DOI:
10.1039/C4NJ01642D
(Paper)
New J. Chem., 2015,
39, 566-575
Novel triazole-based ligands and their zinc(II) and nickel(II) complexes with a nitrogen donor environment as potential structural models for mononuclear active sites†
Received
(in Victoria, Australia)
23rd September 2014
, Accepted 29th October 2014
First published on 29th October 2014
Abstract
Three new 1,2,3-triazole-based ligands with an N,N,N coordination core were prepared using a convergent synthetic protocol starting from racemic 2-amino-1-phenylethanol. They were tested as chelators for biorelevant ZnII or NiII ions. An N,N,N ligand with a terminal amino functionality coordinated the ZnII in a bidentate fashion, not including the triazole nitrogen. The ligand with two pendant 2-pyridyl groups acted as a tridentate ligand without an N2-triazole coordination to ZnII, while the ligand containing one 2-pyridyl group acted as an inverse-click chelator for NiII ions.
Introduction
Mimetics of the active site of an enzyme are useful to better understand the structural features and how a donor array modulates the chemistry around the metal centre in the catalytic site.1 For instance, Rivas et al. have designed ligands that provide N4O, N2S2O and N2O coordination spheres for zinc and have observed that a nitrogen coordination environment is more efficient than mixed nitrogen/sulphur ligation in promoting the amide cleavage,2 which would explain the predominant N-donating ligand environment in many catalytic zinc sites.3 Besides zinc, other late-first-row transition metals, such as iron, copper and nickel, are frequently encountered as metals in metalloproteins.4 Thus, the complexes of ZnII, NiII, FeII, CoII and CuII with tris(pyrazolyl)methane sulfonate (Tpms),5 bis(3,5-dialkylpyrazol-1-yl)acetic acid6 or 3,3-bis(1-methylimidazol-2-yl) propionate7 ligands served as structural models for the N,N,O binding in 2-His-1-carboxylate metalloenzymes. Namely, the facial motif of the triad histidine, histidine and aspartate (or glutamate) is assumed to be essential for the activity of several metalloenzymes.8 Although a great contribution has been made to the design of N,N,O facially coordinating ligands,9 tris(pyrazolyl)borate scorpionate ligands with N,N,N ligation have frequently been used to mimic the 2-His-1-carboxylate active site of several metalloenzymes because they can be relatively easily prepared and modified.10,11 Reinaud and coworkers reported, in many papers over the past decade, on calixarene- and resorcinarene-based supramolecular biomimetic models of the trihistidine mononuclear active site, which not only mimic the first, but also the second and third coordination spheres of the catalytic metal.12 A triazole ring, on the other hand, can be regarded as an aza-analogue of imidazole, a nitrogen donor present in the histidine-containing active sites of many metalloenzymes. 1,2,3-Triazoles also act as N-donors and thus effectively coordinate metals, creating coordination materials with attractive physico-chemical properties.13 Of particular interest are the 1,4-disubstituted 1,2,3-triazole-based mono-, bi- or multidentate chelating ligands, which are employed in many fields, such as drug therapy, bioimaging and sensing, fluorescence, and catalysis.14 This is mainly because both the N2 and N3 atoms of the triazole nucleus can participate in metal coordination, while additional coordination sites can be readily introduced into 1- or 4-substituents using very efficient and selective “click reactions”.15 The majority of the 1,4-disubstituted 1,2,3-triazole ligands use their more Lewis basic N3 atom together with the pendant pyridyl group at position 4 (regular click chelators) to form planar or nonplanar pockets for the complexation of transition metals, such as ReI,16 PdII,17 FeII,18 CuII,19 ZnII,20 and RuII.18,21 There are few 1,2,3-triazole-based chelating ligands that coordinate to the metal through the triazole N3 atom and the sp3N donor (e.g., sec-amino) in the side chain at position 4.20,22 Despite the fact that the N2 atom in a 1,2,3-triazole has a much lower electron density compared to N3,23 numerous stable complexes with the coordinating N2 atom have been prepared using suitable bi(or multi)dentate ligands with the 2-pyridyl group as the nitrogen donor, directly or remotely attached to the N1 atom of the 1,2,3-triazole ring (inverse click chelators).24 When instead of the pyridyl group, an aminoalkyl group is grafted at the N1 position of the 1,2,3-triazole ring, no complexation with metal occurs.22b Kuang et al. reported on utility of pyridyl or quinolinyl containing azides to accelerate the Cu-catalyzed cycloaddition with alkynes. The resulting 1,2,3-triazole ligands were demonstrated to chelate CuII or ZnII ions in a multidentate manner, including the N2 nitrogen of the triazolyl group.25 The successful complexation of pyridyl-containing inverse chelators can be explained by the enhanced electrophilicity of the metal centre, caused by the coordination with the π-acceptor pyridyl group,26 which consequently enables coordination, even of the less-electron-rich N2 atom of the 1,2,3-triazole ring. No such effect occurs involving an amino functionality as a donor.
As a part of our joint interest in designing biomimetic catalyst systems12 and nitrogen ligand-transition metal architectures,27 we noticed the enormous potential of the 1,2,3-triazole-based inverse-click chelators for the formation of NNX (where X stands for O, N) type biometal complexes with one or two free coordination sites for the reversible guest binding. Here we present the synthesis of new ligands containing a 1,2,3-triazole moiety with potential N,N,N coordination sites and their chelation ability for ZnII and NiII ions.
Results and discussion
Ligand design
On the basis of geometrical preferences we envisaged that ligands comprising a 1,2,3-triazole N2 donor and two additional coordination sites in the substituent attached to the N1 atom of the triazole unit could be suitable ligands for coordination to metals in a tridentate fashion. The retrosynthetic approach to the tridentate 1,2,3-triazole-based ligands with non-planar coordination pockets containing three nitrogens or mixed nitrogen/oxygen donors is depicted in Fig. 1. The embedment of a chiral centre is designed for the potential use of these edifices in asymmetric transformations.
 |
| | Fig. 1 Retro-synthesis of potential tridentate 1,2,3-triazole-based ligands. | |
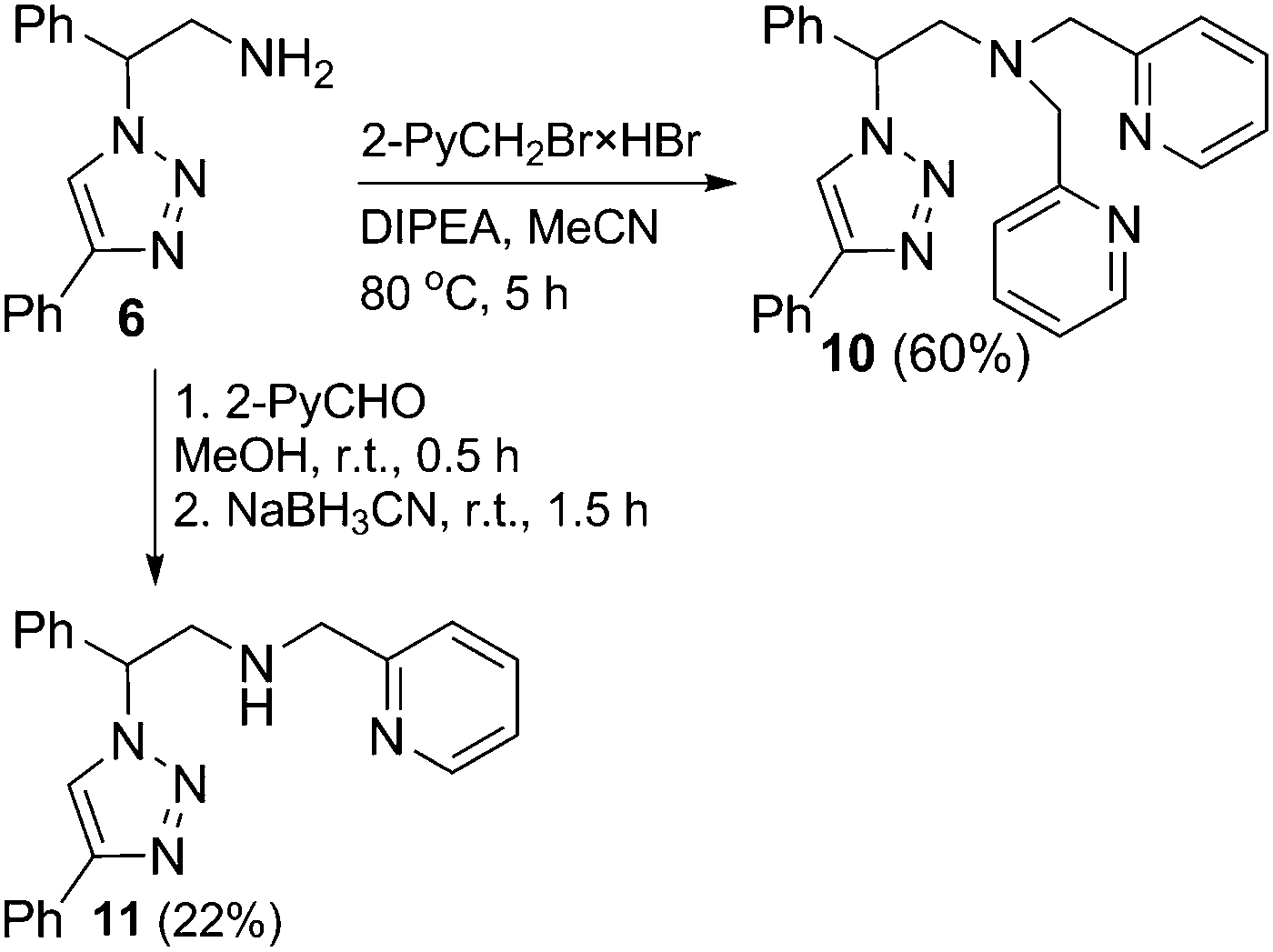
2-Amino-1-phenylethanol (1) was used as a starting material for the synthesis of triazole ligands. First, the hydroxyl group of amino alcohol 1 was transformed into the azido group. This was achieved via carbamate-protected methane sulphonate 3, which underwent a smooth substitution with sodium azide to yield the organic azide 4. A well-established Cu-catalyzed [3+2] cycloaddition of 4 with phenylacetylene selectively furnished 1,4-disubstituted 1,2,3-triazole 5, which was catalytically hydrogenated under a hydrogen pressure to remove the carbamate protection. Thus, the aminoalkyl-1,2,3-triazole 6 with two potential coordination sites (N2 atom of triazole and NH2) was formed in a 5-step synthesis and isolated in an overall yield of 49% (Scheme 1).
 |
| | Scheme 1 Synthesis of 1,2,3-triazole 6. (i) ClCO2Bn (1.2 equiv.), NEt3 (1.5 equiv.), CH2Cl2, r.t., overnight, 81%; (ii) MeSO2Cl (1.2 equiv.), NEt3 (1.5 equiv.), CH2Cl2, 0 °C 0.5 h and r.t., 1 h, 97%; (iii) NaN3 (5 equiv.), DMF, r.t, 3 h, 92%; (iv) PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (1.5 equiv.), CuSO4 (0.01 equiv.), Na-ascorbate (0.1 equiv.), t-BuOH/H2O (1 CH (1.5 equiv.), CuSO4 (0.01 equiv.), Na-ascorbate (0.1 equiv.), t-BuOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), r.t., 20 h, 70%; (v) H2 (60 psi), Pd/C (20 mol%), MeOH, r.t., 8 h, 96%. :1), r.t., 20 h, 70%; (v) H2 (60 psi), Pd/C (20 mol%), MeOH, r.t., 8 h, 96%. | |
The identity of compound 6 was confirmed by NMR and IR spectroscopy, high-resolution mass spectrometry, and elemental analysis. The diastereotopic methylenic protons of 6 show in the 1H NMR spectrum in DMSO-d6 two well-separated doublets of doublets at 3.33 and 3.58 ppm. Correspondingly, the signal of the proton at the chiral centre appears as a doublet of doublets. The characteristic signal of the triazole ring appears at 8.79 ppm.
In the next step, the ligand arm of the triazole 6 was elongated in order to introduce the third coordination donor. Since the direct reaction of 6 with 2-bromoethylamine hydrobromide as an aziridine precursor under basic conditions led to a complex mixture of products, the phthalate-protected reagent 7 was used. The heating of 6 with 7 in the presence of diisopropylethylamine (DIPEA) yielded N-substituted phthalimide 8, which after hydrazinolysis gave the target compound 9 in a 33% yield over two steps (Scheme 2).
 |
| | Scheme 2 Synthesis of N,N,N ligand 9. | |
Due to the positive effect of pyridyl groups in inverse-click 1,2,3-triazole-based chelators, a potential hybrid sp2N^sp3N^sp2N ligand was also designed. When triazole 6 was reacted with a slight excess of 2-picolyl bromide hydrobromide, both the dipyridyl and monopyridyl products 10 and 11 were obtained, together with starting triazole 6, while the employment of 2.1 equivalents of 2-picolyl bromide hydrobromide selectively afforded the dipyridyl product 10 (Scheme 3). The selective synthesis of 11 was achieved by the reductive amination of 6. An in situ formed imine of 6 and pyridine-2-carboxaldehyde was reduced with sodium cyanoborohydride and the desired product 11 was isolated by chromatography in a low yield of 22% (Scheme 3). The NMR spectroscopy investigation in DMSO-d6 revealed that both picolyl groups in the ligand 10 are equivalent and that their methylenic protons display a well-resolved AB spin system (two doublets at 3.81 and 3.92 ppm, J = 14.5 Hz). Similarly, the methylenic protons of the picolyl group in 11 in DMSO-d6 are also magnetically nonequivalent.
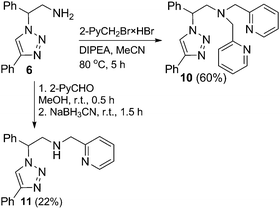 |
| | Scheme 3 Synthesis of the triazole-based ligands with the pendant 2-pyridyl group. | |
In order to extend the above-mentioned methodology, the synthesis of the potential heteroleptic N,N,O-donor ligand 13 was also derived from aminoalkyl-1,2,3-triazole 6. The pendant hydroxyl group with geometry suitable for tridentate ligation, together with two nitrogen donors, was installed by the reaction of 6 with 2-chloroethyl chloroformate. A basic hydrolysis of the resulting carbamate 12 gave the desired product 13 in a good isolated yield (70% from 6) (Scheme 4).
 |
| | Scheme 4 Synthesis of the N,N,O ligand. | |
Metal complexes
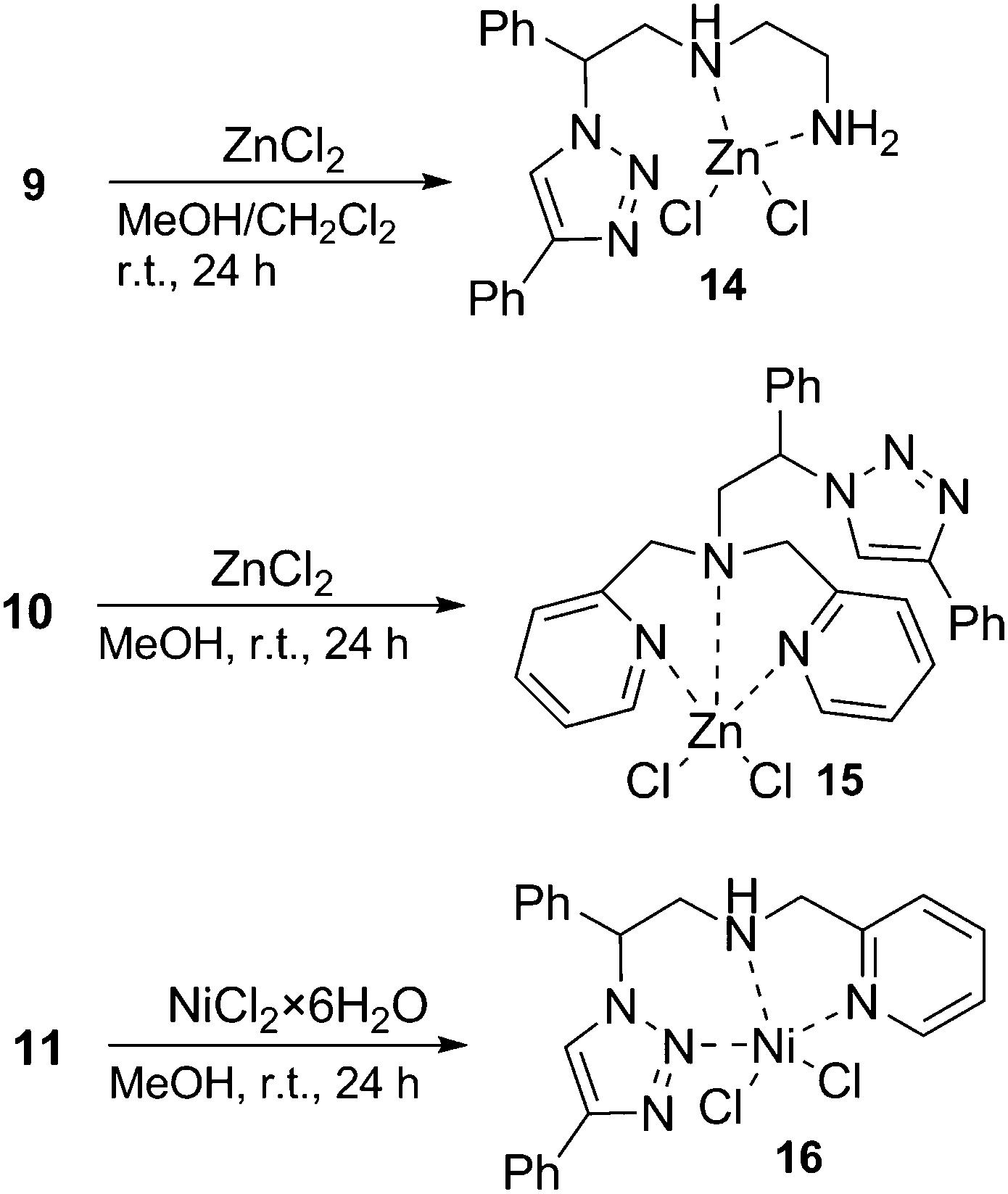
The reaction of ligand 9 with 1 equivalent of ZnCl2 in the mixture of methanol and dichloromethane at room temperature over 24 hours, followed by the recrystallization of the crude residue from the EtOH/MeOH 1:1 mixture led to the formation of white crystals of product 14 (Scheme 5). The elemental analysis indicated that ligand 9 formed a zinc(II) complex with a 1:1 metal:ligand stoichiometry, and the formation of the complex was additionally confirmed by the Maldi-Tof HRMS study (m/z = 406.1). In the 1H NMR spectrum of 14 in DMSO-d6 all three amino protons appear as one broad singlet at approximately 4 ppm, strongly (for about 2 ppm) shifted downfield compared to the free ligand 9, indicating that both the primary and secondary amino groups undergo an interaction with the metal atom. In addition, recognizable downfield shifts in the 1H NMR resonances of all the methylenic protons and of the proton at the chiral centre were observed. However, the resonance of the proton of the triazole unit of the complex 14 remained virtually unchanged, suggesting that the triazole ring is not involved in the zinc coordination. Indeed, a single-crystal X-ray diffraction study of 14 revealed that the zinc ion is embedded in a tetrahedral coordination sphere formed by two nitrogen donors from the ligand side chain and two chlorine anions (Fig. 2). The overall conformation of the potentially tridentate ligand molecule is defined by a strong intramolecular hydrogen bond N20–H20⋯N2 [2.32 Å, 2.951 (5) Å, 126°] forming a hydrogen-bonded six-membered ring. In addition to this intramolecular H-bond, there is the intermolecular hydrogen bond N23–H23B⋯N3 [2.29 Å, 3.109 (4) Å, 151°] observed in the structure of 14, connecting two molecules related by a crystallographic inversion centre, and thus forming the molecular dimers as the basic molecular building blocks of the crystal structure. This means that both available triazole nitrogens (N2 and N3) act as hydrogen-bond acceptors rather than being involved in the Zn(II) coordination.
 |
| | Scheme 5 Synthesis of metal complexes. | |
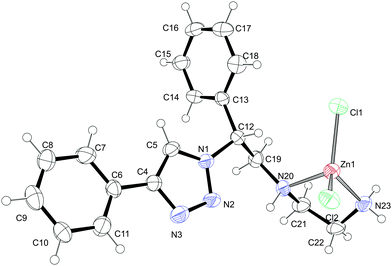 |
| | Fig. 2 Crystal structure of 14. Selected bond lengths [Å] and angles [°]: Zn1–N23 2.029(6), Zn1–N20 2.110(6), Zn1–Cl2 2.221(2), Zn1–Cl1 2.227(2), N23–Zn1–N20 87.0(3), N23–Zn1–Cl2 110.9(2), N20–Zn1–Cl2 109.98(17), N23–Zn1–Cl1 115.9(2), Cl1–Zn1–N20 107.97(18), Cl1–Zn1–Cl2 119.93(9). | |
We assumed that the dipyridyl compound 10 could act as a scorpionate-like tridentate ligand involving a triazole N2 atom and two pyridine nitrogens as the donors. Ligand 10 was mixed with 1 equivalent of ZnCl2 in methanol and after cooling, colourless crystals of 15 were formed (Scheme 5). While the HRMS of the product gave only the signal for dissociated ligand 10, its coordination to the zinc centre was well supported by the NMR analysis. The 1H NMR spectrum in DMSO-d6 of 15 shows a single set of resonances for all pyridine hydrogen atoms appearing as broadened signals (without a fine structure) and are all shifted downfield relative to the free ligand 10. Moreover, methylenic protons of the 2-picolyl group appear as two very broad singlets, also slightly shifted at 3.87 and 4.08 ppm, respectively. These results indicate that the two picolyl groups are equivalent and thus both bind to the metal in a symmetric fashion. Similarly, the 13C NMR signals of pyridyl carbons (except 6′-C) and the methylenic carbon of the picolyl group are broadened. The broadening of the 1H and 13C picolyl signals suggests a fluxional structure, probably due to the weak interaction between the pyridyl nitrogens and the zinc ion. Interestingly, the signal of a proton at the chiral centre in ligand 10 changed its fine structure from a doublet of doublets to a not-well-resolved doublet once 10 coordinated to zinc and is also remarkably shifted downfield (Δδ ≈ 0.3 ppm) in comparison to the free ligand. On the other hand, the resonance of the triazole proton in complex 15 is practically the same as that for the proton in the free ligand 10 (8.72 and 8.71 ppm), indicating the absence of triazole coordination to the metal atom. A detailed insight into the geometry and the binding mode of the zinc atom to ligand 10 was obtained by single crystal X-ray analysis.
The complex 15 is monomeric and features a five coordinated zinc centre in a distorted trigonal bipyramidal geometry (Fig. 3). The distortion of a trigonal bipyramid can be best described by the structural parameter τ (0 for an ideal square pyramid and 1 for an ideal trigonal bipyramid),28 which in our case adopts the value of 0.72. The ligand 10 is tridentatly coordinated to the Zn metal centre in fac-geometry with Zn–N bonding distances with N1 and N2 pyridyl atoms of 2.068(3) and 2.074(4) Å, respectively, and one longer distance with the amino N3 atom [2.418(3) Å]. The Zn–Cl bonding distances are 2.2630(11) and 2.3110(12) Å. The N–Zn–N angles are 117.56(14)°, 75.90(11)° and 74.78(12)° and these deviate from the values expected for an ideal trigonal bipyramid. The lattice structure is stabilized by weak CH⋯Cl and CH⋯π interactions. Although the compound 15 has five aromatic rings, no significant π–π interactions can be observed in the crystal structure.
 |
| | Fig. 3 Crystal structure of 15. Selected bond lengths [Å] and angles [°]: Zn1–N1 2.068(3), Zn1–N2 2.074(4), Zn1–N3 2.418(3), Zn1–Cl1 2.2630(11), Zn1–Cl2 2.3110(12), N1–Zn1–N2 117.56(14), N1–Zn1–N3 75.90(11), N2–Zn1–N3 74.78(12), N1–Zn1–Cl1 109.38(10), N1–Zn1–Cl2 101.94(9), N2–Zn1–Cl2 96.45(10), N2–Zn1–Cl1 124.94(10), Cl1–Zn1–N3 91.10(8), Cl2–Zn1–N3 168.12(8), Cl1–Zn1–Cl2 100.58(5). | |
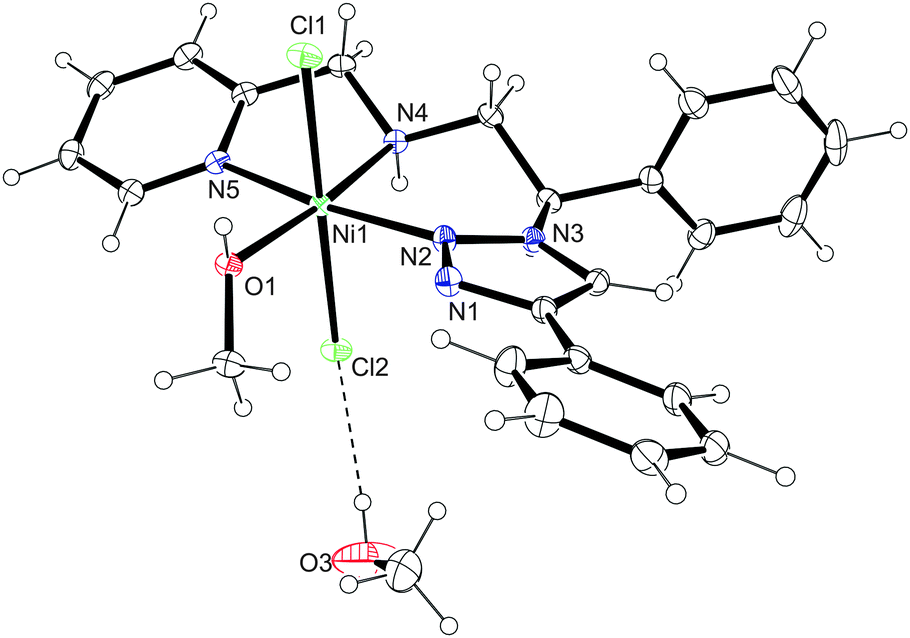
The nickel complex with ligand 11 was formed by reacting the ligand with 1 equivalent of NiCl2·6H2O (Scheme 5). Blue crystals, separated from the mother liquor by filtration, revealed a broad and ill-defined 1H NMR spectrum, which is in agreement with the paramagnetic nature of the 3d8 species. The evidence for the formation of a nickel complex with the ligand 11 was provided by the IR spectrum, significantly altered with respect to the one obtained for the free ligand. The metal coordination was further supported by mass spectrometry (ESI+), which showed a signal corresponding to a [(11)NiCl]+ ion. The coordination of ligand 11 to Ni was finally confirmed by the single crystal X-ray analysis. The asymmetric unit consists of two crystallographically independent complexes (molecule A and B) and two methanol solvate molecules (Fig. 4). The octahedral geometry in both molecules (A, B) is slightly distorted, with maximum deviations from the ideal angles of 90 and 180° ranging from 0.08(5)° to 10.71(6)° and from 2.94(5)° to 9.60(6)°, respectively. Ligand 11 is tridentately coordinated to the Ni metal centre in the mer-geometry with Ni–N bonding distances 2.0488(15)–2.0733(15) for molecule A and 2.0457(15)–2.0492(15) for molecule B. The Ni–O bonding distances are 2.0858(13)–2.0902(13) Å and the Ni–Cl distances are 2.4289(5)–2.4698(5) Å for both molecules. Hydrogen bonding between the coordinated methanol molecule and the NH group with the chlorido ligand connects molecules A to a 1D chain (Fig. 5), as well as molecules B (Table 1). The chains are mutually connected by π–π stacking [3.827(2) and 3.8765(12) Å] and weak CH⋯N and CH⋯π interactions.
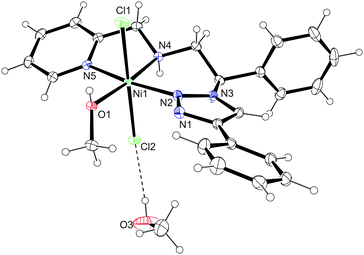 |
| | Fig. 4 Crystal structure of [16(MeOH)]·MeOH, only molecule A is presented. Selected bond lengths [Å] and angles [°] for molecule A: Ni(1)–N(2) 2.0733(15), Ni(1)–N(4) 2.0488(15), Ni(1)–N(5) 2.0718(15), Ni(1)–O(1) 2.0902(13), Ni(1)–Cl(1) 2.4311(5), Ni(1)–Cl(2) 2.4289(5), N(2)–Ni(1)–O(1) 92.92(5), N(2)–Ni(1)–Cl(1) 89.92(5), N(2)–Ni(1)–Cl(2) 88.22(5), N(4)–Ni(1)–N(2) 91.11(6), N(4)–Ni(1)–N(5) 79.29(6), N(4)–Ni(1)–O(1) 174.97(6), N(4)–Ni(1)–Cl(1) 92.36(5), N(4)–Ni(1)–Cl(2) 89.94(5), N(5)–Ni(1)–N(2) 170.40(6), N(5)–Ni(1)–O(1) 96.66(6), N(5)–Ni(1)–Cl(1) 90.49(5), N(5)–Ni(1)–Cl(2) 91.73(5), O(1)–Ni(1)–Cl(1) 84.65(4), O(1)–Ni(1)–Cl(2) 93.18(4), Cl(2)–Ni(1)–Cl(1) 177.063(18); selected bond lengths [Å] and angles [°] for molecule B: Ni(2)–N(7) 2.0465(15), Ni(2)–N(9) 2.0492(15), Ni(2)–N(10) 2.0457(15), Ni(2)–O(2) 2.0858(13), Ni(2)–Cl(3) 2.4353(5), Ni(2)–Cl(4) 2.4698(5), N(7)–Ni(2)–N(9) 91.77(6), N(7)–Ni(2)–O(2) 93.04(6), N(7)–Ni(2)–Cl(3) 89.67(5), N(7)–Ni(2)–Cl(4) 86.60(5), N(9)–Ni(2)–O(2) 173.74(6), N(9)–Ni(2)–Cl(3) 89.30(5), N(9)–Ni(2)–Cl(4) 91.57(5), N(10)–Ni(2)–N(7) 171.60(6), N(10)–Ni(2)–N(9) 81.42(6), N(10)–Ni(2)–O(2) 93.42(6), N(10)–Ni(2)–Cl(3) 95.13(4), O(2)–Ni(2)–Cl(3) 94.72(4), N(10)–Ni(2)–Cl(4) 88.66(5), O(2)–Ni(2)–Cl(4) 84.73(4), Cl(3)–Ni(2)–Cl(4) 176.195(19). | |
 |
| | Fig. 5 Chain formation of molecules A in [16(MeOH)]·MeOH. Dashed lines indicate the hydrogen bonds. Hydrogen atoms that are not involved in the motif shown have been omitted for clarity [symmetry codes (i) x + 1, y, z; (ii) x − 1, y, z]. | |
Table 1 Hydrogen bonds for [16(MeOH)]·MeOH; distances [Å] and angles [°], [symmetry codes (i) x + 1, y, z; (ii) x − 1, y, z]
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
∠(DHA) |
| O1–H1A⋯Cl2i |
0.836(9) |
2.491(15) |
3.2271(14) |
147(2) |
| N4–H4B⋯Cl1ii |
0.93 |
2.38 |
3.2123(16) |
148.5 |
| O2–H2A⋯Cl3ii |
0.828(10) |
2.513(14) |
3.2643(14) |
151(2) |
| N9–H9B⋯Cl4i |
0.93 |
2.43 |
3.2205(16) |
142.4 |
| N9–H9B⋯Cl3 |
0.93 |
2.77 |
3.1636(16) |
106.7 |
| O3–H3A⋯Cl2 |
0.84 |
2.36 |
3.196(2) |
173.6 |
| O4–H4A⋯Cl4 |
0.84 |
2.34 |
3.1577(19) |
165.6 |
Finally, chelation of N,N,O ligand 13 was also attempted, but we did not succeed in preparing and isolating the Zn or Ni complexes of the ligand 13 so far, regardless of the environment (basic or neutral) that was used in the preparation or the counteranion (chloride or acetate). The study of the coordination ability of the ligand 13 as well as potential utility of our metal complexes is currently ongoing and will be the subject of our forthcoming report.
Conclusions
The synthesis of potential tridentate ligands containing a 1,2,3-triazole moiety with either nitrogen or mixed nitrogen and oxygen donor sites was achieved by employing the convergent protocol starting from 2-amino-1-phenylethanol. Their coordination ability was tested with two biorelevant metals to mimic a histidine-carboxylate active site of metallopeptidases. An all-nitrogen-donor ligand containing a terminal amino functionality was coordinated to the ZnII ion in a bidentate fashion, not including a triazole N2 atom. A 1,2,3-triazole-based ligand containing two 2-picolyl groups was coordinated to the ZnII ion through tertiary and two pyridine nitrogens, while the N,N,N ligand with one 2-picolyl arm acted as an inverse-click chelator for the NiII ion, since an N2 atom of the triazole ring together with the amino and pyridyl nitrogens was successfully coordinated to the metal. However, the coordination of the N,N,O ligand with a terminal hydroxyl group was not achieved under neutral or under basic conditions. The synthesized complexes may have potential as bioinspired catalysts and due to the possibility of introducing a chiral centre they may also be used in asymmetric transformations.
Experimental section
General considerations
The reagents and solvents were used as received from commercial suppliers. The reagent 7 was prepared according to the reported procedure.29 Melting points were determined on a Kofler micro hot stage. The NMR spectra were recorded at 302 K either on a Bruker Avance DPX 300 or Avance III 500 MHz spectrometer operating at 300 MHz (or 500 MHz) and 75.5 MHz (or 125 MHz) for 1H and 13C. The 1H NMR spectra are referenced with respect to TMS as the internal standard. The 13C NMR spectra are referenced against the central line of the solvent signal (DMSO-d6 septet at δ = 39.5 ppm, CDCl3 triplet at δ = 77.0 ppm). The coupling constants (J) are given in Hz. The multiplicities are indicated as follows: s (singlet), d (doublet), t (triplet), m (multiplet) and br (broadened). IR spectra were obtained with a Bruker ALPHA FT-IR spectrophotometer or Bio-Rad FTS 3000MX. MS spectra were recorded with an Agilent 6224 Accurate Mass TOF LC/MS instrument, VG-Analytical AutoSpec Q instrument or 4800 MALDI TOF/TOF Analyzer, Applied Biosystems. Elemental analyses (C, H, N) were performed using a Perkin-Elmer 2400 Series II CHNS/O Analyzer. TLC was carried out on Fluka silica gel TLC-cards. Merck silica gel 60 PF254 containing gypsum was used to prepare chromatotron plates. Radial chromatography was performed using a Harrison Research, model 7924T chromatotron.
X-ray diffraction analysis
Single-crystal X-ray diffraction data were collected using a Agilent Technologies SuperNova Dual diffractometer with an Atlas detector and Mo-Kα radiation (λ = 0.71073 Å) at room temperature (14 and 15) and 150 K ([16(MeOH)]·MeOH). The data were processed using CrysAlis Pro.30 The structures were solved by direct methods using the program SHELXS-97 (ref. 31) (15) or SIR97 (ref. 32) (14 and [16(MeOH)]·MeOH) and refined on F2 using full-matrix least-square procedures (SHELXL-97).31 Single crystals of 14 were of an extremely poor quality and the data collection was difficult. Hence the somewhat ill-refined structure. However, the main goal was to establish the zinc binding mode which is undoubtedly and fully achieved. All our later attempts to produce the better quality single crystals were unsuccessful. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were treated as riding atoms in geometrically idealized positions, except the hydrogen atoms attached to O1 and O2 atoms in [16(MeOH)]·MeOH that were refined fixing the bond lengths and isotropic temperature factors. Crystallographic data are listed in Table 2.
Table 2 Crystal-data and structure-refinement details of 14–16
| |
14
|
15
|
[16(MeOH)]·MeOH |
|
R = ∑||Fo| − |Fc||/∑|Fo|.
wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2.
S = {∑[(Fo2 − Fc2)2]/(n/p)}1/2 where n is the number of reflections and p is the total number of parameters refined.
|
| Formula |
C18H21Cl2N5Zn |
C28H26Cl2N6Zn |
C24H29Cl2N5NiO2 |
|
M
r
|
443.67 |
582.82 |
549.13 |
|
T (K) |
293(2) |
293(2) |
150(2) |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
|
a (Å) |
7.927(1) |
8.8955(9) |
6.42170(10) |
|
b (Å) |
10.6150(11) |
14.5691(11) |
11.6331(2) |
|
c (Å) |
12.920(2) |
21.5718(19) |
34.4621(8) |
|
α (°) |
78.84(1) |
90 |
89.715(2) |
|
β (°) |
74.08(2) |
91.451(8) |
85.211(2) |
|
γ (°) |
70.62(1) |
90 |
82.580(2) |
| Volume (Å3) |
979.90(17) |
2794.8(4) |
2543.95(8) |
|
Z
|
2 |
4 |
4 |
|
D
c [Mg m−3] |
1.504 |
1.385 |
1.434 |
|
μ [mm−1] |
1.538 |
1.098 |
1.004 |
|
F(000) |
456 |
1200 |
1144 |
| Crystal size (mm) |
0.60 × 0.30 × 0.20 |
0.50 × 0.10 × 0.10 |
0.40 × 0.15 × 0.10 |
| Reflections collected |
10999 |
20508 |
20188 |
| Reflections unique (Rint) |
6397 (0.060) |
6399 (0.0581) |
11625 (0.0170) |
| Parameters |
235 |
334 |
625 |
|
R, wR2 [I > 2σ(I)]a |
0.0989, 0.3133 |
0.0595, 0.1418 |
0.0348, 0.0764 |
|
R, wR2 (all data)b |
0.1881, 0.3425 |
0.1035, 0.1641 |
0.0399, 0.0793 |
| GOF, Sc |
1.049 |
1.071 |
1.110 |
Syntheses
Benzyl (2-hydroxy-2-phenylethyl)carbamate (2).
To the cooled (0 °C) mixture of 2-amino-1-phenylethanol (7.36 g, 53.65 mmol), triethylamine (11.2 mL, 80.24 mmol) and CH2Cl2 (80 mL), solution of benzyl chloroformate (9.2 mL, 64.45 mmol) in CH2Cl2 (20 mL) was slowly added. The resulting mixture was allowed to warm to r.t. and stirred over night. Then water (100 mL) was added and after separation, the aqueous phase was extracted with CH2Cl2 (50 mL). Combined organic layers were washed with water (100 mL) and dried (Na2SO4). After removal of the solvent, the solid residue was suspended in petroleum ether and filtered off to give 11.717 g (81%) of a white crystalline product. Rf = 0.61 (1:10 MeOH/CH2Cl2); m.p. 109–111 °C (ref. 33, 120–121 °C). 1H NMR (300 MHz, CDCl3): δ = 2.66 (br, 1H, OH), 3.32 (m, 1H, CHaHb-CH), 3.56 (m, 1H, CHaHb-CH), 4.84 (dd, J = 3.1, 7.5 Hz, 1H, CH), 5.11 (s, 2H, CH2-Ph), 5.13 (br, 1H, NH), 7.28–7.41 (m, 10H, Ar) ppm.
2-[(Benzyloxycarbonyl)amino]-1-phenylethylmethane sulfonate (3).
To the cooled (0 °C) mixture of 2 (3.772 g, 13.90 mmol), triethylamine (2.9 mL, 20.78 mmol) and CH2Cl2 (70 mL), methanesulfonyl chloride (1.3 mL, 16.80 mmol) was slowly added. The resulting mixture was stirred at the same temperature for 0.5 h and then at r.t. for 1 h. Then water (100 mL) was added and after separation, the organic phase was successively washed with HCl (40 mL, 1 M), NaHCO3 (50 mL, sat.), and brine (50 mL) and then dried (Na2SO4). After removal of volatiles, 4.695 g (97%) of yellow oil was obtained. 1H NMR (500 MHz, CDCl3): δ = 3.55 (m, 1H, CHaHb-CH), 3.69 (m, 1H, CHaHb-CH), 5.10 (d, J = 12 Hz, 1H, CHaHb-Ph), 5.13 (d, J = 12 Hz, 1H, CHaHb-Ph), 5.24 (s, 3H, Me), 5.65 (m, 1H, CH), 6.07 (br, 1H, NH), 7.30–7.48 (m, 10H, Ar) ppm. IR (neat): ν = 1743, 1350, 1233, 1216, 1170, 966, 914, 698 cm−1. HRMS (ESI+) calcd for C16H16O2N ([M-OSO2Me]+): 254.1181; found: 254.1177.
Benzyl (2-azido-2-phenylethyl)carbamate (4).
The mixture of 3 (4.695 g, 13.44 mmol), used directly from the previous reaction, NaN3 (4.369 g, 67.21 mmol) and DMF (15 mL) was stirred at r.t. for 3 h. Then water (100 mL) was added and extracted with Et2O (3 × 80 mL). The combined organic layers were washed with brine (2 × 80 mL) and dried (Na2SO4). After removal of volatiles, 3.674 g (92%) of orange oil was obtained. Rf = 0.59 (3:5 petroleum ether/EtOAc). 1H NMR (300 MHz, CDCl3): δ = 3.32 (m, 1H, CHaHb-CH), 3.55 (m, 1H, CHaHb-CH), 4.70 (dd, J = 3.0, 6.0 Hz, 1H, CH), 5.02 (br, 1H, NH), 5.10 (d, J = 12.3 Hz, 1H, CHaHb-Ph), 5.14 (d, J = 12.3 Hz, 1H, CHaHb-Ph), 7.30–7.40 (m, 10H, Ar) ppm. 13C NMR (75.5 MHz, CDCl3): 46.3, 65.5, 66.9, 126.9, 128.0, 128.1, 128.5, 128.7, 128.9, 136.2, 136.8, 156.2 ppm. IR (NaCl) ν = 3419, 3334, 3063, 3033, 2939, 2103, 1708, 1522, 1455, 1250, 1147, 1067, 989, 755, 699 cm−1. HRMS (ESI+) calcd for C16H17N4O2 ([M + H]+): 297.1352; found 297.1346.
Benzyl [2-phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethyl]carbamate (5).
To the mixture of 4 (1.689 g, 5.70 mmol), phenylacetylene (0.873 g, 8.55 mmol), and t-BuOH/H2O (20 mL, 1:1), freshly prepared aqueous solutions of sodium ascorbate (570 μL, 1 M) and CuSO4·5H2O (57 μL, 1 M) were added. The resulting mixture was stirred at r.t. for 20 h, then evaporated, and water (50 mL) was added and extracted with CH2Cl2 (3 × 50 mL). Combined organic layers were dried (Na2SO4). After removal of volatiles, the solid residue was suspended in Et2O (50 mL) and filtered off to give 1.59 g (70%) of the product. Rf = 0.41 (1:1 petroleum ether/EtOAc); m.p. 152–154 °C. 1H NMR (300 MHz, CDCl3): δ = 4.09 (m, 1H, CHaHb-CH), 4.22 (m, 1H, CHaHb-CH), 5.05 (d, J = 12.3 Hz, 1H, CHaHb-Ph), 5.11 (d, J = 12.3 Hz, 1H, CHaHb-Ph), 5.45 (br, 1H, NH), 5.76 (dd, J = 4.2, 9.0 Hz, 1H, CH), 7.26–7.44 (m, 13H, Ar), 7.68 (s, 1H, CH triazol), 7.79 (m, 2H, Ar) ppm. 13C NMR (75.5 MHz, DMSO-d6): 44.4, 63.5, 65.3, 120.8, 125.1, 127.0, 127.5, 127.7, 127.8, 128.2, 128.5, 128.6, 128.9, 130.7, 136.9, 137.3, 146.4, 156.2 ppm. IR (KBr) ν = 3395, 3078, 2934, 1699, 1523, 1458, 1425, 1261, 1204, 1157, 1076, 998, 932, 909, 854, 745, 693, 544, 515 cm−1. HRMS (ESI+) calcd for C24H23N4O2 ([M + H]+): 399.1821; found 399.1835. C24H22N4O2 (398.46): calcd C 72.34, H 5.57, N 14.06; found C 72.15, H 5.50, N 13.97.
2-Phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethan-1-amine (6).
The mixture of 5 (2.22 g, 5.57 mmol), MeOH (250 mL), and Pd/C (1.18 g, 1.11 mmol; 10 wt%) was hydrogenated under H2 (60 psi) at r.t. for 8 h. Then the catalyst was filtered off, the filtrate was concentrated and the residue was passed through a short column of silicagel with MeOH as the eluent. After the removal of the solvent, 1.41 g (96%) of a white product was obtained. Rf = 0.35 (1:10 MeOH/CH2Cl2); m.p. 108.5–110.5 °C. 1H NMR (300 MHz, DMSO-d6): δ = 3.22 (br, 2H, NH2), 3.33 (dd, J = 5.1, 13.5 Hz, 1H, CHaHb-CH), 3.58 (dd, J = 9.3, 13.5 Hz, 1H, CHaHb-CH), 5.74 (dd, J = 5.4, 9.2 Hz, 1H, CH), 7.29–7.48 (m, 8H, Ar), 7.86 (m, 2H, Ar), 8.79 (s, 1H, CH triazole) ppm. 13C NMR (75.5 MHz, DMSO-d6): 46.1, 67.5, 120.8, 125.1, 126.8, 127.7, 128.0, 128.7, 128.8, 130.8, 138.6, 146.3 ppm. IR (KBr) ν = 3451, 3366, 3283, 1607, 1456, 1218, 1075, 760, 691 cm−1. HRMS (ESI+) calcd for C16H17N4 ([M + H]+): 265.1453; found 265.1459. C16H16N4 (264.33): calcd C 72.70, H 6.10, N 21.20; found C 72.50, H 6.14, N 20.99.
2-{2-[2-Phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethylamino]ethyl}isoindoline-1,3-dione (8).
A solution of 6 (622 mg, 2.353 mmol), N-(2-bromoethyl)phthalimide (598 mg, 2.354 mmol), DIPEA (608 mg, 4.70 mmol), and DMSO (5 mL) was stirred at 100 °C for 14 h. To the cold reaction mixture brine (30 mL) was added and extracted with EtOAc (3 × 30 mL). Combined organic layers were washed with brine (2 × 30 mL) and dried (Na2SO4). After removal of volatiles, the oily residue was subjected to radial chromatography (SiO2; MeOH/CH2Cl2 1:100, then 50:1, then 20:1) giving 532 mg (52%) of orange oil. 1H NMR (300 MHz, CDCl3): δ = 1.40 (br, 1H, NH), 2.98 (t, J = 6.0 Hz, 2H, CH2), 3.38 (dd, J = 5.1, 12.9 Hz, 1H, CHaHb-CH), 3.81 (m, 3H, CHaHb-CH and CH2), 5.72 (dd, J = 5.1, 8.7 Hz, 1H, CH), 7.24–7.43 (m, 8H, Ar), 7.65 (m, 2H, Ar), 7.73–7.82 (m, 5H, Ar and CH triazole) ppm. 13C NMR (75.5 MHz, CDCl3): 37.3, 47.2, 52.7, 65.3, 119.8. 123.2, 125.7, 126.9, 128.0, 128.6, 128.7, 129.0, 130.6, 132.0, 133.8, 137.5, 147.5, 168.4 ppm. IR (KBr): ν = 3419, 1770, 1713, 1612, 1396, 767, 719 cm−1. HRMS (ESI+) calcd for C26H24N5O2 ([M + H]+): 438.1925; found 438.1918.
N-[2-Phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethyl]ethane-1,2-diamine (9).
The mixture of 8 (115 mg, 0.263 mmol), hydrazine hydrate (107 mg, 2.137 mmol), and ethanol (8 mL) was refluxed for 3 h. Then volatiles were evaporated and 1 M aqueous solution of HCl (5 mL) was added. Upon cooling 2,3-dihydrophathalazine-1,4-dione was filtered off. The filtrate was basified with 40% aqueous solution of NaOH to pH ≈ 12 and extracted with Et2O (3 × 10 mL). Combined organic layers were washed with water (5 mL) and dried (Na2SO4). After the removal of the solvent 52 mg (64%) of a white solid was obtained. Rf = 0.26 (1:5 MeOH/CH2Cl2); m.p. 113–115 °C (petroleum ether/EtOAc). 1H NMR (500 MHz, DMSO-d6): δ = 1.96 (br, 3H, NH, NH2), 2.54 (m, 4H, CH2-CH2), 3.24 (dd, J = 5.0, 13.0 Hz, 1H, CHaHb-CH), 3.57 (dd, J = 9.5, 13.0 Hz, 1H, CHaHb-CH), 5.87 (dd, J = 5.0, 9.5 Hz, 1H, CH), 7.30–7.47 (m, 8H, Ar), 7.86 (m, 2H, Ar), 8.81 (s, 1H, CH triazole) ppm. 13C NMR (125.7 MHz, DMSO-d6): 41.2, 51.8, 52.9, 64.4, 120.8, 125.1, 126.9, 127.8, 128.1, 128.7, 128.8, 130.8, 138.7, 146.2 ppm. IR (KBr): ν = 3409, 3338, 3087, 2936, 1618, 1559, 1458, 766, 697 cm−1. HRMS (ESI+) calcd for C18H22N5 ([M + H]+): 308.1875; found 308.1888. C18H21N5 (307.39): calcd C 70.33, H 6.89, N 22.78; found C 70.30, H 6.67, N 22.57.
2-Phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-N,N-bis(pyridin-2-ylmethyl)ethanamine (10).
The mixture of 6 (100 mg, 0.378 mmol), 2-picolylbromide hydrobromide (200 mg, 0.791 mmol), DIPEA (195 mg, 1.509 mmol), and MeCN (5 mL) was stirred at 80 °C for 5 h. Then volatiles were evaporated, water was (10 mL) added and extracted with DCM (3 × 10 mL). Combined organic layers were dried (Na2SO4) and evaporated. The solid residue was subjected to column chromatography (SiO2, MeOH/CH2Cl2 1:20) to give 101 mg (60%) of a white product. Rf = 0.55 (1:10 MeOH/CH2Cl2); m.p. 127.5–129 °C (petroleum ether/EtOAc). 1H NMR (500 MHz, DMSO-d6): δ = 3.33 (m, 1H, CHaHb-CH), 3.67 (dd, J = 10 Hz, 13.5 Hz, 1H, CHaHb-CH), 3.81 (d, J = 14.5 Hz, 2H, 2 × Py-CHaHb), 3.92 (d, J = 14.5 Hz, 2H, 2 × Py-CHaHb), 5.93 (dd, J = 5 Hz, 10 Hz, 1H, CH), 7.07 (m, 2H, Py), 7.22 (ddd, J = 1, 5, 7.5 Hz, 2H, Py), 7.31–7.41 (m, 6H, Ar), 7.46 (m, 2H, Ar), 7.59 (ddd, J = 2, 7.5, 7.5 Hz, 2H, Py), 7.83 (m, 2H, Ph), 8.48 (ddd, J = 1, 2, 5 Hz, 2H, Py), 8.72 (s, 1H, CH triazole) ppm. 13C NMR (75.5 MHz, CDCl3): δ = 58.5, 60.6, 63.1, 118.9, 122.2, 123.4, 125.6, 127.0, 128.0, 128.5, 128.7, 128.9, 130.7, 136.5, 137.4, 147.3, 148.8, 158.8 ppm. IR (neat): ν = 3144, 3061, 3011, 2829 1590, 1432, 1362, 1028, 767, 696 cm−1. HRMS (ESI+) calcd for C28H27N6 ([M + H]+): 447.2297; found 447.2292. C28H26N6 (446.55): calcd C 75.31, H 5.87, N 18.82; found C75.58, H 5.68, N 18.87.
2-Phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)-N-(pyridin-2-ylmethyl)ethanamine (11).
To the mixture of 6 (106 mg, 0.401 mmol) and MeOH/CH3CO2H (3 mL/0.1 mL), a solution of 2-pyridinecarboxaldehyde (86 mg, 0.803 mmol) in MeOH (1 mL) was added. The resulting mixture was stirred at r.t. for 0.5 h, then NaCNBH3 (101 mg, 1.607 mmol) was added portion wise over a period of 1 h and afterwards stirred for additional 0.5 h. The reaction mixture was evaporated and the mixture of NaOH (2 mL, 35%) and water (10 mL) was added, and extracted with DCM (4 × 10 mL). Combined organic layers were dried (Na2SO4) and evaporated. The residue was purified by radial chromatography (SiO2; MeOH/CH2Cl2 50:1) giving 32 mg (22%) of a yellowish product. Rf = 0.63 (1:10 MeOH/CH2Cl2); m.p. 116–119 °C (petroleum ether/EtOAc). 1H NMR (500 MHz, DMSO-d6): δ = 2.58 (br, 1H, NH), 3.28 (dd, J = 5, 13 Hz, 1H, CHaHb-CH), 3.61 (dd, J = 9.5, 13 Hz, 1H, CHaHb-CH), 3.87 (AB, 2H, Py-CH2), 5.96 (dd, J = 5, 9.5 Hz, 1H, CH), 7.22 (ddd, J = 1, 4.5, 7.5 Hz, 1H, Py), 7.31–7.47 (m, 9H, Py and Ar), 7.70 (ddd, J = 2, 7.5, 7.5 Hz, 1H, Py), 7.86 (m, 2H, Ar), 8.47 (ddd, J = 1, 2, 5 Hz, 1H, Py), 8.82 (s, 1H, CH triazole) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ = 52.6, 53.9, 64.3, 120.9, 121.8, 121.9, 125.1, 126.9, 127.8, 128.2, 128.7, 128.9, 130.8, 136.5, 138.6, 146.2, 148.8, 159.9 ppm. IR (neat): ν = 3299, 3120, 3085, 2907, 2821, 1589, 1478, 1428, 841, 759, 688 cm−1. HRMS (ESI+) calcd for C22H22N5 ([M + H]+): 356.1870; found 356.1870. C22H21N5 (355.44): calcd C 74.34, H 5.96, N 19.70; found C 73.94, H 6.11, N 19.49.
2-Chloroethyl 2-phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethylcarbamate (12).
To a solution of 6 (368 mg, 1.392 mmol) and pyridine (156 μL, 1.94 mmol) in CH2Cl2 (10 mL), 2-chloroethyl chloroformate (157 μL, 1.52 mmol) was added. The resulting mixture was stirred at r.t. for 2.5 h, afterwards water (15 mL) was added. Layers were separated and the water phase was extracted with CH2Cl2 (3 × 10 mL). Combined organic layers were dried (Na2SO4) and evaporated to give 467 mg (90%) of a white solid. Rf = 0.59 (1:20 MeOH/CH2Cl2); m.p. 138–140 °C. 1H NMR (300 MHz, CDCl3): δ = 3.61 (t, J = 5.7 Hz, 2H, CH2), 4.09 (m, 1H, CHaHb-CH), 4.21 (m, 1H, CHaHb-CH), 4.28 (m, 2H, CH2), 5.58 (t, J = 4.7 Hz, 1H, NH), 5.75 (dd, J = 4.7, 9.3 Hz, 1H, CH), 7.23–7.45 (m, 8H, Ar), 7.70 (s, 1H, CH triazole), 7.79 (m, 2H, Ar) ppm. 13C NMR (75.5 MHz, CDCl3): δ = 41.8, 45.2, 64.7, 64.9, 120.5, 125.7, 126.7, 128.3, 128.8, 129.0, 129.2, 130.2, 136.8, 148.1, 156.0 ppm. IR (KBr): ν = 3401, 1710, 1520, 1263, 1240, 764, 692 cm−1. HRMS (ESI+) calcd for C19H20ClN4O2 ([M + H]+): 371.1269; found 371.1270.
2-{[2-Phenyl-2-(4-phenyl-1H-1,2,3-triazol-1-yl)ethyl]amino}ethan-1-ol (13).
To the mixture of 12 (467 mg, 1.26 mmol), directly used from the previous reaction, and EtOH (20 mL), solution of KOH (680 mg, 12.12 mmol) in water (6 mL) was added and refluxed for 1.5 h. Afterwards the mixture was evaporated under reduced pressure and the solid residue was suspended in water (15 mL) and vigorously stirred. The white solid was filtered off, thoroughly washed with water and dried to give 302 mg (78%) of the product. Rf = 0.18 (1:20 MeOH/CH2Cl2); m.p. 109–112 °C (MeOH/H2O). 1H NMR (300 MHz, DMSO-d6): δ = 1.82 (br, 1H, NH), 2.64 (m, 2H, CH2), 3.28 (dd, J = 4.9, 13.1 Hz, 1H, CHaHb-CH), 3.41 (dt, J = 4.9, 4.9 Hz, 2H, CH2-OH), 3.60 (dd, J = 9.6, 13.1 Hz, 1H, CHaHb-CH), 4.44 (t, J = 4.9 Hz, 1H, OH), 5.89 (dd, J = 4.9, 9.6 Hz, 1H, CH), 7.25–7.50 (m, 8H, Ar), 7.86 (m, 2H, Ar), 8.79 (s, 1H, CH triazole) ppm. 13C NMR (75.5 MHz, DMSO-d6): 51.1, 53.0, 60.3, 64.4, 120.8, 125.1, 126.9, 127.8, 128.1, 128.7, 128.8, 130.8, 138.7, 146.2 ppm. IR (KBr): ν = 3409, 3273, 3081, 2896, 2870, 1487, 1457, 1230, 1076, 913, 764, 693 cm−1. HRMS (ESI+) calcd for C18H21N4O ([M + H]+): 309.1710; found 309.1706. C18H20N4O (308.38): calcd C 70.11, H 6.54, N 18.17; found C 70.18, H 6.41, N 17.87.
Complex [Zn(9)Cl2] (14).
The mixture of 9 (134 mg, 0.44 mmol) in CH2Cl2 (6 mL) and ZnCl2 (63 mg, 0.46 mmol) in MeOH (5 mL) was stirred at room temperature. The white precipitate was filtered off, washed with cold MeOH and dried to give 165 mg (85%) of an analytically (CHN and NMR) pure complex. Colourless crystals suitable for the single crystal X-ray analysis were obtained by a slow evaporation from the acetonitrile solution at the room temperature. 1H NMR (500 MHz, DMSO-d6): δ = 2.58–2.78 (m, 4H, CH2-CH2), 3.35 (m, 1H, CHaHb-CH), 3.85 (dd, J = 10, 13.5 Hz, 1H, CHaHb-CH), 4.05 (br, 3H, NH, NH2), 6.13 (dd, J = 3.5, 10 Hz, 1H, CH), 7.32–7.47 (m, 8H, Ar), 7.87 (m, 2H, Ar), 8.82 (s, 1H, CH triazole) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ = 38.7, 48.5, 52.5, 63.3, 121.7, 125.1, 126.6, 127.9, 128.5, 128.90, 128.92, 130.7, 138.0, 146.6 ppm. IR (neat): ν = 3321, 3231, 3149, 1593, 1457, 1442, 1431, 1226, 1153, 1122, 1078, 1016, 983, 761, 727, 694 cm−1. MALDI-TOF [C18H21ClN5Zn]+ ([M − Cl]+): (406.07); found 406.07. C18H21Cl2N5Zn (443.71): calcd C 48.72, H 4.77, N 15.78; found C 48.73, H 4.71, N 15.52.
Complex [Zn(10)Cl2] (15).
The mixture of 10 (150 mg, 0.336 mmol), ZnCl2 (46 mg, 0.337 mmol), and MeOH (3 mL) was stirred for 1 day at r.t. and then cooled. After approximately 1 week white crystals were filtered off, washed with MeOH and dried to give 165 mg (84%) of an analytically (CHN and NMR) pure complex. Crystals suitable for X-ray analysis were obtained from methanol solution at 4 °C. 1H NMR (500 MHz, DMSO-d6): δ = 3.29 (dd, J = 3.5, 15 Hz, 1H, CHaHb-CH), 3.90 (dd, J = 9, 14.5 Hz, 1H, CHaHb-CH), 3.98 (br, 2H, 2 × Py-CHaHb), 4.13 (br, 2H, 2 × Py-CHaHb), 6.46 (d, J = 6 Hz, 1H, CH), 7.27 (br, 2H, Py), 7.29–7.39 (m, 6H, Ar), 7.48 (m, 2H, Ar), 7.47 (br, 2H, Py), 7.84 (m, 2H, Ar), 7.95 (br, 2H, Py), 8.72 (s, 1H, CH triazole), 8.92 (br, 2H, Py) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ = 55.8 (br), 56.4 (br), 60.4 (br), 121.1, 124.1 (br), 124.2 (br), 125.2, 126.7, 128.1, 128.5, 128.9, 129.0, 130.4, 138.5, 140.0 (br), 146.9, 148.6, 154.7 (br) ppm. IR (neat): ν = 3127, 3103, 3064, 3035, 2940, 1607, 1572, 1486, 1444, 1429, 1025, 764, 697, 648 cm−1. HRMS (ESI+) calcd for C28H27N6 ([M − ZnCl2 + H]+): 447.2292; found 447.2290. C28H26Cl2N6Zn (582.86): calcd C 57.70, H 4.50, N 14.42; found C 57.61, H 4.27, N 14.55.
Complex [Ni(11)Cl2] (16).
The mixture of 11 (77 mg, 0.217 mmol), NiCl2·6H2O (52 mg, 0.219 mmol), and MeOH (2 mL) was stirred for 1 day, then treated with Et2O (1 mL) and cooled. After approximately one week blue crystals were filtered off, washed with MeOH/Et2O (2:1) and dried to give 56 mg (53%) of the product. Crystals suitable for X-ray analysis were obtained from MeOH/Et2O (2:1) solution at 4 °C. IR (neat): ν = 3160, 2918, 1608, 1481, 1456, 1441, 1333, 1160, 1083, 1024, 975, 961, 932, 769, 724, 696 cm−1. HRMS (ESI+) calcd for C22H21ClN5Ni ([M − Cl]+): 448.0833; found 448.0833. C22H21Cl2N5Ni·H2O·0.5MeOH: calcd C 52.06, H 4.85, N 13.49; found C 52.08, H 4.41, N 13.72.
Acknowledgements
We thank the Ministry of Higher Education, Science and Technology of the Republic of Slovenia and the Slovenian Research Agency for financial support (P1-0230-0103, P1-0230-0175) and Ministry of Science, Education and Sports of the Republic of Croatia (grant 098-1191344-2943) for financial support. Dr D. Žigon (Center for Mass Spectroscopy, “Jožef Stefan” Institute, Ljubljana, Slovenia) is gratefully acknowledged for the mass measurements. This work was partially supported by the infrastructure of the EN-FIST Centre of Excellence, Trg Osvobodilne fronte 13, SI-1000 Ljubljana, Slovenia. We also thank S. Marković, N. Dragoš and J. Bobnar for their laboratory assistance.
Notes and references
-
(a) J. Suh, Acc. Chem. Res., 1992, 25, 273 CrossRef CAS;
(b) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Jr., Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed;
(c) F. G. Mutti, M. Gullotti, L. Casella, L. Santagostini, R. Pagliarin, K. K. Andersson, M. F. Iozzi and G. Zoppellaro, Dalton Trans., 2011, 40, 5436 RSC.
- J. C. M. Rivas, E. Salvagni, R. Prabaharan, R. T. M. de Rosales and S. Parsons, Dalton Trans., 2004, 172 RSC.
-
(a) W. N. Lipscomb and N. Sträter, Chem. Rev., 1996, 96, 2375 CrossRef CAS PubMed;
(b) E. L. Hegg and J. N. Burstyn, Coord. Chem. Rev., 1998, 173, 133 CrossRef CAS.
-
Handbook of Metalloproteins, ed. A. Messerschmidt, R. Huber, T. Poulos and K. Wieghardt, Wiley, Chichester, 2001, vol. 1 and 2 Search PubMed.
- E. T. Papish, M. T. Taylor, F. E. Jernigan III, M. J. Rodig, R. R. Shawhan, G. P. A. Yap and F. A. Jové, Inorg. Chem., 2006, 45, 2242 CrossRef CAS PubMed.
- A. Beck, B. Weibert and N. Burzlaff, Eur. J. Inorg. Chem., 2001, 521 CrossRef CAS.
- P. C. A. Bruijnincx, I. L. C. Buurmans, S. Gosiewska, M. A. H. Moelands, M. Lutz, A. L. Spek, G. van Koten and R. J. M. Klein Gebbink, Chem. – Eur. J., 2008, 14, 1228 CrossRef CAS PubMed.
-
(a) E. L. Hegg and L. Que, Jr., Eur. J. Biochem., 1997, 250, 625 CAS;
(b) S. J. Lange and L. Que, Jr., Curr. Opin. Chem. Biol., 1998, 2, 159 CrossRef CAS;
(c) L. Que, Jr., Nat. Struct. Biol., 2000, 7, 182 CrossRef PubMed.
-
(a) C. Dowling, V. J. Murphy and G. Parkin, Inorg. Chem., 1996, 35, 2415 CrossRef CAS PubMed;
(b) P. Ghosh and G. Parkin, J. Chem. Soc., Dalton Trans., 1998, 2281 RSC;
(c) A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda and A. Lara-Sánchez, Dalton Trans., 2004, 1499 RSC , and references therein;
(d) M. Ortiz, A. Díaz, R. Cao, R. Suardíaz, A. Otero, A. Antiñolo and J. Fernández-Baeza, Eur. J. Inorg. Chem., 2004, 3353 CrossRef CAS;
(e) K. Kervinen, P. C. A. Bruijnincx, A. M. Beale, J. G. Mesu, G. van Koten, R. J. M. Klein Gebbink and B. M. Weckhuysen, J. Am. Chem. Soc., 2006, 128, 3208 CrossRef CAS PubMed;
(f) A. Otero, J. Fernández-Baeza, J. Tejeda, A. Lara-Sánchez, M. Sánchez-Molina, S. Franco, I. López-Solera and A. M. Rodríguez, Inorg. Chem., 2009, 48, 5540 CrossRef CAS PubMed;
(g) M. Honrado, A. Otero, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2014, 33, 1859 CrossRef CAS.
- H. Vahrenkamp, Acc. Chem. Res., 1999, 32, 589 CrossRef CAS.
-
(a) S. Trofimenko, J. C. Calabrese and J. S. Thompson, Inorg. Chem., 1987, 26, 1507 CrossRef CAS;
(b) S. Trofimenko, Chem. Rev., 1993, 93, 943 CrossRef CAS;
(c) N. Kitajima and Y. Moro-oka, Chem. Rev., 1994, 94, 737 CrossRef CAS;
(d) N. Kitajima and W. B. Tolman, Prog. Inorg. Chem., 1995, 43, 419 CrossRef CAS;
(e)
S. Trofimenko, Scorpionates: The Coordination Chemistry of Polypyrazolylborate Ligands, Imperial College Press, London, 1999 Search PubMed;
(f) G. Parkin, Chem. Commun., 2000, 1971 RSC.
-
(a) A. Višnjevac, J. Gout, N. Ingert, O. Bistri and O. Reinaud, Org. Lett., 2010, 12, 2044 CrossRef PubMed;
(b) J. Gout, A. Višnjevac, S. Rat, A. Parrot, A. Hessani, O. Bistri, N. Le Poul, Y. Le Mest and O. Reinaud, Inorg. Chem., 2014, 53, 6224 CrossRef CAS PubMed;
(c) J. Gout, A. Višnjevac, S. Rat, O. Bistri, N. Le Poul, Y. Le Mest and O. Reinaud, Eur. J. Inorg. Chem., 2013, 5171 CrossRef CAS ; and the references therein.
- G. Aromí, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485 CrossRef PubMed.
- H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675 RSC.
-
J. D. Crowley and D. McMorran, in Top. Heterocycl. Chem., ed. J. Košmrlj, Springer-Verlag, Berlin, 2012, vol. 28, p. 31 Search PubMed.
- T. Y. Kim, A. B. S. Elliot, K. J. Shaffer, C. J. McAdam, K. C. Gordon and J. D. Crowley, Polyhedron, 2013, 52, 1391 CrossRef CAS PubMed.
-
(a) D. Wang, D. Denux, J. Ruiz and D. Astruc, Adv. Synth. Catal., 2013, 355, 129 CrossRef CAS;
(b) K. J. Kilpin and J. D. Crowley, Polyhedron, 2010, 29, 3111 CrossRef CAS PubMed.
- Y. Li, J. C. Huffman and A. H. Flood, Chem. Commun., 2007, 2692 RSC.
- P. M. Guha, H. Phan, J. S. Kinyon, W. S. Brotherton, K. Sreenath, J. T. Simmons, Z. Wang, R. J. Clark, N. S. Dalal, M. Shatruk and L. Zhu, Inorg. Chem., 2012, 51, 3465 CrossRef CAS PubMed.
- J. T. Simmons, J. R. Allen, D. R. Morris, R. J. Clark, C. W. Levenson, M. W. Davidson and L. Zhu, Inorg. Chem., 2013, 52, 5838 CrossRef CAS PubMed.
-
(a) B. S. Uppal, A. Zahid and P. I. P. Elliott, Eur. J. Inorg. Chem., 2013, 2571 CrossRef CAS;
(b) J. T. Fletcher, B. J. Bumgarner, N. D. Engels and D. A. Skoglund, Organometallics, 2008, 27, 5430 CrossRef CAS;
(c) C. Richardson, C. M. Fitchett, F. R. Keene and P. J. Steel, Dalton Trans., 2008, 2534 RSC.
-
(a) H. Struthers, B. Spingler, T. L. Mindt and R. Schibli, Chem. – Eur. J., 2008, 14, 6173 CrossRef CAS PubMed;
(b) A. Maisonial, P. Serafin, M. Traïkia, E. Debiton, V. Théry, D. J. Aitken, P. Lemoine, B. Viossat and A. Gautier, Eur. J. Inorg. Chem., 2008, 298 CrossRef CAS.
- A. Bastero, D. Font and M. A. Pericàs, J. Org. Chem., 2007, 72, 2460 CrossRef CAS PubMed.
- Selected examples:
(a) D. Urankar, A. Pevec and J. Košmrlj, Eur. J. Inorg. Chem., 2011, 1921 CrossRef CAS;
(b) I. Bratsos, D. Urankar, E. Zangrando, P. Genova-Kalou, J. Košmrlj, E. Alessio and I. Turel, Dalton Trans., 2011, 40, 5188 RSC;
(c) B. Pinter, A. Demšar, D. Urankar, F. De Proft and J. Košmrlj, Polyhedron, 2011, 30, 2368 CrossRef CAS PubMed;
(d) J. D. Crowley, P. H. Bandeen and L. R. Hanton, Polyhedron, 2010, 29, 70 CrossRef CAS PubMed;
(e) K. J. Kilpin, E. L. Gavey, C. J. McAdam, C. B. Anderson, S. J. Lind, C. C. Keep, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2011, 50, 6334 CrossRef CAS PubMed;
(f) C. B. Anderson, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Organometallics, 2013, 32, 788 CrossRef CAS;
(g) D. Urankar, B. Pinter, A. Pevec, F. De Proft, I. Turel and J. Košmrlj, Inorg. Chem., 2010, 49, 4820 CrossRef CAS PubMed;
(h) W. S. Brotherton, P. M. Guha, H. Phan, R. J. Clark, M. Shatruk and L. Zhu, Dalton Trans., 2011, 40, 3655 RSC;
(i) Y. Fu, H. Li, X. Chen and J. Qin, Inorg. Chem. Commun., 2011, 14, 268 CrossRef CAS PubMed;
(j) E. Amadio, M. Bertoldini, A. Scrivanti, G. Chessa, V. Beghetto, U. Matteoli, R. Bertani and A. Dolmella, Inorg. Chim. Acta, 2011, 370, 388 CrossRef CAS PubMed.
- G.-C. Kuang, H. A. Michaels, J. T. Simmons, R. J. Clark and L. Zhu, J. Org. Chem., 2010, 75, 6540 CrossRef CAS PubMed.
-
(a) A. Hofmann, D. Jaganyi, O. Q. Munro, G. Liehr and R. Van Eldik, Inorg. Chem., 2003, 42, 1688 CrossRef CAS PubMed;
(b) C. F. Weber and R. Van Eldik, Eur. J. Inorg. Chem., 2005, 4755 CrossRef CAS.
- F. Požgan, L. Toupet and P. H. Dixneuf, Dalton Trans., 2011, 40, 6619 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
-
T. O. Soine and M. R. Buchdahl, Organic Syntheses 1963, Coll., vol. 4, p. 106; 1952, vol. 32, p. 18.
-
CrysAlis PRO, Agilent Technologies, Yarnton, 2011 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- J. Xu and S. Xu, Synthesis, 2004, 276 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.