
Bile acid hydrazides: gelation, structural, physical and spectroscopic properties†
Vandana S.
Pore
*a,
Sandip G.
Agalave
a,
Shrikant G.
Pharande
a,
Prashant A.
Patil
b and
Amol S.
Kotmale
a
aOrganic Chemistry Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pashan, Pune 411 008, India. E-mail: vs.pore@ncl.res.in; Fax: +91 2025902629; Tel: +91 2025902320
bPolymer Science and Engineering Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pashan, Pune 411 008, India
First published on 27th October 2014
Abstract
Synthesis and gelation properties of a series of novel bile acid hydrazides are presented. These compounds are found to undergo self-assembly leading to organogelation in certain organic solvents. Compound 1 was found to be the most “effective” gelator in this series. The properties of this gel have been thoroughly investigated by conventional methods typical for molecular gel studies. Sol–gel transition temperature (Tg) of chloroform gels of compounds 1 and 3 was found to increase with increase in the chain length. Sol–gel transition was probed using the isothermal time test and results show that there is instantaneous increase in both the moduli after shear melting, which suggests that the kinetics of formation of the network was very fast. IR and NMR studies revealed hydrogen bonding between amidic carbonyl in the side chain and hydroxyl groups of cholic acid.
1. Introduction
Synthesis of supramolecular assemblies is the fundamental step to new materials.1–3 In recent years low molecular mass organic molecules, capable of gelation with various liquids, have attracted considerable attention.4 The self-assembly of small organic molecules to form three-dimensional networks creates matrices that can hold solvent molecules, thereby forming gels. This gives systems with new properties based on a three-dimensional network held together by various multiple non-covalent interactions such as hydrogen bonding, hydrophobic interaction, dispersion forces and π–π stacking. There are several building blocks known for their good gelation properties such as steroids, saccharides, nucleobases, porphyrins etc. Sol–gel systems have been used as active components of sensors, drug delivery systems, membranes, etc. due to their interesting properties.5–7 Evolution in Low Molecular weight Organo Gel (LMOG) research paves the way for its developments in high-tech materials and biomaterials.8,9Design of new molecules capable of self-assembly leading to gelation is very complicated due to the challenging specification of conditions and structural requirements for a gelator. Structural features known to promote one-dimensional aggregation include amidic, carbamate, urea or oxalamide groups combined with aliphatic or aromatic molecules with a large surface area. Many efficient gelators of organic solvents (organogelators) and water (hydrogelators) have actually been prepared by their structural combination.10
Recent advances in the field of steroidal supramolecular gels and their potential applications have been summarized by Sievänen and co-workers.11 Bile acids and their derivatives, because of their unique structural features, represent fascinating compounds from a supramolecular point of view.12 Bile acids are natural compounds consisting of a facially amphiphilic steroid nucleus with a hydrophobic β-side due to angular methyl groups and a hydrophilic α-side due to hydroxyl groups. The amphiphilicity, structure rigidity and acid–base characteristics of the bile acid molecules make them useful building blocks in biomedical applications. Furthermore, the overall polarity profile of these compounds can be tailored and the self-assembling characteristics adjusted, which makes them potential components of supramolecular gels. Numerous organo- and hydrogels composed of bile acids/salts and their derivatives have been reported and investigated.13 Recently, there was a review article by Maitra and Sajisha on functional soft materials synthesized in bile acid-based supramolecular hydrogel matrices.14 Dukh and co-workers have synthesized bile acid amides–phenanthroline hybrids and studied their gelation and metal co-ordination properties.15 Noponen et al. have reported synthesis and gelation properties of bile acid–amino acid conjugates.16 There is report on cholic acid hydrazide-modified dextran nanoparticles for in vitro drug release.17 Lotowski and Guzmanski have synthesized cholic acid dimers with hydrazide as a spacer.18 Hydrazones prepared from bile acid hydrazide have been reported to show good antimicrobial activity.19
Hydrazine is a useful building block in organic synthesis of pharmaceuticals and pesticides. The antitubercular drug isoniazid contains the hydrazine moiety. There is a recent report on supramolecular organogels based on a hydrazide derivatives.20 Very recently aroylhydrazine–amide receptors with a hydrazine spacer have been reported as novel colorimetric chemosensors.21 Polymers containing the hydrazine spacer have been synthesized and used for encapsulation of anticancer drugs.22
Bile acid derived gelators can interact specifically with their transporter proteins and/or possess specific functions such as controlled and organ targeted drug release via enterohepatic circulation. Alkyl and amide derivatives of bile acids are potential gelating agents.13a,c–e In continuation of our work on bile acids,23 we wish to report here synthesis of novel bile acid conjugates containing the hydrazine spacer. Interestingly, few conjugates showed sol–gel transition in few organic solvents. One of these gels has been investigated for structural, thermal and morphological studies. It was hypothesized that the synergic effect of both moieties of the conjugate may lead to the formation of a new kind of supramolecular structures.
2. Results and discussion
A series of bile acid hydrazides was synthesized by reacting cholic acid with respective hydrazides using the literature procedure24 with some modifications (Fig. 1). All the compounds were obtained in moderate chemical yields and were characterized by conventional physical and spectroscopic methods. The attempts to obtain single crystals of these compounds remained unsuccessful. This may be due to the highly amorphous nature of these molecules and hence the melting points were not sharp. All the synthesized compounds were tested for their gelation behaviour in 10 different solvents and the results are summarized in Table 1. | ||
| Fig. 1 Chemical structures of compounds 1–6. | ||
| Solvent | Results | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| a TGb, transparent gel (at room temperature); TGc, transparent gel (after heating); I, insoluble; S, solution; CG, cloudy gel (after heating); P, precipitate, C, coagulation; WGb, weak gel (at room temperature); WGc, weak gel (after heating at boiling temperature of solvent). | ||||||
| Methanol | S | S | S | S | S | S |
| DCM | TGb | C | I | C | WG | I |
| CHCl3 | TGb | C | TGc | C | P | P |
| CCl4 | TGb | C | WGc | I | C | I |
| THF | S | S | S | S | S | S |
| CH3CN | S | I | S | P | I | I |
| Benzene | CG | I | TGc | I | I | I |
| Toluene | CG | I | TGc | I | I | I |
| n-Hexane | I | I | I | C | I | I |
DMF–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
P | S | S | S | S | S |
Three out of six compounds were found to form gels depending upon the solvent. Gels were obtained from 2% (w/v) solutions and appeared as opaque (cloudy) or transparent or weak depending on the compound and solvent in question. Regarding the number of solvents gelled, compound 1 seems to be the most “effective” gelator in this series. It formed gel in five different solvents. Hence further study was carried out on compound 1.
Gelation ability, efficiency and properties change dramatically with small structural variations in the gelator molecule. It was observed that the molecule with shorter chain length than compound 1 (compound 2) did not form gel with any of the tested solvents while compounds having longer chain length than compound 1 (compound 3) formed gel in chloroform or carbon tetrachloride solution after heating at boiling temperature of the solvent. Introduction of an aromatic group in the alkyl tail deteriorates the gelation ability. The compound containing different substitution groups in the aromatic ring also behaved differently. The compound with the 4-chlorobenzoyl moiety (compound 5) could form weak gel only in dichloromethane while other aromatic compounds such as 4 and 6 did not give gels. The driving force for aggregation of bile acid gelators is hydrogen bonding between the amide group and the hydroxyl groups and also intermolecular hydrogen bonding between amidic carbonyls of one molecule with amidic NH of another molecule. It is reported that compounds lacking one of these groups did not produce gels in some solvents.13a There is a report in which compounds lacking all the hydroxyl groups due to formation of ketones gave organogels.13d There is another report in which lithocholic acid having one and cholic acid having three hydroxy groups formed gels but deoxycholic acid with two hydroxy groups did not give gels.13e We also found that deoxycholic acid in which hydroxyl group at C-7 is absent, did not give gel in any of the tested solvents.
Interestingly, gel to sol phase transition can be induced by shaking the gel vigorously, in contrast, resting the solution results in gelation again (Fig. 2). Ultrasonic treatment is another effective way to trigger the phase transition of the gel. In this case, the liquid is a viscous suspension, rather than a clear solution (Fig. 2). Melting points of 2% w/v benzene and chloroform gels of compound 1 and 3 were determined with Differential Scanning Calorimetry (DSC). Benzene gels of compounds 1 and 3 showed two endothermic peaks (please see DSC figure in ESI†). The first endothermic peak at around 8 °C is due to the melting point of benzene and the second endothermic peak at around 84 °C and 87 °C is due to melting of gel network structures in compounds 1 and 3 respectively. The chloroform gel of compound 5 was found to be unstable and it was not possible to find its sol–gel transition temperature. Sol–gel transition temperatures (Tg) of the chloroform gel of compound 1 show two peaks at 65 and 71 °C while that of compound 3 shows a single peak at 68 °C. Melting point of the chloroform gel of compound 1 was also determined by visually monitoring the flow of gel in a thermally controlled bath13i and was found to be 62 °C. It is worth mentioning that the benzene and chloroform gels are so stable that their melting does not occur below the boiling point of the pure solvents. As expected, the gel is thermoreversible in nature. Gel melts upon heating and forms upon cooling the solution (Fig. 2). This shear-stress-triggered reversible sol–gel phase transition phenomenon was further studied by rheological techniques (Fig. 6 to 9).
 | ||
| Fig. 2 Phase transition of gel of compound 1. | ||
2.1. Morphology
The morphology of gels derived from compound 1 in two different solvents (benzene and chloroform) was investigated by environmental scanning electron microscopy (E-SEM). (a) Gels in benzene and chloroform, (b) xerogels obtained after sonication, (c) xerogels obtained after complete drying and (d) dried sample were studied. The influence of the solvent on gel formation is well recognized as depending on the solvent, it leads to different shapes of the aggregates giving different microstructures (see ESI†). The E-SEM micrograph did not reveal any regular fibrous structures. It only showed three-dimensional structures constructed from thin micro flakes of different sizes and mostly rounded shapes in benzene gel (Fig. 3, and ESI†) suggesting that the gel microstructure was very fragile and collapsed upon solvent evaporation as well as due to sonication. E-SEM analysis of chloroform gels showed that fibers driven by weak intermolecular interactions were formed, as can be seen from Fig. 3. It can be suggested that hydrogen bonding interactions may be responsible for the fiber formation, as the molecules contain an amide group and hydroxyl groups. Such type of intermolecular hydrogen bonding has been observed in previous studies of gelation properties of bile acid amides.13a Hydrogen bonding may be one of the driving forces, but also hydrophobic as well as van der Waals interactions must be taken into account. However it is not fully understood how these fibers are constructed from the gelator molecules.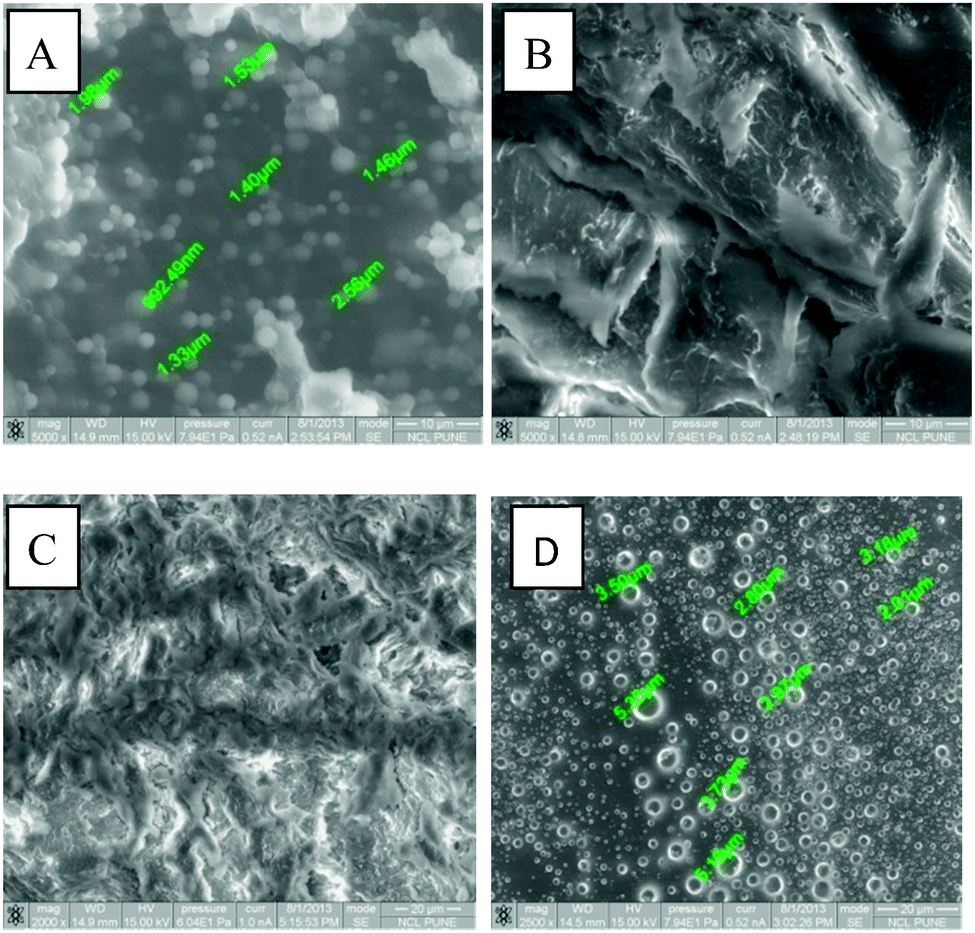 | ||
| Fig. 3 E-SEM images of gels of 2% (w/v) compound 1 (A) in benzene; (B) in CHCl3; E-SEM images of xerogels of 2% (w/v) compound 1 (C) in benzene; (D) in CHCl3 (scale bars 10 μm). | ||
2.2. Spectroscopy
 | ||
| Fig. 4 Variable-temperature 1H NMR of compound 1 (2% w/v in CDCl3). | ||
 | ||
| Fig. 5 Concentration dependent 1H NMR of compound 1. | ||
2.3. Rheology
All the experiments were carried out on 2% w/v of the benzene gel of compound 1 at 15 °C.The main purpose of this test is to identify the linear viscoelastic regime for the gel. All further experiments were performed in linear viscoelastic range. The results of strain sweep tests are shown in Fig. 6. At low strain values, storage modulus (G′) was higher than loss modulus (G′′) indicating the solid like behaviour of the gel. As the strain percentage was increased to 3%, the storage modulus and loss modulus started dropping, indicating the yielding of the gel and after 12% strain, G′′ crossed over G′ (G′′ > G′) indicating the liquid like behaviour.
 | ||
| Fig. 6 Storage and loss moduli as a function of strain%. | ||
 | ||
| Fig. 7 Shear stress ramp test. | ||
 | ||
| Fig. 8 Shear melting and the isothermal time test. | ||
 | ||
| Fig. 9 Storage and loss moduli as a function of frequency. | ||
3. Conclusions
In the present work, we have presented novel cholic acid hydrazides gelators which form thermoreversible organogels in different solvents. These are new supramolecular gelators with potential high-tech applications. IR and NMR study of these gels confirm intermolecular hydrogen bonding due to OH and NH protons which is responsible for gelation. Gel shows solid like behaviour over the entire frequency range. Sol gel transition was probed using an isothermal time test and results show that instantaneous rise in both the moduli after shear melting suggesting that the kinetics of formation of the network is very fast. The DSC study shows that benzene gel to sol conversion takes place at 84 °C while chloroform gel to sol conversion takes place at 65 °C both of which are above the boiling points of their solvents. It is very interesting and uncommon that this type of small organogelators can gelate some solvents at room temperature. The potential applications of these gels are under further investigation. These molecules are under biological testing, and as bile acid conjugates, they are expected to show good biological activity.4. Experimental section
4.1. General methods
All chemicals were obtained from commercial sources and used as received without further purification. All the reactions were carried out under a nitrogen atmosphere. Column chromatography was carried out by using silica gel (60–120 mm, Merck). All reactions were monitored by TLC with silica gel coated plates; spots were visualized by UV light and/or with dipping in a phosphomolybdic acid solution and charring on a hot plate. All the liquid state 1D-NMR spectra (1H, 13C NMR) were recorded in CDCl3 (for compound 1), CDCl3 + CD3OD (for compounds 5 and 6) and CD3OD (for compounds 2, 3 and 4) on AC 200 MHz and AV-500 MHz Bruker NMR spectrometers. Chemical shifts are reported in ppm. Coupling constants J are reported in Hz. The temperature dependent 1H NMR experiments for compound 1 (2% w/v) were carried out in CDCl3 by increasing temperature in 5 steps (25 °C, 30 °C, 35 °C, 40 °C and 45 °C successively) and for concentration dependent 1H NMR experiments for compound 1 were carried out in CDCl3 by using different NMR tubes of concentrations 0.2%, 0.4%, 0.8%, 1.2%, 1.7% and 2.5%. The 1H chemical shifts were referenced to TMS as an internal standard. Infrared (IR) spectra were recorded on a liquid cell with KBr plates. Only diagnostic bands are reported on a cm−1 scale. The ESI ion trap mass spectra were measured by a Finnigan MAT LCQ mass spectrometer. The HRMS spectra were acquired on a thermoscientific Q exactive spectrometer. An environmental scanning electron microscope Quanta 200 3D by FEI was used for obtaining morphology of gels. Rheological properties of gels were studied using a MCR 301 Rheometer (Anton Paar) with a cup and bob geometry. The gel was loaded in a cup and sufficient delay time was given in order to form gel in the cup. A DSC study of the gel sample was performed on TA instruments Q 100 Differential Scanning Calorimeter. A sealed pan was used for the measurement.4.2. Synthesis
Cholic acid hydrazides were synthesized by reacting cholic acid with respective hydrazides using a literature procedure with some modifications (Fig. 1).24To a solution of cholic acid (1 mmol), acid hydrazide (1 mmol), and EDCI (1.2 mmol) dissolved in DCM (10 mL), DMAP (1.2 mmol) was added in one portion at room temperature. The reaction mixture was stirred at room temperature for 8 h. After completion of reaction, the reaction mixture was extracted with 10% methanol in DCM. The organic layer was washed with saturated aqueous NaHCO3, brine, and was dried over Na2SO4. The crude product upon purification by column chromatography on silica gel yielded the pure product.
4.3. Gelation test
In the gelation experiments, a known weight (2 wt%) of a potential gelator and a measured aliquot of liquid were placed into a capped glass sample vial and the system was heated in an oil bath until the solid was dissolved, then, the solution was cooled slowly to room temperature in air, and finally the sample vial was inverted to look at if the solution inside could still flow. The sample was defined as a gel, if no flow was observed. When a gel was formed at this stage, it was denoted as “G”. It is to be noted that some transparent gels can be obtained at room temperature, which were denoted as “TG”. In some cases, a gel remains cloudy so this kind of system has been referred to as “cloudy gels (CG)”. For systems in which only solution remained until the end of the tests, they were referred to as solution (S). When the gelator of a system appeared as a precipitate or crystals, the system was denoted as “P”. The system, in which the potential gelator could not be dissolved even at the boiling point of the solvent, was called an insoluble system (I) (please see Table 1). Gelator 1 dissolved in DCM, CHCl3 and CCl4 at room temperature and forms transparent gel after some time while gelator 1 dissolved in benzene and toluene near to their boiling point and forms cloudy gel upon cooling solution. Gelator 3 forms clear solution upon heating the solution to the boiling point.Acknowledgements
The authors thank CSIR, New Delhi, for financial support under ORIGIN (CSC-0108) and OSDD (HCP-0001). SGA (31/11(802)/2013-EMR-I) and PAP (31/11(442)/2008-EMR-I) thank CSIR, New Delhi, for a senior research fellowship. We acknowledge the help from Central NMR facility and centre for material characterization division of CSIR, NCL.References
- J.-M. Lehn, Supramolecular Chemistry. Concepts and perspectives, VCH, Weinheim, Germany, 1995 Search PubMed.
- (a) D. Venkataraman, S. Lee, J. Zhang and J. S. Moore, Nature, 1994, 371, 591–593 CrossRef CAS; (b) G. R. Desiraju, Crystal Engineering. The Design of Organic Solids, Elsevier, Amsterdam, The Netherlands, 1989 Search PubMed.
- J.-H. Furhop and J. Köning, in Membranes and Molecular Assemblies: The Synkinetic Approach, RSC Monographs in Supramolecular Chemistry, ed. J. Fraser Stoddart, 1994 Search PubMed.
- Some recent examples on small organic gelators include the following: (a) P. Terech, Ber. Bunsen-Ges., 1998, 102, 1630–1643 CrossRef CAS; (b) P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed; (c) K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinomori, M. Takeuchi, S. Shinkai and D. N. Reinhoudt, Chem. – Eur. J., 1999, 5, 2722–2729 CrossRef CAS; (d) R. Oda, I. Huc and S. J. Candau, Angew. Chem., Int. Ed., 1998, 37, 2689–2691 CrossRef CAS; (e) T. Kato, G. Kondo and K. Hanabusa, Chem. Lett., 1988, 193–194 Search PubMed; (f) J. van Esch, S. De Feyter, R. M. Kellog, F. De Schryver and B. L. Feringa, Chem. – Eur. J., 1997, 3, 1238–1243 CrossRef CAS; (g) J.-E. S. Sohna and F. Fages, Chem. Commun., 1997, 327–328 RSC; (h) F. Placin, M. Colomes and J.-P. Desvergne, Tetrahedron Lett., 1997, 38, 2665–2668 CrossRef CAS.
- L. C. Klein, Filters and Membranes by the Sol-Gel Process, in Sol-Gel Technology: For Thin Films, Fibers, Preforms, Electronics and Specialty Shapes, ed. L. C. Klein, Noyes Publications, Park Ridge, NJ, 1988, pp. 382–399 Search PubMed.
- A. L. Graham, C. A. Carlson and P. L. Edmiston, Anal. Chem., 2002, 74, 458–467 CrossRef CAS.
- G. Wang and A. D. Hamilton, Chem. – Eur. J., 2002, 8, 1954–1961 CrossRef CAS.
- (a) S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766–1776 CrossRef CAS PubMed; (b) Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef CAS PubMed; (c) K. K. Kartha, S. S. Babu, S. Srinivasan and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 4834–4841 CrossRef CAS PubMed.
- (a) B. E. Yoldas, M. J. Annen and J. Bostaph, Chem. Mater., 2000, 12, 2475–2484 CrossRef CAS; (b) M. Giorgetti, S. Passerini, W. H. Smyrl and M. Berrettoni, Chem. Mater., 1999, 11, 2257–2264 CrossRef CAS; (c) S. Passerini, F. Coustier, M. Giorgetti and W. H. Smyrl, Electrochem. Solid-State Lett., 1999, 2, 483–485 CrossRef CAS PubMed.
- F. Fages, F. Vögtle and M. Žinić, Top. Curr. Chem., 2005, 256, 77–131 CrossRef CAS.
- H. Svobodova, V. Noponen, E. Kolehmainen and E. Sievänen, RSC Adv., 2012, 2, 4985–5007 RSC.
- E. Virtanen and E. Kolehmainen, Eur. J. Org. Chem., 2004, 3385–3399 CrossRef CAS.
- See for example: (a) H. M. Willemen, T. Vermonden, A. T. M. Marcelis and E. J. R. Sudhölter, Langmuir, 2002, 18, 7102–7106 CrossRef CAS; (b) K. Nakano, Y. Hishikawa, K. Sada, M. Miyata and K. Hanabusa, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2002, 389, 11–16 CrossRef CAS; (c) A. Valkonen, M. Lahtinen, E. Virtanen, S. Kaikkonen and E. Kolehmainen, Biosens. Bioelectron., 2004, 20, 1233–1241 CrossRef CAS PubMed; (d) V. Noponen, H. Belt, M. Lahtinen, A. Valkonen, H. Salo, J. Ulrichová, A. Galandakova and E. Sievänen, Steroids, 2012, 77, 193–203 CrossRef CAS PubMed; (e) V. Noponen, Nonappa, M. Lahtinen, A. Valkonen, H. Salo, E. Kolehmainen and E. Sievänen, Soft Matter, 2010, 6, 3789–3796 RSC; (f) P. Babu, N. M. Sangeetha and U. Maitra, Macromol. Symp., 2006, 241, 60–67 CrossRef CAS; (g) Nonappa and U. Maitra, Soft Matter, 2007, 3, 1428–1433 RSC; (h) Nonappa and U. Maitra, Org. Biomol. Chem., 2008, 6, 657–669 RSC; (i) H. M. Willemen, T. Vermonden, A. T. M. Marcelis and E. J. R. Sudhölter, Eur. J. Org. Chem., 2001, 2329–2335 CrossRef CAS.
- S. Sajisha and U. Maitra, Chimia, 2013, 67, 44–50 CrossRef PubMed.
- M. Dukh, D. Šaman, J. Kroulík, I. Černý, V. Pouzar, V. Král and P. Drašar, Tetrahedron, 2003, 59, 4069–4076 CrossRef CAS.
- V. Noponen, Nonappa, M. Lahtinen, A. Valkonen, H. Salo, E. Kolehmainen and E. Sievänen, Soft Matter, 2010, 6, 3789–3796 RSC.
- X. B. Yuan, H. Li, X. X. Xiao-Xia Zhu and H. J. Woo, J. Chem. Technol. Biotechnol., 2006, 81, 746–754 CrossRef CAS.
- Z. Lotowski and D. Guzmanski, Monatsh. Chem., 2006, 137, 117–124 CrossRef CAS.
- A. J. M. Rasras, T. H. Al-Tel, A. F. Al-Aboudi and R. A. Al-Qawasmeh, Eur. J. Med. Chem., 2010, 45, 2307–2313 CrossRef CAS PubMed.
- M. Bielejewski, J. Kowalczuk, J. Kaszynska, A. Łapinski, R. Luboradzki, O. Demchuk and J. Tritt-Goc, Soft Matter, 2013, 9, 7501–7514 RSC.
- S. Lia, H. Lia, C. Chena, X. Yuea, X. Caoa and S. Keb, Supramol. Chem., 2013, 25, 384–392 CrossRef.
- A. R. Diwan and A. L. Onton, WO 2009011702 A1, Allexcel, Inc., 2009.
- (a) D. B. Salunke, B. G. Hazra, V. S. Pore, M. K. Bhat, P. B. Nahar and M. V. Deshpande, J. Med. Chem., 2004, 47, 1591–1594 CrossRef CAS PubMed; (b) B. G. Hazra, V. S. Pore, S. K. Dey, S. Datta, M. P. Darokar, D. Saikia, S. P. S Khanuja and A. P. Thakur, Bioorg. Med. Chem. Lett., 2004, 14, 773–777 CrossRef CAS PubMed; (c) N. G. Aher and V. S. Pore, Synlett, 2005, 2155–2158 CAS; (d) D. B. Salunke, B. G. Hazra, R. G. Gonnade, M. M. Bhadbhade and V. S. Pore, Tetrahedron, 2005, 61, 3605–3612 CrossRef CAS PubMed; (e) V. S. Pore, N. G. Aher, M. Kumar and P. K. Shukla, Tetrahedron, 2006, 62, 11178–11186 CrossRef CAS PubMed; (f) D. B. Salunke, D. S. Ravi, V. S. Pore, D. Mitra and B. G. Hazra, J. Med. Chem., 2006, 49, 2652–2655 CrossRef CAS PubMed; (g) N. G. Aher, V. S. Pore and S. P. Patil, Tetrahedron, 2007, 63, 12927–12934 CrossRef CAS PubMed; (h) B. G. Hazra, N. S. Vatmurge, V. S. Pore, F. Shirazi, P. S. Chavan and M. V. Deshpande, Bioorg. Med. Chem. Lett., 2008, 18, 2043–2047 CrossRef PubMed; (i) N. S. Vatmurge, B. G. Hazra, V. S. Pore, F. Shirazi, M. V. Deshpande, S. Kadreppa and S. Chattopadhyay, Org. Biomol. Chem., 2008, 6, 3823–3830 RSC; (j) S. N. Bavikar, D. B. Salunke, B. G. Hazra, V. S. Pore, R. H. Dodd, J. Thierry, F. Shirazi, M. V. Deshpande, S. Kadreppa and S. Chattopadhyay, Bioorg. Med. Chem. Lett., 2008, 18(20), 5512–5517 CrossRef CAS PubMed; (k) N. G. Aher, V. S. Pore, N. N. Mishra, P. K. Shukla and R. G. Gonnade, Bioorg. Med. Chem. Lett., 2009, 19, 5411–5414 CrossRef CAS PubMed.
- S. H. Lee, H. J. Seo, S.-H. Lee, M. E. Jung, J.-H. Park, H.-J. Park, J. Yoo, H. Yun, J. Na, S. Y. Kang, K.-S. Song, M.-a. Kim, C.-H. Chang, J. Kim and J. Lee, J. Med. Chem., 2008, 51, 7216–7233 CrossRef CAS PubMed.
- Dispersions characterization testing and measurements by Eric Kissa surfactant science series vol. 84, chapter 14, 1999.
- Q. D. Nguyen and D. V. Boger, Annu. Rev. Fluid Mech., 1992, 24, 47–88 CrossRef.
- S. A. Madbouly and J. U. Otaigbe, Macromolecules, 2005, 38, 10178–10184 CrossRef CAS.
- V. M.-F. Lai and C.-Y. Lii, J. Food Sci., 2002, 67, 672–681 CrossRef CAS PubMed.
- J.-Y. Xiong, J. Narayanan, X.-Y. Liu, T. K. Chong, S. B. Chen and T.-S. Chung, J. Phys. Chem. B, 2005, 109, 5638–5643 CrossRef CAS PubMed.
- Z. Gong, Y. Yang, L. Huang, X. Chen and Z. Shao, Soft Matter, 2010, 6, 1217–1223 RSC.
- X.-J. Wu, Y. Wang, W. Yang, B.-H. Xie, M.-B. Yang and W. Dan, Soft Matter, 2012, 8, 10457 RSC.
- V. Meuniver, T. Nicolai, D. Durand and A. Parker, Macromolecules, 1999, 32(8), 2610–2616 CrossRef.
- R. K. Richardson, G. Robinson, S. B. R. Murphy and S. Todd, Polym. Bull., 1981, 4(9), 541–546 CrossRef CAS.
- S. Ikeda and K. Nishinari, J. Agric. Food Chem., 2001, 49, 4436–4441 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data and SEM images. See DOI: 10.1039/c4nj01352b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |