
From Ni-based nanoprecursors to NiO nanostructures: morphology-controlled synthesis and structure-dependent electrochemical behavior
Gang
Cheng
*a,
Yinan
Yan
b and
Rong
Chen
*a
aSchool of Chemistry and Environmental Engineering, Wuhan Institute of Technology, Xiongchu Avenue, Wuhan, 430073, P. R. China. E-mail: gchenglab@163.com; rchenhku@hotmail.com
bNational Engineering Research Center for Nanotechnology, Shanghai, 200241, P. R. China
First published on 7th November 2014
Abstract
Phase transformation and morphology tailoring of NiO nanostructures have been successfully achieved using Ni-based precursors (Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2) synthesized by a facile urea-controlled hydrothermal route. The as-synthesized NiO nanomaterials were characterized by X-ray diffraction (XRD), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), and nitrogen adsorption analyses. The electrochemical performance of the as-prepared NiO nanostructures was evaluated by CV and galvanostatic charge–discharge tests. NiO nanoparticles with larger surface areas exhibited higher electrochemical performance than nanobelts and nanospheres when used as the supercapacitor electrodes. The discharge capacity of NiO nanoparticles can retain 609.5 F g−1 after 500 cycles at a charging–discharging current density of 5 A g−1.
1. Introduction
Supercapacitors have attracted considerable attention in the past decade as they have the potential to satisfy the demand of high power density in many advanced technologies.1,2 Nickel oxide (NiO) is regarded as one of promising supercapacitor materials because of its excellent electrochemical properties and potential applications in electrodes.3–7 In particular, engineering NiO materials at the nanoscale offers unique properties resulting in high performance electrodes for energy storage devices. Consequently, considerable efforts have been made in recent years to fulfill the future requirements of electrochemical energy storage using NiO-based nanomaterials. At the same time, the study on the intrinsic relationship between the structure/size and property has engendered an urgent need for adjustable synthetic strategies,8,9 where the particle size and morphology of NiO nanomaterials can be precisely controlled with designed functionalities.Being one of the most commonly used methods, thermal decomposition of the Ni(OH)2 intermediate has been widely employed to synthesize NiO nanostructures.10,11 Cui et al. fabricated lotus-root-like NiO nanosheets and flower-like NiO microspheres through calcination treatment of Ni(OH)2 nanosheets and microsphere precursors, respectively.12 However, such a synthesis technique for preparing nanostructured NiO materials, especially achieving control of the morphology and structure, still remains a challenge because the morphology of the as-synthesized Ni(OH)2 is mainly in the form of two dimensional and three dimensional nanostructures, including nanosheets,5 nanoflakes,13 and hierarchical nanostructures.14 Although much attention has been paid on structural control of Ni(OH)2 nanomaterials using ionic liquids, surfactants, templates,15–18 most of the methods suffered from drawbacks such as complicated shape-control processes because of the involvement of other additives. On the other hand, substantial efforts have been made toward the development of choosing a new suitable Ni-based nanoprecursor with a proper transformation process to selectively prepare NiO with special shapes and structures.19–21 However, there are few publications relevant for tunable fabrication of NiO nanostructures and investigation of the effect of structure/morphology on the electrochemical performance.
Herein, we have successfully synthesized Ni-based nanostructure precursors including Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2 by a facile urea-controlled hydrothermal approach. At the same time, we have achieved phase transformation and morphology tailoring of NiO nanomaterials from the as-synthesized Ni-based precursors. In addition, the electrochemical performances of the as-prepared NiO nanostructures with different morphologies were also evaluated.
2. Experimental section
All of the reagents were of analytical grade and were used without further purification. De-ionized water was used for all experiments.The Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2 nanostructure precursors were synthesized by a facile urea-controlled hydrothermal method. In a typical synthesis, 2 mmol of NiSO4·(NH4)2SO4·6H2O and an appropriate amount of urea were added to 30 mL of deionized water, and then sonicated until a transparent solution was formed. After that, the homogeneous solution was transferred into a 50 mL Teflon-lined autoclave and a temperature of 180 °C was maintained for 18 h. After cooling down to room temperature, the products were collected and washed with de-ionized water and ethanol several times by centrifugation. The final product was dried for 12 h at 80 °C. The mole ratios of NiSO4·(NH4)2SO4·6H2O with respect to the urea were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:6, and 1:15, which are referred to as P1, P2, and P3, respectively.
:1, 1:6, and 1:15, which are referred to as P1, P2, and P3, respectively.
NiO nanomaterials with different morphologies were prepared by calcining Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2 nanostructure precursors at 450 °C for 2 h in air at a ramping rate of 4 °C min−1.
The as-synthesized Ni-based nanostructures were characterized by powder X-ray diffraction (XRD), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). XRD was performed using a D8 Advance (Bruker, Germany) at room temperature in specular reflection mode with Cu/Kα radiation at a scanning rate of 0.03° s−1 in the 2θ range from 20° to 90°. SEM images were taken on a field-emission electron microscope S4800 operating at an acceleration voltage of 5 kV. TEM images were recorded on a JEOL 2010 electron microscope at an accelerating voltage of 200 kV. The Brunauer–Emmett–Teller (BET) specific surface areas of the powders were determined by nitrogen adsorption in a Micromeritics ASAP 2020 nitrogen adsorption apparatus (USA). TGA measurements were recorded on linseis STAPt1600 system under N2 purge from room temperature to 800 °C at a heating rate of 10 °C min−1.
Electrochemical performance was recorded using a computer-controlled CH660D electrochemical work station (CHI instruments Inc., USA) equipped with two compartments and three electrode cells. A platinum gauze was used as the current collector for the working electrode, and platinum foil and a saturated calomel electrode was used as the auxiliary electrode and reference electrode, respectively. For preparing the working electrode, the electro-active material (NiO, 85 wt%), acetylene black (10 wt%), and poly(tetrafluoroethylene) (5 wt%) were firstly mixed together, then a few drops of ethanol were added to form a homogeneous slurry and finally the slurry was pressed and pasted on the platinum gauze with a mass of 5 mg. All electrochemical measurements were carried out in a 6 M KOH solution as the electrolyte.
3. Results and discussion
The phase purity and crystal structure of the products obtained with different mole ratios of NiSO4·(NH4)2SO4·6H2O/urea were examined by XRD patterns and SEM images. Fig. 1a shows the XRD pattern of the sample (P1) prepared when the mole ratio of NiSO4·(NH4)2SO4·6H2O/urea is 1:1. All of the diffraction peaks could be perfectly indexed to pure Ni(SO4)0.3(OH)1.4 (JCPDS Card No. 41-1424). No other diffraction peaks were detected, demonstrating the high purity of Ni(SO4)0.3(OH)1.4. The morphologies of the as-prepared Ni(SO4)0.3(OH)1.4 sample (P1) were studied by SEM. As shown in Fig. 1b and c, a large quantity of belt-like structures with a typical length of several micrometers and a width of about 100 nm were obtained. When decreasing the mole ratio of NiSO4·(NH4)2SO4·6H2O/urea to 1:6, as shown in Fig. 1d, the XRD pattern of sample P2 shows that the as-fabricated products belong to the crystal phase of Ni(HCO3)2 (JCPDS No. 15-782). The broad diffraction peaks indicate that the sample is composed of a large scale of fine Ni(HCO3)2 nanocrystals. Fig. 1e and f show that the prepared Ni(HCO3)2 consists of many microspheres, which are assembled with small nanoparticles. While the mole ratio of NiSO4·(NH4)2SO4·6H2O/urea is adjusted to 1:15, as depicted in the XRD pattern (Fig. 1g) and SEM images (Fig. 1h and i), it can be clearly seen that many Ni(HCO3)2 nanoparticles were fabricated.
 | ||
| Fig. 1 XRD patterns (a, d, and g) and SEM images (b, c, e, f, h, and i) of the as-synthesized products (P1, P2, and P3) with different mole ratios of NiSO4·(NH4)2SO4·6H2O/urea: P1 (a–c), P2 (d–f), and P3 (g–i). | ||
Based on the above results, Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2 nanostructure precursors can be selectively prepared in the mixed NiSO4·(NH4)2SO4·6H2O–urea–H2O system via tuning the amounts of urea. It is well known that the decomposition of urea results in the release of CO32− and NH4+, which could generate HCO3− and OH− through the hydrolysis process. In the present reaction system, it was proposed that at lower urea amounts of 2 mmol (the mole ratio of NiSO4·(NH4)2SO4·6H2O/urea is 1:1), the generation of OH− is predominant, leading to the formation of Ni(SO4)0.3(OH)1.4. Yang et al.21 and Wen et al.20 also pointed out that the gradual release of OH− from hydrolysis of the acetate plays a key role in the formation of one dimensional (needle-like and belt-like) Ni(SO4)0.3(OH)1.4 nanostructures. When the amount of urea is increased to 12 mmol (the mole ratio of NiSO4·(NH4)2SO4·6H2O/urea to 1:6), HCO3− would be produced, resulting in the precipitation of Ni(HCO3)2. Interestingly, the product is not well-crystallized according to the XRD pattern, which might be because not enough CO2 would react with CO32− forming HCO3−, resulting in a poor-crystallized Ni(HCO3)2 phase.22 While the mole ratio of NiSO4·(NH4)2SO4·6H2O/urea is adjusted to 1:15, the generation of large amounts of HCO3− predominantly occurs during ureolysis, resulting in the formation of a well-crystallized Ni(HCO3)2 phase.
In brief, the morphology tailoring of NiO nanostructures involves the calcining of Ni-based nanostructure precursors at 450 °C for 2 h. Fig. 2 shows the XRD patterns of the as-obtained samples synthesized by a calcination treatment of Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2 nanostructure precursors. Their diffraction peaks correspond to the (222), (311), (220), (200), and (111) planes, respectively. All the diffraction peaks could be perfectly indexed to the face-centered cubic phase (space group Fm3m) NiO (JCPDS Card No. 73-1523). No other diffraction peak was detected, indicating that pure NiO crystals have been successfully fabricated.
 | ||
| Fig. 2 XRD patterns of the obtained samples synthesized by calcining different Ni-based precursors: (a) P1, Ni(SO4)0.3(OH)1.4 nanobelts, (b) Ni(HCO3)2 nanospheres, and (c) Ni(HCO3)2 nanoparticles. | ||
Fig. 3 shows the TGA curves of Ni(SO4)0.3(OH)1.4 nanobelts, Ni(HCO3)2 nanospheres, and Ni(HCO3)2 nanoparticles. It was observed that NiO was transformed by calcination treatment of the Ni(HCO3)2 phase at 450 °C for 2 h. Zhang et al.23 found that after heat treatment of Ni(SO4)0.3(OH)1.4 at 500 °C NiO was obtained, retaining the same morphology as that of Ni(SO4)0.3(OH)1.4. As the heat treatment temperature increased to 750 °C, the belt-like Ni(SO4)0.3(OH)1.4 was completely destroyed and transformed into agglomerated NiO nanoparticles with a random orientation but better crystallinity. However, in the present system, the Ni(SO4)0.3(OH)1.4 nanobelts still have a weight loss when the calcining temperature is higher than 450 °C, which is in good agreement with Wen et al.20 From the XRD result, it was observed that the peaks of NiO products prepared by calcining of Ni(SO4)0.3(OH)1.4 have a slight shift, which might be due to the lower calcining temperature of Ni(SO4)0.3(OH)1.4.20 A further study on tunable synthesis of Ni(SO4)0.3(OH)1.4 nanostructures and their transformation to NiO nanomaterials by the calcining process is still in progress.
 | ||
| Fig. 3 TGA curves of Ni(SO4)0.3(OH)1.4 nanobelts, Ni(HCO3)2 nanospheres, and Ni(HCO3)2 nanoparticles. | ||
Electron microscopy was used to further characterize the changes in morphology and structure associated with calcinations of the as-prepared Ni-based nanostructure precursors. It is clearly seen that the as-synthesized NiO powders retained the dimensional structure and morphology of the Ni-based precursors based on the SEM images and TEM images (Fig. 4). As shown in Fig. 4a and b, plenty of NiO nanobelts were obtained when using belt-like Ni(SO4)0.3(OH)1.4 products as the precursor. While employing spherical Ni(HCO3)2 nanostructures as the precursor, as displayed in Fig. 4c and d, NiO nanospheres composed of small nanoparticles were fabricated. Fig. 4e and f show that a large number of NiO nanoparticles with a diameter of 30 nm were prepared with the calcination treatment of Ni(HCO3)2 nanoparticles at 450 °C for 2 h. On the basis of the above observation, NiO nanostructures with different morphologies have been successfully synthesized by using different Ni-based nanostructure precursors prepared via a urea involved hydrothermal method, as shown in Scheme 1. In this synthesis, Ni-based nanoprecursors were employed as hard templates, directing the formation of different shaped NiO nanostructures by calcining treatment.
 | ||
| Fig. 4 SEM (a–e) images and TEM images (f) of the obtained samples synthesized by calcining different Ni-base precursors: (a and b) P1, Ni(SO4)0.3(OH)1.4 nanobelts, (c and d) Ni(HCO3)2 nanospheres, and (e and f) Ni(HCO3)2 nanoparticles. | ||
 | ||
| Scheme 1 Illustration for the synthesis of NiO nanostructures with different morphologies from the obtained Ni-based nanoprecursors prepared via a urea-controlled hydrothermal method. | ||
Fig. 5 shows the nitrogen adsorption–desorption isotherms of different NiO nanostructures to determine the BET specific surface areas by nitrogen adsorption–desorption measurements. The BET surface areas of the as-prepared NiO nanobelts, nanospheres, and nanoparticles were 44.2, 18.1, and 58.5 m2 g−1, respectively. The as-synthesized NiO materials demonstrated different BET surface areas, which is attributed to the differences in their structures, as shown by the SEM and TEM images. The pore size distribution of three kinds of NiO was determined by using the Barrett–Joyner–Halenda (BJH) method. It can be observed that NiO nanoparticles contained small mesopores, which may be ascribed to the aggregation of nanoparticles during the calcination process. The BET specific surface areas of the NiO nanoparticles demonstrate that the nanoparticle structure of NiO has a relatively high surface-to-volume ratio, which is helpful for the electrochemical performance.24,25
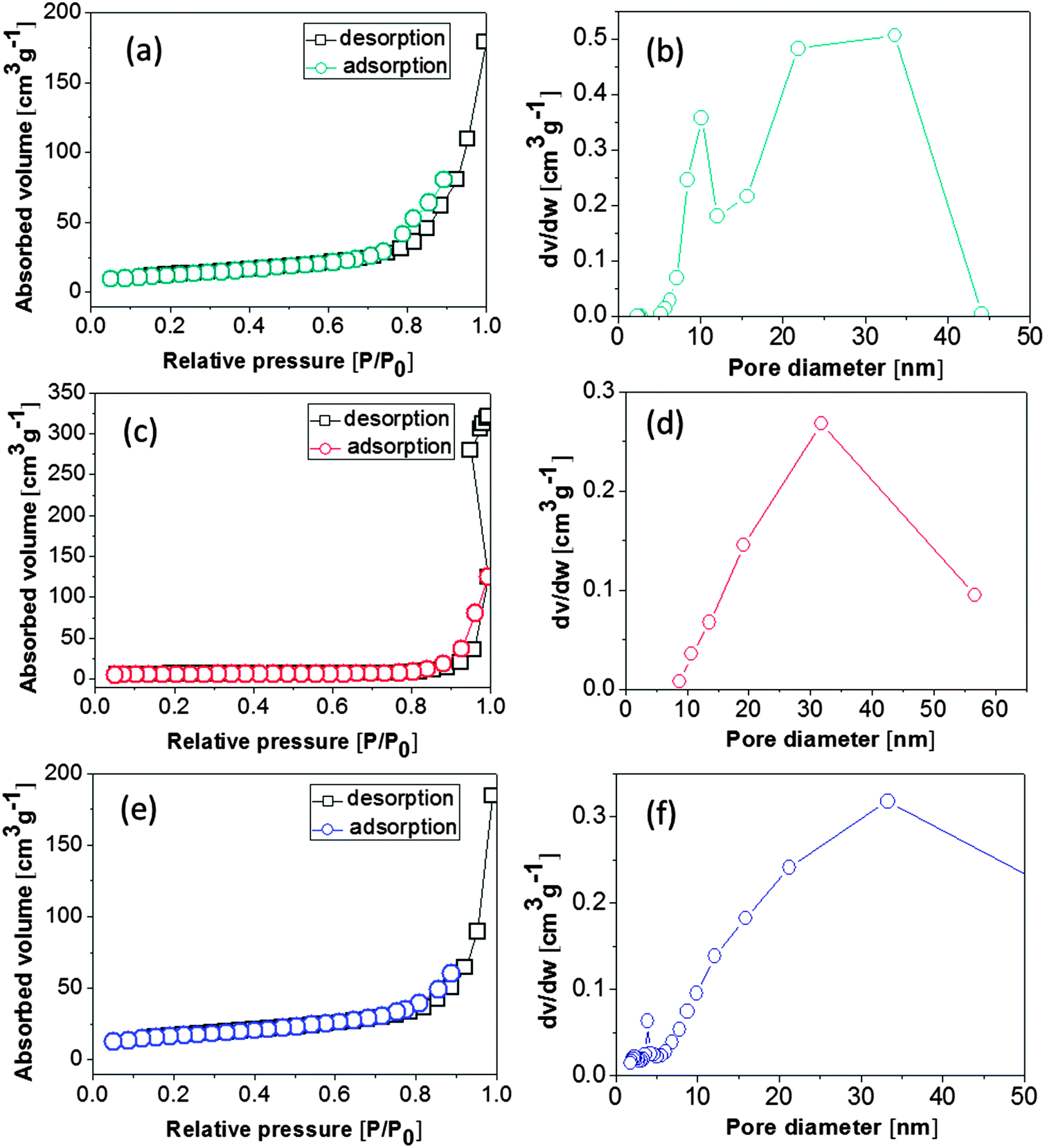 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms (a, c and e) and pore size distribution (b, d and f) of NiO samples: (a and b) nanobelts, (c and d) nanospheres, and (e and f) nanoparticles. | ||
The capacity performances of the as-synthesized NiO nanobelts, nanoparticles, and nanospheres were investigated in 6 M KOH. Fig. 6a shows the cyclic voltammetry (CV) curves scanned at 10 mV s−1 of three different kinds of NiO electrodes. Faradic pseudo-activity induced capacitance was reflected by a pair of strong symmetry redox peaks in each CV curve. It was observed that the anodic peaks were in the range of 0.25–0.4 V, which could be attributed to the reduction of NiO to NiOOH, while the catholic peaks at 0.0–0.1 V are related to the conversion of NiOOH back to NiO. The charge storage of NiO arises from the following redox equation:5,9,26
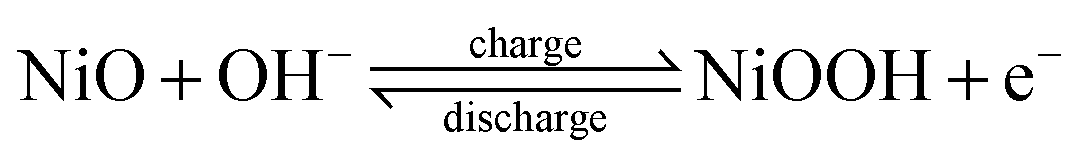
 | ||
| Fig. 6 The cyclic voltammetry curves (a) and galvanic charge–discharge curves (b) of NiO nanobelts, nanospheres, and nanoparticles, (c) cyclic voltammetry of NiO nanoparticles at different scan rates, (d) cyclic voltammetry of NiO nanoparticles at different current densities, and (e) the capacitance stability of NiO nanoparticles at a constant current density of 5 A g−1. | ||
The galvanic charge–discharge curves at a current density of 5 A g−1 within the voltage range of −0.2–0.5 V are shown in Fig. 6b. The flat stage in the charge–discharge curve clearly demonstrates that the pseudo-capacitance was induced by faradaic charges. The specific capacitances of NiO nanospheres, nanobelts, and nanoparticles calculated from CV curves are 45.2, 556.2, and 609.5 F g−1, respectively. The duration of the galvanic charge–discharge cycle is in good agreement with CV measurements.
In our experiment, judging from the XRD pattern in Fig. 2, NiO nanobelts derived from the calcination of Ni(SO4)0.3(OH)1.4 have poor crystallization compared to another two samples derived from the calcination of Ni(HCO3)2. Consequently, as reflected in electrochemical figures, the as-made samples from calcination of Ni(HCO3)2 show better symmetric features in the cyclic voltammetry curve. The catholic shift is due to the different crystallinities of calcinated samples, where ions are charged and discharged continuously and the electrolyte diffuses, inducing ohmic resistance and polarization. The crystallinity alteration resulting in different redox peak positions was also observed in previously published papers.5,27
The capacitive performances of nickel oxide are affected by main factors such as the particle size, surface area and morphologies.24,25 As for the pseudo-capacitive materials, the shrinked particle size and the enlarged surface area are substantially helpful in augmenting the amount of electrons in valence-reversible faradaic reaction. Among the three different kinds of nanostructures, NiO nanoparticles show the largest BET surface area, which could lead to improved electrolyte permeability and facilitate the enhancement of electron pathways, thus potentially improving the pseudocapacitance.27 For current corrective acetylene black, it was difficult to go into the inner space of the big NiO spheres, resulting in a low electron-transfer efficiency induced by uniform dispersion of acetylene black in nanospheres, which greatly compromised the electrochemical performance of nanospheres. As reflected in the cyclic voltammetry and galvanic charge–discharge, the curves of nanobelts and nanoparticles cover a bigger area than that of nanospheres, which is in good agreement with the sequence of surface area analysis.
On the basis of these morphological studies, cyclic voltammogram tests on the NiO nanoparticles as active materials for supercapacitors were performed at different scan rates to observe the CV curve change, as shown in Fig. 6c. A gradual increase in the current density was observed in the redox CV curves with an increase in the scan rate, indicating that the electrochemical process was surface confined. In addition, within the increase in the scan rate, the anodic peak potential and catholic peak potential shift positively and negatively, respectively, while the capacitance decreases as scan rates increased higher than 50 mV s−1. As shown in Fig. 6d, the specific capacitance of NiO nanoparticles at different current densities is provided, in which the current density is 0.5 A g−1, 1 A g−1, 5 A g−1 and 10 A g−1, respectively. From the experimental results, it can be concluded that the increase in the current density decreases the performance of specific capacitance of samples (calculated as 770.5 F g−1, 680.4 F g−1, 609.5 F g−1 and 524.4 F g−1, respectively).
The cyclic stability of NiO nanoparticles after 5000 repeated cycles is shown in Fig. 6e. It was found that there was no capacity shrinks in the first 500 cycles, while there was a drastic capacity decrease from 500 to 3000 cycles. It was observed that the capacity was stabilized by 3750 cycles and only ∼62% of the original specific capacitance was retained, which might be due to the detachment of NiO nanoparticles from the current collectors. It was previously reported that the capacitance of NiO nanomaterials is generally between 69.8 and 285 F g−1.28–31 The above results suggest that the as-synthesized NiO nanoparticles have relatively high supercapacitances and excellent capacitance retention.
4. Conclusion
In summary, by tuning the amounts of urea, Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2 nanostructure precursors were selectively synthesized in water via a facile hydrothermal method. After thermal treatment, different nanostructured Ni(SO4)0.3(OH)1.4 and Ni(HCO3)2 can be converted into NiO nanobelts, nanospheres, and nanoparticles, which shows different BET surface areas. The electrochemical measurements of three kinds of different NiO nanostructure-based supercapacitors revealed that NiO nanoparticles exhibit pseudo-capacitive properties with high capacitance and good capacitance retention, which probably benefit from the structural features of being nanoparticulate in size and having large surface areas.Conflicts of interest
The authors declare no competing financial interest.References
- H. Jiang, J. Ma and C. Li, Adv. Mater., 2012, 24, 4197–4202 CrossRef CAS.
- X. Lu, D. Zheng, T. Zhai, Z. Liu, Y. Huang, S. Xie and Y. Tong, Energy Environ. Sci., 2011, 4, 2915–2921 CAS.
- L. Feng, Y. Zhu, H. Ding and C. Ni, J. Power Sources, 2014, 267, 430–444 CrossRef CAS PubMed.
- B. Wang, J. S. Chen, Z. Wang, S. Madhavi and X. W. Lou, Adv. Energy Mater., 2012, 2, 1188–1192 CrossRef CAS.
- K. K. Purushothaman, I. Manohara Babu, B. Sethuraman and G. Muralidharan, ACS Appl. Mater. Interfaces, 2013, 5, 10767–10773 CAS.
- G. Zhang, L. Yu, H. E. Hoster and X. W. Lou, Nanoscale, 2013, 5, 877–881 RSC.
- W. Yu, X. Jiang, S. Ding and B. Q. Li, J. Power Sources, 2014, 256, 440–448 CrossRef CAS PubMed.
- Z. Yang, F. Xu, W. Zhang, Z. Mei, B. Pei and X. Zhu, J. Power Sources, 2014, 246, 24–31 CrossRef CAS PubMed.
- S. K. Meher, P. Justin and G. Ranga Rao, Nanoscale, 2011, 3, 683–692 RSC.
- Y. Wang, Q. Zhu and H. Zhang, Chem. Commun., 2005, 5231–5233 RSC.
- X. Wan, M. Yuan, S.-l. Tie and S. Lan, Appl. Surf. Sci., 2013, 277, 40–46 CrossRef CAS PubMed.
- Y. Cui, C. Wang, S. Wu, G. Liu, F. Zhang and T. Wang, CrystEngComm, 2011, 13, 4930–4934 RSC.
- S. Vijayakumar, S. Nagamuthu and G. Muralidharan, ACS Appl. Mater. Interfaces, 2013, 5, 2188–2196 CAS.
- X. Tian, C. Cheng, L. Qian, B. Zheng, H. Yuan, S. Xie, D. Xiao and M. M. F. Choi, J. Mater. Chem., 2012, 22, 8029–8035 RSC.
- T. Alammar, O. Shekhah, J. Wohlgemuth and A.-V. Mudring, J. Mater. Chem., 2012, 22, 18252–18260 RSC.
- J. H. Pan, Q. Huang, Z. Y. Koh, D. Neo, X. Z. Wang and Q. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 6292–6299 CAS.
- J. Li, F. Luo, Q. Zhao, Z. Li, H. Yuan and D. Xiao, J. Mater. Chem. A, 2014, 2, 4690–4697 CAS.
- R. Wang, Q. Li, D. Xie, H. Xiao and H. Lu, Appl. Surf. Sci., 2013, 279, 129–136 CrossRef CAS PubMed.
- S. Ding, T. Zhu, J. S. Chen, Z. Wang, C. Yuan and X. W. Lou, J. Mater. Chem., 2011, 21, 6602–6606 RSC.
- W. Wen, J.-M. Wu, L.-L. Lai, G.-P. Ling and M.-H. Cao, CrystEngComm, 2012, 14, 6565–6572 RSC.
- D. Yang, P. Liu, Y. Gao, H. Wu, Y. Cao, Q. Xiao and H. Li, J. Mater. Chem., 2012, 22, 7224–7231 RSC.
- Y. Yan, G. Cheng, P. Wang, D. He and R. Chen, RSC Adv., 2014, 4, 49303–49307 RSC.
- K. Zhang, J. Wang, X. Lu, L. Li, Y. Tang and Z. Jia, J. Phys. Chem. C, 2009, 113, 142–147 CAS.
- S. Chen, W. Xing, J. Duan, X. Hu and S. Z. Qiao, J. Mater. Chem. A, 2013, 1, 2941–2954 CAS.
- R. B. Rakhi, N. A. Alhebshi, D. H. Anjum and H. N. Alshareef, J. Mater. Chem. A, 2014, 2, 16190–16198 CAS.
- S. Xiong, C. Yuan, X. Zhang and Y. Qian, CrystEngComm, 2011, 13, 626–632 RSC.
- S.-I. Kim, J.-S. Lee, H.-J. Ahn, H.-K. Song and J.-H. Jang, ACS Appl. Mater. Interfaces, 2013, 5, 1596–1603 CAS.
- Y.-g. Wang and Y.-y. Xia, Electrochim. Acta, 2006, 51, 3223–3227 CrossRef CAS PubMed.
- X. Zhang, W. Shi, J. Zhu, W. Zhao, J. Ma, S. Mhaisalkar, T. Maria, Y. Yang, H. Zhang, H. Hng and Q. Yan, Nano Res., 2010, 3, 643–652 CrossRef CAS PubMed.
- A. I. Inamdar, Y. Kim, S. M. Pawar, J. H. Kim, H. Im and H. Kim, J. Power Sources, 2011, 196, 2393–2397 CrossRef CAS PubMed.
- Y.-z. Zheng, H.-y. Ding and M.-l. Zhang, Mater. Res. Bull., 2009, 44, 403–407 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |