
Mechanism and application of halogen bond induced fluorescence enhancement and iodine molecule cleavage in solution†
Wen-Sheng
Zou
a,
Sen
Lin
b,
Jia-Yuan
Li
a,
Hong-Qing
Wei
c,
Xiao-Qin
Zhang
a,
Dong-Xu
Shen
a,
Jun-Qin
Qiao
a,
Hong-Zhen
Lian
*a,
Dai-Qian
Xie
*b and
Xin
Ge
b
aState Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry & Chemical Engineering and Center of Materials Analysis, Nanjing University, 22 Hankou Road, Nanjing 210093, China. E-mail: hzlian@nju.edu.cn; Fax: +86 25 8332 5180; Tel: +86 25 8368 6075
bKey Laboratory of Mesoscopic Chemistry (Ministry of Education), School of Chemistry and Chemical Engineering, Nanjing University, 22 Hankou Road, Nanjing 210093, China. E-mail: dqxie@nju.edu.cn; Fax: +86 25 8368 6553; Tel: +86 25 8359 6383
cSchool of Materials Science and Engineering, Shaanxi Normal University, Xian 710062, China
First published on 9th October 2014
Abstract
In this paper, a strong halogen bond (XB) donor (iodine) and photoinduced electron transfer (PET) molecule (ciprofloxacin, Cip) were selected with the objective to investigate halogen bonding under weakly alkaline conditions. A series of experimental characterization techniques was employed to elucidate the interaction mechanism of the XB, in combination with theoretical calculations. It is found that new UV-Vis absorption peaks and the fluorescence enhancement with the mixing of Cip and iodine are attributed to the disruption of the PET charge separation process through the halogen bonding interaction. The 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the XB complex (I2:Cip) was attested using a modified Benesi–Hildebrand method. 1H NMR spectra showed that the iodine molecule can interact with three nitrogen atoms of Cip to form three XBs. FT-IR spectra indicated that the nitrogen atom of the imino group is the preferential interaction site of the XB. Notably, direct analysis in real time-mass spectrometry (DART-MS) gave a distinct quasi-molecular ion of the supramolecular complex (Cip + I) in solution. Meanwhile, density functional theory (DFT), taking into account the dispersion energy, revealed that the formation of an I⋯N XB not only disrupts the PET charge separation process of Cip to enhance fluorescence but also induces the cleavage of an iodine molecule (I–I) to produce a triiodine anion (I3−) XB. This explained why I3− was observed in UV-Vis and DART-MS as well as in the crystal, and how the fourth iodine atom involved in the self-assembly of the XB existed stably. Moreover, a developed optosensor based on halogen bonding has been successfully used to analyze commercial Cip·HCl capsules, suggesting the potential applicability of halogen bonding in real pharmaceutical analyses.
:1 stoichiometry of the XB complex (I2:Cip) was attested using a modified Benesi–Hildebrand method. 1H NMR spectra showed that the iodine molecule can interact with three nitrogen atoms of Cip to form three XBs. FT-IR spectra indicated that the nitrogen atom of the imino group is the preferential interaction site of the XB. Notably, direct analysis in real time-mass spectrometry (DART-MS) gave a distinct quasi-molecular ion of the supramolecular complex (Cip + I) in solution. Meanwhile, density functional theory (DFT), taking into account the dispersion energy, revealed that the formation of an I⋯N XB not only disrupts the PET charge separation process of Cip to enhance fluorescence but also induces the cleavage of an iodine molecule (I–I) to produce a triiodine anion (I3−) XB. This explained why I3− was observed in UV-Vis and DART-MS as well as in the crystal, and how the fourth iodine atom involved in the self-assembly of the XB existed stably. Moreover, a developed optosensor based on halogen bonding has been successfully used to analyze commercial Cip·HCl capsules, suggesting the potential applicability of halogen bonding in real pharmaceutical analyses.
Introduction
The halogen bond (XB), an emerging non-covalent intermolecular interaction analogous to a hydrogen bond (HB),1 occurs between organic halogen atoms (Lewis acids) and those atoms containing lone electron pairs or anionic character (Lewis bases) in organic solvents, crystals or gas phases.2 This interaction is currently named as a XB, as suggested by Legon at the end of the 1990s, to emphasize the similarity with a HB,3 and this nomenclature was recommended by IUPAC in 2013.4 But relative to a HB, the XB is a much stronger non-covalent interaction. For instance, the interaction energy of the strongest F–H⋯F HB is 28.0 kJ mol−1, whereas that of the weakest Cl⋯Cl XB is ∼180 kJ mol−1, and moreover, the stabilization energy of the strongest I2⋯I− XB interaction is much greater. Thus, in some cases, XBs can dominate over HBs when the recognition pattern controlling the self-assembly process is based on either hydrogen bonding or halogen bonding.5 So far, the best part of the endeavors in studying the XB has focused on the organic solid phase involved in materials science,6 drug design,7 and halogenated biomolecules or drugs as ligands with their protein receptors,8 and so on. Meanwhile, the role of XBs in solid phase chemical sensing and molecular recognition has also attracted some of attention in the past decade.9The charge transfer (CT) model seemingly works well in the explanation of experimental absorption spectroscopy findings, the establishment of thermodynamic parameters, and determination of the bonding constants of XB complexes, as described by Kochi’s group.10 The directionality and geometry of a XB, as well as the peculiarity of the halogen–halogen interaction,11 were confirmed by single crystal data.10,12 The CT between XB donor and acceptor species is a well recognized pattern and CT theory gives a good explanation for the excited states of a XB complex. In other CT models, hydrogen bonding is most frequently utilized to assemble organic molecules in solid phases, and also plays an important role in stabilizing supramolecular aggregates in solution,13 while metal ion coordination is generally the key feature of inorganic supramolecular systems,14 and metal ions also act as electron acceptors in photoinduced electron transfer (PET) molecules at all times to inhibit the PET process in solution and cell imaging.15 Compared with the above two usual CT interactions, the study on XB CT in solution is only in its infancy, with very few analytical applications, though the XB CT interaction in solution has been a strong focus of several research groups.1,2,6,16 This is probably because the flaws of a high concentration-dependence or low equilibrium constant have greatly limited the application of XBs in solution.10,16d,17 Recently, the reports regarding XB recognition in solution all employed theoretical calculations or NMR to elucidate the existence of halogen bonding character.18 On the other hand, the weak non-covalent interaction in solution seems to be of great importance, as the interaction largely affects the direction and rate of chemical and biological reactions.19 XBs involving carbonyl or hydroxyl oxygen as the acceptors are particularly interesting in biochemistry, because they are the most common XB types in protein–ligand interactions. Therefore, the development of halogen bonding in solution will not only improve the rate of biological reactions but also facilitate the understanding of the 3-dimensional structures of biomacromolecules.
Ciprofloxacin (Cip, Fig. 1) is a typical organic molecule with a PET pathway from an acceptor to fluorophores across a spacer, which has been experimentally supported by using another representative fluoroquinolone norfloxacin as the model compound.20 Herein, a simple system including an iodine molecule (strong XB donor) and Cip (XB acceptor) is employed to investigate the XB in solution. Various experimental characterization techniques including UV-Vis, fluorescence, FT-IR and 1H NMR spectroscopy, and mass spectrometry, and X-ray diffraction were utilized to decode the interaction mechanism of the XB, in combination with theoretical calculations. It is found that 2 moles of I2 bound 1 mole of Cip, the triiodine anion (I3−) was produced in alkaline solution, and the fourth iodine atom was involved in the XB self-assembly. Consequently, the formation of the I⋯N XB can not only disrupt the PET charge separation process of Cip but also induce the cleavage of an iodine molecule. Moreover, several commercial medicines (Cip·HCl capsules) were used to evaluate the reliability of a developed protocol based on halogen bonding, and the analytical results were in good agreement with the labeled values.
 | ||
| Fig. 1 Molecular structure of Cip and the native positions of protons and nitrogen atoms. | ||
Results and discussion
In order to elucidate the interaction mechanism of Cip and I2via a XB, a diversity of characterization methods were utilized in combination with theoretical calculations. Meanwhile, the XB based optosensor was applied to assay Cip·HCl capsules to assess the viability of using halogen bonding in real pharmaceutical analysis. The detailed experimental conditions, operation procedures and computational methods are listed in the Experimental section.Fluorescence of an ethanolic solution of Cip with iodine titration
Very weak fluorescence signals of Cip were observed in ethanolic solution (5 μg mL−1) over a pH range from 11.5 to 9.05. However, when a very small amount of iodine solution (1 × 10−2 M [I2]) was injected into the above solution at pH 9.05, significant variation in the excitation and emission wavelengths of Cip was observed with tremendous fluorescence enhancement. Fig. 2 depicts the excitation and emission spectra of Cip in ethanol before and after the addition of iodine. The excitation and emission wavelengths of Cip in weakly alkaline solution are located at 276 nm and 433 nm, respectively. With iodine solution being injected, the excitation wavelength blue-shifted to 273 nm while the emission wavelength red-shifted to 438 nm. The fluorescence intensity was enhanced once iodine was added, implying that the charge separation process of Cip was disrupted. The protonation of Cip in an aqueous solution gave the same result (see Fig. S1 in the ESI†).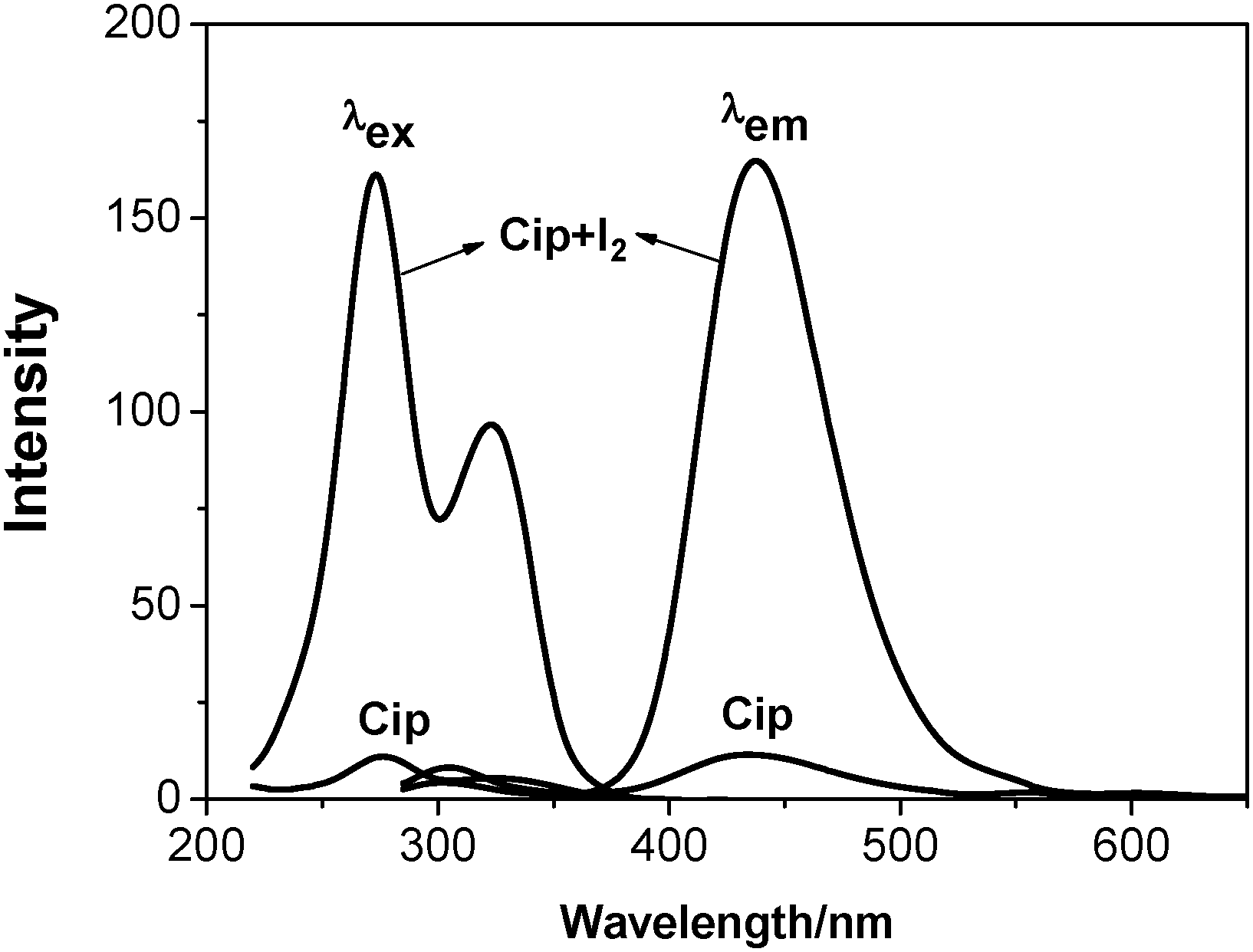 | ||
| Fig. 2 Excitation and emission spectra of Cip before (Cip) and after the addition of iodine (Cip + I2) at pH 9.05. [Cip] = 5 μg mL−1, [I2] = 2 × 10−5 M. | ||
UV-Vis absorption of an ethanolic solution of Cip with iodine titration
The UV-Vis spectra were obtained at the same time as the fluorescence spectra were recorded, while titrating iodine into an ethanolic solution of Cip. The bold curve in Fig. 3 corresponds to the absorption of Cip in alkaline solution, with the peak absorption values located at 277 nm and 326 nm. The peak absorption values of all the other curves at 300 nm and 357 nm are typical of the absorption of the triiodine anion,21 suggesting that the cleavage of an I–I bond was induced by halogen bonding once iodine was added.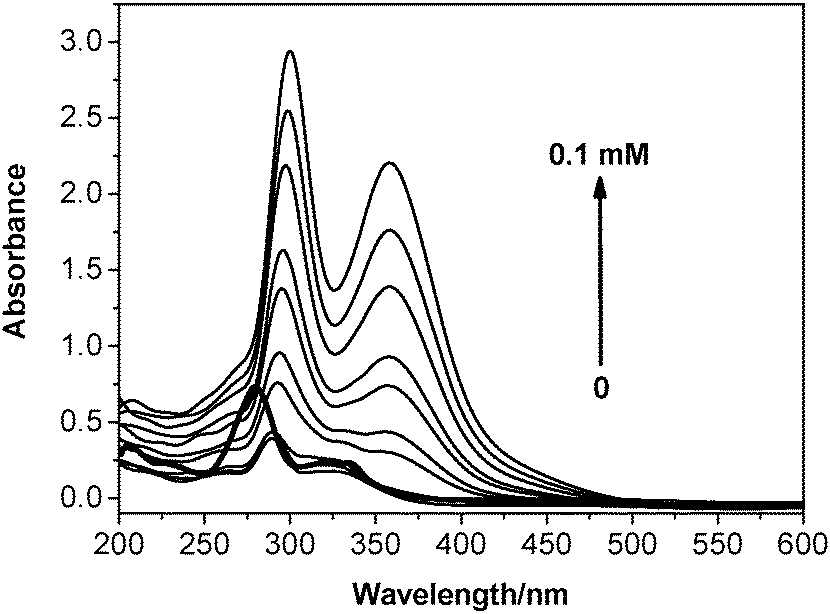 | ||
| Fig. 3 Evolution of the UV-Vis spectra with increasing iodine concentration from 0 to 0.1 mM in the system containing a Cip concentration of 5 μg mL−1 at pH 9.05. The bold curve corresponds to the absorption of 5 μg mL−1 of Cip. | ||
According to the characteristics of the fluorescence enhancement during the protonation of Cip (see Fig. S1 in the ESI†), the above fluorescence enhancement by and large demonstrated that the interaction site of the XB acceptor is most possibly located at the imino group of Cip.22 Although the XB interaction model is as simple as that of the HB, with regard to the generation of a triiodine anion after adding iodine into an ethanolic solution of Cip, a series of questions concerning the other cleaved iodine (that is, the fourth I atom) still requires answering. Is there an iodine cation (I+) in the investigated system? If not, with what valency does the cleaved iodine atom stably exist? How does the cleaved iodine atom interact with Cip? To solve these problems, 1H NMR and FT-IR spectroscopy, and mass spectrometry, and X-ray diffraction on the single crystal structure of the XB complex, as well as theoretical calculations, were employed to investigate the interaction mechanism of the XB.
Stoichiometry of the complexes of Cip with iodine
Generally, the stoichiometry of the complexes of a two-component or multicomponent system can be determined by the Benesi–Hildebrand method if the general assumption is met.23 Herein, the Benesi–Hildebrand formulation should be modified appropriately in order to better describe the specific XB complex system. If D is taken to represent the electron donor, and A the electron acceptor, then the equilibrium can be written as| D + nA = D·An | (1) |
The equilibrium constant is then expressed as
| K = [D·An]/[D]·[A]n | (2) |
| logK = log[D·An] − (log[D] + nlog[A]) | (3) |
| logK = log{[D·An]/[D]} − nlog[A] | (4) |
| log{[D·An]/[D]} = logK + nlog[A] | (5) |
Given that the original fluorescence intensity of D is I0, the fluorescence intensity of the resulting complex and maximal fluorescence intensity are Ii and Imax, respectively. Ci represents the volume of added iodine solution, and
| log(Ii − I0)/(Imax − Ii) = logK+nlogCi | (6) |
The plot of the log(Ii − I0)/(Imax − Ii) versus logCi was linear with correlation coefficients of greater than 0.9972, and n was 1.948. So, the 2:1 complex formed between the donor I2 and acceptor Cip is confirmed (see Fig. S2 in the ESI†).
1H NMR spectroscopy of the three XBs between Cip and iodine
NMR spectra can provide valuable information on molecular interactions. Although the concentration needed in NMR measurements is so high that it is beyond the UV-Vis absorption limit and fails to ensure the same species form, NMR spectroscopy can still provide some clues to explain coordination or aggregation behavior.24In order to confirm the existence of a XB between iodine and Cip, 1H NMR spectra in deuterated methanol were obtained. Fig. 1 labels the native positions of the protons and nitrogen atoms of a Cip molecule, and Fig. 4 shows the corresponding locations of the protons in the 1H NMR spectra. Spectra A and B of Fig. 4 correspond to the 1H NMR spectra of the basic Cip molecule and mixture of Cip and iodine (Cip + I2), respectively. The two highest intensities around 4.87 ppm and 3.33 ppm are ascribed to residual H2O and solvent, respectively. Table 1 lists the corresponding changes in chemical shift of the various protons of Cip before and after coordination with iodine to summarize the NMR behavior of XB-induced electron transfer. The imino group of Cip is an electron-rich group and interacts with protons or other electron-deficient molecules easily to form CT complexes, leading to the change in the emission spectrum, i.e. red/blue-shift or fluorescence enhancement etc.22 Protons 2 and 3 shifting from upfield to downfield indicated that nitrogen atom a of the imino group is coordinated with iodine. Generally, a nitrogen atom containing a lone electron pair neighboring an aromatic ring conjugates with the aromatic ring through p–π conjugation. Such an interaction inevitably increases the charge density of the involved atoms. The lessened chemical shifts of protons 5 and 6 proved that nitrogen b interacted with iodine. Of course, p–π conjugation also contributed to the change of the chemical shifts of protons 2 and 3. Under high acidity, three nitrogen atoms of Cip could be protonated.25 In the 1H NMR experiment, owing to the inherent demand for a high Cip concentration and the solubility of iodine being much higher than that of Cip in methanol, the added iodine was apt to precipitate Cip from the solvent, which made the iodine excessive in the mixed system and then caused it to interact with nitrogen c of Cip by halogen bonding, analogous to protonation. The changes in the shifts of protons 1, 4 and 7 were analyzed similarly. Therefore, the ultra-high concentration of iodine relative to that of Cip facilitated the formation of three XBs, leading to a deshielding effect and corresponding proton shift to downfield.
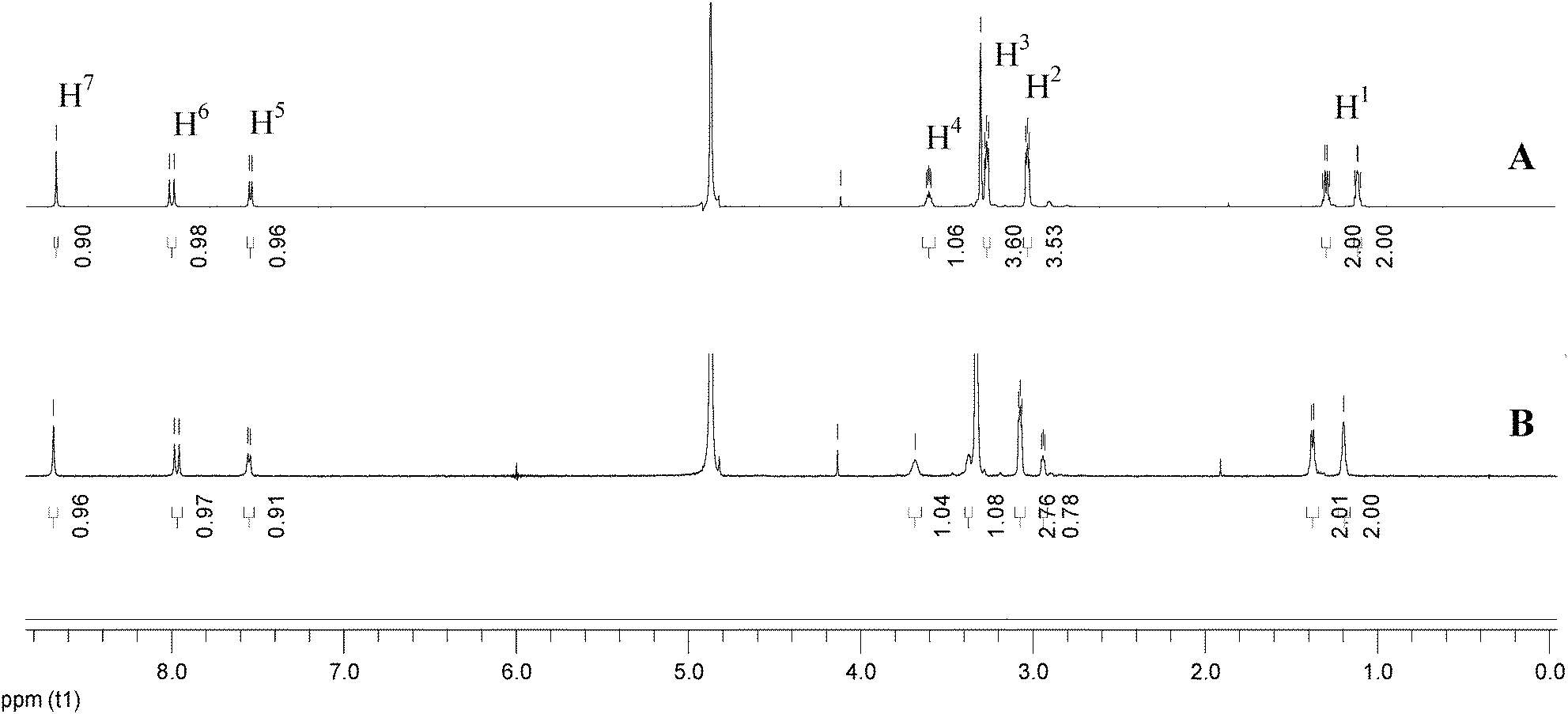 | ||
| Fig. 4 Comparison of the 1H NMR spectra of Cip (A) and the mixture of Cip and iodine (Cip + I2) (B) in deuterated methanol solution. | ||
| H1 | H2 | H3 | H4 | H5 | H6 | H7 | |
|---|---|---|---|---|---|---|---|
| δ(Cip) | 1.171/1.352 | 3.061 | 3.352 | 3.626 | 7.509 | 7.961 | 8.622 |
| δ(Cip + I2) | 1.197/1.378 | 3.074 | 3.330 | 3.683 | 7.550 | 7.971 | 8.686 |
| Δδ | 0.026 | 0.013 | 0.022 | 0.057 | 0.041 | 0.010 | 0.064 |
Preferential interaction site inference through FT-IR spectroscopy
FT-IR spectra of Cip, I2, I3− and Cip + I2 are compared in Fig. 5 to learn the bonding character and preferential interaction site between Cip and iodine. The curve of Cip + I2 had not only the characteristic transmittance peaks of Cip at 1469, 1379 and 1272 cm−1, but also those of I3− at 1393 and 1122 cm−1, which indicated the presence of a triiodine anion in the system. The assembly of iodine onto Cip resulted in the disappearance of the bending vibration of N–H at 1025 cm−1 and characteristic peak around 1578 cm−1, and the decrease of the stretching vibration of N–H at 3445 cm−1, suggesting that the imino group of Cip is the preferential interaction site.26 Moreover, the curve of Cip + I2 also displayed some differences in band number, intensity and position from that of Cip in terms of N–H vibration in the region of 3450–3200 cm−1, and in N–H deformation in the regions of 1500–1000 cm−1 and 850–700 cm−1,27 which also clearly proved, as expected, that the imino group was involved in the formation of the XB complex.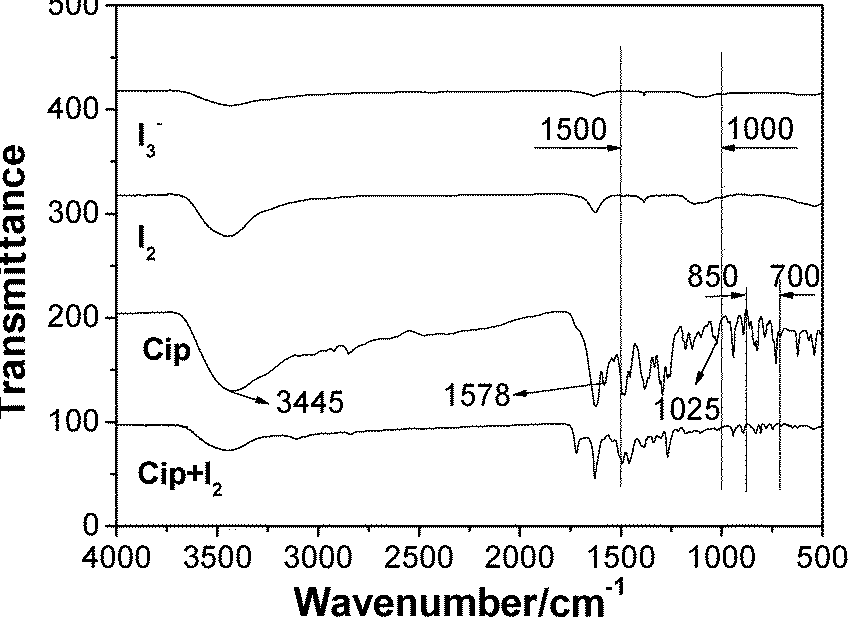 | ||
| Fig. 5 FT-IR for Cip, I2, I3− and Cip + I2. | ||
Single crystal structure
The crystallographic method is a much more powerful tool to assess the undoubted coordination sites, and then to verify the metal ion coordination that inhibits the molecule-based PET mechanism.28 In the present work, the crystallographic method was also employed to obtain useful information about Cip interacting with iodine. The packing diagram along the a, b and c axes, crystal data and structure refinement are shown in Fig. S3A, B and C, and Table S1 (see ESI†), respectively. The X-ray analysis revealed that Cip and iodine form a dimer complex related by a symmetric plane in the unit cell, as shown in Fig. 6. In the crystal, there was an intermolecular HB between the O3 and H2D atoms, and the distance and angle of O3H2DO2 were 1.62 Å and 147°, respectively. Another HB was located between the H3B and I1 atoms, and the distance and angle of N3H3B2DI1 were 2.27 Å and 167°, respectively. The distances were within the sum of van der Waals radii (3.9 Å), indicating the existence of two HBs in the crystal. The I⋯I− separation of 2.7964(10) Å was far shorter than the sum of van der Waals radii (I⋯I, 4.3 Å). The angle of I3–I2⋯I1 was 176.81(3)°, nearly linear and close to the theoretical range of 165(±8)° for a XB angle.29 In addition, the interatom distances of I2N2 and I3N1 were 4.292 Å and 3.894 Å, respectively, longer than the sum of van der Waals radii (I⋯N, 3.53 Å), suggesting that no N⋯I XB exists in the present system.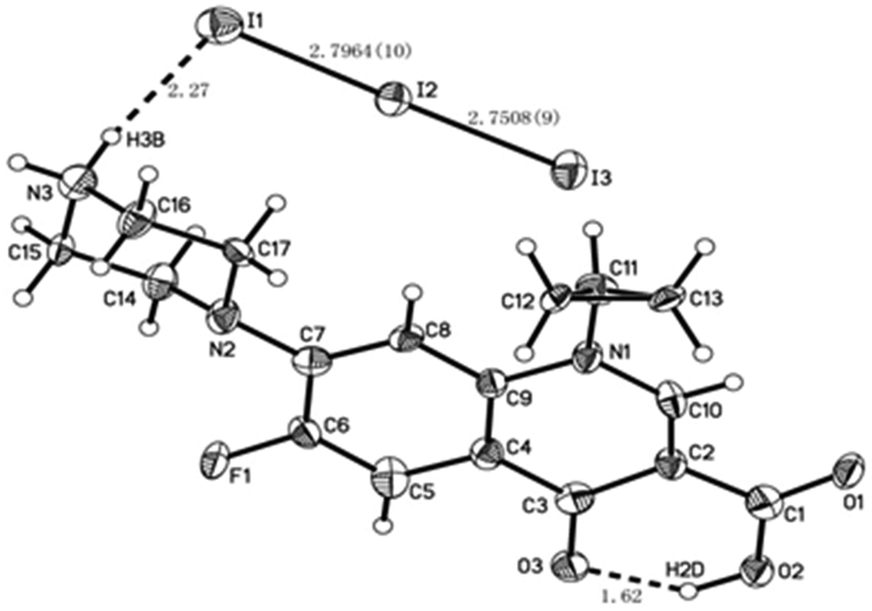 | ||
| Fig. 6 ORTEP drawing of the Cip and iodine structure in a crystal constructed of Cip and iodine. | ||
Study on the mass spectrometry of the XB complex
This is the first attempt to utilize mass spectrometry (MS) for XB study, mainly in order to acquire a straightforward piece of evidence for the XB complex (Cip + I) that did not emerge in all the above experiments, that is, the molecular ion in the mass spectrum of Cip + I, when Cip was mixed with I2 in alkaline solution.The invention of electrospray ionization mass spectrometry (ESI-MS) provided an efficient way to separate analytes from the solution-phase matrix, and so to transfer free ions from solution at atmospheric pressure into the high-vacuum environment required for analysis by MS.30 In ESI-related techniques, analyte molecules are first desorbed or sampled from solid or liquid samples, and then transported into the mass spectrometer in evaporating charged solvent droplets. As an ambient desorption ionization technique for MS, ESI allows for the direct analysis of ordinary objects in the open atmosphere of the laboratory or in their natural environment.31 Based on a soft ionization technique, ESI-MS is often employed to obtain structural information of complexes with non-breakage of weak interactions32 and, moreover, has found use in the field of supramolecular chemistry.33
We used ESI-MS to observe the mass spectrum of Cip + I. When the ESI ionization mode was set to negative ion (Fig. S4A in the ESI†), the I3− ion of m/z 381.3 was found in the mixture containing Cip·HCl and I2, again suggesting the possible existence of a XB between Cip and iodine that disturbed the PET charge separation process of Cip and induced I–I bond cleavage. This is consistent with the results of the UV-Vis spectra and single crystal structure. The peak of m/z 366.3 seemed to represent a quasi-molecular ion [M + Cl]− of Cip while the peak of m/z 458.0 represents the [M + I]− ion of Cip. However, since it is common that quasi-molecular ions of [M + X]− (X is a halogen atom, including I) are observed in negative ion mode even though no halogen is spiked into the sample solution, especially for compounds containing an oxygen atom, the formation of a XB between Cip and I2 is not unquestionable based on the ESI-MS experiment. While the ESI mode was set to positive ion mode, the mass spectra of Cip·HCl and the mixture containing Cip·HCl and I2 had the same base peak at m/z 332.3, that is, the [M + H]+ of Cip (Fig. S4B in the ESI†). The possible reason was that the affinity between the proton and nitrogen atom was much stronger than that between the iodine and nitrogen atoms.
Although ESI-MS is successful with moderately polar to polar molecules that can be dissolved in aqueous solvents, the mass spectra are often complex due to the formation of solvent clusters and multiple-ionization products, such as multiply-charged ions, as well as alkali metal cations and halogen anion adducts ([M + X]−). Therefore, it was not possible to ascertain the formation of molecular ions from the XB complex in the Cip + I2 system utilizing ESI-MS. In addition, as mentioned above, a XB is similar to a HB in terms of their interactions,3,4 therefore, a competitive interaction must occur in a system in which HBs and XBs co-exist. The above fluorescence experiments reflected that donors of HBs (proton) and XBs (iodine) competed with one and the same acceptor (Cip), and that halogen bonding dominated over hydrogen bonding in a weakly alkaline medium (see Fig. 2), whereas hydrogen bonding dominated in an acidic medium (see Fig. S1 in the ESI†). The usual positive ESI ion source of a mass spectrometer is a device generating protons, and so the ionization chamber is equivalent to a highly acidic environment, where it is probably hard for a XB to exist stably. On the other hand, adduct ions of [M + X]− in negative ion mode ESI were not proven to be molecular ions of the XB complex produced in alkaline solution.
Direct analysis in real time (DART) is a new atmospheric pressure chemical ionization (APCI)-related ionization technique that was developed by Cody et al. in 2005.34 Ionization of analytes using DART begins with an electric discharge in a stream of nitrogen or helium gas that produces a plasma of electrons, ions, and metastable species. When helium is used, the ionization mechanism involves the reaction of excited-state helium with water in the atmosphere to produce protonated water clusters, followed by proton transfer to the analyte. Another potential ionization mechanism that has been reported involves Penning ionization, in which a metastable species ionizes a neutral one to produce a radical cation and an electron. In the negative ion mode, negative ions of analytes have been proposed to form from reactions with negatively charged oxygen–water cluster ions.31 DART is compatible with many mass analyzer units. A sample placed in the stream of the He plasma is ionized, generally via protonation, followed by desorption, and the resultant ions make their way through the intake orifice of the analyzer. A slightly offset second orifice (in the analyzer) ensures that only ions, focused by ion guide lenses, make it through to the analyzer and detector.35,36 With the introduction of DART, it became possible to analyze samples directly in their native condition, bypassing most elements of the analytical system and transferring ions into the mass spectrometer without any sample-manipulation or sample-preparation steps.31,34,35,37 Most importantly, DART exposes the sample to a stream of excited gas and does not require a liquid solvent for electrospraying, like in ESI. It is believed that the electronic or vibronic excited-state species (metastable helium atoms or nitrogen molecules) are the working reagents in DART.34 Therefore, the DART technique is able to ionize small polar and non-polar molecules, and the mass range can be extended by heating the sample with a stream of gas. Moreover, the mass spectra are generally simple, dominated by the molecular ions that are mostly the singly-charged protonated or deprotonated molecules or the radical cations (through Penning ionization of the molecules with low ionization potentials). Alkali metal cation adducts and multiply-charged ions are generally not observed.31 These specific merits of DART-MS likely open a new road to XB study.
In order to confirm the formation of a XB between Cip and I2 in solution by MS, pure Cip instead of Cip·HCl was used in the following DART-MS experiment. The mass spectrum of the mixture of alkaline Cip ethanolic solution and I2 ethanolic solution in negative ion mode is given in Fig. 7A. The peaks of m/z 126.9, 253.8 and 380.8 represented I−, I2− and I3−, respectively, and the peak of m/z 458.0 was distinct. Very interestingly, no peak of m/z 458.0 was observed in the mass spectrum of alkaline Cip ethanolic solution and gaseous I2 as the dopant in negative ion mode, however, the peaks of I−, I2− and I3− still emerged (see Fig. 7B). These phenomena mean that the quasi-molecular ion [M + I]− is ascribed to the XB complex of Cip and I2 in alkaline solution, rather than the adduct of Cip and I2 produced in ESI-MS. The simultaneous existence of I−, I2− and I3− in the mass spectrum of alkaline Cip ethanolic solution with gaseous I2 is presumably attributed to the disproportionation reaction of I2 in the alkaline environment of the negative ion ESI chamber. All the other peaks in the original DART mass spectra were from the corresponding solutions and air blank, because the experiments were conducted in a totally open atmosphere in an analytical laboratory. Consequently, the XB between Cip and I2 is definitely formed prior to ESI in the alkaline solution but not in the gas phase, which reasonably supports the conclusion that the halogen bonding interaction inhibits the PET charge separation of Cip and induces I–I cleavage.
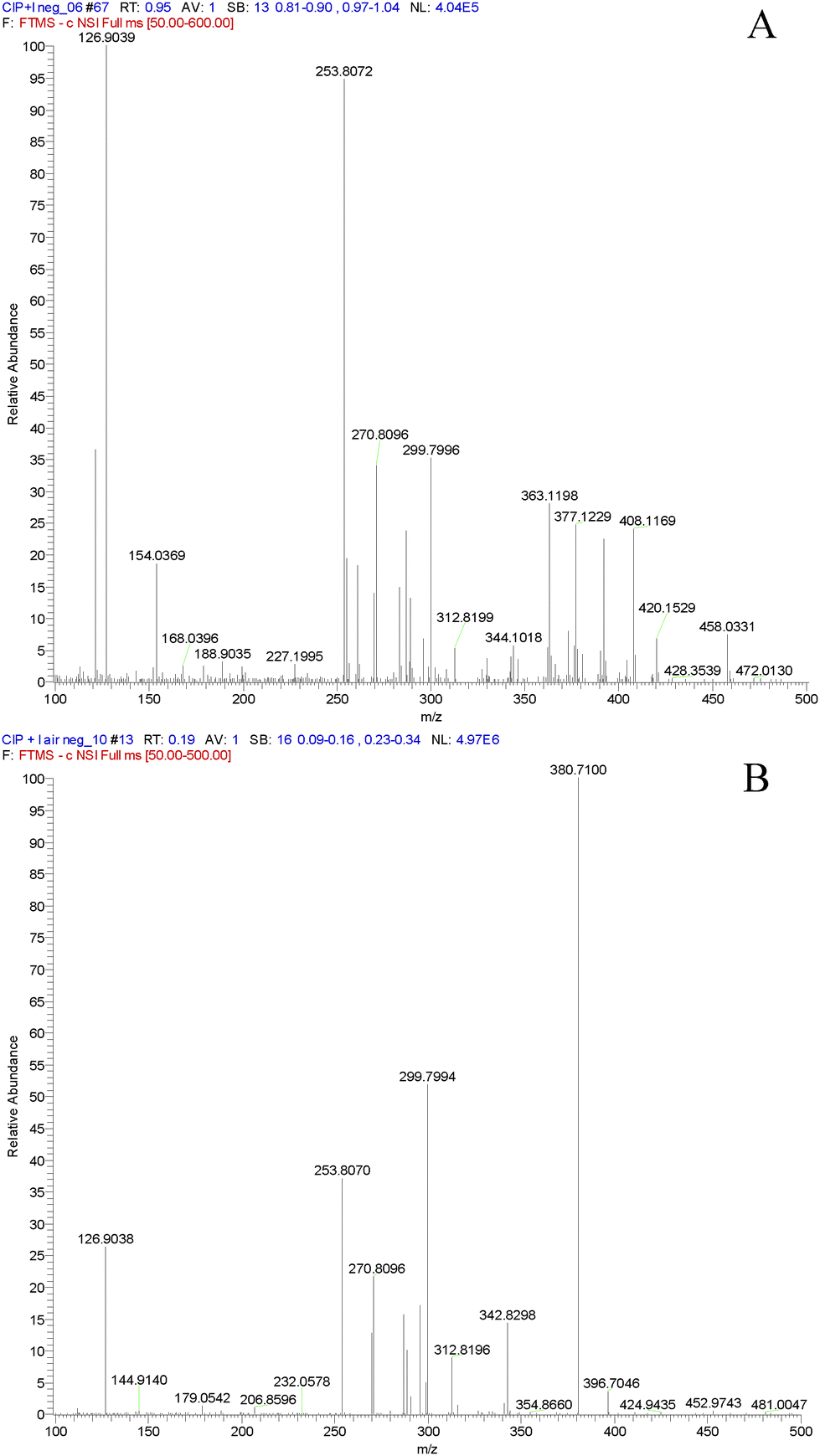 | ||
| Fig. 7 DART-MS spectra of the mixture of alkaline Cip ethanolic solution and iodine ethanolic solution (A) and alkaline Cip ethanolic solution with gaseous iodine indrawn (B) in negative ion mode. Concentration: 100 μg mL−1 each. Full scan: 100–500 amu. | ||
Theoretical results
Fig. 8 shows the theoretically optimized complexes when the proton and iodine species each approach the nitrogen atom of Cip. For the proton species, the distance of H⋯N (1.033 Å) was almost equal to the H–N bond length (1.023 Å), implying that the covalent bond between the proton and the nitrogen atom was fully formed (see Fig. 8-left). It is not surprising, because a proton is more prone to interact with the XB acceptor.16f Therefore, an ion with classical halogen bonding is not observed in the current MS, especially in positive ion mode. While, for the iodine molecule, after full optimization, a novel “L-type” structure was given between two iodine molecules and a XB was formed between I4 and a nitrogen atom of Cip (I4⋯N, 2.472 Å) (see Fig. 8-right). As shown by the I⋯I distances in Fig. 8-right, the XB can help induce the cleavage of the involved iodine. On one hand, the I4⋯N XB facilitated the elongation of the I4–I3 bond (from 2.884 to 3.078 Å). On the other hand, the interaction between the I3 atom and I1I2 led to the formation of a I1I2⋯I3 transition state complex (I2⋯I3, 3.450 Å), which reveals the structural characteristic of I3−. It is to be noted that the absorption peak of I3− was also detected and the stoichiometry was 2:1 for iodine and Cip in the present experimental work, indicating a very good agreement between the theoretical and experimental findings.
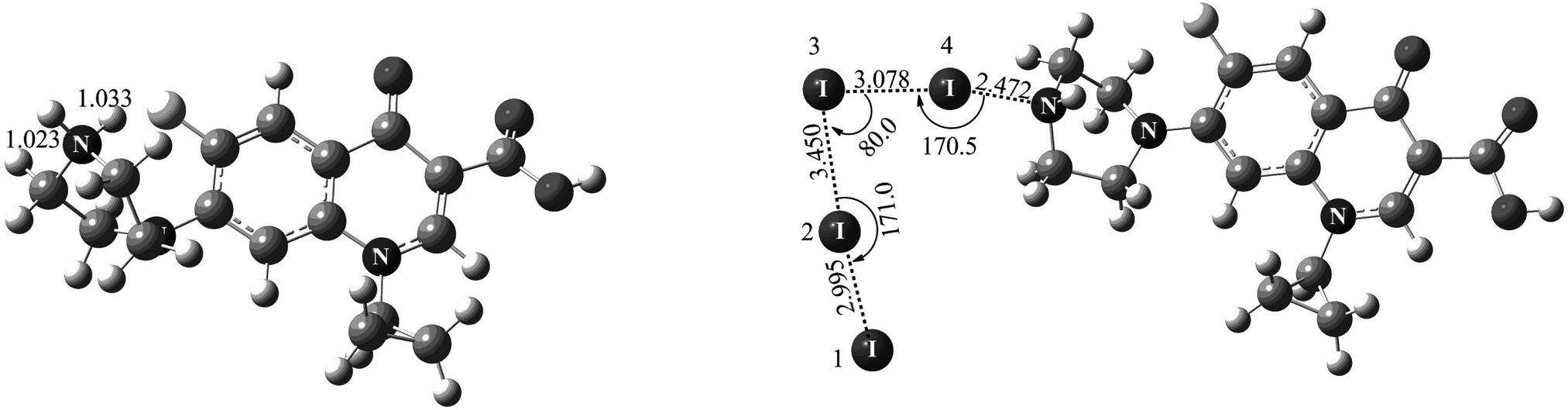 | ||
| Fig. 8 Optimized complexes when a proton (left) and iodine (right) near the nitrogen atom. Only N and I atoms are labelled. The units for distance and angle are angstroms (Å) and degrees (°), respectively. | ||
With the aim of ascertaining the charge character in the XB, Mülliken charges were also taken into account in our calculations. The calculated Mülliken charges of I1, I2, I3 and I4 participating in the formation of the complex were −0.147, −0.046, −0.109 and −0.022, respectively. So the I1I2⋯I3 complex (I3−) displayed an electronegative character, further confirming the experimental observations. Because the proton and nitrogen atom possess positive and negative charges respectively, it was very facile for the proton to connect with the N atom of Cip. Besides, when a proton approached Cip, the charge of the proton became more negative, indicating that part of the positive charge had transferred from the proton to Cip. As far as we know, blue-shift type XBs are rare in common experiments.16d,38 In order to figure out whether I⋯N in our system is a blue- or red-shift type XB, bond length and vibrational frequency analyses were carried out. As expected, the calculated results showed that the bond lengths of I1I2 and I3I4 were elongated by 0.111 and 0.194 Å, respectively, relative to that of I2 (2.884 Å). In addition, compared to the calculated frequency of I2 (181.37 cm−1), the vibrational frequencies of I1I2 and I3I4 decreased by 30.47 and 39.32 cm−1, respectively, indicating that I⋯N was a red-shift type XB.
Analytical application
Iodine is of great importance in halogen bonding, just as the proton is in hydrogen bonding, and we truly hope that the XB can reach the altitude of the HB in real-world applications as soon as possible. For this purpose, commercial Cip·HCl capsules were used to evaluate the reliability of the developed method based on halogen bonding. The XB-induced fluorescence shows a distinct linear enhancement towards Cip in the concentration ranges of 1.0–5.0 μg mL−1 and 0.05–1.0 μg mL−1 with respective correlation coefficients of 0.9980 and 0.9996. As for the latter, the linear regression equation was Y = 8.89 + 158.29C (where C is the concentration of Cip in μg mL−1 and Y the fluorescence intensity), and the precision for the detection over five replicates of 0.2 μg mL−1 Cip was 3.4% (relative standard deviation, RSD). The limit of detection (LOD), calculated as the concentration of Cip that produced a fluorescence enhancement three times the standard deviation of the blank signal, was 0.01 μg mL−1. The method of XB-induced fluorescence enhancement gave excellent selectivity for detecting Cip in the presence of usually used auxiliary materials, such as starch, talcum powder, lactose, glucose, fructose, stearate, etc., in the Cip·HCl capsule. The fluorescence intensity due to the addition of Cip at 0.2 μg mL−1 was unaffected by 1000-fold excesses of co-existing substances in the capsule. To evaluate the accuracy of the developed method based on halogen bonding, several Cip·HCl capsules purchased from different manufacturers were analyzed. The results are listed in Table S2 in the ESI,† and the content of Cip in these capsules determined by the present method using a simple standard calibration protocol were in good agreement with the certified values, along with quantitative recovery values from 97.2% to 105.4%, demonstrating the potential applicability of the XB-based optosensor for the quantification of Cip in real pharmaceutical samples.Although attempts to validate the applicability of halogen bonding in solution are in their preliminary stages, and halogen bonding has not been used as widely as hydrogen bonding and metal ion coordination in analysis, the XB will attract more and more analysts due to its promising capabilities in a wide range of analytical applications in the near future.
Conclusions
In summary, a simple system containing iodine and ciprofloxacin (Cip) was employed to investigate halogen bond (XB) formation in solution by experimental characterization and theoretical calculations. The comprehensive results indicated that the photoinduced electron transfer (PET) charge separation process of basic Cip is disrupted after the addition of iodine, leading to tremendous fluorescence enhancement, and that the formation of an I⋯N XB induces I–I cleavage and the occurrence of supramolecular complexes (Cip + I) and I3− in solution. This is the first time that XB-based PET has been investigated. The study on the mechanism revealed that the XB donor and acceptor form an inner complex with 2:1 stoichiometry and the XB interaction site lies on the nitrogen atoms of the imino group of the piperazine of Cip. Moreover, it is found that iodine possesses many of the same properties in a XB as a proton does in a hydrogen bond (HB), so a XB has the potential to become not only an indispensable factor to consider for chemical reactions, molecular recognition and medicine design, but also a powerful tool to understand the interaction mechanism of halogen-containing pharmaceuticals with biomolecules. Interestingly, the developed method based on halogen bonding has been successfully utilized to analyze commercial medicines of Cip. The present finding will emphasise the importance of studies that range from examining the physical and chemical properties of XBs to exploring their analytical application in solution, and provide novel insights in the design of new halogen-containing pharmaceuticals. Finally, the endeavors to further explore the exact mechanism of the fluorescence enhancement of Cip and iodine cleavage are currently in progress.
Experimental
Materials
Ciprofloxacin hydrochloric acid (Cip·HCl) with a purity of over 99.5% was kindly gifted by Jiangsu Entry Exit Inspect & Quarantine Bureau (Nanjing, China). Cip stock solution of 100 μg mL−1 was prepared by dissolving this in ethanol. Ciprofloxacin (Cip) with a purity of over 98% was purchased from Tokyo Chemical Industry Company (Tokyo, Japan). Cip stock solution of 100 μg mL−1 was prepared by dissolving this in ethanol with the assistance of NaOH. Iodine used as sublimed was the product of Shanghai Chemical Reagent Factory (Shanghai, China) and prepared as I2 solution of 1 × 10−2 M in ethanol. Purified water was produced using an Elix 5 pure water system (Millipore, Milwood, MA, USA). Deuterated methanol was purchased from Cambridge Isotope Laboratories (Andover, MA, USA). All other reagents were of analytical grade and used without further purification.Preparation
For the preparation of the solid sample for FT-IR, an appropriate amount of Cip·HCl was dissolved in absolute ethanol, and the hydrochloric acid was neutralized by 0.25 M sodium hydroxide in ethanol. Then, iodine solution (double the stoichiometric amount of Cip) was added dropwise, and the resultant solution was placed in the dark. After naturally drying at room temperature, an orange powder was obtained. As for the triiodine anion salt, 1:1 stoichiometric amounts of iodine and KI salt were dissolved in ethanol at the same time. After the reactant dried under vacuum for 48 hours, the powder was obtained.
For the preparation of the sample for 1H NMR spectroscopy, an appropriate amount of Cip·HCl was dissolved in absolute ethanol, and 0.25 M sodium hydroxide ethanol solution, approaching 1.4 times the stoichiometric amount of Cip, was added subsequently. The weakly basic Cip powder was obtained after the resulting solution was dried under vacuum for 24 hours. About 5.0 mg Cip powder was dissolved in 0.5 mL deuterated methanol, and sublimed iodine (nearly double the stoichiometric amount of Cip) was added. This mixture was left to stand for 30 minutes prior to the 1H NMR experiment.
Preparation of the crystalline Cip and iodine compound. Cip (0.02 mg) and iodine (0.056 mg) in 1:2 molar ratio were dissolved in 10 mL mixed solution of 1:1 (v/v) chloroform and ethanol. The beaker was covered by a thin film with holes and then placed in a refrigerator. The violet block crystal was obtained on the bottom of the beaker one month later.
Apparatus
UV-Vis absorption spectra were obtained using a UV-3600 UV-Vis spectrophotometer (Shimadzu, Tokyo, Japan). Fluorescence spectra were recorded on a RF-5301PC (Shimadzu, Tokyo, Japan) fluorescence spectrophotometer with an excitation and emission slit width of 5 nm and photomultiplier tube voltage of 700 V. Fluorescence at an emission wavelength of 435 nm was monitored at an excitation wavelength of 278 nm. 1H NMR spectra were obtained on a DXR-500 NMR 500 MHZ superconducting nuclear magnetic resonance apparatus (Bruker, Switzerland), with the chemical shift expressed using tetramethylsilane as an internal standard. The acidity was measured with a PB-10 pH meter (Sartorius, Germany). FT-IR spectra (4000–400 cm−1) in KBr were recorded on a Nicolet-6700 spectrometer (Nicolet, Madison, WI, USA). Single crystal data were collected at room temperature with a SMART APEX diffractometer (Bruker, Germany) using Cu Kα X-ray radiation (λ = 1.54056 Å), a graphite monochromator and φ and ω scans, and 2700 reflections were collected, with 2100 independent reflections (Rint ≥ 0.0675). Data collection and reduction were performed using SMART and SAINT and absorption correction. The structures were solved by a direct method with SHELXS-97 and refined full-matrix least-squares on F2 with SHELXL-97. CCDC 805819.ESI-MS analyses were carried out using a Surveyor LC system coupled to an LCQ Deca XP plus ion trap mass spectrometer (Thermo Finnigan, San Jose, CA, USA) with an ESI interface. The experiment was performed by infusing individual standard solution (100 μg mL−1) via a syringe pump at a flow rate of 10 μL min−1. The parameters of the interface were as follows: positive/negative ion mode; source collision induced dissociation (SID), 10 V; sheath gas (nitrogen) pressure, 4.0; aux gas (nitrogen) pressure, 0; ion sweep gas (nitrogen) pressure, 0; capillary temperature, 250 °C, spray voltage 4.0 KV; full scan, 150–500 amu.
The DART-MS system employed in this study consisted of a DART-SVP ion source (IonSense, Saugus, MA, USA) coupled to LTQ-Orbitrap XL benchtop orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). A Vapur™ gas ion separator interface (IonSense, Saugus, MA, USA) was used to combine the ion source and the mass spectrometer. A low vacuum in the interface chamber was maintained by a membrane pump (Vacuubrand, Wertheim, Germany). The use of the Vapur™ gas ion separator during DART ionization was essential to maintain a stable vacuum within the operating pressure limits of the mass spectrometer. The distance between the exit of the DART source and the ceramic transfer tube of the Vapur™ was set to 10 mm; and the gap between the ceramic tube and the inlet to the heated capillary of the LTQ-Orbitrap XL was 2 mm. The DART-MS instrument was operated in either positive or negative ionization mode, and the settings of the system parameters were as follows: (i) DART ionization: helium flow: 3 L min−1; gas temperature: 350 °C; discharge needle voltage: 4 KV; grid electrode: ±350 V; (ii) mass spectrometric detection for positive mode: capillary voltage 25 V, tube lens voltage 20 V, capillary temperature 200 °C; for negative mode: capillary voltage −41 V, tube lens voltage −100 V, capillary temperature 200 °C. The sheath, auxiliary and sweep gases were disabled during the DART-MS analysis. The acquisition rate was set to 2 spectra per s, and under this setting the mass resolving power calculated for m/z 200 was 50000 FWHM (full width at half maximum). Samples were analyzed by dipping the end of a melting point tube into the solution of analyte(s) in ethanol, such that approximately 2 μL of sample was sampled, and subsequently inserting the melting point tube into the center of the helium gas stream at the outlet of the source. The concentrations of the sample solutions were 100 μg mL−1 for Cip unless stated otherwise. In addition, to check the XB formation between Cip solution and the XB donor in the gas phase, a small vial of I2 was opened near the DART source.
Computational methods
All the calculations were performed using Gaussian 09 program packages.39 Molecular geometries of the model complexes were fully optimized using density functional theory (DFT) with Grimme's functional including dispersion (B97D).40 The iodine atom was described using the effective core potential basis set of LANL2DZ.41 The basis set of 6-311+G(d,p) was used for all of the other atoms. By allowing all the parameters to be relaxed, the calculations converged to optimized geometries and the required structural parameters. Furthermore, in order to get the correlative vibrational strength, frequency calculations at the same level have also been carried out.Acknowledgements
This research was financially supported by National Basic Research Program of China (973 program, 2009CB421601, 2011CB911003), National Natural Science Foundation of China (21275069, 90913012), National Science Funds for Creative Research Groups (21121091), and Analysis & Test Fund of Nanjing University. Authors sincerely thank Professor Wei Jun Jin, College of Chemistry, Beijing Normal University, for his idea and valuable discussion. Authors thank Dr Jun Zhang and Professor Dr Yi-zhi Li, State Key Laboratory of Coordination Chemistry, Nanjing University, for their help in the collection and decoding of single crystal data. Also, authors thank Miss Xiao-yan Yang for participating in part of the experiments.Notes and references
- P. Metrangolo and G. Resnati, Science, 2008, 321, 918–919 CrossRef CAS PubMed.
- (a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed; (b) P. Politzer, P. Lane, M. C. Concha, Y. G. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305–311 CrossRef CAS PubMed.
- (a) A. C. Legon, Chem. – Eur. J., 1998, 4, 1890–1897 CrossRef CAS; (b) A. C. Legon, Angew. Chem., Int. Ed., 1999, 38, 2686–2714 CrossRef.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- E. Corradi, S. V. Meille, M. T. Messina, P. Metrangolo and G. Resnati, Angew. Chem., Int. Ed., 2000, 39, 1782–1786 CrossRef CAS.
- (a) P. Metrangolo, V. Carcenac, M. Lahtinen, T. Pilati, K. Rissanen, A. Vij and G. Resnati, Science, 2009, 323, 1461–1464 CrossRef CAS PubMed; (b) Q. J. Shen, H. Q. Wei, W. S. Zou, H. L. Sun and W. J. Jin, CrystEngComm, 2012, 14, 1010–1015 RSC; (c) H. Y. Gao, Q. J. Shen, X. R. Zhao, X. Q. Yan, Y. Pan and W. J. Jin, J. Mater. Chem., 2012, 22, 5336–5343 RSC; (d) X. Q. Yan, Q. J. Shen, X. R. Zhao, H. Y. Gao, X. Pang and W. J. Jin, Anal. Chim. Acta, 2012, 753, 48–56 CrossRef CAS PubMed; (e) L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed; (f) J. Marti-Rujas, L. Meazza, G. K. Lim, G. Terraneo, T. Pilati, K. D. M. Harris, P. Metrangolo and G. Resnati, Angew. Chem., Int. Ed., 2013, 52, 13444–13448 CrossRef CAS PubMed; (g) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed; (h) X. Q. Yan, H. Wang, W. D. Chen and W. J. Jin, Anal. Sci., 2014, 30, 365–370 CrossRef CAS.
- (a) J. M. Vargason, B. F. Eichman and P. S. Ho, Nat. Struct. Biol., 2000, 7, 758–761 CrossRef CAS PubMed; (b) Y. Jiang, A. A. Alcaraz, J. M. Chen, H. Kobayashi, Y. J. Lu and J. P. Snyder, J. Med. Chem., 2006, 49, 1891–1899 CrossRef CAS PubMed.
- (a) H. Matter, M. Nazaré, S. Gussregen, D. W. Will, H. Schreuder, A. Baer, M. Uramann, K. Ritter, M. Wagner and V. Vehner, Angew. Chem., Int. Ed., 2009, 48, 2911–2916 CrossRef CAS PubMed; (b) P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed; (c) A. R. Voth, F. A. Hays and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6188–6193 CrossRef CAS PubMed.
- (a) A. Mele, P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2005, 127, 14972–14973 CrossRef CAS PubMed; (b) G. Gattuso, R. Liantonio, P. Metrangolo, F. Meyer, G. Resnati, A. Pappalardo, M. F. Parisi, T. Pilati and I. Pisagatti, Supramol. Chem., 2006, 18, 235–243 CrossRef CAS; (c) K. Tanaka, D. Fujimot, A. Altreuther, T. Oese, H. Irngartinger and F. Toda, J. Chem. Soc., Perkin Trans., 2000, 2115–2120 RSC; (d) K. Tanaka, D. Fujimoto, T. Oeser, H. Irngartinger and F. Toda, Chem. Commun., 2000, 413–414 RSC; (e) A. Farina, S. V. Meille, M. T. Messina, P. Metrangolo, G. Resnati and G. Vecchio, Angew. Chem., Int. Ed., 1999, 38, 2433–2436 CrossRef CAS; (f) A. Cassnati, G. Cavallo, P. Metrangolo, G. Resnati, F. Ugozzoli and R. Ungaro, Chem. – Eur. J., 2009, 15, 7903–7912 CrossRef PubMed; (g) F. Zapata, A. Caballero, N. G. White, T. D. W. Claridge, P. J. Costa, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2012, 134, 11533–11541 CrossRef CAS PubMed; (h) K. Fukuroi, K. Takahashi, T. Mochida, T. Sakurai, H. Ohta, T. Yamamoto, Y. Einaga and H. Mori, Angew. Chem., Int. Ed., 2014, 53, 1983–1986 CrossRef CAS PubMed.
- S. V. Rosokha, I. S. Neretin, T. Y. Rosokha, J. Hecht and J. K. Kochi, Heteroat. Chem., 2006, 17, 449–459 CrossRef CAS.
- (a) E. Bosch and C. L. Barnes, Cryst. Growth Des., 2002, 2, 299–302 CrossRef CAS; (b) C. M. Reddy, M. T. Kirchner, R. C. Gundakaram, K. A. Padmanabhan and G. R. Desiraju, Chem. – Eur. J., 2006, 12, 2222–2234 CrossRef CAS PubMed; (c) C. B. Aakeroy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- (a) H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS; (b) O. Hassel and C. Rømming, Quart. Rev., 1962, 16, 1–18 RSC.
- (a) G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed; (b) O. Ermer and A. Eling, J. Chem. Soc., Perkin Trans., 1994, 925–930 RSC; (c) C. B. Aacheröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–405 RSC.
- J. P. Sauvage, Transition Metals in Supramolecular Chemistry, Wiley, Chichester, 1999 Search PubMed.
- (a) T. Chang, Y. Xu, S. Zhang, W. Zhu, X. Qian and L. Duan, J. Am. Chem. Soc., 2008, 130, 16160–16161 CrossRef PubMed; (b) M. Taki, M. Desaki, A. Ojida, S. Iyoshi, T. Hirayama, I. Hamuchi and Y. Yamamoto, J. Am. Chem. Soc., 2008, 130, 12564–12565 CrossRef CAS PubMed; (c) H. Y. Lee, D. R. Bae, J. C. Park, H. Song, W. S. Han and J. H. Jung, Angew. Chem., Int. Ed., 2009, 48, 1239–1243 CrossRef CAS PubMed; (d) A. Ojida, I. Takashima, T. Kohira, H. Nonaka and I. Hamachi, J. Am. Chem. Soc., 2008, 130, 12095–12101 CrossRef CAS PubMed; (e) F. Qian, C. Zhang, Y. Zhang, W. He, X. Gao, P. Hu and Z. Guo, J. Am. Chem. Soc., 2009, 131, 1460–1468 CrossRef CAS PubMed.
- (a) P. L. Wash, S. H. Ma, U. Obst and J. Rebek Jr, J. Am. Chem. Soc., 1999, 121, 7973–7974 CrossRef CAS; (b) S. C. Blackstock, J. P. Lorand and J. K. Kochi, J. Org. Chem., 1987, 52, 1451–1460 CrossRef CAS; (c) W. J. Liang, Y. Wang, F. Feng and W. J. Jin, Anal. Chim. Acta, 2006, 572, 295–302 CrossRef CAS PubMed; (d) W. S. Zou, J. Han and W. J. Jin, J. Phys. Chem. A, 2009, 113, 10125–10132 CrossRef CAS PubMed; (e) A. C. B. Lucassen, M. Vartanian, G. Leitus and M. E. van der Boom, Cryst. Growth Des., 2005, 5, 1671–1673 CrossRef CAS; (f) R. Cabot and C. A. Hunter, Chem. Commun., 2009, 2005–2007 RSC; (g) S. V. Lindeman, J. Hecht and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 11597–11606 CrossRef CAS PubMed; (h) Q. J. Shen and W. J. Jin, Phys. Chem. Chem. Phys., 2011, 13, 13721–13729 RSC.
- (a) M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646–1653 CrossRef CAS PubMed; (b) T. Shirman, T. Arad and M. E. van der Boom, Angew. Chem., Int. Ed., 2010, 49, 926–929 CrossRef CAS PubMed; (c) M. G. Sarwar, B. Dragisic, S. Sagoo and M. S. Taylor, Angew. Chem., Int. Ed., 2010, 49, 1674–1677 CrossRef CAS PubMed.
- (a) C. J. Serpell, N. L. Kilah, P. J. Costa, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2010, 49, 5322–5326 CrossRef CAS PubMed; (b) M. G. Sarwar, B. Dragisic, S. Sagoo and M. S. Taylor, Angew. Chem., Int. Ed., 2010, 49, 1674–1677 CrossRef CAS PubMed; (c) T. Shirman, T. Arad and M. E. van der Boom, Angew. Chem., Int. Ed., 2010, 49, 926–929 CrossRef CAS PubMed; (d) X. Q. Yan, X. R. Zhao, H. Wang and W. J. Jin, J. Phys. Chem. B, 2014, 118, 1080–1087 CrossRef CAS PubMed.
- (a) T. B. Truong, J. Phys. Chem., 1980, 84, 964–970 CrossRef CAS; (b) S. K. Pal, L. Zhao and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8113–8118 CrossRef CAS PubMed.
- M. C. Cuquerella, M. A. Miranda and F. Bosca, J. Phys. Chem. A, 2006, 110, 2607–2612 CrossRef CAS PubMed.
- (a) Y. J. Wei, C. G. Liu and L. P. Mo, Spectrosc. Spectral Anal., 2005, 25, 86–88 CAS; (b) Y. J. Wei, C. G. Liu, L. P. Mo and Q. L. Wang, Chin. J. Anal. Chem., 2003, 31, 120 CAS.
- S. Mostafa, M. El-Sadek and E. A. Alla, J. Pharm. Biomed. Anal., 2002, 27, 133–142 CrossRef CAS.
- (a) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS; (b) W. B. Person, J. Am. Chem. Soc., 1965, 87, 167–170 CrossRef CAS.
- (a) M. Y. Choi, J. A. Pollard, M. A. Webb and J. L. Mchale, J. Am. Chem. Soc., 2003, 125, 810–820 CrossRef CAS PubMed; (b) J. J. Wu, N. Li, K. A. Li and F. Liu, J. Phys. Chem. B, 2008, 112, 8134–8138 CrossRef CAS PubMed.
- A. Espinosa-Mansilla, A. M. de la Peńa, F. Salinas and D. G. Gŏmez, Talanta, 2004, 62, 853–860 CrossRef CAS PubMed.
- (a) Z. X. Cai and X. P. Yan, Nanotechnology, 2006, 17, 4212–4216 CrossRef CAS PubMed; (b) D. M. Han, G. Z. Fang and X. P. Yan, J. Chromatogr. A, 2005, 1100, 131–136 CrossRef CAS PubMed.
- N. A. Al-Hashimi, Spectrochim. Acta, Part A, 2004, 60, 2181–2184 CrossRef PubMed.
- (a) B. A. Wong, S. Friedle and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 7142–7152 CrossRef CAS PubMed; (b) Z. Xu, K.-H. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601–610 CrossRef CAS PubMed; (c) J. G. Melnick, K. Yurkerwich and G. Parkin, J. Am. Chem. Soc., 2010, 132, 647–655 CrossRef CAS PubMed; (d) Y. Gao, J. Wu, Y. Li, P. Sun, H. Zhou, J. Yang, S. Zhang, B. Jin and Y. Tian, J. Am. Chem. Soc., 2009, 131, 5208–5213 CrossRef CAS PubMed.
- (a) J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS; (b) N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308–4314 CrossRef CAS.
- M. Yamashita and J. B. Fenn, J. Phys. Chem., 1984, 88, 4451–4459 CrossRef CAS.
- A. Venter, M. Nefliu and R. G. Cooks, TrAC, Trends Anal. Chem., 2008, 27, 284–290 CrossRef CAS PubMed.
- (a) N. Shao, J. Jin, H. Wang, J. Zheng, R. Yang, W. Chan and Z. Abliz, J. Am. Chem. Soc., 2010, 132, 725–736 CrossRef CAS PubMed; (b) J. He, Z. Abliz, R. Zhang, Y. Liang and K. Ding, Anal. Chem., 2006, 78, 4737–4740 CrossRef CAS PubMed.
- W. Xia, X.-Y. Hu, Y. Chen, C. Lin and L. Wang, Chem. Commun., 2013, 49, 5085–5087 RSC.
- R. B. Cody, J. A. Laramee and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS PubMed.
- K. Kpegba, T. Spadaro, R. B. Cody, N. Nesnas and J. A. Olson, Anal. Chem., 2007, 79, 5479–5483 CrossRef CAS PubMed.
- L. Song, A. B. Dykstra, H. Yao and J. E. Bartmess, J. Am. Soc. Mass Spectrom., 2009, 20, 42–50 CrossRef CAS PubMed.
- (a) O. P. Haefliger and N. Jeckelmann, Rapid Commun. Mass Spectrom., 2007, 21, 1361–1366 CrossRef CAS PubMed; (b) C. Y. Pierce, J. R. Barr, R. B. Cody, R. F. Massung, A. R. Woolfitt, H. Moura, H. A. Thompson and F. M. Fernandez, Chem. Commun., 2007, 807–809 RSC; (c) G. Morlock and Y. Ueda, J. Chromatogr. A, 2007, 1143, 243–251 CrossRef CAS PubMed; (d) C. Petucci, J. Diffendal, D. Kaufman, B. Mekonnen, G. Terefenko and B. Musselman, Anal. Chem., 2007, 79, 5064–5070 CrossRef CAS PubMed; (e) Y. Li, Anal. Methods, 2013, 5, 6933–6940 RSC.
- J. S. Murray, M. C. Concha, P. Lane, P. Hobza and P. Politzer, J. Mol. Model., 2008, 14, 699–704 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–276 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Proton-dependent fluorescence spectra of Cip in alkaline solution; linear relationship based on the Benesi–Hildebrand method for the stoichiometry of the complex of Cip and iodine; crystal data and structure refinement of the crystal compound of Cip and iodine; electrospray ionization mass spectra of the mixture of Cip and iodine. Analytical results for the determination of Cip based on XB-induced fluorescence in real samples. CCDC 805819. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01396d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |