
Advances in spinel Li4Ti5O12 anode materials for lithium-ion batteries
Xiangcheng
Sun
*a,
Pavle V.
Radovanovic
b and
Bo
Cui
a
aDepartment of Electrical and Computer Engineering and Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, Ontario, Canada. E-mail: sunxc824@gmail.com; Tel: +1-519-496-7918
bDepartment of Chemistry and Waterloo Institute for Nanotechnology, University of Waterloo, Waterloo, Ontario, Canada
First published on 6th October 2014
Abstract
Anode materials of rechargeable lithium-ion batteries have been developed towards the aim of high power density, long cycle life, and environmental benignity. As a promising anode material for high power density batteries for large scale applications in both electric vehicle and large stationary power supplies, the spinel Li4Ti5O12 anode has become more attractive for alternative anodes for its relatively high theoretical capacity (175 mA h g−1), stable voltage plateau of 1.5 V vs. Li/Li+, better cycling performance, high safety, easy fabrication, and low cost precursors. This perspective first introduces recent studies on the electronic structure and performance, synthesis methods, and strategies for improvement including carbon-coating, ion-doping, surface modifications, nano-structuring and optimization of the particle morphology of the Li4Ti5O12 anode. Furthermore, practical applications of the commercial spinel lithium-ion batteries are demonstrated. Finally, the future research directions and key developments of the spinel Li4Ti5O12 anode are pointed out from a scientific and an industrial point of view. In addition, the prospect of the synthesis of graphene–Li4Ti5O12 hybrid composite anode materials for next-generation lithium-ion batteries is highlighted.
 Xiangcheng Sun | Xiangcheng Sun is currently a PhD researcher in the Nanofabrication Group in the Department of Electrical and Computer Engineering, University of Waterloo, Canada. He received his Master’s degree at the Chinese Academy of Sciences in 1997, and his first PhD degree in Materials Science at the University of the State of Mexico in 2000. He has worked as a research scientist for 10 years in the USA, China and Canada before joining the University of Waterloo in January 2011. His current research interests are focused on the development of nanoscale cathode and anode materials for the design of advanced lithium-ion batteries for high-power backup systems and electric vehicles. |
 Pavle V. Radovanovic | Pavle Radovanovic is an Associate Professor of Chemistry and the Canada Research Chair in Spectroscopy of Nanoscale Materials at the University of Waterloo. He obtained his PhD degree in chemistry from the University of Washington (Seattle). After postdoctoral research at Harvard University, he joined the University of Waterloo, and was subsequently promoted to an Associate Professor in 2012. His research program is concerned with nanoscale multifunctionality and his research interests include polymorphism and phase transformations in the nanoscale range, materials for energy, quantum information processing and nanophotonics, and photocatalysis. |
 Bo Cui | Bo Cui is currently an Assistant Professor in the Department of Electrical and Computer Engineering at the University of Waterloo. He received his Bachelor’s degree in physics from Peking University in 1994, and his PhD degree in electrical engineering from Princeton University in 2003. His research is focused on nano- and microstructure fabrication using nanolithography, thin film deposition and etching, as well as bottom-up fabrication techniques. He has published 70 peer-reviewed journal articles and three book chapters, and edited one book. He is the recipient of the 2014 Engineering Research Excellence Award from the University of Waterloo. |
1. Overview of electronic structure and electrochemical properties
Rechargeable lithium-ion batteries (LIBs) with a high energy density and long lifetime have been regarded as promising energy storage and conversion devices for application in electric vehicles (EVs) and smart grids. But the state-of-the-art LIBs based on graphite anode materials can hardly meet the requirements for those high-capacity large-scale applications due to the limitations associated with power density and safety characteristics.1,2 In the last decade, much attention has been paid to the search for alternative anode candidates instead of the conventional commercial graphite. Spinel lithium titanate oxide (Li4Ti5O12, LTO) has been considered the most promising one because of its excellent safety characteristics and long lifetime.3–6 Unlike the graphite anode, which expands up to 10 vol% during charging, the LTO anode can accommodate up to three lithium ions per formula unit, which allows a theoretical capacity of 175 mA h g−1 in the spinel structure with negligible volume change during charging and discharging.4,7 The LTO lattice has a spinel crystal structure with the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and a cubic symmetry.3,8 The lattice constant was calculated to be 0.8364 nm.9 Lithium ions occupy the tetrahedral 8a sites and 1/6 of the octahedral 16d sites, while the rest of the octahedral 16d sites are occupied by tetravalent Ti4+ ions, and the ratio of lithium ions to titanium ions is 1
m space group and a cubic symmetry.3,8 The lattice constant was calculated to be 0.8364 nm.9 Lithium ions occupy the tetrahedral 8a sites and 1/6 of the octahedral 16d sites, while the rest of the octahedral 16d sites are occupied by tetravalent Ti4+ ions, and the ratio of lithium ions to titanium ions is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. The oxygen ions are located at the 32e sites. Thus, the spinel-LTO could be expressed as Li(8a)[Li1/3Ti5/3](16d)O4(32e) in space notation,10,11 where the octahedral 16c sites and the tetrahedral 8b and 48f sites are empty, which is suitable for lithium insertion and extraction.12,13 The crystal units of spinel-LTO and rock-salt-LTO are shown in Scheme 1(a) and (b), respectively.
:5. The oxygen ions are located at the 32e sites. Thus, the spinel-LTO could be expressed as Li(8a)[Li1/3Ti5/3](16d)O4(32e) in space notation,10,11 where the octahedral 16c sites and the tetrahedral 8b and 48f sites are empty, which is suitable for lithium insertion and extraction.12,13 The crystal units of spinel-LTO and rock-salt-LTO are shown in Scheme 1(a) and (b), respectively.
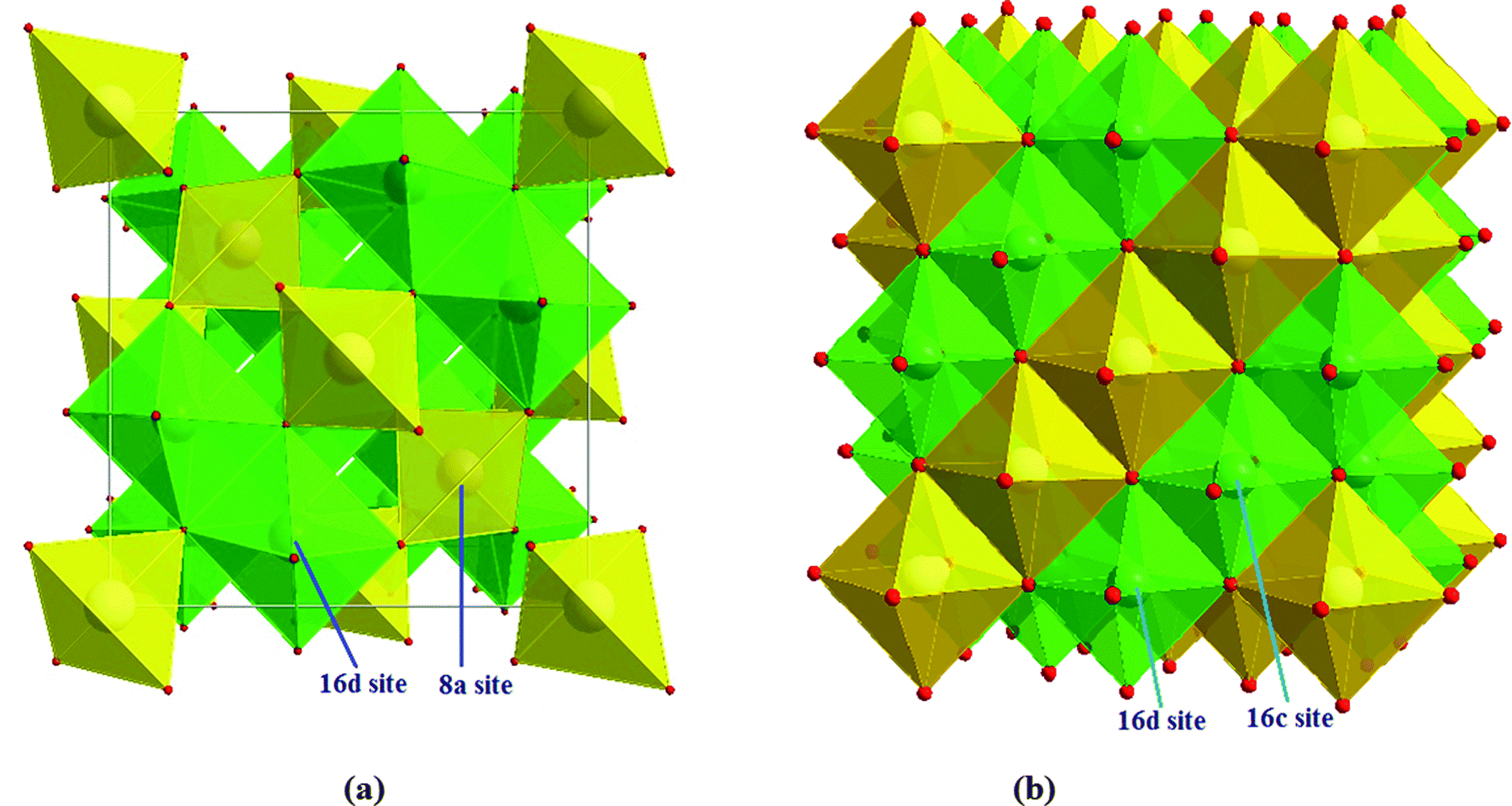 | ||
| Scheme 1 (a) Spinel- and (b) rock-salt-LTO structures. Yellow tetrahedra represent lithium, and green octahedra represent disordered lithium and titanium. | ||
[TiO6] is the main frame for the lithium insertion/extraction. Basically, the Li+ ions occupy the 8a sites forming the spinel structure at the initial stage of discharge. As the lithium ion insertion (discharge) proceeds, three lithium atoms from the 8a sites transfer to 16c sites, and the inserted three lithium ions move to the 16c sites via 8a sites. During extraction (charging), the lithium ions are extracted out from the 16c sites via 8a sites, and the other lithium atoms move back to 8a sites from 16c sites.12 Simultaneously, there is the redox reaction of Ti4+/Ti3+ within the octahedrally coordinated framework, allowing a topotactic transition from the spinel structure of Li4Ti5O12 (spinel-LTO) to the rock-salt structure of Li7Ti5O12 (rock-salt-LTO); in space notation: from Li(8a)[Li1/3Ti5/3](16d)O4(32e) to Li2(16c)[Li1/3Ti5/3](16d)O4(32e).11,14,15
Twenty years ago, Ohzuku and co-authors3 reported that the charge–discharge curve for the LTO electrode as the anode is characterized by a flat and extended voltage plateau at around 1.55 V, which is typical of the phase transition between a Li-rich phase (rock-salt-LTO) and a Li-poor phase (spinel-LTO). Although substantial chemical changes occur during the conversion between the two phases, the corresponding volume change is only 0.2–0.3%. This volume change is associated with the reduction of the lattice constant from 0.8364 nm to 0.8353 nm during the discharge process. Owing to the negligible volume change, this type of materials is referred to as zero-strain insertion materials, indicating their exceptional reversibility performance as anode materials for LIBs.6,16
Three Ti4+ ions are reduced to Ti3+ ions during the two-phase transition from spinel-LTO to rock-salt-LTO.3,4,6,12 The corresponding electrochemical reaction can be described as:
| (spinel-LTO) Li4Ti5O12 + 3Li+ + 3e− ↔ Li7Ti5O12 (rock-salt-LTO) | (1) |
The core–shell model describes a plausible two-phase transition process,17–21 as illustrated in Fig. 1. As the lithium intercalation (discharge) proceeds, the spinel-LTO on the surface of the particle is reduced and transformed to rock-salt-LTO. The shell with the rock-salt structure formed in the process becomes thicker with an increasing depth of lithium insertion. Simultaneously, the core with the spinel structure shrinks. At the end of the discharging process, the entire particle becomes rock-salt-LTO. Conversely, the particle transforms from the rock-salt into the spinel phase during the charging (lithium extraction) process. The two types of LTO crystallographic data are summarized in Table 1.22
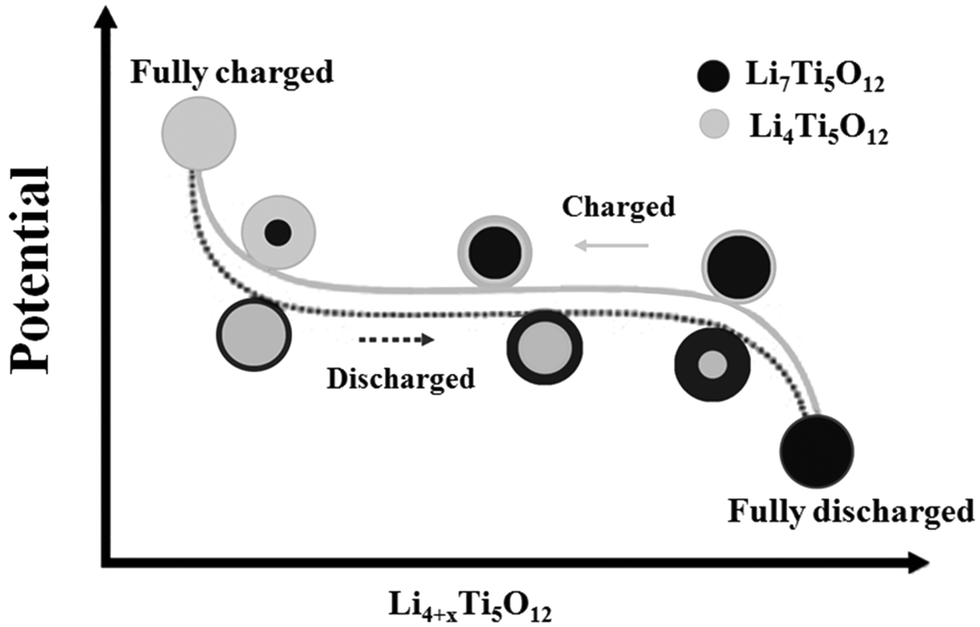 | ||
| Fig. 1 Illustration of phase transformation from spinel-LTO (Li4Ti5O12) and rock-salt-LTO (Li7Ti5O12) during the charging–discharging process. Reproduced with permission from ref. 19. Copyright 2007 Elsevier. | ||
| Anode materials | Wyckoff sites | Atoms | Occupancy | Cell parameters | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Spinel-LTO | 8a | Li1 | 1.0 | 0.125 | 0.125 | 0.125 |
| 16d | Li2 | 0.1667 | 0.5 | 0.5 | 0.5 | |
| 16d | Ti | 0.8333 | 0.5 | 0.5 | 0.5 | |
| 32e | O | 1.0 | 0.2625(1) | 0.2625(1) | 0.2625(1) | |
| Rock-salt-LTO | 8a | Li1 | 1.0 | 0 | 0 | 0 |
| 16d | Li2 | 0.1667 | 0.5 | 0.5 | 0.5 | |
| 16d | Ti | 0.8333 | 0.5 | 0.5 | 0.5 | |
| 32e | O | 1.0 | 0.2576(3) | 0.2576(3) | 0.2576(3) | |
As reported earlier, such a spinel-LTO structure provides a very flat voltage plateau of 1.55 V vs. Li+/Li due to the stable Ti4+/Ti3+ redox couple during the discharge–charge cycling. During the lithium insertion and extraction processes, a solid electrolyte interface (SEI) film is often formed at a potential below 0.8 V versus Li+/Li, which consumes active lithium and results in a decrease in specific capacity. However, the potential of LTO is significantly above the reduction potential of most organic electrolytes. Thus, the formation of an SEI film can be avoided on the particle surface by applying spinel-LTO as the anode in the electrochemical battery cell. It has also been demonstrated that spinel-LTO shows better thermal stability performance than conventional graphite even below 230 °C.23
Many investigations already reported that the spinel-LTO anode can accommodate up to three lithium ions per formula unit at a potential of 1.55 V.3,4,9,24–28 The insertion mechanism is based on the spinel–rock-salt phase transition, allowing the reduction of 3 Ti4+ atoms out of 5, which provides a theoretical capacity of 175 mA h g−1 in the spinel structure.11,16,17,29–40
As stated previously, the lattice parameters remain almost unchanged during lithium insertion and extraction.41 However, some researchers12,42 stated that spinel-LTO can deliver a theoretical capacity of 293–296 mA h g−1 based on the reduction of all Ti4+ in the compound with the discharge voltage extended to approximately 0 V. In the literature,42,43 it is reported that spinel-LTO exhibited excellent electrochemical performance at different discharge voltage ranges, especially when extended to 0 V. The Li-ion insertion can occur at three crystallographic sites, i.e., the (8a), (16c), and (48f) sites, indicating that the Li ions can intercalate into LTO at a low potential (<1.0 V vs. Li/Li+). This low-potential intercalation process would lead to an improvement in the energy density of spinel LTO electrodes related to the numbers of octahedral (16c) and tetrahedral (8a) sites and their occupancies between 2.5 and 0.01 V vs. Li/Li+. Generally, additional Li+ ions are inserted into the lattice and located at the octahedral sites in the intercalation process. Meanwhile, the lithium ions initially located at the tetrahedral sites also migrate to the octahedral sites. The intercalation–de-intercalation process can thus be expressed as:
| Li3(8a)[LiTi54+](16d)O12(32e) + 3e− + 3Li+ ↔ Li6(16c)[LiTi33+Ti24+](16d)O12(32e) | (2) |
| Li6(16c)[LiTi33+Ti24+](16d)O12(32e) + 2e− + 2Li+ ↔ Li2(8a)Li6(16c)[LiTi53+](16d)O12(32e) | (3) |
Here, vacant tetrahedral (8c) sites are available to accommodate lithium ions under 1.0 V vs. Li/Li+, thus enhancing the reversible capacity at a low potential. Accordingly, the second stage of the charging plateau contributes to a significant portion of the total capacity of the LTO anodes. Due to this mechanism, LTO can display a reversible capacity of 215.1 mA h g−1 within 2.5–0.01 V vs. Li/Li+ at 0.2 C, which is much higher than the conventional theoretical capacity of 175 mA h g−1.
It should be noted that the existence of available free octahedral sites in the lattice of spinel-LTO makes it a good lithium-ion conductor.44 However, since the oxidation state of Ti in spinel-LTO is +4, the highest possible valence for Ti, spinel-LTO is a very poor electronic conductor.19 On the contrary, rock-salt-LTO is a good electronic conductor,17 which is attributed to the average oxidation state of Ti which is +3.4. This oxidation state suggests the coexistence of Ti3+ (60%) and Ti4+ (40%) in the lattice, and that rock-salt-LTO is a very poor lithium-ion conductor due to the full occupancy of the (16c) octahedral sites by Li+.37 More details about the physical and chemical properties of spinel-LTO are summarized in Table 2.
| Advantages | • Cheap, safe, and easy to prepare3,4,6,45 |
| • Good lithium-ion mobility, environmentally friendly46 | |
| • Higher lithium-ion mobility than graphite47 | |
| • Zero-strain insertion that provides little volume variation and negligible lattice parameter changes during charge–discharge process3,48 | |
| • Almost free of solid electrolyte interphase (SEI) film, little gas evolution and little electrolyte decomposition12 | |
| • Good Li-ion insertion/extraction reversibility and good structural stability34 | |
| • Stable performance at high temperatures (higher than 60 °C), high-rate discharge performance even at 10 C49 | |
| • High columbic efficiency (>95% at 1 C) and thermodynamically flat discharge profile at 1.55 V (vs. Li/Li+)48 | |
| Disadvantages | • Low electronic conductivity (<10−13 S cm−1 at room temperature)50 |
| • A cubic closely packed oxygen array, Li-ion diffusion coefficient is generally about 10−6 cm2 s−1 (ref. 4 and 7) | |
| • A theoretical specific capacity of 175 mA h g−1, a relative practical specific capacity of 150–160 mA h g−1, compared with graphite (∼300 mA h g−1)51,52 | |
Applied as an anode, spinel LTO can be coupled to high voltage cathodes such as LiCoO2, LiNiO2, LiMn2O4, or LiFePO4, to fabricate lithium-ion batteries. When compared with its counterparts as anode materials, spinel-LTO is cheaper and easier to prepare than alloy-based anodes, and has better electrochemical properties and better safety than carbonaceous anodes in lithium-ion batteries.53 The comparison between spinel-LTO and other anode materials is summarized in Table 3.
| Anode materials | Potential (V) | Capacity (mA h g−1) | Remarks and references |
|---|---|---|---|
| Graphene | 0.05 | 540 | • Superior electronic conductivity compared to graphitic carbon54 |
| • Broad electrochemical window54 | |||
| • High surface area of over 2600 m2 g−1 (ref. 54) | |||
| Spherical graphitized mesocarbon microbeads | 0.05 | 320–330 | • Easy coating55 |
| Pitch-based graphite | 0.05 | 350 | • Largest share in the market55 |
| Carbon-coated natural graphite | 0.05 | 360–365 | • Less decomposition of electrolyte without additives55 |
| Si(Li4.4Si) | 0.5–1 | 4200 | • Maximum swelling: 400%56 |
| Sn | 0.6 | 990 | • Maximum swelling: 360%56 |
| LiC6 | 0.1–0.2 | 372 | • Safety issues1,23 |
| InSb, Cu2Sb | 0.9 | 250–300 | • Poor cyclability1 |
| TiO2-B | 1.5–2.0 | 305 | • Excellent capacity retention during cycling57 |
| TiO2-AB (acetylene black) | 1.5–2.0 | 320 | • Severe capacity fading58 |
| Rutile TiO2 | 1.5–2.1 | 270 | • Poorer electrochemical properties than anatase TiO2 (ref. 58) |
| Anatase TiO2 | 1.5–2.1 | 336 | • Low costs, nontoxicity, low volume expansion (3–4%) during lithium insertion59 |
| • Environmentally friendly59 | |||
| Li2Ti3O7 | 1.3–2.1 | 240 | • Stable and good reversibility in Li insertion and extraction60 |
| • Rapid capacity fading at high current density of 525 mA g−1 (ref. 60) | |||
| LiTi2O4 | 1.17–1.75 | 133.6 | • Good electrochemical reversibility and high rate performance61 |
| • Higher electric conductivity61 | |||
| • Pure LiTi2O4 is difficult to synthesize61 | |||
| Li4Ti5O12 | 1.0–2.0 | 150–160 | • Good structural stability and safety3,4,6 |
| • Better coulombic efficiency >95% at 1 C48 | |||
| • Flat discharge profile at 1.55 V (vs. Li/Li+)48 | |||
| • Stable high-temperature performance even at 60 °C49,51 | |||
| • Good Li-ion insertion/extraction reversibility3,4,6 | |||
| • Free of solid electrolyte interphase (SEI)12 |
Spinel-LTO has many advantages such as an excellent lithium ion insertion and extraction reversibility, negligible volume or structural change during the charge–discharge process, and a flat potential plateau. However, due to its poor electrical conductivity, spinel-LTO is a very poor electronic conductor, which results in a low specific capacity and poor capability for high rate performance. The practical specific capacity is about 150–160 mA h g−1, which is much lower than those of other anode materials such as graphite (350 mA h g−1). Therefore, spinel-LTO might not be suitable for high current applications in its native form. Several effective ways have been proposed and explored to improve the conductivity, and one of the most common strategies is to synthesize nanostructured LTO materials, which will be highlighted in detail in the following section. Other attractive ways are to dope the material with metal or non-metal ions in the Li, Ti or O sites, or to introduce surface modifications via surface coating using conductive species to enhance the surface electrical conductivity.62,63
2. Synthesis methods
The charge–discharge capacity and cycle performance of spinel-LTO are greatly affected by the synthesis method and conditions. The most conventional synthesis methods include solid-state synthesis, hydrothermal method, solvothermal method, sol–gel synthesis, solution-combustion methods, spray pyrolysis, etc.2.1 Solid-state method
The solid-state reaction method is normally conducted by mixing solid materials and annealing the mixture at high temperatures. This synthetic method is used for the synthesis of spinel anode materials because it is simple and easy. LTO powders are synthesized by a solid-state reaction of lithium and titanium salts at 700–1000 °C.12,23,26,37,64–73 The solid precursor materials are ground before their calcination in a furnace. Generally, TiO2 anatase or rutile is used as the titanium source, and Li2CO3 or LiOH as the lithium salt. Solvent (i.e. ethanol) is also used as a dispersant to provide a better environment to homogeneously mix the starting materials.Usually, stoichiometric amounts of TiO2 and LiOH·H2O are first dispersed in n-hexane to assure homogeneity. The mixture is subsequently heated at 800 °C for 24 h in oxygen, after removing the solvent.29 It has been reported that the electrochemical performance is influenced by different synthetic parameters, such as the starting materials (e.g., lithium salt), the calcination temperature, and the calcination time.38,74–76 Hong and co-workers77 studied the influence of the starting materials and the annealing temperature on the rate performance and the capacity retention of LTO prepared under different conditions. They used Li2CO3 and TiO2 (either anatase or rutile), which were mixed with de-ionized water after adding 2 wt% of the ammonium salt of polycarboxylic acid as a dispersant. The powder mixtures were exposed to high energy milling for 3 h, and the dried powders were finally calcined at 700, 800, or 900 °C for 3 h in air. The rate performance and capacity retention were found to have different values. Yao and co-workers37 have reported that LTO prepared using a solid-state reaction can be cycled in the voltage range between 0 and 3.0 V with excellent cyclability and a capacity of about 200 mA h g−1. Rutile TiO2 is more desirable in producing high purity LTO than anatase TiO2, due to the anatase-to-rutile phase transformation, which is found to be more rigid in the solid-state reaction than the intact rutile phase.77–79 The formation mechanism of spinel-LTO, using a solid-state reaction between Li2CO3 and TiO2, for the application in lithium-ion batteries was proposed.78 A schematic model was provided to explain the different spinel-LTO formation mechanisms from pure anatase and anatase/rutile mixed TiO2. According to this model, the anatase/rutile-phase boundaries played an important role in enhancing the solid-state reaction.78 The utilization of rutile TiO2 as a starting phase would be desirable in enhancing the spinel-LTO content, which is the key factor for increasing the specific capacity.77 The specific capacity of spinel-LTO prepared from anatase TiO2 depends significantly on the milling method (i.e., high energy milling, ball milling) and the heat treatment temperature, whereas spinel-LTO prepared from rutile TiO2 showed a uniform capacity of 160 mA h g−1, regardless of the synthesis parameters, owing to their high LTO phase content. Overall, spinel-LTO powder with a specific capacity of 165 mA h g−1 could be synthesized by optimizing the milling method and the starting materials.77 The conventional solid-state method would easily cause aggregation of the LTO particles at the expense of loss of the material’s nanostructure. The desired properties of the LTO particles are difficult to realize as the expected morphology would be destroyed through particle aggregation. A modified synthesis of LTO, based on the solid-state method, has also been developed. Li and co-authors80 demonstrated a microwave reaction system for the preparation of LTO particles with less aggregation. The starting materials Li2CO3 and anatase TiO2 were thoroughly mixed in a stoichiometric ratio. Since Li2CO3 and TiO2 could only absorb a small amount of microwaves at relatively low temperatures, the double crucible system was applied to achieve the high temperature needed for the solid-state reaction, with charcoal used as a heating medium between two porcelain crucibles. The microwave reaction system was placed in the centre of a rotating plate of a modified microwave oven, and irradiated at 500–700 W for 10–15 min. A molten-salt reaction81,82 was also used as a modified solid-state method by introducing salts such as LiCl, NaCl or KCl into the conventional solid-state reaction. A composite LiCl–KCl can dramatically decrease the melting point, and provide a liquid reaction environment, which can accelerate the diffusion and promote the crystal formation.81 The spinel-LTO that was obtained using the eutectic molten-salt method, using the mixtures with a molar ratio of LiCl/KCl = 1.5, achieved a high initial discharge capacity of 169 mA h g−1, a high charge–discharge efficiency of 94% at a 0.2 C rate, and also good rate performance from 0.2 to 5 C.81
2.2 Hydrothermal method
The hydrothermal synthesis is performed in autoclaves at a controlled temperature and/or pressure with the reaction in aqueous solution, followed by a calcination step at temperatures above 500 °C. This method has been widely used for the production of uniform small particles in industry.Many research efforts have been dedicated to improve the hydrothermal method for pure spinel-LTO anode materials.46,83–90 For example, Lai and co-authors87 reported a hydrothermal method to prepare LTO; they employed TiOSO4 and LiOH·H2O as the starting materials and water as the solvent with a certain amount of urea. After thoroughly mixing the starting materials in water containing urea, the resultant solution was transferred into a Teflon-lined autoclave and treated at 180 °C for 24 h. The precipitate was harvested and then calcined at 500 °C for 2 h under an Ar atmosphere. Chen and co-workers46 reported a hydrothermal method to obtain hierarchical layered LTO nanosheets using titanium tetra(isopropoxide) and LiOH as the starting materials. Hydrogen peroxide was used to control the hierarchical morphology of LTO. Typically, 1 ml of 30% hydrogen peroxide was dispersed in 20 ml of 0.4 M LiOH, followed by the addition of 2 mmol titanium tetra(isopropoxide). The solution was transferred into a 30 ml Teflon-lined stainless autoclave and maintained at 130 °C for 12 h. The resulting white precipitate was recovered by centrifugation, thoroughly washed with deionized water, and then dried in an oven at 80 °C. Finally, the as-prepared sample was calcined in a muffle furnace at 550 °C for 6 h in air. Using this method, hierarchical sawtooth-like LTO nanosheets were obtained.
2.3 Solvothermal method
The difference between the solvothermal and the hydrothermal methods is the solvent used in the preparation process. The solvothermal method uses a non-aqueous solution, e.g., ethanol, while the hydrothermal method employs water as the solvent. Since a variety of organic solvents with high boiling points can be selected in the solvothermal method, the temperature can be much higher than that in the hydrothermal method. Besides, the solvothermal method is characterized by better control of the size and shape distributions and a better crystallinity of the LTO nanoparticles relative to the hydrothermal method. The solvothermal method has been an alternative method to prepare LTO with small and uniform particles.60,91–93 For example, lithium hydroxide (LiOH), tetrabutyl titanate and ethanol were used as the starting materials. The reaction system was formed using 0.1 mol tetrabutyl titanate dissolved in 100 ml ethanol, followed by the addition of 0.1 or 0.08 mol LiOH. The undissolved LiOH particles were suspended in the orange-like solution. The alcoholic suspension was solvothermally treated at 140 °C for 24 h in a 150 ml autoclave under autogenous pressure. The pressure generated in the autoclave at 140 °C is ∼747 kPa. After the solvothermal reaction, the produced powders were washed and filtered with ethanol and then deionized water (five times) to eliminate the unreacted reagents, followed by drying at 80 °C for 24 h in air.602.4 Sol–gel method
The sol–gel method is a wet-chemical technique widely used in materials science and ceramic engineering. The synthesis of LTO particles using the sol–gel method results in particles with a nearly cubic morphology and a narrow size distribution, which also exhibit a high initial discharge capacity and good cycling performance.94 Unfortunately, the reaction materials usually include organic acids such as citric acid, which have a high cost, so this method is unsuitable for large-scale applications.A colloidal suspension (sol) acts as the precursor for an integrated network (gel) of either discrete particles or network polymers. Typical precursors are metal alkoxides, metal salts (such as chlorides and acetates), and organic metal compounds which undergo various forms of hydrolysis and polycondensation reactions.51,95–99 The sol–gel method can avoid the problem caused by the solid-state method.100 It is an environmentally friendly process as it uses an aqueous solution. This process can easily control the phase structure, composition homogeneity, crystallite size, monodispersity and microstructure.101 It also has the advantage of low fabrication costs, relatively easy stoichiometry control, high deposition rate, low synthesis temperature, shorter heating time, and better crystallinity.102,103
It is a desirable method to obtain LTO with good homogeneity, uniform morphology, and a narrow size distribution.94 However, it is difficult to control the synthesis conditions in terms of the large quantities of solvents and organic materials such as citric acid, ethylene glycol and polyvinyl alcohol needed, which prevents its extensive application in energy storage.81 Moreover, the sol–gel method also requires a post-synthetic high-temperature calcination at 800 °C or higher temperatures to prepare a pure spinel-LTO phase, which results in an undesirable particle growth.84 In order to obtain spinel-LTO with optimal electrochemical performance, much effort has been invested to optimize the sol–gel synthetic approach. Hao and co-authors51 reported a novel sol–gel method using triethanolamine (TEA) to produce spinel-LTO with an initial discharge capacity of 168 mA h g−1. They also reported that the spinel-LTO synthesized using a Li/Ti atomic ratio 4:5 and a molar ratio of citric acid to total metal ions of R = 1/2, exhibited an initial discharge capacity of 167 mA h g−1 at 23.5 mA g−1.104 The authors further investigated an oxalic acid-assisted sol–gel method to synthesize LTO, which exhibits a capacity of 171 mA h g−1 in the first cycle and of 150 mA h g−1 after 35 cycles under optimal calcination conditions at 800 °C for 20 h.105
Alias and co-authors96 used lithium tert-butoxide and titanium isopropoxide as the starting materials, which were mixed together in ethanol, and the residue was harvested and finally sintered at different temperatures (700–1000 °C) and times (1–5 h). Khomane and co-workers97 employed hexadecyltrimethylammonium bromide (CTAB) as a cationic surfactant to control the crystal growth of LTO. In a typical process, CTAB was dissolved in 100 ml ethanol under magnetic stirring. Four grams of lithium acetate dihydrate was dissolved in the above solution with continued magnetic stirring. Titanium(IV) isopropoxide was added to the above solution dropwise by keeping the molar ratio Li:Ti = 1:1.25. The temperature of the solution was raised to 90 °C and it was stirred continuously to form a gel. The gel was aged at 100 °C for 24 h in air and the precursor was decomposed at 400 °C for 4 h followed by calcination at 800 °C for 12 h in air. The average discharge capacity of the prepared LTO after 20 cycles was ∼160 mA h g−1 at a constant current density of 21.37 mA g−1.
2.5 Solution-combustion methods
The solution-combustion method is a facile and economical technique for the preparation of nanomaterials for a variety of applications such as fuel cells and lithium-ion batteries. It also has the advantage of relatively simple infrastructure requirements, the formation of high-purity products with size and shape control, and stabilization of metastable phases.106 The initial reaction medium is an aqueous or non-aqueous solution, and fuels are used as a C and H source to liberate heat via combustion. The complexes formed with the metal ions could facilitate the homogeneous mixing of the cations in solution.107,108 Prakash et al.109 reported a solution-combustion synthesis of LTO nanopowders using titanyl nitrate [TiO(NO3)2] and LiNO3 as the oxidant precursors and glycine as the fuel. In a typical preparation, an aqueous redox mixture containing stoichiometric amounts of titanyl nitrate, LiNO3, and glycine were put into an alumina crucible and placed into a muffle furnace pre-heated at 800 °C. The synthesis of the LTO nanoparticles, crystallizing in a cubic spinel-phase, was completed in less than one minute. TEM images indicate that the primary particles are agglomerated crystallites of varying sizes between 20 and 50 nm with a three-dimensional (3D) interconnected porous network. It was found that the LTO prepared using the combustion technique is superior in terms of both high rate-capability and capacity retention compared with those of the solid-state method, which can be attributed to the high crystallinity and nanosized particles of LTO. Raja and co-authors110 reported a novel aqueous combustion process using the common amino acid alanine as the fuel to synthesize LTO, which showed a uniform morphology with an average particle size in the range 40–80 nm and an optical band gap at 1.8 eV. Yuan and co-workers111 reported a glycine–nitrate combustion process for the preparation of spinel-LTO, which showed high electrochemical performance and reached a capacity of 125 mA h g−1 at 10 C with a fairly stable cycling performance. In addition, they79 reported a cellulose-assisted combustion process to synthesize pure-phase spinel LTO at reduced temperatures using anatase TiO2 as the titanium source. Compared with the solid-state reaction, the cellulose-assisted process produced LTO with a smaller particle size and higher specific capacity due to the lower synthesis temperature. A capacity of ∼175 mA h g−1 (theoretical value) at a 1 C rate was achieved, and it even reached ∼100 mA h g−1 at a high discharge rate of 10 C with a high cycling stability. It is a promising method for the synthesis of LTO with high rate performance and a high cycling stability.2.6 Other methods
Beside the above-mentioned methods, other methods have also been developed; including a reflux method,119 a continuous flow supercritical method,91 and a sonochemical method.120 Doi and co-authors121 first reported the preparation of uniform nanosize LTO particles using an electrospray deposition method. The spinel-LTO particles have a fairly uniform particle size of ∼12 nm with a distinct crystal structure. Ernst and co-authors122 used the flame spray pyrolysis (FSP) to synthesize spinel-LTO with primary crystallite sizes of 7–30 nm and a high temperature stability. It has been found that the FSP process optimization could be used to further remove impurities. Moreover, FSP-prepared LTO nanoparticles showed a good sintering stability at elevated temperatures and the scalability of the process.
3. Approaches to improve performance
3.1 Lattice ion-doping of LTO
As mentioned previously, since the oxidation state of Ti in spinel-LTO is the highest possible valence (+4), spinel-LTO is a very poor electronic conductor, which leads to a low specific capacity and poor capabilities at high rates. To overcome this drawback, great efforts have been made to increase the electrical conductivity of LTO.A first-principle theoretical approach has been used to calculate the electronic structure of the M-doped LTO (M = Cr, Fe, Ni, Mg).41 It has been found that both Cr and Mg doping improve the electronic conduction of LTO, while Fe or Ni doping is not beneficial for enhancing the electronic conductivity.
Doping with a variety of metal cations including Ag+, Sn2+, K+, Mg2+, Ca2+, Zn2+, Al3+, Ga3+, Cr3+, Co3+, La3+, Y3+, Zr4+, Ru4+, Mo4+, Mn4+, V5+, Ta5+, Nb5+, Sr2+ and Cu2+ into the Li+ and Ti4+ sites45,64,123–159 has been investigated extensively, and the structural properties such as ion valence and distribution, and conduction characteristics were systematically reported. Doping has been realized as an effective way to greatly improve the kinetics of the LTO anode materials in terms of discharge capacity delivery, cycling stability and, especially, rate capability. Several reports of doping LTO with metal cations are highlighted here. Gao and co-workers45 first reported spherical La3+-doped LTO powders with a high tap density synthesized using an outer gel method. The results indicate that the electrochemical performance has been improved by doping La3+ into LTO. The initial discharge capacity is 161.5 mA h g−1 at a 0.1 C rate, and the capacity still reaches 135.4 mA h g−1 after 50 cycles. Yi and co-workers128 reported a micro-sized Li4Ti5−xLaxO12 (0 ≤ x ≤ 0.2) particle synthesized using a simple solid-state method in air. The electronic conductivity and lithium diffusion coefficient of the La-modified LTO were improved. Compared with the V-doped LTO materials such as the Li4Ti5–xVxO12 reported by other researchers, Yu and co-authors126 have prepared a new sample of the V-doped LTO materials by a simple solid-state reaction method; this is typical Li4Ti4.9V0.1O12. An electrochemical impedance spectrum (EIS) indicates that Li4Ti4.9V0.1O12 has a faster lithium-ion diffusivity than pristine LTO. The Li4Ti4.9V0.1O12 exhibits a discharge capacity of 117.3 mA h g−1 at a 5 C rate. Wolfenstine and the co-authors123 reported Ta-doped LTO in the form Li4Ti4.95Ta0.05O12 synthesized using a solid-state method. Ta doping does not affect the lattice structure and particle morphology. The substitution of the Ti sites by Ta can enhance the electronic conductivity of LTO via the generation of a mixed Ti4+/Ti3+ species, which leads to a remarkably high cycling stability at a high charge–discharge rate. Li4Ti4.95Ta0.05O12 exhibits excellent discharge capacities of 116.1 mA h g−1 at 10 C and even 91.0 mA h g−1 at 30 C. Bai and co-authors142 first reported yttrium-doped LTO fabricated using a co-precipitation method followed by a simple sintering process. The results show that Y-modified LTO exhibits a long-term cycling stability over more than 1000 cycles at a high current rate of 10 C, which is ascribed to the increased lattice constant, and the improved electronic and ionic conductivities arising from the Y doping. Li and co-authors145 reported Zr-doped LTO prepared using a solid-state reaction under an air atmosphere. Zr4+ doping was found to reduce the particle size to less than 100 nm and effectively avoid particle agglomeration, contributing to the improvement in rate capability. The optimal composition of Li4Ti4.9Zr0.1O12 yielded a desirable rate capability by maintaining a discharge capacity of 118 mA h g−1 at 20 C. Tian and co-workers150 reported niobium-doped lithium titanate with a composition of Li4Ti4.95Nb0.05O12 prepared using a sol–gel method. The Li4Ti4.95Nb0.05O12 exhibits a high rate capacity with reversible capacities of 135 mA h g−1 at 10 C, 127 mA h g−1 at 20 C and 80 mA h g−1 at 40 C. It can be ascribed to the higher electronic conductivity and the faster lithium-ion diffusivity by Nb5+ doping. Cai and co-workers154 reported that controlled Al3+ doping into the tetrahedral 8a Li+ sites will promote the reduction of Ti4+ to Ti3+ to increase the electronic conductivity of Al-doped LTO, which then showed a better electrochemical performance than the pure LTO anode. Zhu and co-workers155 reported the spinel-type electrode materials Li4−xMgxTi5O12 (x = 0.05, 0.1, 0.15, 0.2) synthesized using a solid-state reaction to improve the electrochemistry and cycle performance. The results show that the electrochemical properties and cycle performance of the Mg2+-doped LTO materials are improved. Zhang and co-workers156 first reported zinc-doped LTO synthesized using the solid-state route. Zn doping is favorable to improve the conductivity and rate performance of LTO.
Huang and co-workers157 also reported that the high-rate discharge capacity and the cycling stability of pristine LTO was significantly improved using Cu doping. The cycling behavior of a Cu-doped LTO composite was studied. As summarized in Table 4, the Cu-doped LTO composite remarkably showed a better reversible capacity and cycling stability than the pristine LTO at high charge–discharge rates.
| Rate (C) | Anode materials | Discharge capacity (mA h g−1)/cycle | |
|---|---|---|---|
| 1st | 10th | ||
| 1 | LTO | 180.6 | 164.2 |
| Cu-doped LTO | 209.2 | 171 | |
| 2 | LTO | 162.2 | 155 |
| Cu-doped LTO | 184.8 | 166.6 | |
| 4 | LTO | 150.7 | 117.3 |
| Cu-doped LTO | 173.4 | 153.6 | |
| 8 | LTO | 98.9 | 72.5 |
| Cu-doped LTO | 165.7 | 144.6 | |
| 10 | LTO | 80.3 | 61.2 |
| Cu-doped LTO | 142.5 | 141.6 | |
Huang and co-authors162 synthesized Li4Ti5O12−xClx (0 ≤ x ≤ 0.3) compounds successfully via high temperature solid-state reaction. The results showed that the Li4Ti5O12−xClx (0 ≤ x ≤ 0.3) compounds have a well-crystallized pure spinel phase. The Li4Ti5O11.8Cl0.2 sample presented the best discharge capacity among all the samples and showed better reversibility and higher cycling stability compared with pristine LTO. When the discharge rate was 0.5 C, the Li4Ti5O11.8Cl0.2 sample presented a superior discharge capacity of 148.7 mA h g−1, while that of pristine LTO was 129.8 mA h g−1. Ma and coauthors163 reported carbon-encapsulated F-doped LTO (C-FLTO) composites obtained using solid-state lithiation at high temperatures. The F-doping of LTO not only increased the electron conductivity of LTO through trivalent titanium (Ti3+) generation, but also increased the robustness of the structure towards repeated lithiation and de-lithiation. The best-performing C–FLTO composite delivered a satisfactory performance with a high charge capacity of 158 mA h g−1 at a 1 C rate with negligible capacity fading after 200 cycles and an extremely high rate performance up to 140 C. A brief summary of some representative LTO anodes doped with typical cations and anions is shown in Table 5.
| Doped LTO | Synthesis methods | Electrochemical performance | Advantages |
|---|---|---|---|
| Cation doping in the Li sites | |||
| Mg-doped130 | • Solid-state reaction with stoichiometric mixtures of Mg(OH)2, TiO2 and LiOH·H2O | • Rate property improved | • Li4−xMgxTi5O12 increases the conductivity by many orders of magnitude |
| Li4−xMgxTi5O12 | • @1000 °C/H2/He for 5 h | • 3% Mg-doped LTO has the best discharge capacity (149 mA h g−1) at 0.1 C | |
| Ca-doped158 | • Solid-state reaction with stoichiometric mixtures of Li2CO3, TiO2 and CaO | • Li3.9Ca0.1Ti5O12 exhibits the best high-rate performance | • Ca-doped LTO shows very high rate capability and excellent stability and reversibility |
| Li4−xCaxTi5O12 | • @850 °C/air for 12 h | • Discharge capacity is 138.7 mA h g−1 at 10 C after 100 cycles | |
| Al-doped159 | • Solid-state reaction with stoichiometric mixtures of LiOH·H2O, TiO2 and Al2O3 | • Good cycling stability at 0.5, 1 and 2 C | • Cycling stability at high rate increased significantly |
| Li4−xAlxTi5O12 | • @ 800 °C/Ar for 5 h | • An optimal composition is Li3.9Al0.1Ti5O12 | |
| Cation doping in the Ti sites | |||
| V-doped148 | • Solid-state method with stoichiometric mixture of TiO2, Li2CO3 and V2O5 | • Li4Ti4.9V0.1O12 has the highest discharge capacity | • No effect on lattice parameter |
| Li4Ti5−xVxO12 | • @ 850 °C/air for 24 h | • Li4Ti4.9V0.1O12 has a discharge capacity of 229 mA h g−1 after 130 cycles | • Improved reversible capacity |
| • Good cycling performance | |||
| Zr-doped145 | • Solid-state reaction using stoichiometric mixture of CH3COOLi·2H2O, TiO2 and Zr(NO3)4·5H2O | • Li4Ti4.9Zr0.1O12 exhibited the best rate capability | • Improved electronic conductivity and Li-ion diffusivity |
| Li4Ti5−xZrxO12 | • @800 °C/air for 10 h | • Better rate capability | |
| Nb-doped150 | • Sol–gel method with stoichiometric amounts of CH3COOLi, Ti(OC4H9)4 and Nb(OH)5 | • Li4Ti4.95Nb0.05O12 has a higher specific capacity and better cycling performance | • Improved electronic conductivity and Li-ion diffusivity |
| Li4Ti5−xNbxO12 | • Gel dried at 120 °C for 10 h | • Specific capacities of 169.1 mA h g−1 at 1 C and 115.7 mA h g−1 at 10 C even after 100 cycles | • Better rate capability |
| • @ 800 °C/Ar for 12 h | • Good crystallinity and high phase purity | ||
| Anions doping in the O site | |||
| Br-doped161 | • Solid-state reaction using stoichiometric mixture of LiOH·H2O, LiBr·H2O, and TiO2 | • Li4Ti5O11.8Br0.2 presented the highest discharge capacity 172 mA h g−1, and 150.2 mA h g−1 after 50 cycles at 0.5 C rate | • Li4Ti5O11.8Br0.2 showed promising anode material for lithium-ion batteries |
| Li4Ti5O12−xBrx | • @900 °C /air for 12 h | ||
| Cl-doped162 | • Solid-state reaction using stoichiometric mixture of | • The Li4Ti5O11.8Cl0.2 presented the best discharge capacity, better reversibility. | • Increased the specific capacity significantly |
| Li4Ti5O12−xClx | • LiOH·H2O, LiCl·H2O, and anatase-TiO2 | • Superior discharge capacity of 148.7 mA h g−1 | • Doped Cl− can increase the amount of Ti3+/Ti4+ mixing charge |
| • @800 °C/air for 12 h | • Improved the lithium-ion diffusion. | ||
With respect to the co-doping method, further insights into structural alternations, interface characteristics, interactions between the co-doping ions and the matrix as well as into the resulting ion distributions are still necessary to evaluate the complicated effects of co-doping on the electrochemical properties of LTO.
It should be pointed out that the conductivity and electrochemical performance of LTO can be improved by doping with some cations and anions. However, it is not feasible to prepare the doped-LTO anode for large scale commercial production. In addition, some ion doping in LTO has demonstrated that the high-rate performances are far from reaching satisfactory common values and further research efforts are still needed. Hence, the research community is still looking for an alternative approach to improve the conductivity and electrochemical performance of LTO using some other techniques and methods.
3.2 Nanostructured LTO
One of the widely used chemical strategies is to synthesize nanostructured LTO materials with various nanoarchitectures such as nanorods, nanowires, and nanotubes.167–170 In this strategy, the electronic conductivity and Li+ ion diffusion coefficient are essentially unchanged. Particularly, these LTO nanostructures can facilitate both the electron and lithium ion transport by reducing their transportation pathways within the LTO particles, and also improve the intercalation kinetics by providing a larger electrode/electrolyte contact area.171–174 Several typical nanostructures, including one-dimensional, two-dimensional, and three-dimensional LTO materials, have been prepared and summarized herein.3.3 Carbon surface modification of LTO
Carbon materials have proven to be an effective means of improving the electrochemical performance of the LTO anode material due to their low cost and high conductivity. So far, tens of literature articles have reported the syntheses and electrochemical properties of various carbon–LTO anode composites. Using different carbon forms (e.g., amorphous carbon, carbon nanotubes, and graphene) and carbon surface modifications on LTO, they generally promote the electron transport among the interconnected LTO particles. This process effectively enhances the electronic conductivity of such carbon–LTO composite anode materials, and thus improves the electrochemical performance in terms of rate capability, lithium storage capacity and capacity retention.168,181–190In addition, the carbon-coating thickness is generally related to the carbon content and surface area. A high carbon content can contribute to a high surface conductivity but also produce thick carbon-coating (i.e. up to 10 nm), which might restrict the transport of lithium ions, in which case it is thus unfavorable for attaining high rate capacities.194–196 Only optimized and uniform carbon-coating can provide good conductivity and fast Li-ion transport in the carbon-coated LTO materials. However, it still remains a major challenge to fabricate carbon-coated LTO materials with optimized carbon content and a desirable coating thickness using facile techniques.63,90,168,181,186–190,192–197 The ultimate goal in many studies is to find a simple and efficient method for the synthesis of these LTO anode materials with optimal and uniform carbon-coatings.182,186,187,191 An enhanced electrochemical performance can be achieved with a synergistic effect between high electric conduction and high ionic transport by optimizing the coated-carbon content and structure.194–198 Here, we summarize several typical studies of the effect of morphologies and structures of the carbon-coated layer on the lithium storage performance of these carbon-coated LTO composites.
Wang and co-authors168 reported LTO nanoparticles with a double conductive surface modification of Ti(III) and carbon, synthesized using a facile solid-state reaction. The initial discharge capacity of the as-prepared C-LTO reaches 160 mA h g−1 at a current density of 0.1 A g−1 between 3.0 and 1.0 V, and maintains 90% of the initial capacity after 100 cycles. After the 10th cycle, the irreversible capacity almost disappears, indicating a charge–discharge efficiency of nearly 100%. Furthermore, a discharge capacity of 75 mA h g−1 is also achieved even at a high current density of 3 A g−1 (about 20 C). Cheng and co-authors182 reported a simple approach to synthesize carbon-coated nanostructured LTO with various morphologies, such as LTO nanorods, hollow spheres and nanoparticles, using a simple carbon pre-coating process. The as-synthesized LTO nanorods exhibit excellent cycling performance between 1.0 and 3.0 V at a rate of 1 C, and maintain 95% of the initial capacity after 1000 cycles. This can be ascribed to the fact that the carbon layer acts as a buffer layer to maintain a good electronic conductivity path. Wang and co-authors185 reported nanocarbon-coated LTO, obtained with a solid-state reaction using the precursors TiO2 and Li2CO3 with added sugar. The sugar does not affect the spinel structure, and it decreases the particle size by inhibiting the agglomeration of particles during the heat treatment. Electrochemical results show that the nanocarbon-coated LTO displays a larger diffusion coefficient for the lithium ions, a higher rate capability and excellent reversibility. Gao and co-authors184 reported a spherical C–LTO composite with a high specific capacity and excellent cycling performance synthesized with a novel technique using carbon black. Yang and co-authors190 reported a C–LTO composite synthesized using a simple solid-state reaction using Super-P–Li conductive carbon black as the reaction precursor. The initial reversible specific capacity of the C–LTO composite at a 0.5 C rate, cycled between 1.0 and 2.0 V, is 174.5 mA h g−1 and after 300 cycles its value remains at 143.9 mA h g−1.
Jung and co-authors199 reported that spherical, high tap density, carbon-coated LTO powders were synthesized using a spray-drying process followed by a facile pitch coating. Structural analyses showed that the carbon layer uniformly coats the LTO particles without producing any crystalline changes. The carbon-coating significantly increases the electrical conductivity of LTO making it an efficient, high-rate electrode for lithium cells. The electrochemical tests in fact confirm that the 3.25 wt% carbon-coated LTO electrode operates at ultra-high rate capacity levels, i.e., 100 C, and has an excellent capacity retention and charge–discharge efficiency for a life extending over 100 cycles. A comparison of structure and electrochemical performance for carbon-free and carbon-coated LTO electrodes is shown in Fig. 2.
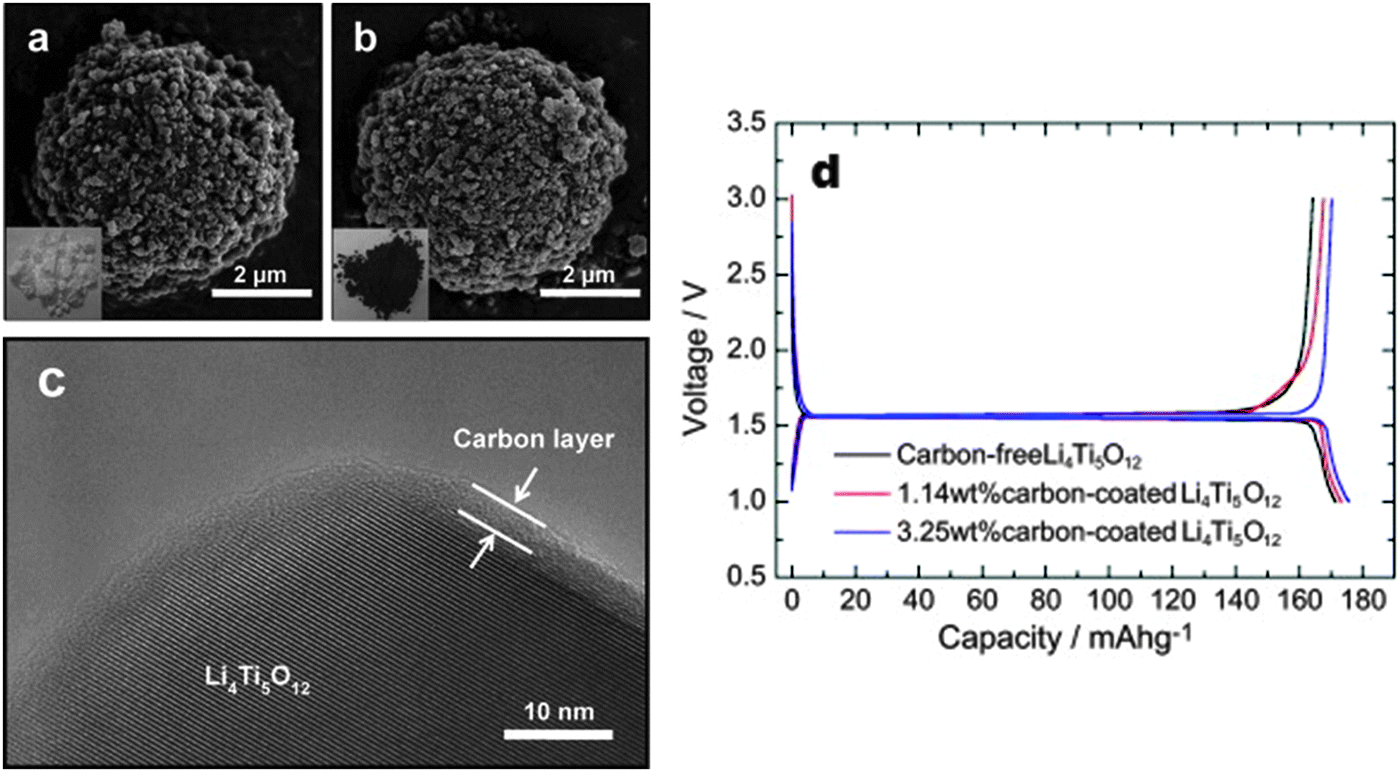 | ||
| Fig. 2 SEM images of (a) carbon-free and (b) carbon-coated LTO powders. (c) High-resolution TEM image of the carbon-coated LTO. (d) Initial charge and discharge curves of carbon-free and of two different carbon-coated LTO electrodes at a rate of 0.1 C. Reproduced with permission from ref. 199. Copyright 2011 Elsevier. | ||
Lin and co-authors200 reported the synthesis of a C–LTO anode composite using a modified one-step solid-state reaction using the original materials of lithium polyacrylate (PAALi) as the lithium and carbon source, and TiO2 as the titanium source. The initial specific capacity of the composite is 130.0 mA h g−1 at a current density of 8.60 mA cm−2, with a capacity loss of 9.0% after 50 cycles. Liu and co-authors201 reported a C–LTO composite synthesized at 800 °C for 15 h under argon, containing 0.98 wt% of carbon. The composite exhibits better electrochemical properties compared with the pristine LTO due to the enhanced electrical conductive network of the carbon-coating on the particle surface. The composite can deliver a capacity of 173.9 mA h g−1 at a 0.1 C rate while that of pure LTO is 165.8 mA h g−1 at 0.1 C. Liu and co-authors201 reported spinel C–LTO powders synthesized successfully with a simple rheological phase method using polyvinylbutyral (PVB) as both template and carbon source. This allows the LTO particles to connect effectively with each other. Compared with pristine LTO, the surface electrical conductivity of the composite is improved significantly due to the in situ formation of carbon-coating, resulting in an enhanced electrochemical performance.
Guo and co-authors202 reported the synthesis of three C–LTO composites with diverse carbon-coating morphologies and structures using a simple solid-state method. The coated-structures were characterized by means of electron microscopy and Raman spectroscopy. Effects of carbon-coating on lithium storage performance, especially on the high rate capability of the C–LTO composites, were investigated. The results revealed that the porous and homogeneous carbon layer was more favorable to achieve the balance between good electron conduction and Li ion diffusion, than the island-like carbon layer and the compact one, and thus the corresponding LTO–C composites exhibited improved electrochemical performance. A schematic illustration for the effect of coated carbon on the electron conduction and Li ion diffusion within three different C–LTO composites is illustrated in Fig. 3. At a low current rate of 0.2 C, the reversible capacity of the C–LTO composite with a porous and homogeneous carbon layer, was as high as 171 mA h g−1. At 5 C, the high capacity of 150 mA h g−1 experienced no loss after 300 cycles. Importantly, at a high charge–discharge rate of 30 C, the reversible capacity still remained at a high level of 82 mA h g−1. Due to the optimal balance between electron conduction and Li ion diffusion, the C–LTO composite with a porous and homogeneous carbon layer exhibited remarkable electrochemical properties including highly reversible capacity, superior rate capability and good cycling stability.
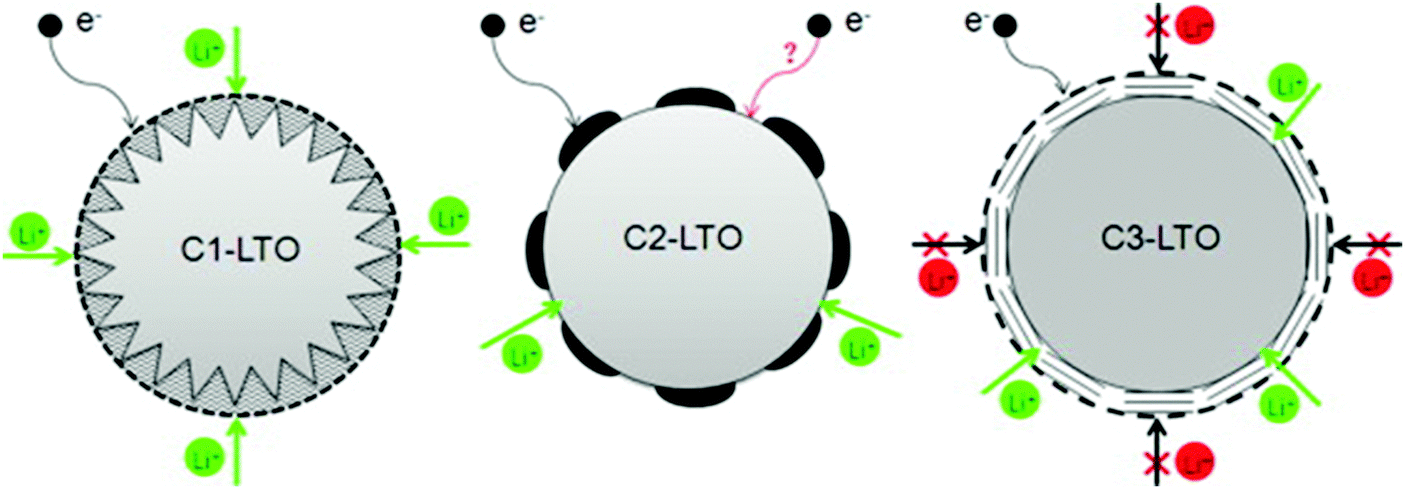 | ||
| Fig. 3 Schematic illustration of the effect of coated carbon on the electron conduction and Li ion diffusion within C1–LTO (porous and homogeneous), C2–LTO (non-homogeneous), and C3–LTO (non-porous coating) composites. Reproduced with permission from ref. 202. Copyright 2014 Elsevier. | ||
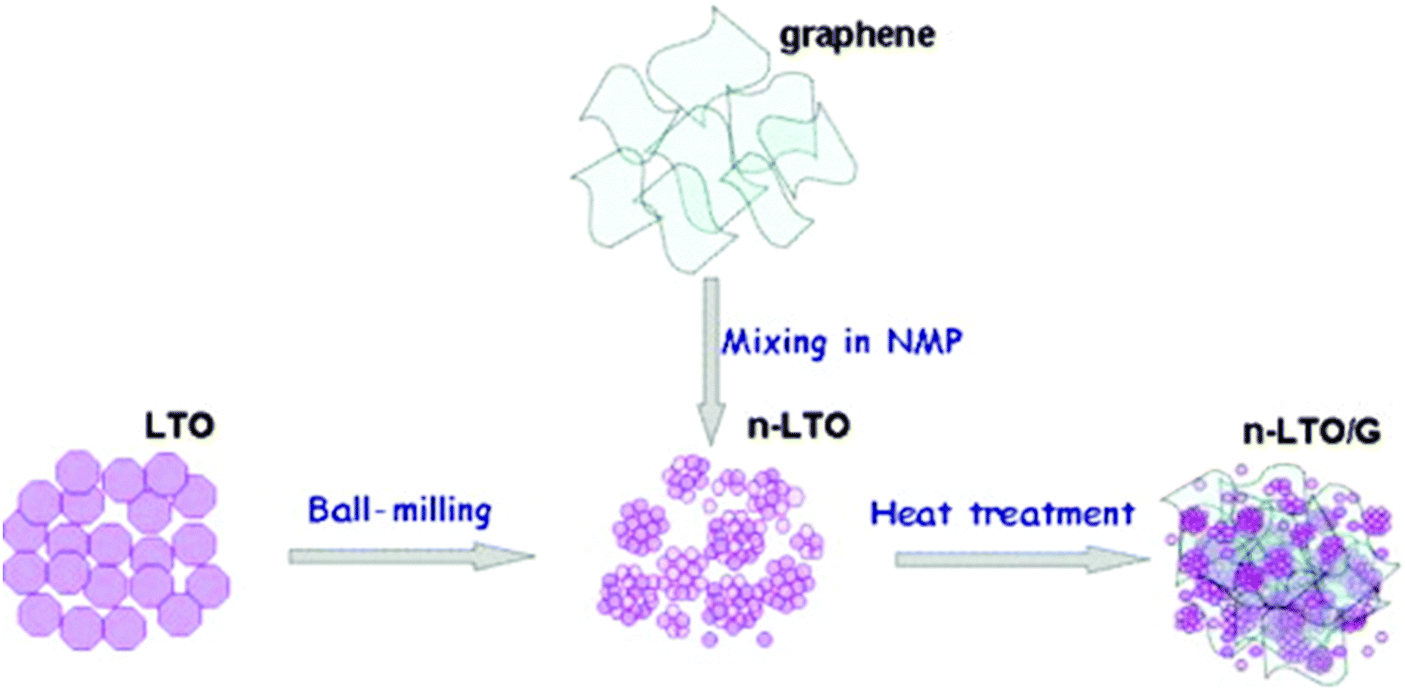
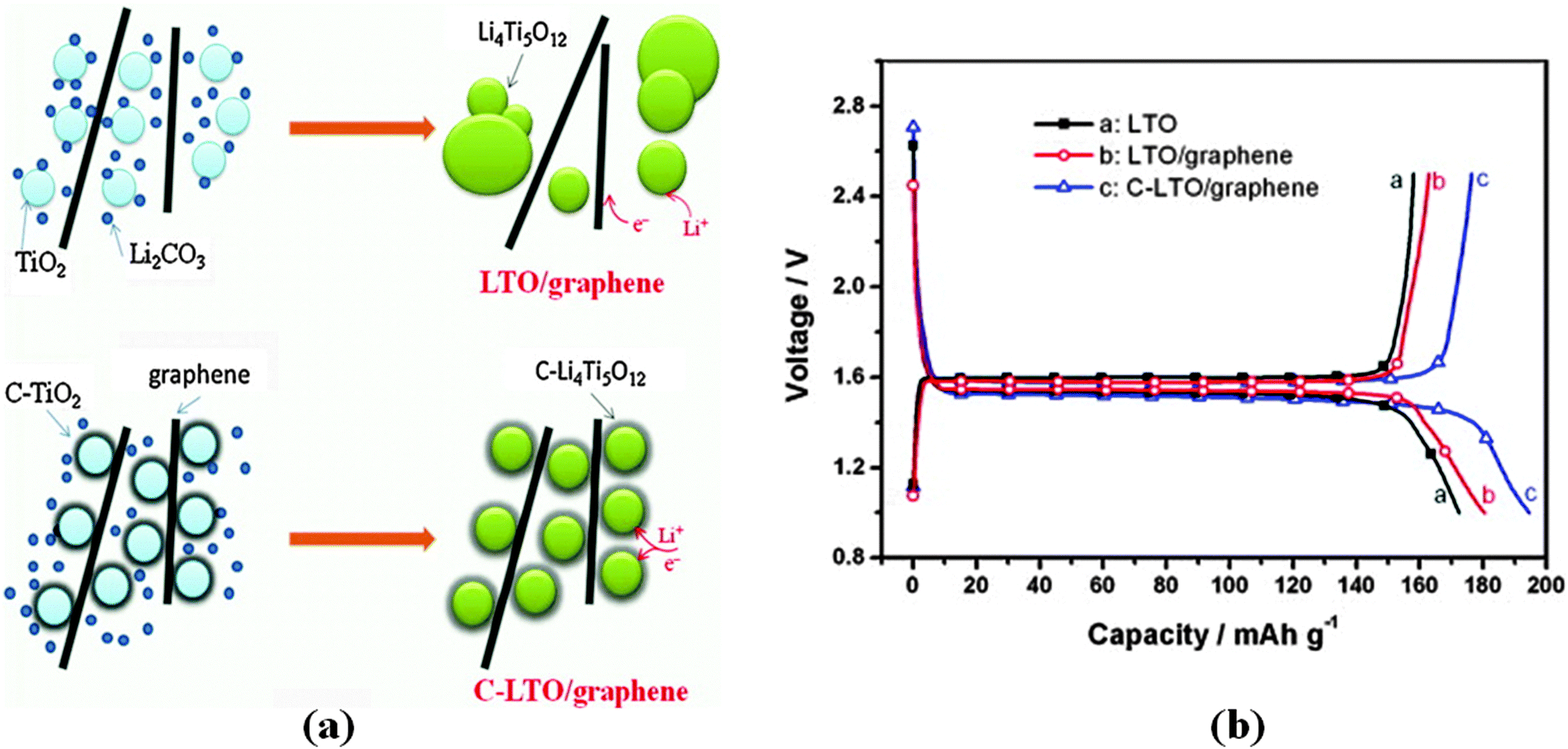
Zhu and co-authors54 reported that the as-prepared graphene-embedded LTO anode material showed improved discharging–charging and cycling properties, particularly at high rates (22 C), which makes the nanocomposite an attractive anode material for applications in electric vehicles. Shen and co-authors205 developed a facile route to fabricate a composite of electrically conductive graphene nanosheets (GNS) anchored with nanocrystalline LTO as an advanced anode material for high performance LIBs. The hybrid nanostructures endow the composites with a high transportation rate for both Li+ and electrons (especially at high rates), because the hybrid nanostructure is capable of effectively utilising the good conductivity, high surface area, and good electrochemical performance of GNS and the good stability of the fine LTO nanoparticles. It is the synergy of these two parts that leads to a high rate capability and stable lithium storage. Shi and co-authors206 reported a simple strategy to prepare a hybrid of LTO nanoparticles well-dispersed on electrically conductive graphene nanosheets as an anode material for high rate LIBs. The preparation process is illustrated in Fig. 4, and the comparison of morphologies for pure nano-LTO and the nano-LTO–G hybrid is shown in Fig. 5. The authors have found that the electron transport was improved by forming a conductive graphene network throughout the insulating LTO nanoparticles. The charge transfer resistance at the particle/electrolyte interface is reduced from 53.9 Ω to 36.2 Ω and the peak currents measured using cyclic voltammetry are increased at each scan rate. The difference between the charge and the discharge plateau potentials becomes much smaller at all discharge rates because of the lowered polarization. With 5 wt% graphene, the hybrid LTO materials deliver a specific capacity of 122 mA h g−1, even at a very high charge–discharge rate of 30 C, and exhibit an excellent cycling performance with the first discharge capacity of 132.2 mA h g−1 and less than 6% discharge capacity loss over 300 cycles at 20 C. The outstanding electrochemical performance of the nano-LTO–graphene hybrid makes it a promising anode material for high-rate LIBs. Oh and co-authors207 reported the synthesis of graphene-wrapped LTO particles using a solid-state reaction. It has been found that the LTO, tightly bound to the graphene, exhibits a remarkable specific capacity of 147 mA h g−1 at a rate of 10 C (175 mA h g−1 at 1 C) after 100 cycles. The improved rate capability was attributed to the enhanced electronic conductivity of each LTO grain via uniform graphene wrapping. Zhang and co-authors208 prepared an LTO–graphene composite (1.86 wt%) electrode material using both ball milling and a simple chemical method. It has been found that the composite displayed a high-rate capacity of 118.7 mA h g−1 at 20 C and a good cycle stability, retaining over 96% of its initial capacity after 50 cycles at 10 C. The excellent electrochemical performance is attributed to a decrease in the charge-transfer resistance due to the fact that the LTO particles uniformly cling to the graphene sheets. Rai and co-authors209 reported the solvothermal preparation of LTO–graphene (15 wt% and 30 wt%) nanocomposite anodes for high-performance lithium-ion batteries. Structure and morphology studies of the nanocomposites revealed that the LTO nanoparticles are embedded onto the graphene nanosheets. The electrochemical performances of the LTO–graphene nanocomposites indicate higher capacities and enhanced cycle performances within the voltage range of 1.0–2.5 V, suggesting that the LTO–graphene nanocomposite is a promising anode material for high-energy density and safe LIBs. Han and co-authors210 reported that strongly coupled LTO–reduced graphene oxide (RGO) nanocomposites were synthesized using the solvothermal treatment of a GO colloidal suspension containing a microcrystalline LTO precursor at elevated temperatures. The solvothermal treatment of the mixture suspension of GO and LTO gives rise not only to the reduction of GO to RGO but also to the attachment of the LTO particles to the flat surface of the RGO 2D nanosheets. The crystal structure and morphology of the LTO particles remain intact after the composite formation with the RGO nanosheets. The LTO–RGO nanocomposites display better anode performance with a larger discharge capacity of 175 mA h g−1, underscoring the merit of RGO hybridization in improving the electrode performance of the bulk metal oxide, as shown in Fig. 6. The obtained LTO–RGO nanocomposites show promising electrode performance, which is superior to those of the pristine LTO microcrystals. The enhancement in electrical conductivity is mainly responsible for the observed improvement in the electrode performance upon composite formation with the RGO nanosheets. Pang and co-authors211 reported a mesoporous LTO–graphene hybrid synthesized using a facile electrostatic adsorption method. Such an electrostatic adsorption method is easy to scale up and will provide a new pathway for the production of various high-rate lithium-ion battery materials. The graphene sheets are strongly attached to the LTO spheres and thus can form an efficient electrically conducting network, permitting the electrode to speed up the charge and discharge processes. The as-obtained LTO–graphene nano-hybrid exhibits a high reversible capacity and outstanding rate performance owing to the synergy of good conductivity, large surface area, flexibility of graphene and good stability of the mesoporous LTO. The discharge capacity of the LTO–graphene hybrid was improved to 124 mA h g−1 at a 20 C rate, which is more than twice the value of the LTO electrode. Xiang and co-authors212 prepared a LTO–graphene composite using a facile sol–gel method. The LTP–graphene composite presents a higher capacity and better cycling performance than LTO at the cut-off of 2.5–1.0 V, especially at high current rates. The cycling stability of the cells with the electrodes using LTO and the LTO–graphene composite at 0.2 C and 10 C were investigated, the discharge capacity of LTO–graphene is slightly higher than the capacity of LTO and the specific capacity retention during 100 cycles is 94%. At 10 C, the 100th discharge capacity of LTO–graphene is 110 mA h g−1, which is much higher than that of LTO (84 mA h g−1). The improvement in capacity retention especially in high rate capacity can be attributed to the introduction of superior conductive graphene. On the one hand, the graphene sheets induce the formation of fine Li4Ti5O12 nanoparticles in the sol–gel synthesis. The reduced size signifies a shortened diffusion distance for the lithium ions and thus the kinetics of the lithium insertion/extraction are accelerated. On the other hand, the electronic conductivity of the Li4Ti5O12 nanoparticles anchored onto the graphene sheets is significantly improved.
 | ||
| Fig. 4 Schematic representation of the preparation of the hybrid of nano-LTO and graphene. Reproduced with permission from ref. 206. Copyright 2014 Elsevier. | ||
 | ||
| Fig. 5 SEM images of nano-LTO (a) and the nano-LTO–graphene hybrid (b). TEM (c) and HRTEM (d) images of the nano-LTO–graphene hybrid. Reproduced with permission from ref. 206. Copyright 2014 Elsevier. | ||
 | ||
| Fig. 6 Schematic illustration of the preparation of the LTO–RGO nanocomposites and graph of the improvement in electrode performance. Reproduced with permission from ref. 210. Copyright 2012 American Chemical Society. | ||
The excellent electrochemical performance of the LTO/graphene electrode could be attributed to the improved electronic conductivity of the graphene sheets. When discharged to 0 V, the LTO–graphene composite exhibited a quite high capacity of over 274 mA h g−1 below 1.0 V, which was beneficial for not only a high energy density but also the safety characteristics of lithium-ion batteries. Ri and the co-authors213 reported the one-step synthesis of an LTO/graphene–ionic liquid (IL) nanostructure using a microwave-assisted hydrothermal reaction, as shown in Fig. 7, in which the ionic liquid of C12H23N2Cl was used as an exfoliating agent to control the microstructure of the graphene sheets, and as an inducer for LTO growth on the graphene sheets. The resultant LTO/graphene–IL exhibited a two-dimensional structure with a total thickness of about 20 nm and with the LTO nanoparticles uniformly anchored on the surface of the thin graphene sheets (2 nm). Such a unique microstructure provided a high electrode/electrolyte contact area for the electron transport, and the nanosize LTO led to a short path for the lithium ion transfer. When LTO/graphene–IL was used as the anode material for a lithium-ion battery, it showed a high-rate performance, as evident from Fig. 8. The LTO–graphene nanostructure exhibits excellent reversibility (159 mA h g−1 at 0.5 C after 100 cycles) and high-rate performance (162 mA h g−1 at 0.2 C, 148.5 mA h g−1 at 20 C).
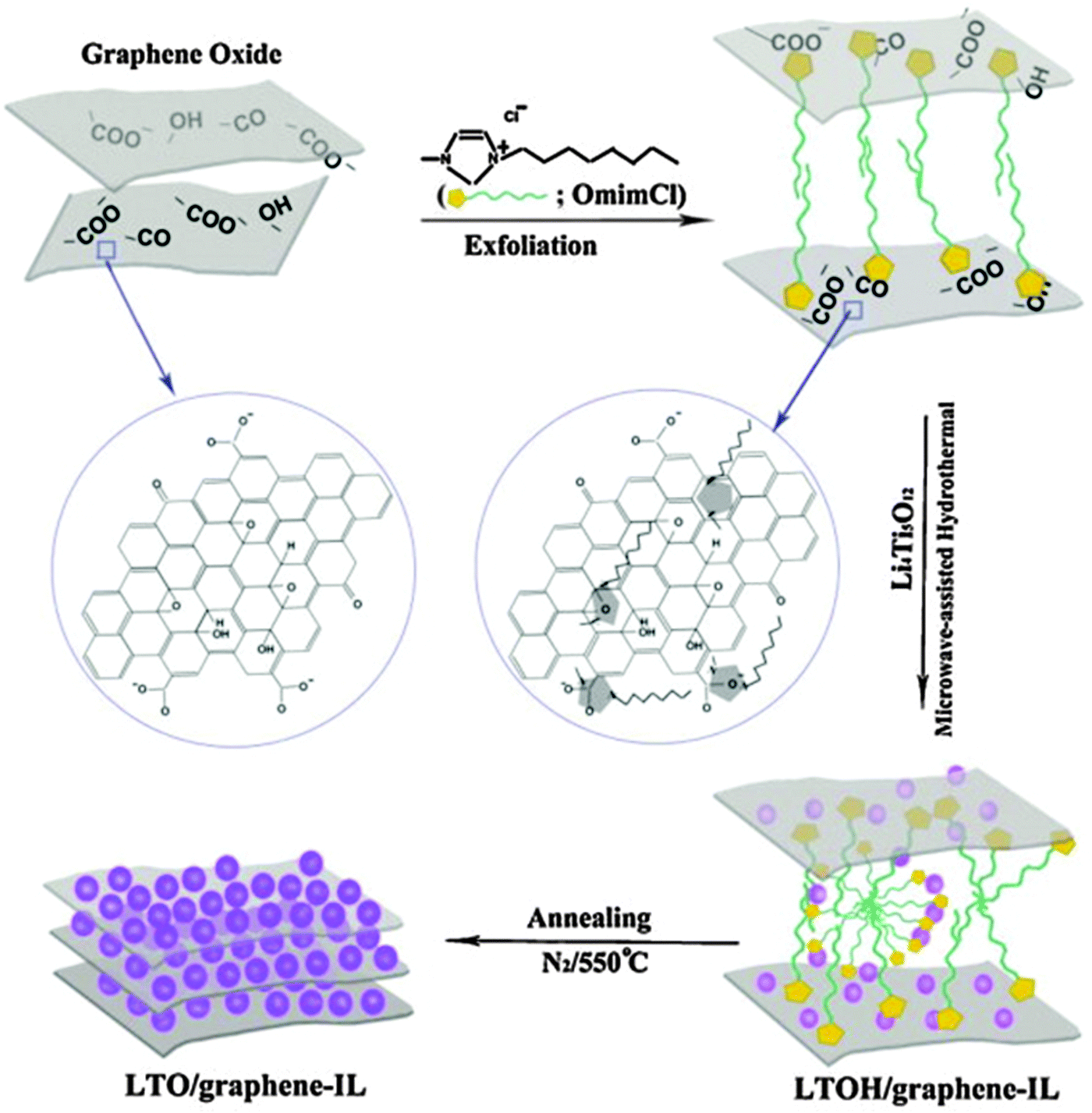 | ||
| Fig. 7 Schematic representation of the preparation of the LTO/graphene–IL nanostructure from the LTO precursors, GO, and the ionic liquid using a microwave-assisted hydrothermal reaction process. Reproduced with permission from ref. 213. Copyright 2013 Elsevier. | ||
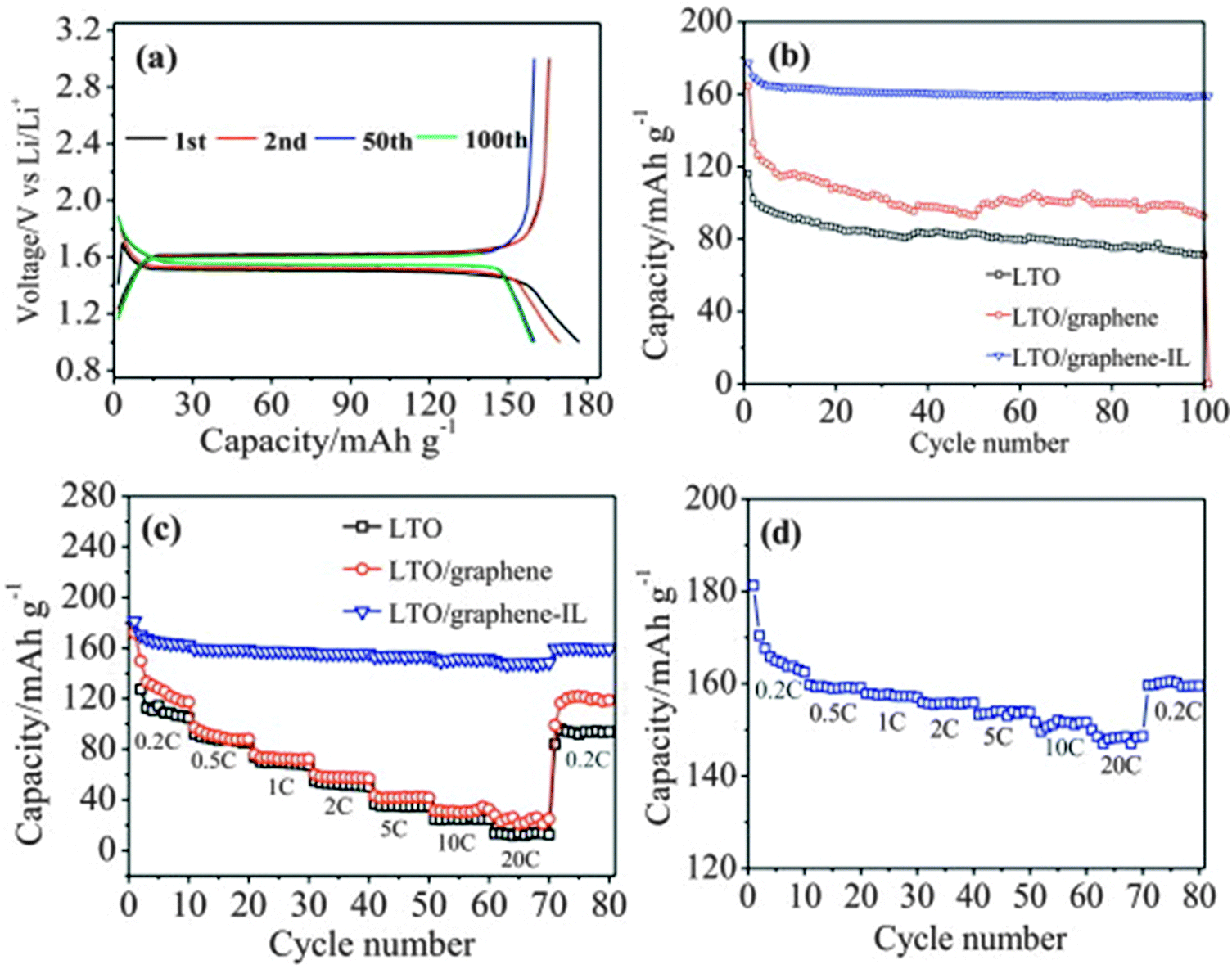 | ||
| Fig. 8 (a) Galvanostatic charge–discharge profiles of the LTO/graphene–IL electrode during 1st, 2nd, 50th and 100th cycle at 0.5 C. (b) Cyclic performance of the LTO, LTO/graphene and LTO/graphene–IL electrodes at 0.5 C. (c) High-rate performance of the LTO, LTO/graphene and LTO/graphene–IL electrodes. (d) High-rate performance of the LTO/graphene–IL electrode. Reproduced with permission from ref. 213. Copyright 2013 Elsevier. | ||
Ni and co-authors214 reported the preparation of LTO–reduced graphene oxide (RGO) composites using a simple strategy, as illustrated in Fig. 9. The as-prepared composites present the LTO nanoparticles uniformly immobilized on the RGO sheets. The LTO–RGO composites possess excellent electrochemical properties with a good cycle stability and high specific capacities of 154 mA h g−1 (at 10 C) and 149 mA h g−1 (at 20 C), much higher than the results found in other literature publications. The superior electrochemical performance of the LTO–RGO composites is attributed to the unique hybrid structure of the conductive graphene network with the uniformly dispersed LTO nanoparticles. Gao and co-authors215 prepared a homogeneous LTO–graphene composite using an in situ solid-state reaction, after carbon pre-coating. Its microstructure is compared with the materials prepared using a similar method but without carbon-coating, which is illustrated in Fig. 10a. The results reveal that the carbon-coating not only effectively confines aggregation and agglomeration of the LTO particles, but also enhances the conjugation between the LTO particles and the graphene sheets. The specific capacities of the pristine LTO, the LTO–graphene composite and then carbon–LTO–graphene composite were measured by charge–discharge tests at a 0.2 C rate over the potential range of 1.0–2.5 V. It can be observed in Fig. 10b that they have similar charge and discharge plateaus, indicating that the small quantity of graphene sheets does not affect the electrochemical reaction process of LTO. All the products display very flat charge–discharge plateaus at around 1.55 V (vs. Li/Li+), demonstrating a characteristic two-phase reaction based on the Ti4+/Ti3+ redox couple. The carbon–LTO–graphene composite delivers a high reversible capacity of 177 mA h g−1, which is even higher than the theoretical capacity of LTO (175 mA h g−1). The increased capacity can be attributed to lithium storage on the graphene sheets. It is clear that carbon pre-coating markedly enhances the lithium storage capability of the LTO–graphene composite. The LTO–graphene composite also presents excellent rate capability and low-temperature performance. Even at 120 C, it still delivers a high capacity of about 136 mA h g−1. In addition, fuel cells using LiNi1/3Co1/3Mn1/3O2 as the cathode material exhibit good rate capability.
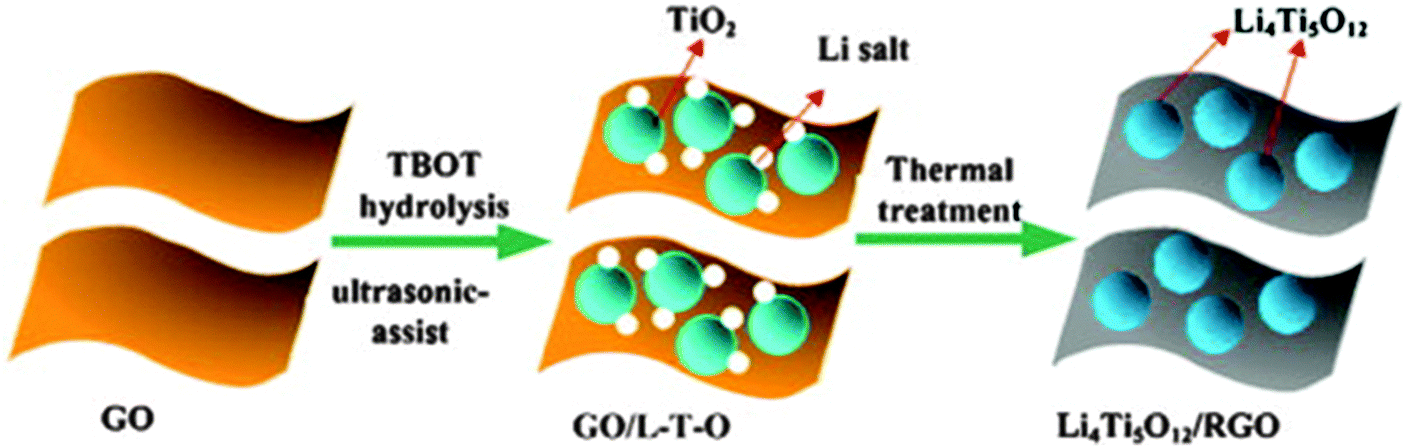 | ||
| Fig. 9 Schematic representation of the fabrication process of the LTO–RGO composites. Note here: TBOT is C16H36O4Ti. Reproduced with permission from ref. 214. Copyright 2014 Elsevier. | ||
 | ||
| Fig. 10 (a) Schematic illustration of the effect of carbon-coating on the LTO–graphene composites. (b) Galvanostatic charge–discharge curves of the LTO, LTO–graphene, and C–LTO–graphene samples at a 0.2 C rate. Reproduced with permission from ref. 215. Copyright 2013 Elsevier. | ||
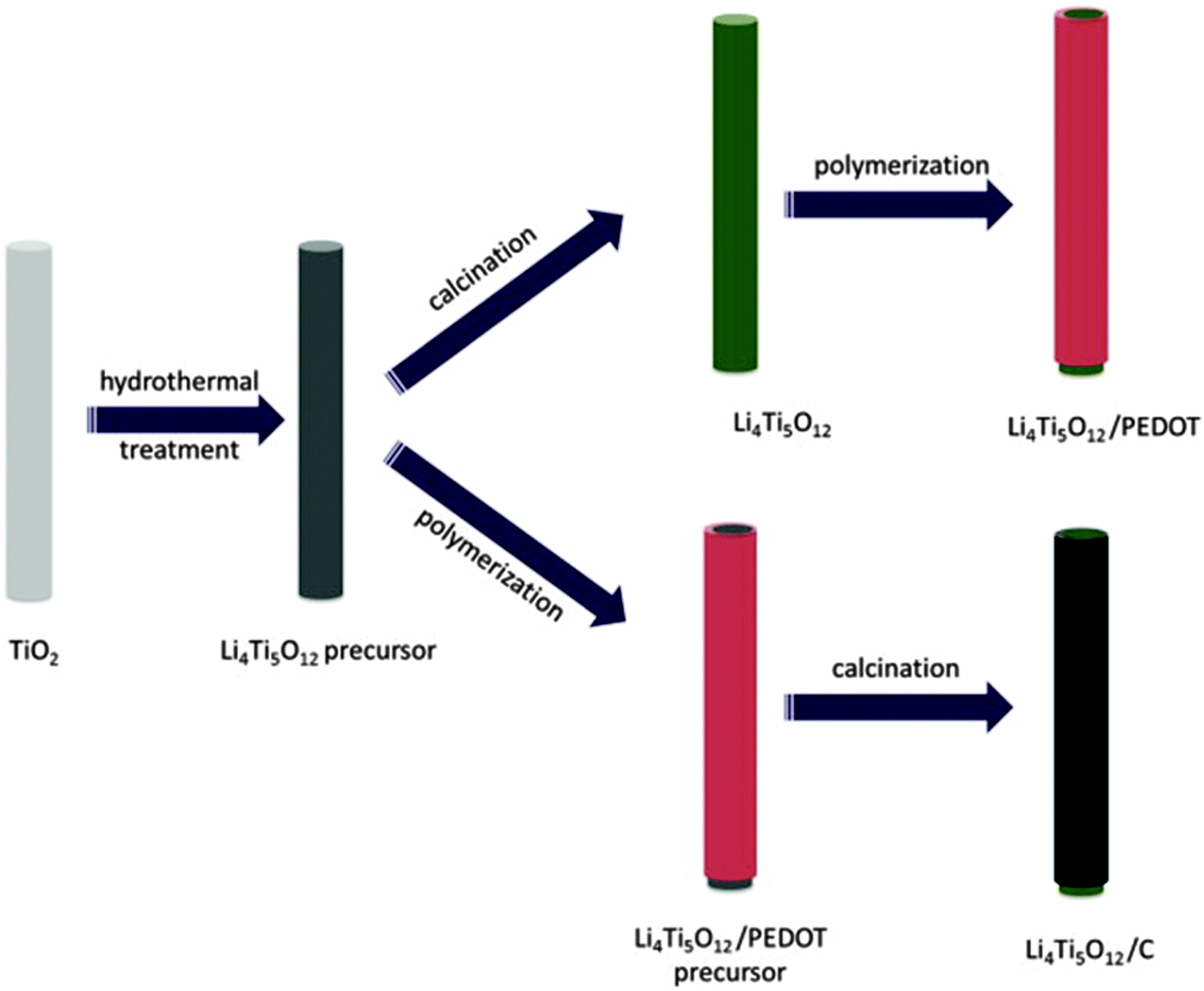 | ||
| Fig. 11 Schematic illustration for the synthesis of the LTO/PEDOT and LTO/C nanorods. Reproduced with permission from ref. 216. Copyright 2014 Elsevier. | ||
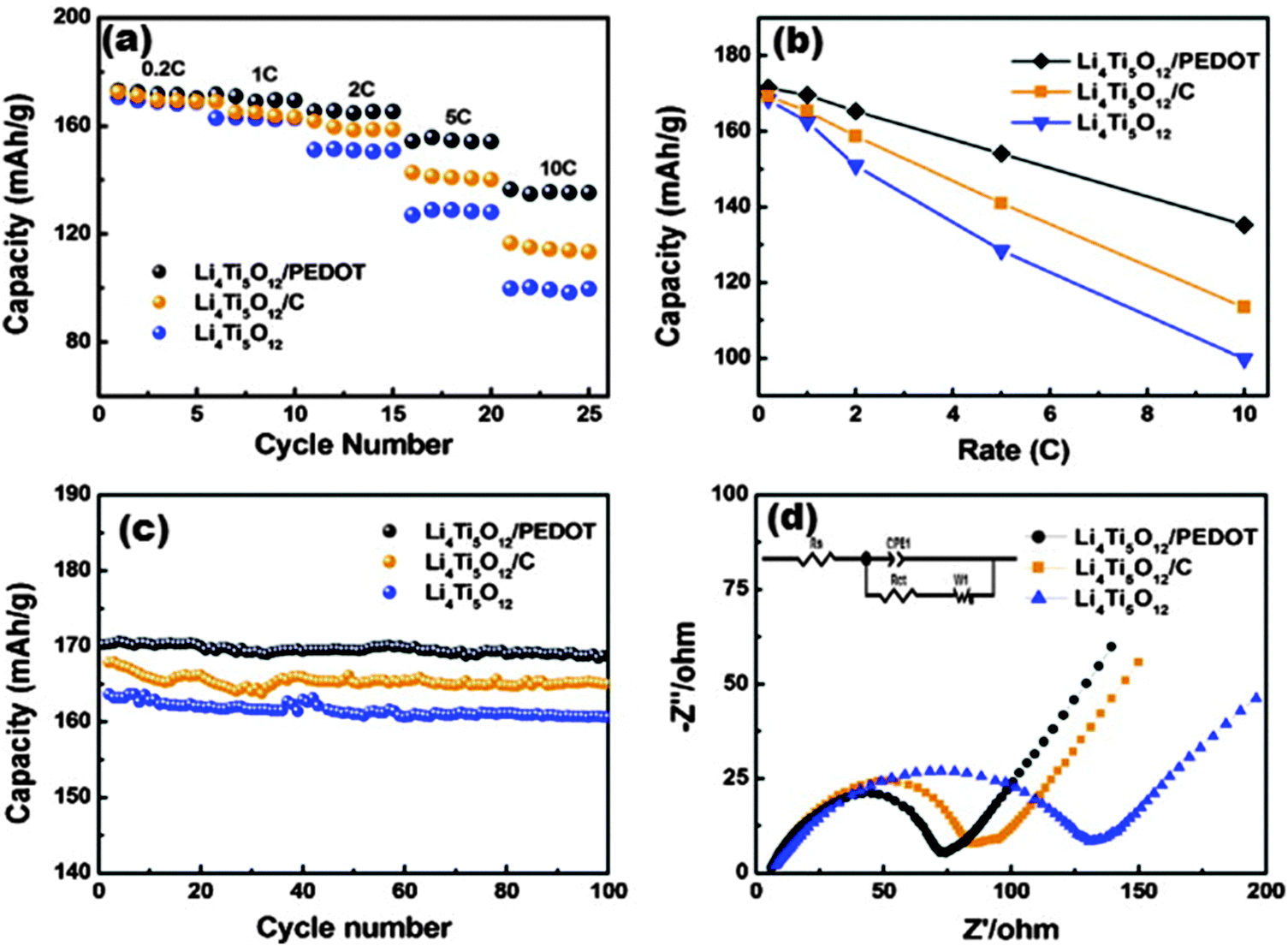 | ||
| Fig. 12 (a) The rate and cycling performances at different rates. (b) Comparison of the specific capacities at different rates. (c) Cycling performance at a 1 C rate. (d) Nyquist plots of the pure LTO, LTO/PEDOT and LTO/C electrodes. Reproduced with permission from ref. 216. Copyright 2014 Elsevier. | ||
4. Application as anodes
A high-power energy storage device is essential for E-transportation such as electric vehicles (EVs), hybrid electric vehicles (HEVs) and plug-in hybrid electric vehicles (PHEVs). Safety is a major consideration for batteries in automotive applications. As discussed previously, the use of LTO as an anode coupled with high voltage cathodes such as LiCoO2 (LCO), LiMn2O4 (LMO), or LiFePO4 (LFP) in a high-power lithium-ion battery has been demonstrated to be feasible in terms of the electrochemical performance, such as excellent cycle life and inherent safety.6,217,218 It was reported that by combining the 1.5 V LTO anode with another cathode, the safety of LIBs can be significantly improved.219 2 V LTO/LCO (LiCoO2) cells exhibit an excellent cycle life as is expected with lithium intercalation/de-intercalation into/out of stable metal oxide structures.28 In this couple, the positive and negative electrodes are based on transition metal oxide electrodes, which have the capability of accommodating a significant amount of lithium within the host electrode structure, as shown in the Fig. 13. By restricting shallow limits of charge and discharge, the structural integrity of the electrodes can be maintained, and this permits a high cycle life to be obtained with this combination.28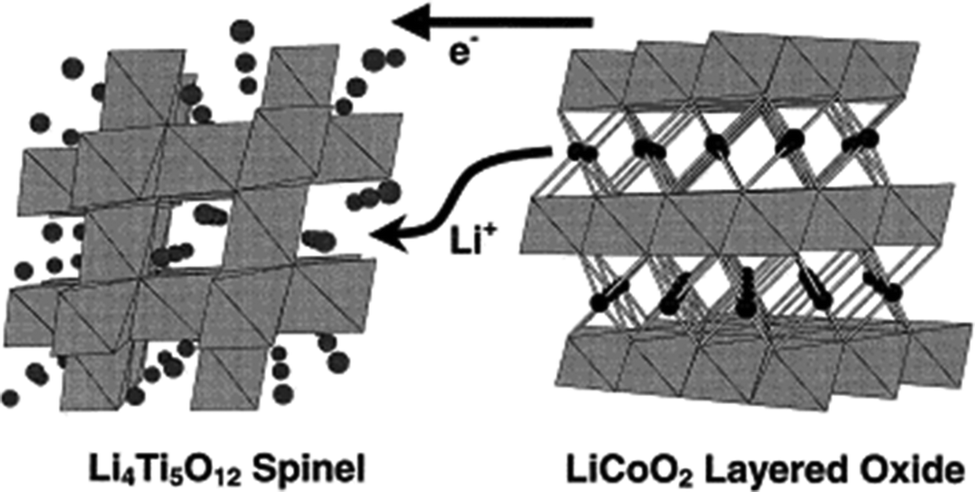 | ||
| Fig. 13 Intercalation into the LTO spinel and de-intercalation out of the LCO layered structure from an LTO/LCO ideal cell couple. Reproduced with permission from ref. 28. Copyright 1999 Elsevier. | ||
Lu and co-authors220 reported the design of a lithium-ion cell of LTO/L333 (Li1.1Ni0.3Co0.3Mn0.3O2). The cell exhibited the improved cycling performance at both room and elevated temperatures. For example, such an LTO/L333 cell at room temperature and at an elevated temperature (55 °C) had the same capacity for 30 cycles at a 10 C rate, and the capacity retention remained stable for over 200 cycles.
Bousquet and co-authors221 reported good cycling performance of the Li-ion cells of 2.5 V LTO/LMO (LiMn2O4) with new stable Li-ion battery electrolytes such as new polyfluorinated boron cluster lithium salts. The capacity retention of such a cell was found to maintain 93% after 160 cycles at a 1 C rate at 50 °C.222,223
It has been reported that 2.5 V LTO/LMO cells223 also showed a very promising cycling performance and a larger capacity at high current rates, which is necessary for the high current pulsing in transportation applications. These LTO/LMO cells, with capacities of ∼2 A h g−1, provided new insights into the commercialization of LTO/LMO batteries.
A cell with a 2.5 V LTO–LMO system224 also showed unprecedented power capability over that of any existing lithium-ion battery system for HEV and other transportation applications. This LTO–LMO system also demonstrated good calendar life, low heat generation during high-rate operation, and excellent low-temperature (−30 °C) performance, as well as unmatched abuse tolerance.
Hu and co-authors225 reported a hybrid battery–supercapacitor LTO/(LiMn2O4 + AC) using an LTO anode and a LiMn2O4/activated carbon (AC) composite cathode. It is demonstrated that the hybrid battery–supercapacitor has advantages in both high rate capability from the hybrid capacitor AC/LTO and high capacity from the secondary battery LTO/LMO. At a 4 C rate, the capacity loss in constant current mode is no more than 7.95% after 5000 cycles, and the capacity loss in constant current–constant voltage mode is no more than 4.75% after 2500 cycles.
Panero and co-authors226 also reported a new type of lithium-ion cell, based on the combination of an LTO anode with a high voltage mixed spinel solid solution Li2Co0.4Fe0.4Mn3.2O8 cathode. This combination provides some promising features that may be used as a starting base for future development and optimization.
In recent years, Li-ion batteries based on an LTO anode have been investigated and developed for load-leveling applications, based on the favorable characteristics emphasized above, including high energy density, long cycle life, stability, and rapid charge. A great deal of research227–231 has been devoted to the new type of 2.5 V lithium-ion cell, based on the combination of a spinel-LTO anode with high-voltage olivine LiMnPO4 (LMPO),228 spinel LiMn0.8Fe0.2PO4 (LMFPO),229 spinel LiMn2O4 (LMO),223,232,233 and spinel LiMn1.5Ni0.5O4 (LMNO)234,235 cathodes. The employment in load-leveling applications has been systemically explored and evaluated for these 2.5 V Li-ion battery cells, including LTO/LMPO, LTO/LMO, LTO/LMNO, and LTO/LMFPO. These Li-ion cells have demonstrated impressive stability, prolonged cycle life and excellent safety features, making them suitable for load-leveling applications. Herein, the redox activity of LTO/LMFPO during the lithiation–delithiation cycling process is shown in Fig. 14.
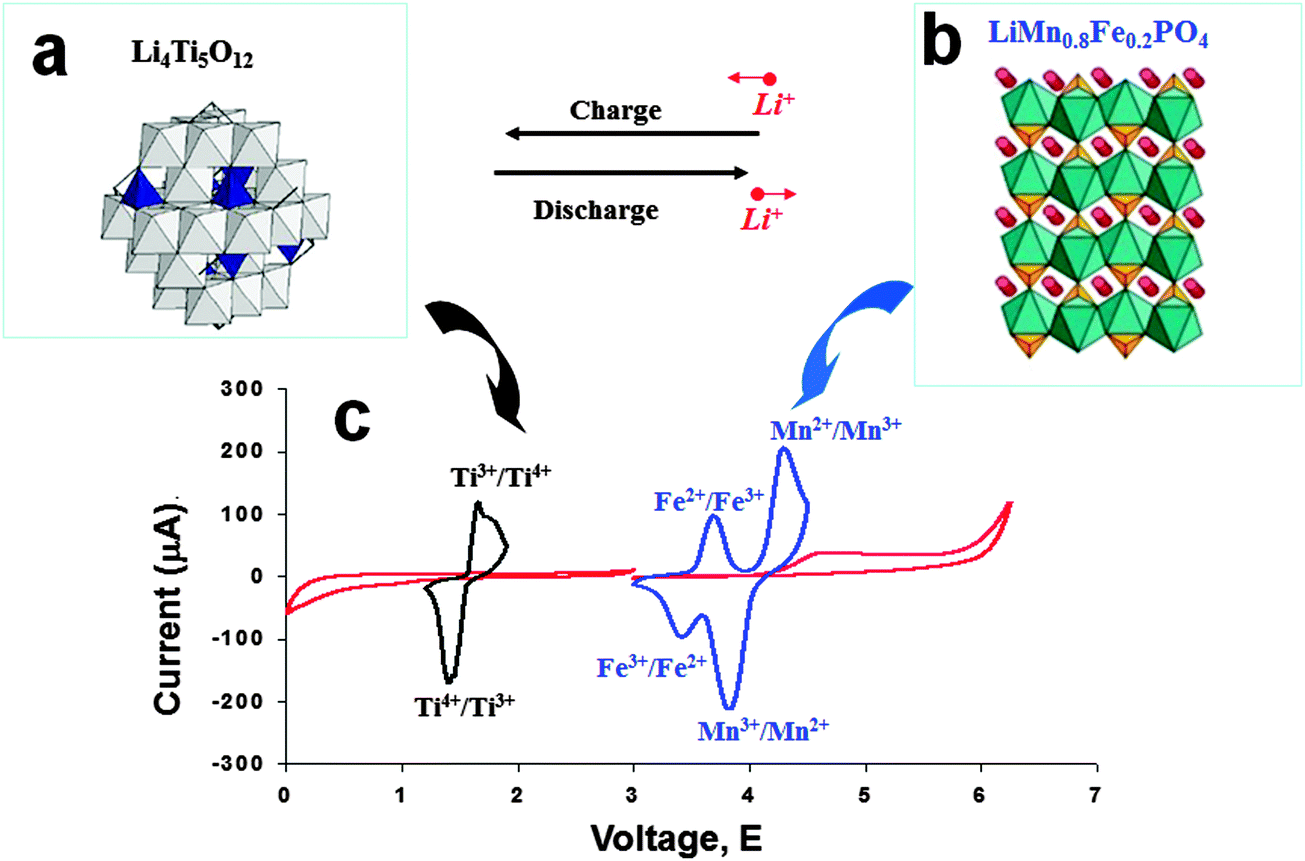 | ||
| Fig. 14 Structure of the LTO spinel anode (a), and the LMFPO (LiMn0.8Fe0.2PO4) cathode (b). (c) Schematic voltammetry response of these electrode materials in an electrolyte. Reproduced with permission from ref. 227. Copyright 2011 The Royal Society of Chemistry. | ||
Sun and co-authors236 reported that commercial battery cells of 2 V LTO/LFP (LiFePO4–PAS (PAS, polyacence–highly conductive polymer)) showed an excellent specific capacity, high relative tap density, long cycle life, and high cycle efficiency at high current rates. It showed no attenuation after 1000 charge–discharge cycles during charging and discharging at a 1 C rate.
Zaghib and co-authors237,238 reported that the LFP/LTO cell has passed successfully the required battery tests for safety use in transportation. LTO/LFP Li-ion cells are safe and can support fast charge–discharge rates without any damage to the cycling life. The impact on electric cars will be important, since it is the most demanding use in terms of performance and safety. The electrochemical performance of the 2 V LTO/LFP 18650-Li-ion batteries has shown them to be fast-charging Li-ion batteries with long shelf lives for power applications in terms of safety and cycling life, which are key issues in E-transportation applications.30,66 The voltage vs. capacity curves for the 18650-cells of LFP/Li, LTO/Li, and LFP/LTO at a 24 C rate are summarized in Fig. 15.
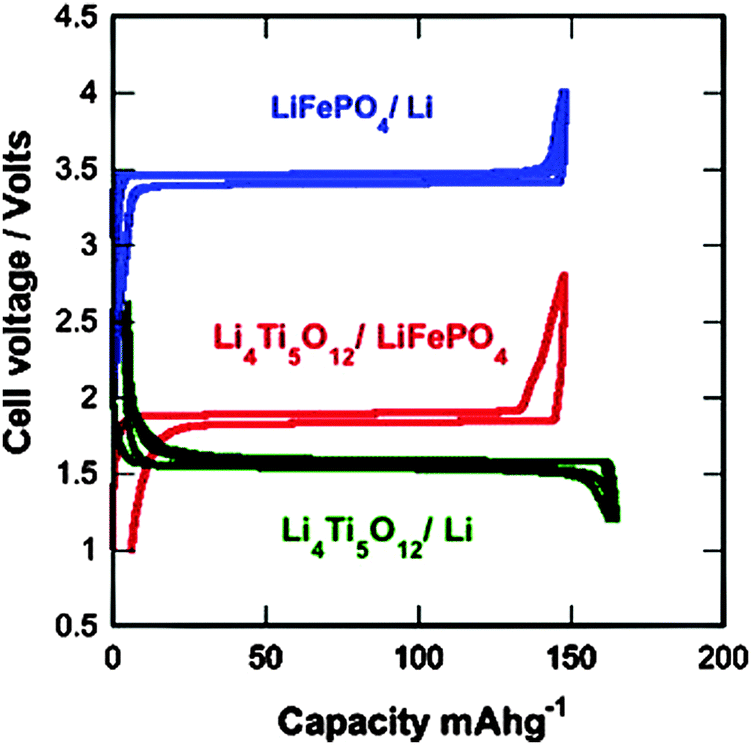 | ||
| Fig. 15 Voltage–capacity cycles for the LFP/Li, LTO/Li, and LFP/LTO-18650-cells at a 24 C rate. Reproduced with permission from ref. 237. Copyright 2011 Elsevier. | ||
Electric cars presented at the World Energy Council (Montréal, September 2010) were equipped with C–LFP/LTO batteries (Fig. 16). They had a weight of 680 kg, a driving range of 32 km with a top speed of 64 km h−1. It has been demonstrated that the charging time was reduced to 5 min with a three-level charger in parallel (500 V, 125 A). This LFP/LTO battery cell technology has been attractive for the fast charging bus Mega Watt battery charging station for EV and PHEV and for energy storage with wind and solar energy.
 | ||
| Fig. 16 E-Cars presented at the World Energy Council (Montréal, September 2010), equipped with C-LFP/LTO battery cells. Reproduced with permission from ref. 237. Copyright 2011 Elsevier. | ||
Ariyoshi and co-authors239 reported the LTO anode combined with the LNMO (LiNi0.5Mn1.5O4) cathode to assemble the 3 V LTO/LNMO cell system, and such a 3 V Li-ion cells showed that 83% of the initial capacity can be stored and delivered even after 1100 cycles, indicating the excellent cyclability and rate capability of the LTO/LNMO cells. Meanwhile, this 3 V LTO/LNMO cell with a dimethyl methylphosphonate (DMMP)-based electrolyte has also demonstrated an acceptable capacity loss after 30 cycles, which makes this 3 V LTO/LNMO cell promising for safe lithium-ion batteries.240
5. Concluding remarks and perspectives
5.1 Concluding remarks
The characteristics of the current lithium-ion batteries have greatly influenced the energy efficiency, reliability, and commercial viability for emerging applications such as high-power backup systems and electric transportation. In fact, the existing lithium-ion batteries have also impacted the reliability of many portable electronic tools such as smart cell phones and laptop computers. So far, lithium-ion batteries have proven to be the most viable solution for electric energy storage. However, the performances of the existing lithium-ion batteries are still not able to meet the increasing demands of these emerging applications. The main scientific objectives are to achieve a rational design of cathode and anode materials with desired properties, such as enhanced rate capability (i.e. power density), high energy density, excellent structural stability, long cycling life, and reasonable costs.This review gives a state of the art overview of spinel-Li4Ti5O12 materials, which have been considered as promising anode materials for lithium-ion batteries due to their attributes of high safety and excellent rate capability.217,241,242 However, a number of challenges still remain in the development of Li4Ti5O12 anode materials for a new generation of lithium-ion batteries to meet the emerging application criteria. Some of the major remaining challenges associated with the existing Li4Ti5O12 anode materials are briefly described below.
(1) Since the improved safety of the Li4Ti5O12 anode is obtained at the expense of energy density due to its high operating voltage vs. Li/Li+ (∼1.5 V), the lithium-ion battery cells with a Li4Ti5O12 anode offer a low theoretical specific energy due to their low cell voltage. Therefore, lithium-ion batteries with Li4Ti5O12 anodes are more likely to be used in the long term for stationary energy storage and hybrid electric vehicles in the transportation sector.
(2) Combined surface coating strategies are needed to create nano-enabled anode materials with improved electronic and ionic conductivities. The combination includes various optimized approaches such as in situ or ex situ active coating and cation or anion doping at nanometer scale. Major technical breakthroughs are needed to tackle the intrinsic insulating character without compromising safety and cost.
(3) Nanostructured Li4Ti5O12 anodes, in the form of nanorods, nanowires, and porous clusters, with carbon-coatings have shown very promising performances. However, little is known about the mechanism of the elementary steps associated with charge and mass transports in the confined dimensions of the Li4Ti5O12 anode (1D, 2D). More sophisticated in situ characterization techniques for probing and mapping those confined Li4Ti5O12 surfaces are needed to gain critical insights into nanoscale phenomena on the nano-Li4Ti5O12 electrode surfaces during cycling.
(4) It is important to mention that safety considerations are as important as performance, since a car accident due to electric vehicle battery safety failure may cause significant damage. High energy density also means high safety risks, and it is the responsibility of the researchers dedicated to this field to ensure that the new lithium-ion batteries meet the safety requirements before they are commercialized.
5.2 New directions
As demonstrated in this perspective review, important future directions and key research and development efforts for spinel-Li4Ti5O12 anode materials for the design of advanced lithium-ion batteries are:(1) From a scientific point of view, more fundamental studies are needed to fully understand and improve electrochemical performance (i.e. rate capability) of the Li4Ti5O12 nanostructures using various parameters such as surface area, crystallinity, and the crystalline orientation. Compared with traditional bulk Li4Ti5O12 materials on the microscale, novel Li4Ti5O12 nanostructured materials have gained more attention as high-rate anodes for rechargeable lithium-ion batteries. Owing to their small size and large surface area, the Li-ion diffusion pathway is reduced and a better contact between the Li4Ti5O12 electrode material and the electrolyte is achieved. Both of these phenomena contribute to the improvement of the rate capability. In addition, more extensive studies of novel Li4Ti5O12 nanoarchitecture anode materials are required. One of the strategies is to create 3D Li4Ti5O12 structures combining microscale templates and nanostructures to maximize the advantages and minimize the disadvantages of the materials at the two scales. The rational design of Li4Ti5O12 nanoarchitecture anodes may lead to higher energy and power densities, significantly enhancing the Li-ion battery performance. Eventually, the fundamental electrochemical kinetic behaviour (ionic or electronic) needs to be evaluated, and the critical factors of lithium intercalation and extraction (e.g., nanostructure size, phase coating, doping, morphology, composition, and mixed valence conduction) need to be investigated systematically.
(2) From a practical point of view, the precursor materials’ availability and cost, a cost-effective synthesis, an improved service life and safety, and a minimal environmental impact are equally important as performance. These features must be in the right balance to achieve the desired battery energy/power density and enhanced charge–discharge rate capability. From the discussion mentioned above, one of the best methods to improve the power performance of Li4Ti5O12 is to increase its electronic conductivity using an effective hybrid surface coating. The hybrid nanocoating of the Li4Ti5O12 anode has played an important role in improving its electrochemical performance.
Nevertheless, the synthesis usually involves low yields and complex procedures, high costs, and toxic precursors, which has limited explorations and applications. The development of controlled, large-scale, low-cost fabrication strategies for Li4Ti5O12 anode nanomaterials with desirable performance is an important challenge in the fabrication of battery materials for wide-spread commercial application. Considering its promise in the design of a new generation of lithium-ion batteries, the graphene–Li4Ti5O12 hybrid nanocomposite has potentially been shown to be a very promising material and might be used for high-rate power lithium-ion batteries due to its super electrochemical performance.
Acknowledgements
All financial support of the President's Award of University of Waterloo at Canada, the Postgraduate Scholarships from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Nano-fellowship from Waterloo Institute for Nanotechnology (WIN) at Canada has greatly been appreciated. P.V.R. thanks Canada Research Chairs program for a general support.Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431 CrossRef CAS PubMed.
- K. Zaghib, M. Simoneau, M. Armand and M. Gauthier, J. Power Sources, 1999, 81, 300 CrossRef.
- K. Nakahara, R. Nakajima, T. Matsushima and H. Majima, J. Power Sources, 2003, 117, 131 CrossRef CAS.
- T.-F. Yi, L.-J. Jiang, J. Shu, C.-B. Yue, R.-S. Zhu and H.-B. Qiao, J. Phys. Chem. Solids, 2010, 71, 1236 CrossRef CAS PubMed.
- K. Mukai, Y. Kato and H. Nakano, J. Phys. Chem. C, 2014, 118, 2992 CAS.
- W. Borghols, M. Wagemaker, U. Lafont, E. Kelder and F. Mulder, J. Am. Chem. Soc., 2009, 131, 17786 CrossRef CAS PubMed.
- P. Kubiak, A. Garcia, M. Womes, L. Aldon, J. Olivier-Fourcade, P.-E. Lippens and J.-C. Jumas, J. Power Sources, 2003, 119, 626 CrossRef.
- G. Wang, D. Bradhurst, S. Dou and H. Liu, J. Power Sources, 1999, 83, 156 CrossRef CAS.
- L. Aldon, P. Kubiak, M. Womes, J. Jumas, J. Olivier-Fourcade, J. Tirado, J. Corredor and C. Pérez Vicente, Chem. Mater., 2004, 16, 5721 CrossRef CAS.
- J. Shu, J. Solid State Electrochem., 2009, 13, 1535 CrossRef CAS.
- N. Jovic, B. Antic, A. Kremenovic, A. Spasojevic-de Bire and V. Spasojevic, Phys. Status Solidi A, 2003, 198, 18 CrossRef CAS.
- M. Wilkening, R. Amade, W. Iwaniak and P. Heitjans, Phys. Chem. Chem. Phys., 2007, 9, 1239 RSC.
- E. M. Sorensen, S. J. Barry, H.-K. Jung, J. M. Rondinelli, J. T. Vaughey and K. R. Poeppelmeier, Chem. Mater., 2006, 18, 482 CrossRef CAS.
- G.-N. Zhu, Y.-G. Wang and Y.-Y. Xia, Energy Environ. Sci., 2012, 5, 6652 CAS.
- S. Scharner, W. Weppner and P. Schmid-Beurmann, J. Electrochem. Soc., 1999, 146, 857 CrossRef CAS PubMed.
- W. Lu, I. Belharouak, J. Liu and K. Amine, J. Electrochem. Soc., 2007, 154, A114 CrossRef CAS PubMed.
- J. Ma, C. Wang and S. Wroblewski, J. Power Sources, 2007, 164, 849 CrossRef CAS PubMed.
- N. Takami, K. Hoshina and H. Inagaki, J. Electrochem. Soc., 2011, 158, A725 CrossRef CAS PubMed.
- N. Takami, H. Inagaki, T. Kishi, Y. Harada, Y. Fujita and K. Hoshina, J. Electrochem. Soc., 2009, 156, A128 CrossRef CAS PubMed.
- J.-F. Colin, V. Godbole and P. Novák, Electrochem. Commun., 2010, 12, 804 CrossRef CAS PubMed.
- X. Yao, S. Xie, C. Chen, Q. Wang, J. Sun, Y. Li and S. Lu, Electrochim. Acta, 2005, 50, 4076 CrossRef CAS PubMed.
- S.-I. Pyun, S.-W. Kim and H.-C. Shin, J. Power Sources, 1999, 81, 248 CrossRef.
- S. Bach, J. Pereira-Ramos and N. Baffier, J. Power Sources, 1999, 81, 273 CrossRef.
- P. P. Prosini, R. Mancini, L. Petrucci, V. Contini and P. Villano, Solid State Ionics, 2001, 144, 185 CrossRef CAS.
- F. Ronci, P. Reale, B. Scrosati, S. Panero, V. Rossi Albertini, P. Perfetti, M. Di Michiel and J. Merino, J. Phys. Chem. B, 2002, 106, 3082 CrossRef CAS.
- A. Jansen, A. Kahaian, K. Kepler, P. Nelson, K. Amine, D. Dees, D. Vissers and M. Thackeray, J. Power Sources, 1999, 81, 902 CrossRef.
- S. Panero, P. Reale, F. Ronci, V. R. Albertini and B. Scrosati, Ionics, 2000, 6, 461 CrossRef CAS.
- P. Reale, S. Panero, B. Scrosati, J. Garche, M. Wohlfahrt-Mehrens and M. Wachtler, J. Electrochem. Soc., 2004, 151, A2138 CrossRef CAS PubMed.
- I. Leonidov, O. Leonidova, L. Perelyaeva, R. Samigullina, S. Kovyazina and M. Patrakeev, Phys. Solid State, 2003, 45, 2183 CrossRef CAS.
- Z. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588 CrossRef CAS PubMed.
- S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, J. Phys. Chem. C, 2011, 115, 16220 CAS.
- K.-C. Hsiao, S.-C. Liao and J.-M. Chen, Electrochim. Acta, 2008, 53, 7242 CrossRef CAS PubMed.
- Y.-B. He, B. Li, M. Liu, C. Zhang, W. Lv, C. Yang, J. Li, H. Du, B. Zhang and Q.-H. Yang, Sci. Rep., 2012, 2, 913 Search PubMed.
- H. Wu, Y. Huang, D. Jia, Z. Guo and M. Miao, J. Nanopart. Res., 2012, 14, 1 Search PubMed.
- X. Yao, S. Xie, H. Nian and C. Chen, J. Alloys Compd., 2008, 465, 375 CrossRef CAS PubMed.
- Y.-H. Jin, K.-M. Min, H.-W. Shim, S.-D. Seo, I.-S. Hwang, K.-S. Park and D.-W. Kim, Nanoscale Res. Lett., 2012, 7, 1 CrossRef PubMed.
- M. Kalbáč, M. Zukalová and L. Kavan, J. Solid State Electrochem., 2003, 8, 2 CrossRef PubMed.
- C. Jiang, Y. Zhou, I. Honma, T. Kudo and H. Zhou, J. Power Sources, 2007, 166, 514 CrossRef CAS PubMed.
- D. Liu, C. Ouyang, J. Shu, J. Jiang, Z. Wang and L. Chen, Phys. Status Solidi B, 2006, 243, 1835 CrossRef CAS.
- H. Ge, N. Li, D. Li, C. Dai and D. Wang, J. Phys. Chem. C, 2009, 113, 6324 CAS.
- C.-T. Hsieh and J.-Y. Lin, J. Alloys Compd., 2010, 506, 231 CrossRef CAS PubMed.
- S. Takai, M. Kamata, S. Fujine, K. Yoneda, K. Kanda and T. Esaka, Solid State Ionics, 1999, 123, 165 CrossRef CAS.
- J. Gao, J. Ying, C. Jiang and C. Wan, Ionics, 2009, 15, 597 CrossRef CAS PubMed.
- J. Chen, L. Yang, S. Fang and Y. Tang, Electrochim. Acta, 2010, 55, 6596 CrossRef CAS PubMed.
- Y. Tang, L. Yang, S. Fang and Z. Qiu, Electrochim. Acta, 2009, 54, 6244 CrossRef CAS PubMed.
- K. Naoi, S. Ishimoto, Y. Isobe and S. Aoyagi, J. Power Sources, 2010, 195, 6250 CrossRef CAS PubMed.
- T.-F. Yi, J. Shu, C.-B. Yue, X.-D. Zhu, A.-N. Zhou, Y.-R. Zhu and R.-S. Zhu, Mater. Res. Bull., 2010, 45, 456 CrossRef CAS PubMed.
- C. Ouyang, Z. Zhong and M. Lei, Electrochem. Commun., 2007, 9, 1107 CrossRef CAS PubMed.
- Y. Hao, Q. Lai, Z. Xu, X. Liu and X. Ji, Solid State Ionics, 2005, 176, 1201 CrossRef CAS PubMed.
- Z. Wen, S. Huang, X. Yang and B. Lin, Solid State Ionics, 2008, 179, 1800 CrossRef CAS PubMed.
- J. Gao, C. Jiang, J. Ying and C. Wan, J. Power Sources, 2006, 155, 364 CrossRef CAS PubMed.
- N. Zhu, W. Liu, M. Xue, Z. Xie, D. Zhao, M. Zhang, J. Chen and T. Cao, Electrochim. Acta, 2010, 55, 5813 CrossRef CAS PubMed.
- M. Yoshio, R. J. Brodd and A. Kozawa, Lithium-Ion Batteries, Springer, 2009 Search PubMed.
- M. Winter and J. O. Besenhard, Electrochim. Acta, 1999, 45, 31 CrossRef CAS.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef PubMed.
- J. Garche, C. K. Dyer, P. T. Moseley, Z. Ogumi, D. A. Rand and B. Scrosati, Encyclopedia of Electrochemical Power Sources, Newnes, 2013 Search PubMed.
- M. M. Rahman, J.-Z. Wang, M. F. Hassan, S. Chou, D. Wexler and H.-K. Liu, J. Power Sources, 2010, 195, 4297 CrossRef CAS PubMed.
- Y. Li, H. Zhao, Z. Tian, W. Qiu and X. Li, J. Alloys Compd., 2008, 455, 471 CrossRef CAS PubMed.
- J. Yang, J. Zhao, Y. Chen and Y. Li, Ionics, 2010, 16, 425 CrossRef CAS.
- Y. Wang, H. Li, P. He, E. Hosono and H. Zhou, Nanoscale, 2010, 2, 1294 RSC.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201 RSC.
- A. Y. Shenouda and K. Murali, J. Power Sources, 2008, 176, 332 CrossRef CAS PubMed.
- W. Lu, I. Belharouak, J. Liu and K. Amine, J. Power Sources, 2007, 174, 673 CrossRef CAS PubMed.
- P. Reale, A. Fernicola and B. Scrosati, J. Power Sources, 2009, 194, 182 CrossRef CAS PubMed.
- A. Guerfi, P. Charest, K. Kinoshita, M. Perrier and K. Zaghib, J. Power Sources, 2004, 126, 163 CrossRef CAS PubMed.
- S. Ji, J. Zhang, W. Wang, Y. Huang, Z. Feng, Z. Zhang and Z. Tang, Mater. Chem. Phys., 2010, 123, 510 CrossRef CAS PubMed.
- K. Kataoka, Y. Takahashi, N. Kijima, H. Hayakawa, J. Akimoto and K.-i. Ohshima, Solid State Ionics, 2009, 180, 631 CrossRef CAS PubMed.
- X. Li, M. Qu, Y. Huai and Z. Yu, Electrochim. Acta, 2010, 55, 2978 CrossRef CAS PubMed.
- X. Li, M. Qu and Z. Yu, Solid State Ionics, 2010, 181, 635 CrossRef CAS PubMed.
- D. Capsoni, M. Bini, V. Massarotti, P. Mustarelli, G. Chiodelli, C. B. Azzoni, M. C. Mozzati, L. Linati and S. Ferrari, Chem. Mater., 2008, 20, 4291 CrossRef CAS.
- H. Ge, N. Li, D. Li, C. Dai and D. Wang, Electrochem. Commun., 2008, 10, 719 CrossRef CAS PubMed.
- G. Wang, J. Xu, M. Wen, R. Cai, R. Ran and Z. Shao, Solid State Ionics, 2008, 179, 946 CrossRef CAS PubMed.
- G. Liu, L. Wen, G. Liu, Q. Wu, H. Luo, B. Ma and Y. Tian, J. Alloys Compd., 2011, 509, 6427 CrossRef CAS PubMed.
- X. Sun, M. Hegde, J. Wang, Y. Zhang, J. Liao, P. V. Radovanovic and B. Cui, ECS Trans., 2014, 58, 79 CrossRef PubMed.
- C.-H. Hong, A. Noviyanto, J. H. Ryu, J. Kim and D.-H. Yoon, Ceram. Int., 2012, 38, 301 CrossRef CAS PubMed.
- J. W. Shin, C. H. Hong and D. H. Yoon, J. Am. Ceram. Soc., 2012, 95, 1894 CrossRef CAS PubMed.
- T. Yuan, R. Cai, P. Gu and Z. Shao, J. Power Sources, 2010, 195, 2883 CrossRef CAS PubMed.
- J. Li, Y.-L. Jin, X.-G. Zhang and H. Yang, Solid State Ionics, 2007, 178, 1590 CrossRef CAS PubMed.
- Y. Bai, F. Wang, F. Wu, C. Wu and L.-Y. Bao, Electrochim. Acta, 2008, 54, 322 CrossRef CAS PubMed.
- K. Teshima, H. Inagaki, S. Tanaka, K. Yubuta, M. Hozumi, K. Kohama, T. Shishido and S. Oishi, Cryst. Growth Des., 2011, 11, 4401 CAS.
- S. C. Lee, S. M. Lee, J. W. Lee, J. B. Lee, S. M. Lee, S. S. Han, H. C. Lee and H. J. Kim, J. Phys. Chem. C, 2009, 113, 18420 CAS.
- Y. Tang, L. Yang, Z. Qiu and J. S. Huang, Electrochem. Commun., 2008, 10, 1513 CrossRef CAS PubMed.
- Y. Tang, L. Yang, Z. Qiu and J. Huang, J. Mater. Chem., 2009, 19, 5980 RSC.
- L. Shen, C. Yuan, H. Luo, X. Zhang, K. Xu and Y. Xia, J. Mater. Chem., 2010, 20, 6998 RSC.
- C. Lai, Y. Dou, X. Li and X. Gao, J. Power Sources, 2010, 195, 3676 CrossRef CAS PubMed.
- J. Chen, L. Yang, S. Fang, S.-I. Hirano and K. Tachibana, J. Power Sources, 2012, 200, 59 CrossRef CAS PubMed.
- J. Liu, X. Li, J. Yang, D. Geng, Y. Li, D. Wang, R. Li, X. Sun, M. Cai and M. W. Verbrugge, Electrochim. Acta, 2012, 63, 100 CrossRef CAS PubMed.
- X. C. Sun, M. Hegde, Y. Zhang, M. He, L. Gu, Y. Wang, J. Shu, P. V. Radovanovic and B. Cui, Int. J. Electrochem. Sci., 2014, 9, 1583 CAS.
- A. Laumann, M. Bremholm, P. Hald, M. Holzapfel, K. T. Fehr and B. B. Iversen, J. Electrochem. Soc., 2011, 159, A166 CrossRef PubMed.
- L. Shen, C. Yuan, H. Luo, X. Zhang, K. Xu and F. Zhang, J. Mater. Chem., 2011, 21, 761 RSC.
- S.-H. Yu, A. Pucci, T. Herntrich, M.-G. Willinger, S.-H. Baek, Y.-E. Sung and N. Pinna, J. Mater. Chem., 2011, 21, 806 RSC.
- C.-M. Shen, X.-G. Zhang, Y.-K. Zhou and H.-L. Li, Mater. Chem. Phys., 2003, 78, 437 CrossRef CAS.
- M.-H. Oh, H.-J. Kim, Y. J. Kim, K. J. Kim and S.-G. Park, ECS Trans., 2007, 3, 101 CAS.
- N. Alias, M. Kufian, L. Teo, S. Majid and A. Arof, J. Alloys Compd., 2009, 486, 645 CrossRef CAS PubMed.
- R. B. Khomane, A. Prakash, K. Ramesha and M. Sathiya, Mater. Res. Bull., 2011, 46, 1139 CrossRef CAS PubMed.
- M. Venkateswarlu, C. Chen, J. Do, C. Lin, T.-C. Chou and B. Hwang, J. Power Sources, 2005, 146, 204 CrossRef CAS PubMed.
- N. Zhang, Z. Liu, T. Yang, C. Liao, Z. Wang and K. Sun, Electrochem. Commun., 2011, 13, 654 CrossRef CAS PubMed.
- Y.-J. Hao, Q.-Y. Lai, J.-Z. Lu, D.-Q. Liu and X.-Y. Ji, J. Alloys Compd., 2007, 439, 330 CrossRef CAS PubMed.
- M. Mohammadi and D. Fray, J. Sol-Gel Sci. Technol., 2010, 55, 19 CrossRef CAS PubMed.
- Y. H. Rho and K. Kanamura, J. Solid State Chem., 2004, 177, 2094 CrossRef CAS PubMed.
- G. Yan, H. Fang, H. Zhao, G. Li, Y. Yang and L. Li, J. Alloys Compd., 2009, 470, 544 CrossRef CAS PubMed.
- Y. Hao, Q.-Y. Lai, D. Liu, Z.-U. Xu and X. Ji, Mater. Chem. Phys., 2005, 94, 382 CrossRef CAS PubMed.
- Y.-J. Hao, Q.-Y. Lai, J.-Z. Lu, H.-L. Wang, Y.-D. Chen and X.-Y. Ji, J. Power Sources, 2006, 158, 1358 CrossRef CAS PubMed.
- A. Mukasyan and P. Dinka, Int. J. Self-Propag. High-Temp. Synth., 2007, 16, 23 CrossRef CAS.
- K. C. Patil, S. Aruna and T. Mimani, Curr. Opin. Solid State Mater. Sci., 2002, 6, 507 CrossRef CAS.
- S. T. Aruna and A. S. Mukasyan, Curr. Opin. Solid State Mater. Sci., 2008, 12, 44 CrossRef CAS PubMed.
- A. Prakash, P. Manikandan, K. Ramesha, M. Sathiya, J. Tarascon and A. Shukla, Chem. Mater., 2010, 22, 2857 CrossRef CAS.
- M. Raja, S. Mahanty, M. Kundu and R. N. Basu, J. Alloys Compd., 2009, 468, 258 CrossRef CAS PubMed.
- T. Yuan, R. Cai, K. Wang, R. Ran, S. Liu and Z. Shao, Ceram. Int., 2009, 35, 1757 CrossRef CAS PubMed.
- G. L. Messing, S. C. Zhang and G. V. Jayanthi, J. Am. Ceram. Soc., 1993, 76, 2707 CrossRef CAS PubMed.
- D. S. Jung, S. B. Park and Y. C. Kang, Korean J. Chem. Eng., 2010, 27, 1621 CrossRef CAS.
- S. H. Ju and Y. C. Kang, J. Phys. Chem. Solids, 2009, 70, 40 CrossRef CAS PubMed.
- S. H. Ju and Y. C. Kang, J. Power Sources, 2009, 189, 185 CrossRef CAS PubMed.
- S. H. Ju and Y. C. Kang, J. Alloys Compd., 2010, 506, 913 CrossRef CAS PubMed.
- S. Yin, L. Song, X. Wang, M. Zhang, K. Zhang and Y. Zhang, Electrochim. Acta, 2009, 54, 5629 CrossRef CAS PubMed.
- T. Yuan, K. Wang, R. Cai, R. Ran and Z. Shao, J. Alloys Compd., 2009, 477, 665 CrossRef CAS PubMed.
- D. Kim, Y. Ahn and J. Kim, Electrochem. Commun., 2005, 7, 1340 CrossRef CAS PubMed.
- S. S. Lee, K.-T. Byun, J. P. Park, S. K. Kim, H.-Y. Kwak and I.-W. Shim, Dalton Trans., 2007, 4182 RSC.
- T. Doi, Y. Iriyama, T. Abe and Z. Ogumi, Chem. Mater., 2005, 17, 1580 CrossRef CAS.
- F. Ernst, H. Kammler, A. Roessler, S. Pratsinis, W. Stark, J. Ufheil and P. Novák, Mater. Chem. Phys., 2007, 101, 372 CrossRef CAS PubMed.
- J. Wolfenstine and J. Allen, J. Power Sources, 2008, 180, 582 CrossRef CAS PubMed.
- X. L. Zhang, G. R. Hu and Z. D. Peng, J. Cent. South Univ., 2013, 20, 1151 CrossRef CAS PubMed.
- T.-F. Yi, H. Liu, Y.-R. Zhu, L.-J. Jiang, Y. Xie and R.-S. Zhu, J. Power Sources, 2012, 215, 258 CrossRef CAS PubMed.
- Z. Yu, X. Zhang, G. Yang, J. Liu, J. Wang, R. Wang and J. Zhang, Electrochim. Acta, 2011, 56, 8611 CrossRef CAS PubMed.
- L. Xing, Chin. J. Inorg. Chem., 2010, 2, 233 Search PubMed.
- T.-F. Yi, Y. Xie, Q. Wu, H. Liu, L. Jiang, M. Ye and R. Zhu, J. Power Sources, 2012, 214, 220 CrossRef CAS PubMed.
- B. Zhang, Z.-D. Huang, S. W. Oh and J.-K. Kim, J. Power Sources, 2011, 196, 10692 CrossRef CAS PubMed.
- C. Chen, J. Vaughey, A. Jansen, D. Dees, A. Kahaian, T. Goacher and M. Thackeray, J. Electrochem. Soc., 2001, 148, A102 CrossRef CAS PubMed.
- R. Cai, S. Jiang, X. Yu, B. Zhao, H. Wang and Z. Shao, J. Mater. Chem., 2012, 22, 8013 RSC.
- D. Wu and Y. Cheng, Ionics, 2013, 19, 395 CrossRef CAS.
- S. Huang, Z. Wen, Z. Gu and X. Zhu, Electrochim. Acta, 2005, 50, 4057 CrossRef CAS PubMed.
- S. Huang, Z. Wen, X. Zhu and Z. Lin, J. Power Sources, 2007, 165, 408 CrossRef CAS PubMed.
- J.-Y. Lin, C.-C. Hsu, H.-P. Ho and S.-H. Wu, Electrochim. Acta, 2013, 87, 126 CrossRef CAS PubMed.
- Z. Wang, G. Chen, J. Xu, Z. Lv and W. Yang, J. Phys. Chem. Solids, 2011, 72, 773 CrossRef CAS PubMed.
- Y.-J. Hao, Q.-Y. Lai, J.-Z. Lu and X.-Y. Ji, Ionics, 2007, 13, 369 CrossRef CAS PubMed.
- W. Long, X. Wang, S. Yang, H. Shu, Q. Wu, Y. Bai and L. Bai, Mater. Chem. Phys., 2011, 131, 431 CrossRef CAS PubMed.
- A. Robertson, L. Trevino, H. Tukamoto and J. Irvine, J. Power Sources, 1999, 81, 352 CrossRef.
- J. Gao, C. Jiang and C. Wan, J. Electrochem. Soc., 2010, 157, K39 CrossRef CAS PubMed.
- Y.-J. Bai, C. Gong, Y.-X. Qi, N. Lun and J. Feng, J. Mater. Chem., 2012, 22, 19054 RSC.
- Y.-J. Bai, C. Gong, N. Lun and Y.-X. Qi, J. Mater. Chem. A, 2013, 1, 89 CAS.
- C. Qiu, Z. Yuan, L. Liu, N. Ye and J. Liu, J. Solid State Electrochem., 2013, 17, 841 CrossRef CAS PubMed.
- K. Abhilash, P. C. Selvin, B. Nalini, P. Nithyadharseni and B. Pillai, Ceram. Int., 2013, 39, 947 CrossRef CAS PubMed.
- X. Li, M. Qu and Z. Yu, J. Alloys Compd., 2009, 487, L12 CrossRef CAS PubMed.
- Y.-R. Jhan and J.-G. Duh, Electrochim. Acta, 2012, 63, 9 CrossRef CAS PubMed.
- V. Nithya, R. Kalai Selvan, K. Vediappan, S. Sharmila and C. W. Lee, Appl. Surf. Sci., 2012, 261, 515 CrossRef CAS PubMed.
- T.-F. Yi, J. Shu, Y.-R. Zhu, X.-D. Zhu, C.-B. Yue, A.-N. Zhou and R.-S. Zhu, Electrochim. Acta, 2009, 54, 7464 CrossRef CAS PubMed.
- Z. Zhong, Electrochem. Solid-State Lett., 2007, 10, A267 CrossRef CAS PubMed.
- B. Tian, H. Xiang, L. Zhang, Z. Li and H. Wang, Electrochim. Acta, 2010, 55, 5453 CrossRef CAS PubMed.
- B. Tian, H. Xiang, L. Zhang and H. Wang, J. Solid State Electrochem., 2012, 16, 205 CrossRef CAS.
- T.-F. Yi, Y. Xie, J. Shu, Z. Wang, C.-B. Yue, R.-S. Zhu and H.-B. Qiao, J. Electrochem. Soc., 2011, 158, A266 CrossRef CAS PubMed.
- W. Wang, H. Wang, S. Wang, Y. Hu, Q. Tian and S. Jiao, J. Power Sources, 2013, 228, 244 CrossRef CAS PubMed.
- R. Cai, T. Yuan, R. Ran, X. Liu and Z. Shao, Int. J. Energy Res., 2011, 35, 68 CrossRef CAS.
- J.-P. Zhu, J.-J. Zhao, H.-W. Yang and G. Yang, Adv. Sci. Lett., 2011, 4, 484 CrossRef CAS PubMed.
- B. Zhang, H. Du, B. Li and F. Kang, Electrochem. Solid-State Lett., 2010, 13, A36 CrossRef CAS PubMed.
- S. Huang, Z. Wen, B. Lin, J. Han and X. Xu, J. Alloys Compd., 2008, 457, 400 CrossRef CAS PubMed.
- Q. Zhang, C. Zhang, B. Li, S. Kang, X. Li and Y. Wang, Electrochim. Acta, 2013, 98, 146 CrossRef CAS PubMed.
- H. Zhao, Y. Li, Z. Zhu, J. Lin, Z. Tian and R. Wang, Electrochim. Acta, 2008, 53, 7079 CrossRef CAS PubMed.
- G. Du, N. Sharma, V. K. Peterson, J. A. Kimpton, D. Jia and Z. Guo, Adv. Funct. Mater., 2011, 21, 3990 CrossRef CAS.
- Y. Qi, Y. Huang, D. Jia, S.-J. Bao and Z. Guo, Electrochim. Acta, 2009, 54, 4772 CrossRef CAS PubMed.
- Y. Huang, Y. Qi, D. Jia, X. Wang, Z. Guo and W. I. Cho, J. Solid State Electrochem., 2012, 16, 2011 CrossRef CAS PubMed.
- Y. Ma, B. Ding, G. Ji and J. Y. Lee, ACS Nano, 2013, 7, 10870 CrossRef CAS PubMed.
- S. Huang, Z. Wen, X. Zhu and X. Yang, J. Electrochem. Soc., 2005, 152, A1301 CrossRef CAS PubMed.
- Y.-Y. Wang, Y.-J. Hao, Q.-Y. Lai, J.-Z. Lu, Y.-D. Chen and X.-Y. Ji, Ionics, 2008, 14, 85 CrossRef CAS PubMed.
- M. Rahman, J.-Z. Wang, M. F. Hassan, S. Chou, D. Wexler and H.-K. Liu, J. Power Sources, 2010, 195, 4297 CrossRef CAS PubMed.
- J. Lim, E. Choi, V. Mathew, D. Kim, D. Ahn, J. Gim, S.-H. Kang and J. Kim, J. Electrochem. Soc., 2011, 158, A275 CrossRef CAS PubMed.
- Y. Wang, H. Liu, K. Wang, H. Eiji, Y. Wang and H. Zhou, J. Mater. Chem., 2009, 19, 6789 RSC.
- A. Jaiswal, C. Horne, O. Chang, W. Zhang, W. Kong, E. Wang, T. Chern and M. Doeff, J. Electrochem. Soc., 2009, 156, A1041 CrossRef CAS PubMed.
- J. Kim and J. Cho, Electrochem. Solid-State Lett., 2007, 10, A81 CrossRef CAS PubMed.
- C. Jiang, M. Ichihara, I. Honma and H. Zhou, Electrochim. Acta, 2007, 52, 6470 CrossRef CAS PubMed.
- L. Yu, H. B. Wu and X. W. D. Lou, Adv. Mater., 2013, 25, 2296 CrossRef CAS PubMed.
- X. Lu, L. Zhao, X. He, R. Xiao, L. Gu, Y. S. Hu, H. Li, Z. Wang, X. Duan and L. Chen, Adv. Mater., 2012, 24, 3233 CrossRef CAS PubMed.
- K.-S. Park, A. Benayad, D.-J. Kang and S.-G. Doo, J. Am. Chem. Soc., 2008, 130, 14930 CrossRef CAS PubMed.
- M. R. Jo, Y. S. Jung and Y.-M. Kang, Nanoscale, 2012, 4, 6870 RSC.
- L. Wang, Q. Xiao, Z. Li, G. Lei, P. Zhang and L. Wu, J. Solid State Electrochem., 2012, 16, 3307 CrossRef CAS PubMed.
- Z. Hong, T. Lan, F. Xiao, H. Zhang and M. Wei, Funct. Mater. Lett., 2011, 4, 389 CrossRef CAS.
- N. He, B. Wang and J. Huang, J. Solid State Electrochem., 2010, 14, 1241 CrossRef CAS.
- D. Shao, J. He, Y. Luo, W. Liu, X. Yu and Y. Fang, J. Solid State Electrochem., 2012, 16, 2047 CrossRef CAS PubMed.
- Y.-S. Lin and J.-G. Duh, J. Power Sources, 2011, 196, 10698 CrossRef CAS PubMed.
- H.-G. Jung, S.-T. Myung, C. S. Yoon, S.-B. Son, K. H. Oh, K. Amine, B. Scrosati and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 1345 CAS.
- L. Cheng, J. Yan, G.-N. Zhu, J.-Y. Luo, C.-X. Wang and Y.-Y. Xia, J. Mater. Chem., 2010, 20, 595 RSC.
- B. Li, C. Han, Y.-B. He, C. Yang, H. Du, Q.-H. Yang and F. Kang, Energy Environ. Sci., 2012, 5, 9595 CAS.
- J. Gao, J. Ying, C. Jiang and C. Wan, J. Power Sources, 2007, 166, 255 CrossRef CAS PubMed.
- G. J. Wang, J. Gao, L. J. Fu, N. H. Zhao, Y. P. Wu and T. Takamura, J. Power Sources, 2007, 174, 1109 CrossRef CAS PubMed.
- L. Cheng, X.-L. Li, H.-J. Liu, H.-M. Xiong, P.-W. Zhang and Y.-Y. Xia, J. Electrochem. Soc., 2007, 154, A692 CrossRef CAS PubMed.
- G.-N. Zhu, C.-X. Wang and Y.-Y. Xia, J. Electrochem. Soc., 2011, 158, A102 CrossRef CAS PubMed.
- H. Ming, J. Ming, X. Li, Q. Zhou, H. Wang, L. Jin, Y. Fu, J. Adkins and J. Zheng, Electrochim. Acta, 2014, 116, 224 CrossRef CAS PubMed.
- H. Li, L. Shen, K. Yin, J. Ji, J. Wang, X. Wang and X. Zhang, J. Mater. Chem. A, 2013, 1, 7270 CAS.
- L. Yang and L. Gao, J. Alloys Compd., 2009, 485, 93 CrossRef CAS PubMed.
- Z. Zhu, F. Cheng and J. Chen, J. Mater. Chem. A, 2013, 1, 9484 CAS.
- L. Zhao, Y. S. Hu, H. Li, Z. Wang and L. Chen, Adv. Mater., 2011, 23, 1385 CrossRef CAS PubMed.
- M. Inagaki, Carbon, 2012, 50, 3247 CrossRef CAS PubMed.
- M. Gaberscek and J. Jamnik, Solid State Ionics, 2006, 177, 2647 CrossRef CAS PubMed.
- R. Dominko, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel, S. Pejovnik and J. Jamnik, J. Electrochem. Soc., 2005, 152, A607 CrossRef CAS PubMed.
- X. C. Sun, M. Hegde, J. Wang, Y. Zhang, J. Liao, P. V. Radovanovic and B. Cui, ECS Trans., 2014, 58, 79 CrossRef PubMed.
- J. Wang, X.-M. Liu, H. Yang and X.-D. Shen, J. Alloys Compd., 2011, 509, 712 CrossRef CAS PubMed.
- H. Luo, L. Shen, K. Rui, H. Li and X. Zhang, J. Alloys Compd., 2013, 572, 37 CrossRef CAS PubMed.
- H.-G. Jung, J. Kim, B. Scrosati and Y.-K. Sun, J. Power Sources, 2011, 196, 7763 CrossRef CAS PubMed.
- Z. Lin, X. Hu, Y. Huai, L. Liu, Z. Deng and J. Suo, Solid State Ionics, 2010, 181, 412 CrossRef CAS PubMed.
- H. Liu, Y. Feng, K. Wang and J. Xie, J. Phys. Chem. Solids, 2008, 69, 2037 CrossRef CAS PubMed.
- X. Guo, H. F. Xiang, T. P. Zhou, X. K. Ju and Y. C. Wu, Electrochim. Acta, 2014, 130, 470 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- L. Shen, C. Yuan, H. Luo, X. Zhang, S. Yang and X. Lu, Nanoscale, 2011, 3, 572 RSC.
- Y. Shi, L. Wen, F. Li and H.-M. Cheng, J. Power Sources, 2011, 196, 8610 CrossRef CAS PubMed.
- Y. Oh, S. Nam, S. Wi, J. Kang, T. Hwang, S. Lee, H. H. Park, J. Cabana, C. Kim and B. Park, J. Mater. Chem. A, 2014, 2, 2023 CAS.
- Q. Zhang, W. Peng, Z. Wang, X. Li, X. Xiong, H. Guo, Z. Wang and F. Wu, Ionics, 2013, 19, 717 CrossRef CAS.
- A. K. Rai, J. Gim, S.-W. Kang, V. Mathew, L. T. Anh, J. Kang, J. Song, B. J. Paul and J. Kim, Mater. Chem. Phys., 2012, 136, 1044 CrossRef CAS PubMed.
- S. Y. Han, I. Y. Kim, K. Y. Jo and S.-J. Hwang, J. Phys. Chem. C, 2012, 116, 7269 CAS.
- S. Pang, Y. Zhao, C. Zhang, Q. Zhang, L. Gu, X. Zhou, G. Li and G. Cui, Scr. Mater., 2013, 69, 171 CrossRef CAS PubMed.
- H. Xiang, B. Tian, P. Lian, Z. Li and H. Wang, J. Alloys Compd., 2011, 509, 7205 CrossRef CAS PubMed.
- S. G. Ri, L. Zhan, Y. Wang, L. Zhou, J. Hu and H. Liu, Electrochim. Acta, 2013, 109, 389 CrossRef CAS PubMed.
- H. Ni, W.-L. Song and L.-Z. Fan, Electrochem. Commun., 2014, 40, 1 CrossRef CAS PubMed.
- X. Guo, H. F. Xiang, T. P. Zhou, W. H. Li, X. W. Wang, J. X. Zhou and Y. Yu, Electrochim. Acta, 2013, 109, 33 CrossRef CAS PubMed.
- X. Wang, L. Shen, H. Li, J. Wang, H. Dou and X. Zhang, Electrochim. Acta, 2014, 129, 283 CrossRef CAS PubMed.
- C. Sandhya, B. John and C. Gouri, Ionics, 2014, 20, 601 CrossRef CAS PubMed.
- E. Ferg, R. Gummow, A. De Kock and M. Thackeray, J. Electrochem. Soc., 1994, 141, L147 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419 CrossRef CAS PubMed.
- W. Lu, J. Liu, Y. K. Sun and K. Amine, J. Power Sources, 2007, 167, 212 CrossRef CAS PubMed.
- C. M. Ionica-Bousquet, D. Muñoz-Rojas, W. J. Casteel, R. M. Pearlstein, G. GirishKumar, G. P. Pez and M. R. Palacín, J. Power Sources, 2010, 195, 1479 CrossRef CAS PubMed.
- I. Belharouak, Y.-K. Sun, W. Lu and K. Amine, J. Electrochem. Soc., 2007, 154, A1083 CrossRef CAS PubMed.
- I. Belharouak, G. M. Koenig, T. Tan, H. Yumoto, N. Ota and K. Amine, J. Electrochem. Soc., 2012, 159, A1165 CrossRef CAS PubMed.
- K. Amine, I. Belharouak, Z. Chen, T. Tran, H. Yumoto, N. Ota, S. T. Myung and Y. K. Sun, Adv. Mater., 2010, 22, 3052 CrossRef CAS PubMed.
- X. Hu, Z. Deng, J. Suo and Z. Pan, J. Power Sources, 2009, 187, 635 CrossRef CAS PubMed.
- S. Panero, D. Satolli, M. Salomon and B. Scrosati, Electrochem. Commun., 2000, 2, 810 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- S. K. Martha, O. Haik, V. Borgel, E. Zinigrad, I. Exnar, T. Drezen, J. H. Miners and D. Aurbach, J. Electrochem. Soc., 2011, 158, A790 CrossRef CAS PubMed.
- V. Borgel, G. Gershinsky, T. Hu, M. G. Theivanayagam and D. Aurbach, J. Electrochem. Soc., 2013, 160, A650 CrossRef CAS PubMed.
- H. Wu, I. Belharouak, H. Deng, A. Abouimrane, Y.-K. Sun and K. Amine, J. Electrochem. Soc., 2009, 156, A1047 CrossRef CAS PubMed.
- S. Li, C. Chen and J. Dahn, J. Electrochem. Soc., 2013, 160, A2166 CrossRef CAS PubMed.
- I. Belharouak, G. M. Koenig Jr and K. Amine, J. Power Sources, 2011, 196, 10344 CrossRef CAS PubMed.
- A. Du Pasquier, C. C. Huang and T. Spitler, J. Power Sources, 2009, 186, 508 CrossRef CAS PubMed.
- N. M. Hagh and G. G. Amatucci, J. Power Sources, 2010, 195, 5005 CrossRef CAS PubMed.
- S. Li, C. Chen, X. Xia and J. Dahn, J. Electrochem. Soc., 2013, 160, A1524 CrossRef CAS PubMed.
- L. Q. Sun, R. H. Cui, A. F. Jalbout, M. J. Li, X. M. Pan, R. S. Wang and H. M. Xie, J. Power Sources, 2009, 189, 522 CrossRef CAS PubMed.
- K. Zaghib, M. Dontigny, A. Guerfi, P. Charest, I. Rodrigues, A. Mauger and C. Julien, J. Power Sources, 2011, 196, 3949 CrossRef CAS PubMed.
- K. Zaghib, M. Dontigny, A. Guerfi, J. Trottier, J. Hamel-Paquet, V. Gariepy, K. Galoutov, P. Hovington, A. Mauger, H. Groult and C. M. Julien, J. Power Sources, 2012, 216, 192 CrossRef CAS PubMed.
- K. Ariyoshi, S. Yamamoto and T. Ohzuku, J. Power Sources, 2003, 119–121, 959 CrossRef CAS.
- H. F. Xiang, Q. Y. Jin, R. Wang, C. H. Chen and X. W. Ge, J. Power Sources, 2008, 179, 351 CrossRef CAS PubMed.
- M. Reddy, G. Subba Rao and B. Chowdari, Chem. Rev., 2013, 113, 5364 CrossRef CAS PubMed.
- W. W. Lee and J.-M. Lee, J. Mater. Chem. A, 2014, 2, 1589 CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |