a–NCS | 1.980 | 12.2/2.4 |
| 2b–NCS | 1.980 | 12.7 | 4.1 | 4b–NCS | 1.980 | 12.2/2.4 |
| 2a–NCSe | 1.979 | —b | 9.8 | 4a–NCSe | 1.978 | 12.3/2.5 |
| 2b–NCSe | 1.980 | —b | 10.2 | 4b–NCSe | 1.979 | 12.1/2.5 |
| 2a–dca | 1.981 | —b | 7.3 | 4a–dca | 1.980 | 12.4/2.2 |
| 2b–dca | 1.981 | —b | 7.1 | 4b–dca | 1.981 | 12.3/2.3 |
| 2a–tcm | 1.983 | —b | 8.8 | 4a–tcm | 1.982 | 12.0/2.7 |
| 2b–tcm | 1.983 | —b | 9.3 | 4b–tcm | 1.983 | 11.8/2.8 |
| 2a–ttt | 1.981 | 10.5 (<0.5)c | 2.3 | —d | ||
| 2b–ttt | 1.981 | 10.4 (1.7)c | 5.4 | —d | ||
| 2c–ttt | 1.980 | 10.0 (1.9)c | 5.5 | —d | ||
| 2a–CN | 1.986(2)f | —b | 29 | 4a–CN | 1.988 | 10.0/—g |
| 2b–CN | 1.977 | 12.1 | 4.4 | 4b–CN | 1.988 | 9.8/—g |
| 1.960 | —b | 9.8 | ||||
| 2c–CN | 1.978 | 8.9 | 1.9 | 4c–CN | 1.988 | 9.6/—g |
| 1.961 | —b | 7.2 | ||||
Interesting solution behaviour showed complexes bearing a ttt ligand. When freshly precipitated 2a–ttt was extracted into THF at 0 °C, dark greenish-brown solution was obtained. The ESR spectrum of this solution revealed the presence of two species, the former being the expected complex 2a–ttt, and latter is 2a–NCS. At the start of the measurement the band of 2a–ttt is dominating, but its intensity quickly decreases and after 20 minutes at ambient temperature single resonance of 2a–NCS is observed. In the course of the time the band of 2a–NCS gradually weakens due to oxidation by released elemental sulphur (see Scheme 2). THF solutions of 2a–ttt are unstable at 0 °C and change to a deep red, ESR-silent mixture within 100 minutes. When the synthesis was carried out in meTHF at −20 °C, only one signal corresponding to 2a–ttt was observed at this temperature.
Ring-substituted complexes 2b–ttt and 2c–ttt are thermally more stable, but heating above 40 °C causes rapid decomposition to thiocyanate species as well. The central line of 2b–ttt and 2c–ttt, as well as 47,49Ti satellites, show significant splitting to triplet which was attributed to the interaction of SOMO electron with 14N having nuclear spin I = 1, see Fig. 8. Thus, we suppose that in these complexes the ttt anion serves as a bidentate S,N-chelating ligand.
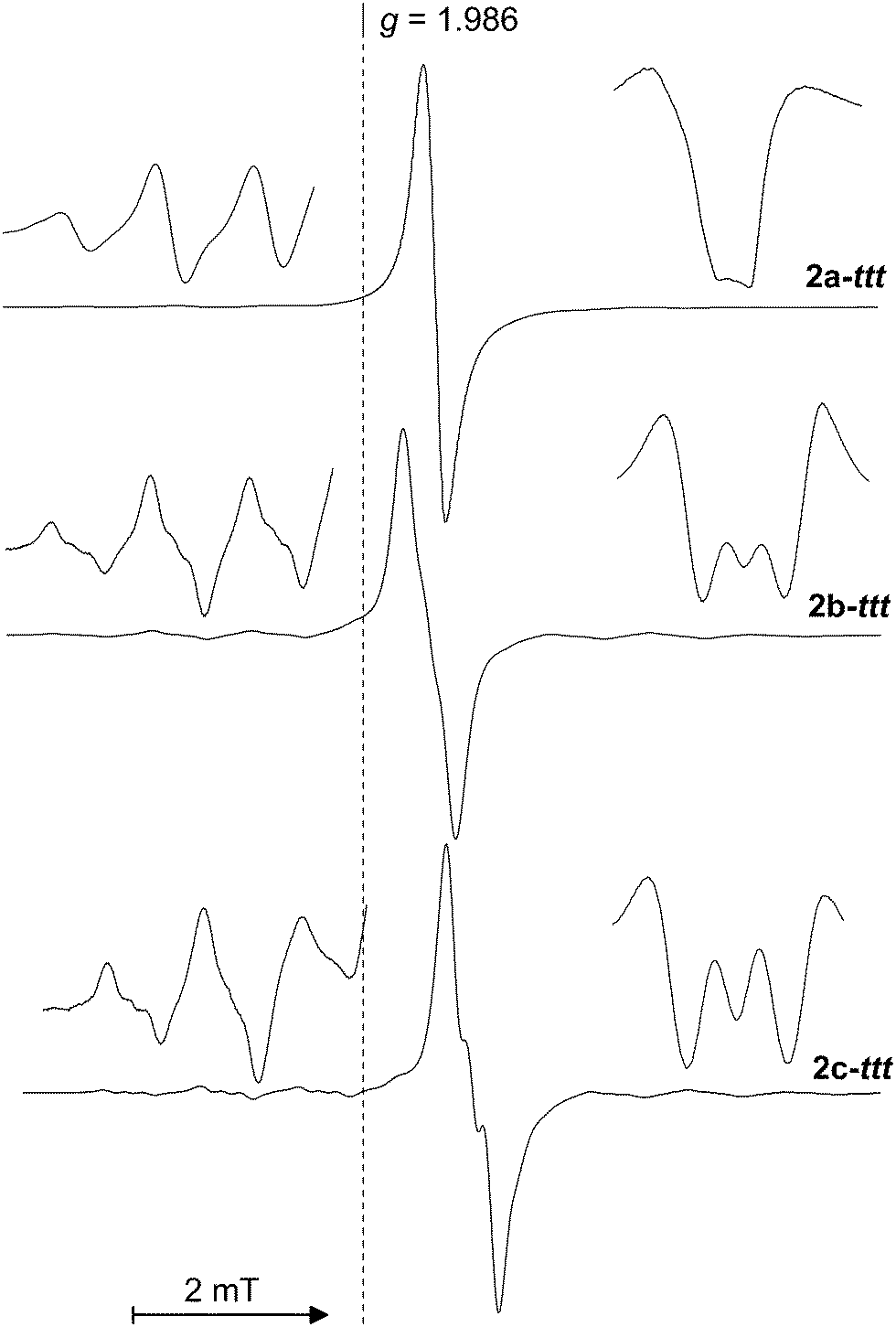 | ||
| Fig. 8 ESR spectra of 2–ttt species prepared in meTHF at 253 K. Left insets show the detail of low-field titanium satellites; right insets show the pattern of the second derivative of the central resonance line. | ||
We observed that aTi increases upon going from bulky 2c–ttt to the unsubstituted complex 2a–ttt, whereas coupling to the nitrogen nucleus decreases in this series. Low coupling constant, together with signal overlapping disabled accurate determination of the aN value from ESR spectra of 2a–ttt, but from the analysis of the second derivative it is evident that aN in 2a–ttt has to be smaller than 0.5 G. We believe that the lowering of aN with decreasing bulkiness of the Cp ligand in the studied thiatriazolate compounds is caused by competitive solvation of the titanium atom which weakens intramolecular coordination of the thiatriazolthiolate ligand via a nitrogen atom.
During our study we found that THF solutions of 2–NCS, 2–NCSe, 2–dca and 2–tcm, readily dissolve additional pseudohalide salt giving homogeneous mixtures. ESR spectra of these solutions are entirely different from those of parent compounds and significant splitting to quintet is observable (e.g.Fig. 9). This ESR pattern is due to interaction of the SOMO electron with two equivalent 14N nuclei in the anionic complex of the formula [TiCp2Y2]− (4–Y), see Scheme 3. Observed coupling constants aTi and aN inhere in very narrow ranges of 12.2 ± 0.3 G and 2.5 ± 0.3 G, respectively. A similar ESR spectrum has been previously attributed to anionic species [TiCp2(NCS)2]− generated in situ by electrochemical reduction of [TiCp2(NCS)2] solutions.30
 | ||
| Fig. 9 ESR spectra of 4b–Y in meTHF. Left insets show the detail of low-field titanium satellites; right insets show the pattern of the second derivative of the central resonance line. | ||
 | ||
| Scheme 3 Observed equilibrium between neutral and anionic pseudohalide species in the solution. | ||
When the solid residue, taken from vacuum evaporation of THF solutions of 4–Y, was extracted with nonpolar solvent (e.g. toluene, benzene, hexane), the extract showed only a broadened signal corresponding to species 2–Y. The equilibrium between 2–Y and 4–Y could be shifted toward anionic species not only by increasing the solvent polarity, but also by the addition of dibenzo-18-crown-6 ether (18-C-6). Using 18-C-6 allowed us to obtain benzene or toluene solutions containing anionic species 4–Y having ESR spectra identical to those measured in THF or meTHF.
The THF solutions of 2a–CN, 2b–CN and 2c–CN dissolve one additional equivalent of KCN as well. Resulting mixtures give a narrow and very intense singlet line (giso = 1.988; ΔH = 1.3, 2.2 and 1.4 G, respectively) surrounded by 47,49Ti satellites. The absence of any significant interaction with 14N nuclei is not surprising as the cyanide anion coordinates to titanium predominantly via the carbon atom. We also tried to use 13C-enriched cyanide for the synthesis, but no interaction with the 13C nucleus (I = ½) was observed, probably due to a low coupling constant.
The ESR spectra of 2–ttt solutions did not change after the addition of sodium thiatriazolthiolate and solely the interaction with one 14N nucleus is evident suggesting that S,N-chelated species remain unchanged. The presence of 18-C-6 in these mixtures does not have any effect on the spectra pattern.
Structure in frozen solutions
Solid state ESR spectra of some bis(cyclopentadienyl)–titanium(III) complexes have been studied previously and the dimeric nature of halides, including 1a is well described.26–28 The frozen-solution ESR spectra of 1b in meTHF exhibits three g features corresponding to rhombic species of solvate (Table 2, Fig. 10). The toluene glass ESR spectrum of 1b consists of a strong central signal surrounded with broad bands characteristic for triplet state species of an approximately axial symmetry. From the outermost features of this spectrum the zero-field splitting (ZFS) parameter D was determined. The presence of the triplet state has also been confirmed by the observation of formally forbidden half-field transition at g = 3.97. From ZFS parameters the distance between the unpaired electrons in the species could be estimated following the equation:where Dd is dipolar contribution of D and Rcal is the distance between interacting electrons (in Å).31 For most of the known titanium(III) dimeric species of cylindrical symmetry, the dipolar contribution is dominating and thus the parameter D derived from frozen glass ESR spectra give reasonable Ti⋯Ti distances.32 The ESR parameters of triplet species g∥ = 1.994, g⊥ = 1.975, |D| = 0.0387 cm−1 and E ≈ 0 cm−1, according to the equation above, give Rcal = 4.06 Å which is in good agreement with the crystallographic Ti⋯Ti distance of 4.005(2) Å. We must note some uncertainty in the determination of ZFS parameters for 1b due to very broad features of triplet state bands. Spectra of sterically more crowded 1c in both meTHF and toluene glass are almost identical showing three g components of rhombic tensor. In frozen meTHF, an additional signal of minor species is observable (Fig. 10). The high-field g-tensor value of 1.902 is due to the non-solvated complex, whereas minor species at g = 1.951 corresponds to solvent adduct. These observations are in accordance with solution ESR spectra and with the fact that 1c is monomeric in the solid state.33
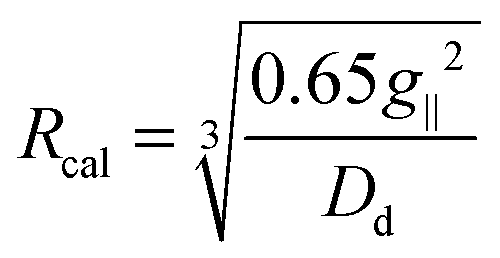
| Compd | g 1, g2, g3 | g avg | |D| [cm−1] | |E| [cm−1] | g (ΔMS = 2) | R cal [Å] |
|---|---|---|---|---|---|---|
| a R cal is the Ti⋯Ti distance calculated from a purely dipole–dipole interaction with Dd ≈ D. b MeTHF. c Toluene. d Not observed. e Only one, unresolved band was observed, for details see the text. f Value obtained from the powder spectrum. g The g values (gz, gxy and gxyz, respectively) of the quartet state. | ||||||
| 1b | 1.999, 1.978, 1.953b | 1.977b | 0.0386c | ∼0c | 3.98 | 4.06 |
| 1.994, 1.975, 1.975c | 1.981c | |||||
| 1c | 1.997, 1.984, 1.951b | 1.961b | — | — | —d | |
| 1.997, 1.984, 1.902c | 1.977c | |||||
| 2a–NCS | 2.000, 1.983, 1.958b | 1.980 | — | — | —d | |
| 2b–NCS | 1.998, 1.982. 1.960b | 1.980 | — | — | —d | |
| 2a–NCSe | 2.000, 1.986. 1.959b | 1.982 | — | — | —d | |
| 2b–NCSe | 2.000, 1.986, 1.958b | 1.981 | — | — | —d | |
| 2a–ttt | 1.997, 1.984, 1.962b | 1.981 | — | — | —d | |
| 2b–ttt | 1.999, 1.985, 1.960b | 1.981 | — | — | —d | |
| 2c–ttt | 1.999, 1.986, 1.956b | 1.980 | — | — | —d | |
| 2a–dca | 1.983, 1.981, 1.978c | 1.981 | 0.0077 | 0.0018 | 3.96 | 6.92 |
| 2b–dca | 1.986, 1.982, 1.980c | 1.983 | 0.0076 | 0.0017 | 3.97 | 6.95 |
| 2a–tcm | 1.985, 1.981, 1.979c | 1.982 | 0.0084 | 0.0015 | 3.97 | 6.73 |
| 2b–tcm | 1.988, 1.986, 1.979c | 1.984 | 0.0082 | 0.0017 | 3.97 | 6.79 |
| 2a–CN | 1.986e | — | — | 3.91f | ||
| 2b–CN | 1.999, 1.977, 1.954b | 1.977b | 0.0189c | ∼0c | 3.92f | 5.16 |
| 1.998, 1.978, 1.972c,g | 1.983c | |||||
| 2c–CN | 1.999, 1.981, 1.953b | 1.978b | — | — | —d | |
| 1.999, 1.981, 1.902c | 1.961c | |||||
 | ||
| Fig. 10 ESR spectra of chloride complexes 1b (top: in toluene, centre: in meTHF) and 1c (bottom: in meTHF) at 130 K. The band of ΔMS = 2 transition observed in spectra of 1b is situated in the arbitrary magnetic field. | ||
Compounds 2–NCS and 2–NCSe in meTHF or toluene glass showed simple spectra consisting of three bands due to rhombic doublet species. The solid powders of 2–NCS and 2–NCSe give only an asymmetric signal of ΔMS = 1 transitions corresponding to the rhombic g-tensor as well. Although an early investigation of 2a–NCS based on magnetic susceptibility measurements7 predicted its oligomeric structure, we observed no significant interaction between unpaired electrons for thiocyanate and selenocyanate complexes during our ESR experiments. Expectedly, the spectra of 2–ttt in meTHF glass showed simple, three-component rhombic g-tensor. Notwithstanding that the S,N-chelate coordination of thiatriazolthiolate is likely in these compounds, no hyperfine splitting due to 15N coupling was observed in frozen glass ESR spectra.
Complexes 2–dca and 2–tcm exhibit ESR spectra of a rhombic triplet state, characteristic for binuclear Ti(III) compounds. The six-line pattern of ΔMS = 1 transition is accompanied by a low-intense band of formally forbidden ΔMS = 2 transition at midfield, see Fig. 11. In the spectra of 2a–dca, 2b–dca and 2a–tcm, additional lines of the rhombic doublet state due to contamination with mononuclear Ti(III) impurity were also detected. When coordinating solvent is used, the central signal of monomeric species, probably solvate, dominates and resolving of triplet features is rather difficult. From ESR spectra measured in toluene glass, ZFS parameters were determined (Table 2) and the D value was used for the calculation of Ti⋯Ti distances.26 Calculated distances are markedly shorter (ca. by 0.8 Å) than observed crystallographic Ti⋯Ti separations. This poor agreement with experimental values could be due to neglecting of pseudodipolar contribution to D parameter or due to enhanced interaction between unpaired electrons through the delocalized π-bond system of bridging pseudohalide ligands.
 | ||
| Fig. 11 The ESR spectra of dimeric 2–dca and 2–tcm in toluene at 150 K. Left insets show the pattern of ΔMS = 2 transition (situated at arbitrary magnetic field). The signals of monomeric impurities are denoted with asterisks. | ||
The behaviour of cyanide complexes
Unsubstituted derivative 2a–CN is poorly soluble and its solutions in toluene, meTHF or CH2Cl2 give one broad ESR band at ambient temperature. Upon cooling, the band broadens and at 130 K only one, slightly dissymmetric signal at g = 1.986 (ΔH ∼ 45 G) is observable. When solid 2a–CN is boiled in THF, the resulting extract gives a fluid spectrum bearing an additional signal of solvated species superimposed on the principal broad band. The appearance of glassy ESR spectra is invariable in all these solvents. In the powder spectrum, a broad intense band at g ∼ 1.99 is accompanied by weak resonance in the half-field region. This ESR behaviour indicates that 2a–CN forms oligomers not only in the solid state, but also in the solution. As glassy spectra in various solvents shows only unresolved broad bands, and we cannot accurately determine the nuclearity of observed paramagnetic species. These oligomers are very stable, resulting in poor solubility of 2a–CN even in coordinating solvents at elevated temperatures.Compound 2b–CN has a completely different ESR pattern. When dissolved in coordinating solvent (THF, meTHF) the main signal at 1.977 in the fluid ESR spectrum corresponds to the solvate moiety and in frozen glass it gives a simple rhombic pattern with gavg = 1.977. In toluene solution, the most intense, broad band is due to the non-solvated complex. The X-ray diffraction analysis of 2b–CN showed that this compound is trimeric in the solid state and we expect this species also in the frozen glass matrix during ESR experiments. Trinuclear titanocene(III) complex has three ESR active states: a quartet and two doublet states. If we assume axial symmetry of the molecule and its quartet ground state, five ΔMS = 1 transitions are expected in the respective resonance fields:31

![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) ) with ZFS parameters |D| = 0.0189 cm−1 and |Z| = 0 cm−1. Parameter D has been also used for the calculation of Ti⋯Ti distance. Similarly as in the case of dicyanamide and tricyanomethanide complexes, the calculated value Rcal is shorter (by ca. 0.24 Å) than metal–metal separations observed in the crystal of 2b–CN pointing to the additional interaction between electrons via the π-system of the cyanide bridge. The trimeric molecule of 2b–CN is significantly less stable than oligomers of 2a–CN and it could be easily dissociated by the addition of a coordinating solvent.
) with ZFS parameters |D| = 0.0189 cm−1 and |Z| = 0 cm−1. Parameter D has been also used for the calculation of Ti⋯Ti distance. Similarly as in the case of dicyanamide and tricyanomethanide complexes, the calculated value Rcal is shorter (by ca. 0.24 Å) than metal–metal separations observed in the crystal of 2b–CN pointing to the additional interaction between electrons via the π-system of the cyanide bridge. The trimeric molecule of 2b–CN is significantly less stable than oligomers of 2a–CN and it could be easily dissociated by the addition of a coordinating solvent.
 | ||
| Fig. 12 ESR spectra of cyanide complexes 2a–CN (top), 2b–CN (middle) and 2c–CN (bottom) measured in toluene at 130 K, the bands of half-field transitions are situated in the arbitrary magnetic field. Dashed curve corresponds to computer simulation of the 2b–CN quartet state; bands are designated in accordance with the text. | ||
Ring disubstituted compound 2c–CN does not form oligomers due to high steric congestion caused by bulky Me3Si groups. In the solution, we observed only minor signals of solvated species and a broad, intense band of non-solvated one. The ratio of these species in the solution could be varied both by the addition of coordinating solvent (THF, meTHF) or by the temperature change. From glass ESR spectra, parameters corresponding to rhombic doublet states of both solvate and non-solvate moieties were obtained (Table 2).
Conclusions
In this paper we have described the preparation and characterization of titanocene(III) pseudohalide compounds of the type [Ti(′Cp)2Y]. Synthesised compounds give characteristic ESR spectra from which the coordination mode of the respective pseudohalide ligand could be deduced. In polar organic solvents (e.g. THF), anionic species of the formula [Ti(′Cp)2Y2]− should be prepared by the reaction of titanocene(III) chloride with an excess pseudohalide salt. Studied cyanides, tricyanomethanides and dicyanamides tend to aggregate giving oligomeric structures with bridging pseudohalide ligands. In the case of thiocyanate, selenocyanate and thiatriazolthiolate complexes, no significant interaction between unpaired electrons was observed and thus we suppose that these compounds are monomeric in the solution and in the solid state. The thiatriazolthiolate ligand serves as an S,N-chelator for the titanium(III) centre yielding a thermally unstable four-membered chelate ring. The solid state structure of six compounds has been studied by spectroscopic methods and by X-ray diffraction analysis. We have found that titanocene(III) tricyanomethanides and dicyanamides are dinuclear compounds having ESR spectra typical for triplet species of rhombic symmetry. The monosubstituted cyanide complex 2b–CN crystallizes as a highly symmetric trimeric molecule with a quartet ground state. Additionally, this compound showed an interesting crystal packing motif consisting of staggered triangular molecules stacked in columns along c-axis. Regarding this structure and quartet ground state of trimeric molecules, significant magnetic anisotropy of crystalline 2b–CN could be expected. Such materials with these properties are intensively studied in the present time in the field of material science e.g. for the construction of spintronic devices. Herein we also demonstrated the utilization of both solution and solid state ESR spectroscopy for the investigation and looking for these new and promising compounds.Experimental section
Materials and methods
All preparative reactions and manipulations were carried out under an inert atmosphere of argon on a Schlenk line or in vacuum-sealed ampoules (single-crystal growth). All solvents used were dried and deoxygenated with appropriate drying agents, distilled under argon and degassed before use. Starting chloride complexes 1a, 1b and 1c have been synthesized by published methods and purified by vacuum sublimation.33,34 Pseudohalide alkali salts and dibenzo-18-crown-6 were obtained from Aldrich and used as received. Unsubstituted complexes 2a–NCSe, 2a–tcm and 2a–dca were prepared from 1a and appropriate pseudohalide salt by the method described previously for 2a–CN, 2a–NCS.7 NaCS2N3·4H2O (Nattt) was prepared according to the literature,12 crystallized from acetone and dried under vacuum at −20 °C.Caution! All studied thiatriazolate compounds, including starting sodium salt, are extremely unstable in the solid state and their heating, friction or careless manipulation causes violent explosion. The preparation of dry, solid samples in the amounts larger than 100 mg is strongly dissuaded.
Spectroscopic measurements
IR spectra were recorded in the 4000–350 cm−1 region on a Nicolet 6700 FTIR spectrometer using ATR technique (single-bounce Si crystal, argon atmosphere, resolution 2 cm−1). Raman spectra were measured on the Nicolet iS-50 FTIR spectrometer with an integrated Raman module (excitation laser 1064 nm, 0.1 mW, 500 scans, resolution 2 cm−1) in vacuum-sealed capillaries. Electronic absorption spectra were run on a Black-Comet C-SR-100 concave grating spectrometer equipped with a dip probe (region 200–1080 nm, variable optical pathway 1.0–10.0 mm). Solution ESR spectra were measured in 50 μl quartz micropipettes (1 mm inner diameter) on a Miniscope MS300 X-band spectrometer at 230 K. Hyperfine and super-hyperfine splitting constants aTi and aN, respectively, were determined by analysis of experimental spectra and verified by computer simulations performed in PEST Winsim software downloaded from http://www.niehs.nih.gov/. For all fluid solution spectra identical settings for simulations (a line width of 1.6–1.8 G, pure Gaussian line shape, natural abundance of 47,49Ti isotopes and no corrections for the tumbling effect) were used. The differences between aiso values obtained from the best fit between simulated and experimental were negligible proving their correct assignment. ESR spectra in frozen glasses were recorded at 150 K in quartz micropipettes (1 mm inner diameter) or in 4 mm quartz tubes. From the solid-state spectra, the zero-field splitting parameters were determined and the correctness of spectra analysis was verified by computer simulation in WINEPR Simfonia software (Bruker Analytische Messtechnik GmbH). Triplet and quartet ESR spectra were simulated as the first derivatives with a lineshape ratio L/G of 0.5, an anisotropic linewidth of 8–9 G and spherical integration angles ϕ and θ of 200°; ZFS parameters and g-tensor values were taken from experimental spectra (as listed in Table 2). Although the ESR spectra are not essentially sensitive to contamination with diamagnetic species (originating from partial oxidation of titanocene(III) compound by the air), the magnetic susceptibility is significantly influenced by the presence of such diamagnetic contaminants. Therefore, we decided not to report dubious magnetic parameters in this article, as it is difficult for us to avoid oxidation of the studied Ti(III) compounds during the preparation of samples for the magnetic susceptibility measurements. Elemental analyses were carried out on a Flash 2000 CHNS Analyzer (ThermoFisher Scientific). Crystalline samples for elemental analyses were initially dried under vacuum (10−2–10−3 Pa) at 40 °C for 12 hours to remove residual solvents and subsequently vacuum-sealed in weighted capillaries. Slightly lower values of CHN analysis could be due to partial oxidation of titanium(III) compounds during the preparation of samples for the measurement.Crystal structure determination
The structure of 2a–tcm contains a THF molecule which is completely disordered. This THF molecule does not interact with any part of the molecule of interest and the disorder is treated by splitting of all atoms to two positions (occupancy 60/40). In the structure of 1b, the minor positional disorder of the Me3Si and Cp groups is observed, reflected in the biggest maxima of about 2.5 electrons close to the Si2 atom. This atom is split into two positions (occupancy 9/1). In the structure of 2b–CN, all the C and N atoms in all three disordered cyano groups are refined to two positions with equivalent occupancy.All treatment procedures were performed with SADI, DFIX, RIGU and EXYZ instructions from the SHELXL97 and 2014 software.35 The X-ray data for crystals of 1b, 2a–tcm, 2b–tcm, 2b–dca, 2b–CN and 3a were obtained at 150 K using an Oxford Cryostream low-temperature device on a Nonius KappaCCD diffractometer with Mo Kα radiation (λ = 0.71073 Å), a graphite monochromator, and the ϕ and χ scan mod; relevant crystallographic data are listed in Table 3. Data reductions were performed with DENZO-SMN.36 The absorption was corrected by integration methods.37 Structures were solved by direct methods (Sir92).38 and refined by full matrix least-square based on F2 (SHELXL-97).35 Hydrogen atoms were mostly localized on a difference Fourier map, however, to ensure uniformity of treatment of crystal, all hydrogen were recalculated into idealized positions (riding model) and assigned temperature factors Hiso(H) = 1.2Ueq (pivot atom) or of 1.5Ueq (methyl). H atoms in methyl, methylene moiety and hydrogen atoms in delocalized systems were placed with C–H distances of 0.96, 0.97 and 0.93 Å, respectively. Crystallographic data for structural analysis have been deposited with the Cambridge Crystallographic Data Centre, CCDC Nos. 1019534, 1019535, 1019536, 1019537, 1019538 and 1019539 for 2a–tcm, 3a, 2b–CN, 2b–dca, 2b–tcm and 1b respectively.
| Compound | 1b | 2a–tcm | 2b–tcm | 2b–dca | 2b–CN | 3a |
|---|---|---|---|---|---|---|
| a R int = ∑|Fo2 − Fo,mean2|/∑Fo2. b GOF = [∑(w(Fo2 − Fc2)2)/(Ndiffrs − Nparams)]1/2 for all data. c R(F) = ∑||Fo| − |Fc||/∑|Fo| for observed data; wR(F2) = [∑(w(Fo2 − Fc2)2)/(∑w(Fo2)2)]1/2 for all data. | ||||||
| Empirical formula | C32H52Cl2Si4Ti2 | C28H20N6Ti2, C4H8O | C40H52N6Si4Ti2, C7H8 | C36H52N6Si4Ti2 | C51H78N3Si6Ti3 | C22H20N2OS2Ti2, 2C4H8O |
| MW | 715.80 | 608.40 | 917.17 | 777.00 | 1045.40 | 632.53 |
| Crystal system | Triclinic | Monoclinic | Triclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P1 | P21/c | P1 | P1 | P1 | P21/c |
| a (Å) | 8.8280(4) | 13.8001(10) | 11.4750(6) | 7.5980(4) | 14.5932(12) | 9.8420(5) |
| b (Å) | 9.4270(8) | 14.1070(8) | 11.6289(10) | 11.5230(6) | 14.9250(11) | 19.9841(14) |
| c (Å) | 12.2471(14) | 14.996(1) | 18.8461(13) | 25.0561(17) | 15.5400(8) | 15.2830(13) |
| α (°) | 71.689(6) | 90 | 92.434(7) | 77.795(5) | 74.368(7) | 90 |
| β (°) | 78.624(6) | 93.400(5) | 99.597(5) | 89.851(6) | 83.551(8) | 99.947(7) |
| γ (°) | 89.462(5) | 90 | 96.700(5) | 84.089(5) | 61.471(6) | 90 |
| Z | 1 | 4 | 2 | 2 | 2 | 4 |
| V (Å3) | 947.08(14) | 2908.4(3) | 2457.8(3) | 2132.3(2) | 2863.2(4) | 2960.7(4) |
| D calc/g cm−3 | 1.255 | 1.389 | 1.239 | 1.210 | 1.213 | 1.419 |
| μ (mm−1) | 0.709 | 0.585 | 0.460 | 0.518 | 0.568 | 0.714 |
| Reflections used | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 438 438 |
25316 |
44176 |
46211 |
52499 |
24232 |
| Independent (Rint)a | 4284 (0.087) | 6626 (0.063) | 11058 (0.078) | 9732 (0.083) | 12979 (0.039) | 6567 (0.051) |
| Observed [I > 2σ(I)] | 3288 | 4675 | 8491 | 6690 | 11577 |
4893 |
| Parameters refined | 190 | 415 | 532 | 433 | 587 | 352 |
| Max/min Δρ/eÅ−3 | 1.13/−0.43 | 0.67/−0.37 | 0.42/−0.42 | 0.28/−0.34 | 0.45/−0.36 | 1.66/−0.58 |
| GOFb | 1.061 | 1.161 | 1.110 | 1.121 | 1.129 | 1.172 |
| R(F)/wR(F2)c | 0.0677/0.1924 | 0.0549/0.1177 | 0.045/0.106 | 0.055/0.115 | 0.0324/0.0815 | 0.050/0.104 |
Synthesis of unsubstituted pseudohalide complexes
Synthesis of trimethylsilyl-substituted complexes
References
- (a) P. Jensen, D. J. Price, S. R. Batten, B. Moubaraki and K. S. Murray, Chem. – Eur. J., 2000, 6, 3186–3195 CrossRef CAS; (b) D. R. Turner, A. S. R. Chesman, K. S. Murray, G. B. Deacon and S. R. Batten, Chem. Commun., 2011, 47, 10189–10210 RSC; (c) S. Tanase and J. Reedijk, Coord. Chem. Rev., 2006, 250, 2501–2510 CrossRef CAS PubMed.
- D.-F. Weng, Z.-M. Wang and S. Gao, Chem. Soc. Rev., 2011, 40, 3157–3181 RSC.
- (a) M. R. Razali, A. Urbatsch, G. B. Deacon and S. R. Batten, Polyhedron, 2013, 64, 352–364 CrossRef CAS PubMed; (b) S. Khan, S. Roy, K. Bhar, R. Ghosh, C.-H. Lin, J. Ribas and B. K. Ghosh, Inorg. Chim. Acta, 2013, 398, 40–45 CrossRef CAS PubMed.
- J. Honzicek, M. Erben, I. Cisarova and J. Vinklarek, Inorg. Chim. Acta, 2005, 358, 814–819 CrossRef CAS PubMed.
- (a) J. Honzicek, J. Vinklarek, M. Erben, L. Strizik, I. Cisarova and Z. Padelkova, J. Organomet. Chem., 2009, 694, 4250–4255 CrossRef CAS PubMed; (b) J. Honzicek, M. Erben, L. Cisarova and J. Vinklarek, Appl. Organomet. Chem., 2005, 19, 100–101 CrossRef CAS; (c) J. Honzicek, M. Erben, I. Cisarova and J. Vinklarek, Appl. Organomet. Chem., 2005, 19, 102–103 CrossRef CAS.
- J. N. Smith and T. M. Brown, Inorg. Chem., 1972, 11, 2697–2701 CrossRef CAS.
- R. Coutts and P. C. Wailes, Inorg. Nucl. Chem. Lett., 1967, 3, 1–5 CrossRef CAS.
- M. Ruhmann, A. Spannenberg, A. Villinger, A. Schulz and T. Beweries, Z. Anorg. Allg. Chem., 2013, 639, 1717–1721 CrossRef CAS.
- P. Schinnerling and U. Thewalt, J. Organomet. Chem., 1992, 431, 41–45 CrossRef CAS.
- (a) A. Cangönül, M. Behlendorf, A. Gansäuer and M. van Gastel, Inorg. Chem., 2013, 52, 11859–11866 CrossRef PubMed; (b) S. Maurelli, E. Morra, S. Van Doorslaer, V. Busico and M. Chiesa, Phys. Chem. Chem. Phys., 2014, 16, 19625–19633 RSC; (c) J. Pinkas, R. Gyepes, I. Císařová, J. Kubišta, M. Horáček and K. Mach, Organometallics, 2014, 33, 3399–3413 CAS.
- S. A. Giddings, Inorg. Chem., 1967, 6, 849–850 CrossRef CAS.
- M.-J. Crawford, T. M. Klapötke, P. Klüfers, P. Mayer and P. S. White, J. Am. Chem. Soc., 2000, 122, 9052–9053 CrossRef CAS.
- J. L. Burmeister, E. A. Deardorff, A. Jensen and V. H. Christiansen, Inorg. Chem., 1970, 9, 58–63 CrossRef CAS.
- M. Erben, I. Cisarova, J. Honzicek and J. Vinklarek, Inorg. Chim. Acta, 2009, 362, 2480–2486 CrossRef CAS PubMed.
- M. Pavlista, R. Bina, Z. Cernosek, M. Erben, J. Vinklarek and I. Pavlik, Appl. Organomet. Chem., 2005, 19, 90–93 CrossRef CAS.
- U. Thewalt and W. Nuding, J. Organomet. Chem., 1996, 512, 127–130 CrossRef CAS.
- M. Erben, J. Merna, O. Hylsky, J. Kredatusova, A. Lycka, L. Dostal, Z. Padelkova and M. Novotny, J. Organomet. Chem., 2013, 725, 5–10 CrossRef CAS PubMed.
- J. L. Burmeister, E. A. Deardorf and C. E. Vandyke, Inorg. Chem., 1969, 8, 170–172 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Theory and Applications in Inorganic Chemistry, Wiley, 2008 Search PubMed.
- K. W. Hipps and A. T. Aplin, J. Phys. Chem., 1985, 89, 5459–5464 CrossRef CAS.
- K. Dahl, G. M. Sando, D. M. Fox, T. E. Sutto and J. C. Owrutsky, J. Chem. Phys., 2005, 123, 084504 CrossRef PubMed.
- (a) M. Klahn, P. Arndt, A. Spannenberg, A. Gansäuer and U. Rosenthal, Organometallics, 2008, 27, 5846–5851 CrossRef CAS; (b) R. Jungst, D. Sekutowski, J. Davis, M. Luly and G. Stucky, Inorg. Chem., 1977, 16, 1645–1655 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- (a) S. Köcher, B. Walfort, G. Rheinwald, T. Rüffer and H. Lang, J. Organomet. Chem., 2008, 693, 3213–3222 CrossRef PubMed; (b) W. Frosch, S. Back, H. Müller, K. Köhler, A. Driess, B. Schiemenz, G. Huttner and H. Lang, J. Organomet. Chem., 2001, 619, 99–109 CrossRef CAS.
- (a) C. P. Raptopoulou, N. Tzavellas and N. Klouras, Z. Anorg. Allg. Chem., 1996, 622, 1387–1391 CrossRef CAS; (b) J. Honzicek, J. Vinklarek, M. Erben and I. Cisarova, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1090–m1091 CAS.
- E. Samuel, J. F. Harrod, D. Gourier, Y. Dromzee, F. Robert and Y. Jeannin, Inorg. Chem., 1992, 31, 3252–3259 CrossRef CAS.
- E. Samuel and J. Vedel, Organometallics, 1989, 8, 237–241 CrossRef CAS.
- K. Mach and J. B. Raynor, J. Chem. Soc., Dalton Trans., 1992, 683–688 RSC.
- (a) M. Erben, A. Růžička, M. Picka and I. Pavlík, Magn. Reson. Chem., 2004, 42, 414–417 CrossRef CAS PubMed; (b) M. Erben, J. Merna, S. Hermanova, I. Cisarova, Z. Padelková and M. Dusek, Organometallics, 2007, 26, 2735–2741 CrossRef CAS.
- J. Losada and M. Moran, J. Organomet. Chem., 1984, 276, 13–19 CrossRef CAS.
- (a) L. C. Francesconi, D. R. Corbin, D. N. Hendrickson and G. D. Stucky, Inorg. Chem., 1979, 18, 3074–3080 CrossRef CAS; (b) P. Neuhaus and W. Sander, Angew. Chem., Int. Ed., 2010, 49, 7277–7280 CrossRef CAS PubMed.
- K. Mach, V. Varga, G. Schmid, J. Hiller and U. Thewalt, Collect. Czech. Chem. Commun., 1996, 61, 1285–1294 CrossRef CAS.
- M. F. Lappert, W. P. Leung, R. A. Bartlett and P. P. Power, Polyhedron, 1989, 8, 1883 CrossRef CAS.
- R. S. P. Coutts, P. C. Wailes and R. L. Martin, J. Organomet. Chem., 1973, 47, 375–382 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, 2008 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- P. Coppens, in Crystallographic Computing, ed. F. R. Ahmed, C. P. Hall and C. P. Huber, Munksgaard, Copenhagen, 1970, pp. 255–270 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1994, 27, 1045–1050 CrossRef CAS.
Footnote |
| † CCDC 1019534–1019539. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01404a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |