
Peralkylated imidazolium carbonate ionic liquids: synthesis using dimethyl carbonate, reactivity and structure†
C.
Maton
a,
K.
Van Hecke
b and
C. V.
Stevens
*a
aDepartment of Sustainable Chemistry and Technology, Ghent University, Coupure Links 653, 9000 Ghent, Belgium. E-mail: chris.stevens@ugent.be; Fax: +32 (0)9 264 62 21
bDepartment of Inorganic and Physical Chemistry, Ghent University, Krijgslaan 281 S3, 9000 Ghent, Belgium
First published on 16th October 2014
Abstract
Tri- and tetra-alkylimidazoles are quaternised into their corresponding ionic liquids with dimethyl carbonate. Upon metathesis of the obtained methyl carbonate salts, only gaseous by-products are generated. These methyl carbonate salts can be transformed into hydrogen carbonate salts by reaction with water. The salts containing a carbonate anion are very alkaline, which results in a hydrogen/deuterium exchange on the anion and some of the cation protons, depending on the substitution. Moreover, the crystalline 1-ethyl-3,4,5-trimethylimidazolium hydrogen carbonate formed carboxylate species upon dissolution. In particular, the carboxylate was able to regenerate the carbene and in the presence of chloroform, this led to the formation of the chloride salt.
Introduction
The interest in the synthesis of ionic liquids by the quaternisation of organic bases with dimethyl carbonate (DMC) was initiated some 20 years ago by the work of Albert and Mori.1,2 This methylating agent is environment friendly because of both its synthesis and biodegradability.3 DMC is synthesised from CO and MeOH over a Cu(I) catalyst, a method developed by Enichem, Italy.4 Furthermore, the quaternisation of neutral building blocks with DMC leads to methyl carbonate ionic liquids. By addition of the Brønsted acid of an anion of choice, the methyl carbonate anion decomposes into MeOH and CO2 and the ionic liquid with the conjugate base as an anion is obtained (Scheme 1). The quaternisation proceeds via a nucleophilic alkylation reaction (SN,Al), which is performed at temperatures above 120 °C. At lower temperatures, a reversible acylation reaction is initiated by a nucleophilic attack on the carbonyl centre of dimethyl carbonate (SN,Ac). Therefore, the alkylation reaction is most often performed in an autoclave or a sealed vial, in which the autogenous pressure keeps DMC in the liquid phase (Tb of DMC: 90 °C).3 | ||
| Scheme 1 Quaternisation of imidazoles with DMC with formation of the carbonate salt and metathesis towards a library of imidazolium salts. | ||
The group of Holbrey and Rogers reported on the formation of zwitterionic carboxylates in high yields during reaction of 1-methylimidazole 4 with DMC.5 Their formation is attributed to an SN,Al mechanism and deprotonation of imidazolium cation 5 by the [MeCO3]− anion. N,N′-Dimethylimidazolium carboxylate 6 is a solid and sufficiently stable to precipitate from solution (Scheme 2).
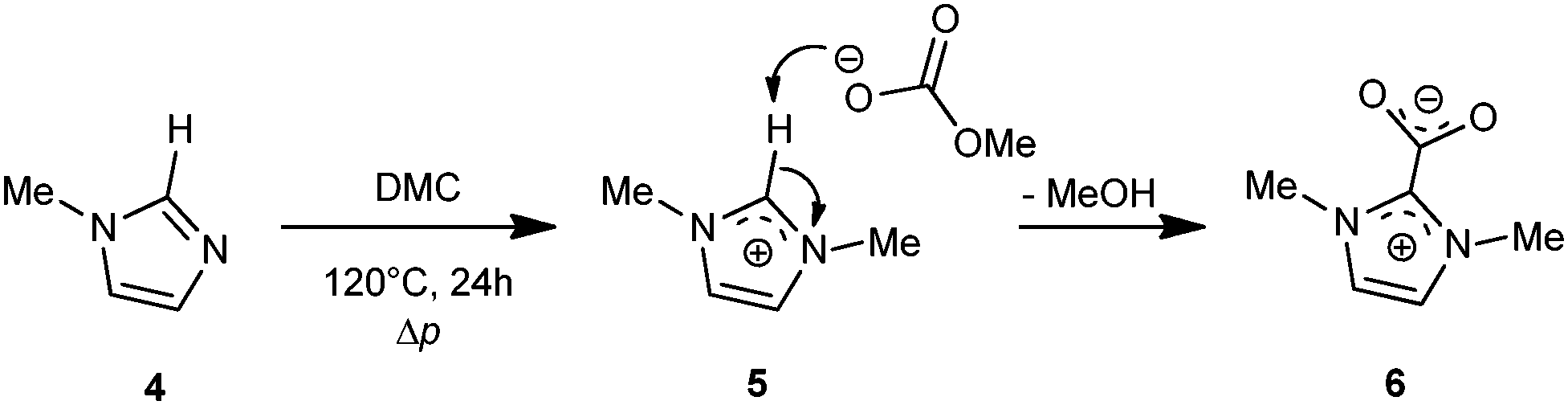 | ||
| Scheme 2 Formation of the zwitterionic carboxylate (6) by reaction of 1-methylimidazole with dimethyl carbonate.5 | ||
In general, imidazolylidene carbenes have very high affinity for CO2 and therefore, 2H-imidazolium ionic liquids combined with basic anions (e.g. [C4mim][OAc]) are excellent for the chemisorption of carbon dioxide.6–9 The zwitterionic carboxylates themselves are also of particular interest in ionic liquid synthesis, as they have already been applied as such in synthesis,10 but also as easy to handle pre-catalysts11 and as CO2 transfer reagents in carboxylation reactions.12–14 Furthermore, the carboxylates have been used as intermediates in the synthesis of hydrogen carbonate ionic liquids.15 Alternatively, these hydrogen carbonate salts can be prepared by oxidation of formate salts over a Pd catalyst.16 A library of hydrogen carbonate ionic liquids is currently commercially available to be applied in Carbonate Based Ionic Liquid Synthesis (CBILS©).
In this research, the application of dimethyl carbonate in the quaternisation of fully substituted imidazoles was investigated. First of all, the reaction conditions, i.e. temperature, reagent stoichiometry and reaction times, were optimised to obtain a complete conversion. Subsequently, the composition of the reaction mixtures as well as the structure of the reaction products was analysed. Some of these reaction products showed remarkable reactivity. Finally, the application of the carbonate ionic liquids in metathesis reactions was examined.
Results and discussion
Quaternisation of 2-alkylimidazoles
High temperatures were required since alkylation demands temperatures above 120 °C (preferably above 160 °C), and alkylated imidazoles are amongst the least reactive organic bases. Therefore, thick wall pressure vials were used, which were submerged in an oil bath. Since the temperature resistance of the screw caps was limited to 180 °C and exceeding this temperature led to the rupture and explosion of the pressure vials, the reaction temperature was set to 170 °C (CAUTION).2-Alkyl-1-ethyl-4,5-dimethyl imidazoles (7a and 7b, Scheme 3) were synthesised using a previously reported procedure.17 They were completely converted into the corresponding ionic liquids upon pressurised heating with 3 equiv. of DMC for 24 hours at 170 °C, although a crude reaction mixture was obtained. Addition of a small amount of MeOH (1.5 equiv.) improved the contact between the neutral imidazole and DMC. Together with the addition of a Lewis acid catalyst, the formation of side products could be suppressed. Thus, K10 Montmorillonite clay was introduced into the reaction mixture as a catalyst without activation. The K10 clay could be successfully recovered by filtration, circumventing contamination of the ionic liquids. After filtration, the catalyst was dried and recycled, and proved equally active as in the first runs.
 | ||
| Scheme 3 Quaternisation of tetraalkylimidazoles. | ||
After the reaction, very dark mixtures were obtained and to reduce this colouration lower temperatures and shorter reaction times were investigated. Evaluation of the reaction temperature during quaternisation of 7b showed that at 150 °C, after 3 and 6 hours, 15 and 22% of the end product were obtained respectively. At a reaction temperature of 170 °C, after 3 and 6 hours, 65 and 75% of end product 8b were formed. Hence, to obtain a complete conversion of both 1-ethyl-2,4,5-trimethylimidazole 7a and 2-isopropyl-4,5-trimethylimidazole 7b, the mixtures were heated for 24 hours at 170 °C. The product could then be isolated by filtration and by washing CH3CN solution with Et2O or by aqueous extraction from EtOAc solution.
In the 13C NMR spectrum of the filtered and mildly evaporated reaction mixture, obsolete MeOH [MeCO3]− (59, 157 ppm) and [OMe]− (59 ppm) were observed. The [MeCO3]− salts (8) were found not to be stable without the presence of methanol for all cations investigated. Upon evaporation, mixtures of methyl carbonate 8 and hydrogen carbonate 9 ([HCO3]− signal at 160 ppm) were formed, and eventually completely transformed into the hydrogen carbonate salt over time. Upon addition of water and subsequent evaporation, the equilibrium shifts toward hydrogen carbonate salt 9 or hydroxide salt 11, since MeOH has a lower boiling point. After evaporation at 100 °C at 0.5 mbar, none of the carboxylate signals were visible in the 13C NMR spectrum. Therefore, it is anticipated that in this case carbon dioxide is lost and results in the formation of methoxide/hydroxide salts.
Quaternisation of 2H-imidazoles
1-Ethyl-2H-4,5-dimethylimidazole (7c) was found to be completely converted to different products upon quaternisation with DMC at 170 °C in a thick wall pressure vial. Here, no reaction was observed at 140 °C, while at 145 °C, 40% was converted after 4 hours and at 170 °C, the conversion was completed after 4 hours. In contrast to the 2-alkyl analogues 7a and 7b, no improvement on the reaction outcome was observed upon addition of a solvent or catalyst.A solid fraction of the residue obtained after solvent evaporation was found to be soluble only in chlorinated solvents, protic solvents, and hot acetonitrile. Recrystallisation in a CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone (3:1) mixture led to consecutive crop yields (up to 4 times, total yield 40–50%). Analysis of the mixture by 1H NMR analysis in CDCl3 revealed the presence of 2 major compounds, hydrogen carbonate salt 9c and carboxylate 12. In CD3OD, methyl carbonate 8c and an amount (<10%) of carboxylate 12 were observed, while in D2O only hydrogen carbonate salt 9c was observed (Scheme 5).
:acetone (3:1) mixture led to consecutive crop yields (up to 4 times, total yield 40–50%). Analysis of the mixture by 1H NMR analysis in CDCl3 revealed the presence of 2 major compounds, hydrogen carbonate salt 9c and carboxylate 12. In CD3OD, methyl carbonate 8c and an amount (<10%) of carboxylate 12 were observed, while in D2O only hydrogen carbonate salt 9c was observed (Scheme 5).
Upon analysis of the crystals by 1H NMR analysis in CDCl3, the ratio of the compounds (9c and 12) was always ca. 3:1, respectively, and was stable over time (72 hours). Upon mild heating of the CDCl3 solution (60 °C, 2 hours), the amount of carboxylate 12 decreased, to eventually disappear. The group of Rogers have observed earlier the presence of two components (the carboxylate and the methyl carbonate salt) in DMSO-d6 NMR spectra of [C4mim][MeCO3], and attributed it to the presence of these two compounds in the reaction mixture.18
Nonetheless, single crystal X-ray diffraction data showed that the obtained crystals consist entirely of the pure 1-ethyl-3,4,5-trimethylimidazolium hydrogen carbonate salt ([C2m3im][HCO3], 9c). The compound crystallised in the centro-symmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit of the structure consists of one [C2m3im]+ cation and one [HCO3]− anion. The [HCO3]− anions form dimers by hydrogen bonding around crystallographic inversion centers (Fig. 2). However, the crystals did not contain the crystal water reported by the group of Rogers.10,15 The hydrogen atom bound to O1 does not show an elongated bond (0.95(3) Å) or a disorder, as often noticed for carbonate dimers.15
. The asymmetric unit of the structure consists of one [C2m3im]+ cation and one [HCO3]− anion. The [HCO3]− anions form dimers by hydrogen bonding around crystallographic inversion centers (Fig. 2). However, the crystals did not contain the crystal water reported by the group of Rogers.10,15 The hydrogen atom bound to O1 does not show an elongated bond (0.95(3) Å) or a disorder, as often noticed for carbonate dimers.15
Given the formation of the pure [C2m3im][HCO3] salt (9c) as a crystalline solid, carboxylate zwitterion 12 observed in CDCl3 solution is formed in situ by thermodynamic stabilisation and water expulsion (Scheme 6). Since no CO2 liberation occurs upon dissolution of the crystalline [C2m3im][HCO3] and most of the carbonate anions can be regenerated from the carboxylate, it seems that imidazolylidene carbene immediately forms a carboxylate. As the chances of dissolved CO2 and a free carbene colliding are very slim, the decomposition of H2CO3 might be promoted by the nucleophilic carbene19 to form the carboxylate at once (negligible lifetime of compound 13). This can be supported by the calculated energies of formation of carboxylates from carbenes and CO2, which show no activation energy barrier at all.20
Although the carboxylate is thermodynamically favoured over the hydrogen carbonate15 and even more stable when the N-substituents are not bulky and allow for a planar p-orbital overlap,20 it was not possible to isolate the carboxylate as a solid. This is attributed to the hygroscopicity of the carboxylate, forming the more energetically favoured hydrogen carbonate salt as it is able to lose the lattice energy upon solidification.
In the case of the 2H-imidazolium salts, a conversion of methyl carbonate 8c into hydrogen carbonate salt 9c was found, analogous to the case of the 2-alkylimidazolium salts (Scheme 4). The solid product 9c is removed from the equilibrium and its formation is dependent on the reaction with air moisture or water present in the reaction or recrystallisation solvents, explaining the successive low crop yields of crystalline [C2m3im][HCO3]. The [OH]− and [OMe]− anions were not experimentally observed in combination with the 2H-imidazolium cation. Firstly, the 2H-imidazolium hydroxides are unstable (non-existing)21 and secondly, alkoxide anions are unlikely to be formed in the presence of imidazolylidene carbene,20 although [C4mim][OMe] was earlier reported by Seddon and Earle.22
 | ||
| Scheme 4 Solvent dependent conversion of the anion in peralkylated imidazolium carbonate ionic liquids. (8–11a: R = Me, 8–11b: R = i-Pr). | ||
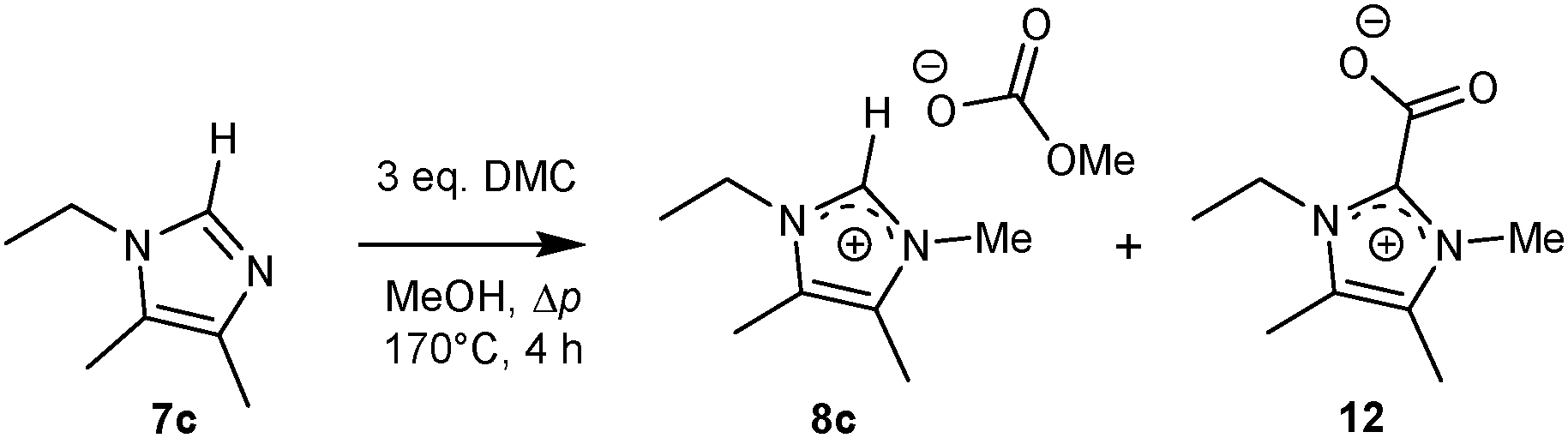 | ||
| Scheme 5 Quaternisation of 1-ethyl-2H-4,5-dimethylimidazole 7c with dimethyl carbonate and formation of the carboxylate zwitterion 12. | ||
 | ||
| Scheme 6 Acid−base equilibrium between hydrogen carbonate imidazolium salt (9c) and the unstable carbene (13) which is trapped as the corresponding carboxylate (12). | ||
Reactivity of the carbonate salts
The 1H NMR analysis of any of the tetra- or penta-alkylimidazolium [HCO3]− salts in deuterated solvents (D2O, CD3OD, CDCl3 or CD3CN) did never reveal the [HCO3]− proton due to hydrogen/deuterium (H/D) exchange with the solvent (Scheme 7). | ||
| Scheme 7 H/D exchange reaction of the hydrogen carbonate anion in deuterated chloroform. | ||
Aromatic 1H NMR signals of the 2H-imidazolium carbonate salts (8c and 9c) (indicating a C2 proton) were only found for the entries containing starting imidazoles (at 7.30 ppm) and for salts to which a Brønsted acid was added (after metathesis).
The spectra of freshly prepared or concentrated carbonate ionic liquids in CDCl3 solutions did reveal the aromatic signal (integrating for less than one proton). Therefore, the absence of the aromatic singlets is attributed to hydrogen/deuterium exchange of C2–H with the deuterated solvent, promoted by the basicity of the anions (Scheme 8). The loss of this singlet is experimentally confirmed in CDCl3 (pKa: ±15),23 but also in CD3CN (pKa: ±31),24 although they have been reported in the literature at 9.6 ppm in DMSO-d6 (pKa: ±35).15,24 Remarkably, the signals of the imidazolium 2-methyl group of compounds 8a and 9a were also often not visible or reduced in 1H NMR spectra and diffuse in 13C NMR spectra. In the 1-ethyl-2-isopropyl-3,4,5-trimethylimidazolium cation, none of the hydrogens were exchanged.
 | ||
| Scheme 8 Equilibrium of the carbonate ionic liquid with carbene and fast H/D-exchange in deuterated solvents. | ||
In the case of the solid 2H-imidazolium hydrogen carbonate salt (9c), mild heating and/or extensive evaporation of the CDCl3 solution lead to the reduction of the carboxylate and [HCO3]− signals in the 13C NMR spectrum. The signals eventually disappear completely, indicating the loss of CO2. After evaporation of the CDCl3 mixture, the residue was again a solid and could be recrystallised. The crystals obtained were analysed by single crystal X-ray diffraction and are found to consist of the [C2m3im][Cl] salt.
Compound 18c crystallised in the centro-symmetric space group P21/c. The asymmetric unit of the structure consists of one [C2m3im]+ cation and one [Cl]− anion (Fig. 1). The ethyl group is found to be almost planar with the imidazolium ring (C1–N1–C7–C8 torsion angle of −8.5(2)°).
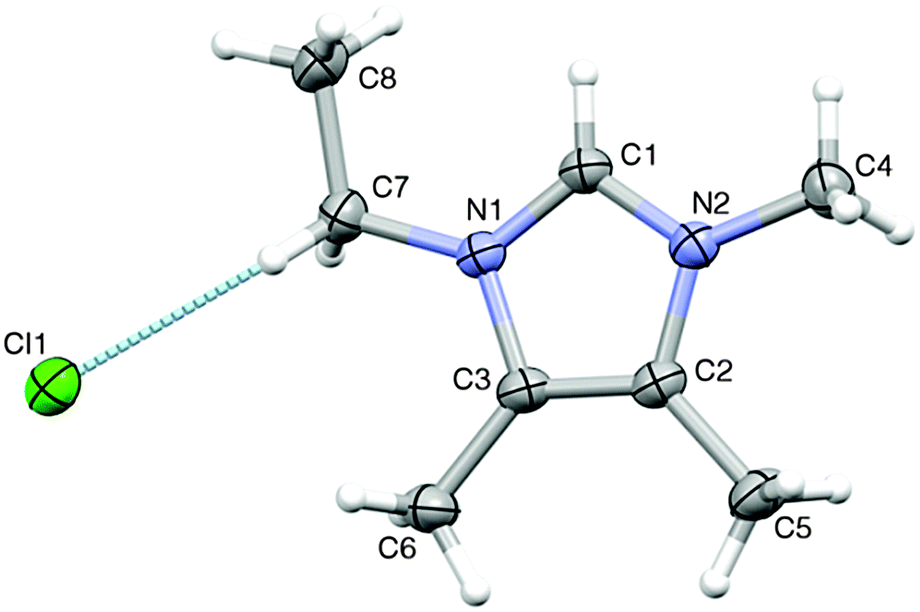 | ||
| Fig. 1 Asymmetric unit in the crystal structure of 18c, showing thermal displacement ellipsoids at the 30% probability level and the atom labelling scheme. The hydrogen bond between the [C2m3im]+ cation and the [Cl]− anion is indicated. | ||
 | ||
| Fig. 2 Hydrogen-bonded [C2m3im][HCO3] dimers in the crystal structure of 9c, showing thermal displacement ellipsoids at the 30% probability level and the atom labelling scheme of the asymmetric unit. Hydrogen bonds are indicated. | ||
The formation of the chloride salt is explained by the alkaline reaction of the salts on (deutero)chloroform residues originating from NMR sampling or recrystallisation (Scheme 9). Upon abstraction of the proton of CHCl3, one chloride is expelled to form dichlorocarbene, which forms consecutively the unstable dichloromethanol and formyl chloride. The latter decomposes rapidly into carbon monoxide (CO) and hydrogen chloride. Overall, three [OH]− anions are converted to three [Cl]− anions, with formation of water and carbon monoxide.25
 | ||
| Scheme 9 Formation of the 1-ethyl-2H-3,4,5-trimethylimidazolium chloride salt (18c). | ||
In contrast to the H/D exchange, the formation of the [Cl]− salt 18c is not readily established, i.e.13C NMR spectra of carbonate salts can be recorded in CDCl3. In contrast, they demand evaporation and heating. The proton transfer required during carboxylate formation and H/D exchange in CDCl3 proves that both the imidazolium cation and CDCl3 can be deprotonated.
The 2-methylimidazolium and 2-isopropylimidazolium chloride salts (18a and 18b) could not be retrieved in the same way. Although for the [C2C1m3im]+ salt, some decomposition of the cation was observed, the [HCO3]− signal remained visible in 13C NMR spectra. It seems that imidazolylidene carbene is necessary to accomplish degradation of chloroform. Therefore it is proposed that during the heating process, carbonate decomposition is aided by carboxylate formation, eventually leading to the basic carbene, prior to chloroform decomposition.
The chloride 18c and carbonate 8c salts could be distinguished by their large difference in melting points and by infrared absorption spectra. Furthermore, once the [Cl]− salt is formed, the aromatic singlet could appear in the 1H NMR spectrum after shaking the CDCl3 solution with a drop of water (Scheme 8). This is not the case for the carbonate salts, as this would lead to CHCl3 formation.
N-Protonated imidazoles
When the reaction of 1-ethyl-2-isopropyl-4,5-dimethylimidazole (7b) with dimethyl carbonate was stopped after 3 or 6 hours, 15% to 75% of the end product was observed in the 1H NMR spectra (vide supra). These spectra also revealed the presence of protonated imidazoles (19), which are proposed to be formed by neutralisation of MeOCO2H, originating from the hydrolysis of DMC (Scheme 10). Formation of MeOCO2H during work-up could be excluded since the protonated imidazoles were not always found in experiments where unreacted imidazole was still present. This indicates that (i) elevated temperatures are needed for the substitution reaction of DMC by water, (ii) a significant amount of DMC is consumed by water present in the mixture and (iii) protonated imidazoles can react further by the presence of the basic anions in the reaction mixture.26,27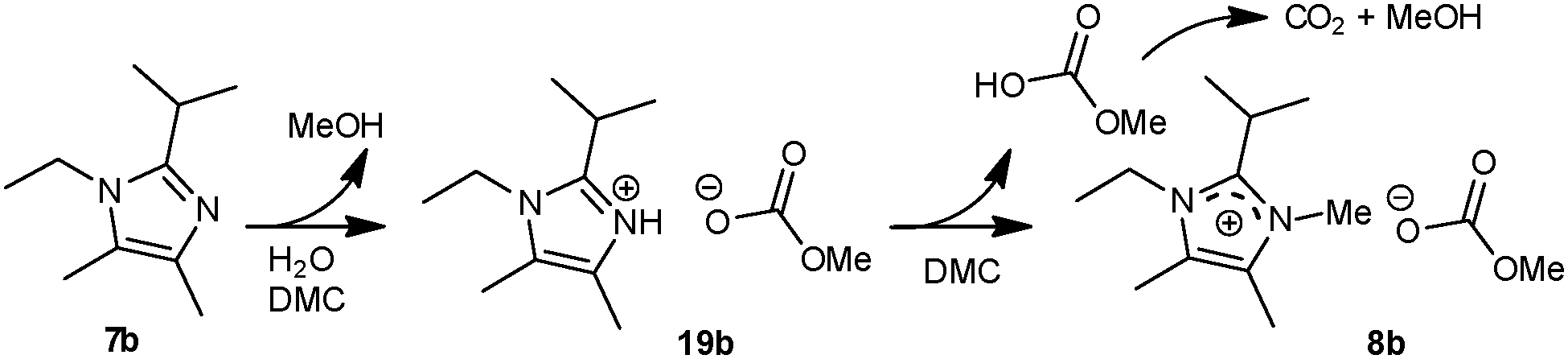 | ||
| Scheme 10 Formation of imidazolium hydrogen methyl carbonate 19bvia hydrolysis of DMC. The protonated imidazole can be methylated to form the N-methyl imidazolium methyl carbonate salt (8b). | ||
The proposed hypothesis was verified with imidazole 7a by reacting 1-ethyl-2,4,5-trimethyl imidazolium hydrochloride salt (20) with dimethyl carbonate (Scheme 11). Although analogous protonated imidazolium salts could not be quaternised with methyl iodide, these salts could be successfully quaternised using dimethyl carbonate. Here, this is probably due to the presence of catalytic amounts of water, since 19a can be formed by an anion exchange between 20 and 21. The reaction to form 18a proceeded very clean, quantitatively and was completed in 3 hours, giving an opaque powder, whose single crystal X-ray diffraction data could be recorded. The compound crystallised in the centro-symmetric space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . The asymmetric unit of the structure consists of one [C2C1m3im]+ cation and one [Cl]− anion (Fig. 3). The fast formation of this chloride 18a suggests that other alkylating species might have been formed (e.g. MeCl), or that a strong (auto)catalytic effect of the imidazolium species is present. This catalytic effect of imidazolium salts was earlier demonstrated in the alkylation with dialkylcarbonates of ammonium halide and nitrate salts.28
. The asymmetric unit of the structure consists of one [C2C1m3im]+ cation and one [Cl]− anion (Fig. 3). The fast formation of this chloride 18a suggests that other alkylating species might have been formed (e.g. MeCl), or that a strong (auto)catalytic effect of the imidazolium species is present. This catalytic effect of imidazolium salts was earlier demonstrated in the alkylation with dialkylcarbonates of ammonium halide and nitrate salts.28
 | ||
| Scheme 11 Quaternisation and in situ metathesis of 20via neutralisation by the methyl carbonate anion. | ||
 | ||
| Fig. 3 Asymmetric unit in the crystal structure of 18a, showing thermal displacement ellipsoids at the 50% probability level and the atom labelling scheme. Hydrogen bonds between the [C2C1m3im]+ cation and the [Cl]− anion are indicated. | ||
In contrast to the previous structure of 18c, the ethyl group is oriented almost perpendicular to the imidazolium ring (C1–N1–C8–C9 torsion angle of 95.8(2)°), due to steric hindrance by the presence of the extra methyl group.
Upon opening of the sealed pressure vial, a substantial amount of CO2 evolved, independent of the cation type. Since the carbonate anion was found in substantial amounts, CO2 does not originate from anion decomposition into CO2 and [OMe]−, as stated by Tundo.29 Later, Holbrey et al. proposed that the anion decomposes upon protonation only,5 which can be confirmed by the present work, i.e. carbonate anions do not degrade during the reaction or over time.
Metathesis of CILs
After crystallisation and filtration of 1-ethyl-2H-3,4,5-trimethylimidazolium hydrogen carbonate ([C2m3im][HCO3]) salts, they could be applied successfully in metathesis reactions with the Brønsted acid aqueous HNTf2 (80%) or the ammonium dicyanamide salt. Hereby, CO2 gas evolved and the temperature of the mixtures slightly increased. To ensure complete reaction, the carbonate salts were stirred with a small excess of the metathesis agent in distilled water and heated to 50 °C overnight. 1H NMR analysis of these salts showed that the imidazolium ring proton signals were completely regenerated.The most straightforward metathesis reaction is with aqueous HNTf2. The use of HNTf2, which is cheaper with regard to its lithium salt, is also more environmentally benign as carcinogenic quaternisation agents can be substituted by dimethyl carbonate and no stoichiometric amounts of lithium halide salts are formed as by-products. Here, a small excess of the super acid leads to rapid and complete conversion, while the remainder of the acid can be evaporated.
Dicyanamide salts could not be obtained via the corresponding Brønsted acid. Therefore, ammonium dicyanamide was synthesised by percolating a NaN(CN)2 solution through an acidic ion exchange resin with high sodium affinity, which was neutralised with an ammonium solution prior to the exchange reaction (Scheme 12).30 After evaporation of the collected aqueous solution, NH4N(CN)2 was obtained as a white powder in good purity (as analysed by elemental analysis). After metathesis in aqueous medium, the excess ammonium dicyanamide could be precipitated from a solution of the evaporated residue at −18 °C.
 | ||
| Scheme 12 Metathesis with ammonia to form ammonium dicyanamide (NH4N(CN)2) via an ion exchange resin. | ||
Conclusion
Completely substituted imidazoles can successfully be methylated in a green way using dimethyl carbonate. K10 Montmorillonite is found to be an excellent catalyst, which allows complete conversion without formation of side products. Still, very high temperature is needed, which leads to very intense colouration. Purification is straightforward in the case of the 2H-imidazolium salts, this product is solid and can be recrystallised to obtain a colourless compound. In the case of liquid 2-substituted imidazolium salts, the product can be washed with Et2O in CH3CN or with EtOAc in demineralised water.The carbonate anions are stable and do not readily decompose under ambient conditions. The formation of anions by loss of carbon dioxide was observed (for 2-alkyl-imidazolium salts), unless a carboxylate can be formed (for [C2m3im][HCO3]). This carboxylate is formed from a carbene, which reacts immediately with the mutually formed H2CO3. The carbene derived from [C2m3im][HCO3] has a very high affinity for CO2 and the resulting carboxylate is thermodynamically favoured, although unstable in contact with air or nucleophilic and protic solvents. The presence of carboxylate in the reaction mixture was confirmed by 1H NMR analysis in CD3OD, while the amount is strongly dependent on the solvent. Most likely, the carbonate salts are stabilised by H-bonding. Thus, the formation of carboxylate is more pronounced in chloroform and is anticipated to be the crucial step in chloroform decomposition to form the chloride anion. The formation of chloride salts with chloroform is only found for [C2m3im][HCO3]. Nonetheless, it was found that the 2-alkyl-imidazolium hydrogen salts can be methylated, leading to methylated salts combined with the right anion at once.
The carbonate salts are reactive species, which will transform when dissolved in different solvents or left open in ambient air. Thus, [imidazolium][MeCO3] forms [imidazolium][HCO3] when comes in contact with air moisture or water present in the solvents. Since this conversion is possible with all of the imidazolium cation types, the carboxylate is not necessary. The conversion of [MeCO3]− to the [HCO3]− anion is observed in water, while in the presence of methanol, the methyl carbonate anion is stable. In the case of the extracted 2-alkyl-imidazolium salts, [OH]− of [OMe]− can also be present, which makes NMR based quantification difficult during metathesis.
Experimental section
Procedure for the synthesis of ammonium dicyanamide (23)
A glass column was packed with 2.74 g of Amberlite IR120 in distilled water (column height: 3.5 cm). The resin was slowly percolated with 1 M NH4Cl solution (200 mL) until the effluent stream pH, monitored by pH indicator paper, had increased to that of the influent. Subsequently, the column was rinsed with aq. dest. (75 mL) until the effluent samples did no longer show precipitation upon addition of AgNO3. Sodium dicyanamide (2 mmol, 178 mg) was dissolved in aq. dest. (40 mL) and MeOH (20 mL), and this solution was percolated very slowly over the column. The percolated solution and the rinsing water (aq. dest., 20 mL) were subjected to lyophilisation to yield ammonium dicyanamide. Elemental analysis calcd (%) for C2H4N4: C 28.57, H 4.80, N 66.64; found: C 27.83, H 4.49, N 64.82; yield: 95%; white powder. At elevated temperatures, decomposition rather than fusion occurs.Procedure for the synthesis of 1-ethyl-2-isopropyl-3,4,5-trimethylimidazolium hydrogen carbonate [C2Ci3m3im][HCO3] (9a)
A 14 mL flame-dried glass pressure vial equipped with a PTFE stirring bar was filled with MeOH (5 mmol, 160 mg), dimethyl carbonate (30 mmol, 2.7 g), 1-ethyl-2-isopropyl-4,5-dimethylimidazole (10 mmol, 1.67 g), and Montmorillonite K10 clay (0.83 g). The pressure vial was flushed with nitrogen, closed and submerged in a preheated oil bath at 170 °C. After 24 hours, the vial was allowed to cool and opened, the mixture was diluted in methanol and filtered over a filter paper (Euro Scientific, 90 mm) on a sintered glass filter. The solvents were removed by rotary evaporation at room temperature. The resulting oil was added to 10 mL of demineralised water and stirred at 50 °C. After 3 hours, the mixture was cooled and water and methanol were removed via lyophilisation or rotary evaporation. The different spectral data sets of the 1-ethyl-2-isopropyl-3,4,5-trimethylimidazolium carbonate salts were obtained by dissolving these crystals in different solvents. As the liquid analogues are extremely hygroscopic, density and viscosity were not measured.1-Ethyl-3,4,5-trimethylimidazolium hydrogen carbonate [C2m3im][HCO3] (9c + 12)
1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = (maj.): δ = 1.49 (t, 3J(H,H) = 7.2 Hz, 3H; CH2CH3), 2.23 (s, 3H; CH3C4/5), 2.24 (s, 3H; CH3C4/5), 3.87 (s, 3H; NCH3), 4.19 (t, 3J(H,H) = 7.2 Hz, 2H; CH2CH3), 10.07 ppm (s; C2H), (min.): δ = 1.42 (t, 3J(H,H) = 7.1 Hz, 3H; CH2CH3), 2.24 (s, 3H; CH3C4/5), 2.26 (s, 3H; CH3C4/5), 3.97 (s, 3H; NCH3), 4.51 ppm (q, 3J(H,H) = 7.1 Hz, 2H; CH2CH3); 13C NMR (75 MHz, CDCl3, 25 °C, TMS): (maj.): δ = 8.29 (CH3C4/5), 8.38 (CH3C4/5), 15.31 (CH2CH3), 33.53 (NCH3), 42.22 (CH2), 125.90 (C4/5), 127.03 (C4/5), 136.30–136.59 (m, C2), 160.86 ppm ([HCO3]−), (min.): δ = 8.52 (CH3C4/5), 8.73 (CH3C4/5), 15.76 (CH2CH3), 33.24 (NCH3), 41.65 (NCH2), 124.44 (C4/5), 125.79 (C4/5), 141.58 (C2), 156.04 ppm (CO2); IR (ATR): δ = 625, 985, 1203, 1342, 1377, 1570, 1617, 2341, 2360, 3386 (broad) cm−1; MW (g mol−1): 200.24; yield: 52%, colourless crystals, Tm (°): 41.1-Ethyl-3,4,5-trimethylimidazolium hydrogen carbonate [C2m3im][HCO3] (9c)
1H NMR (300 MHz, D2O, 25 °C, ACN): δ = 1.43 (t, 3J(H,H) = 7.3 Hz, 3H; CH2CH3), 2.21 (s, 3H; CH3C4/5), 2.23 (s, 3H; CH3C4/5), 3.71 (s, 3H; NCH3), 4.08 ppm (q, 3J(H,H) = 7.3 Hz, 2H; NCH2); 13C NMR (75 MHz, CDCl3, 25 °C, TMS): δ = 7.78 (CH3C4/5), 7.92 (CH3C4/5), 14.65 (CH2CH3), 33.46 (NCH3), 42.41 (NCH2), 127.16 (C4/5), 128.06 (C4/5), 132.92 (C2), 161.61 ppm (HCO3).1-Ethyl-3,4,5-trimethylimidazolium methyl carbonate [C2m3im][CH3CO3] (8c)
1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 1.52 (t, 3J(H,H) = 7.4 Hz, 3H; CH2CH3), 2.24 (s, 3H; CH3C4/5), 2.26 (s, 3H; CH3C4/5), 3.55 (s, 3H; CH3CO3), 3.88 (s, 3H; NCH3), 4.20 (q, 3J(H,H) = 7.4 Hz, 2H; CH2CH3), 10.43 ppm (s; C2H); 13C NMR (75 MHz, CDCl3, 25 °C, TMS): δ = 8.23 (CH3C4/5), 15.19 (CH2CH3), 33.44 (NCH3), 42.19 (NCH2), 52.28 (CH3O), 126.05 (C4/5), 127.11 (C4/5), 136.28 (C2), 158.76 ppm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O).
1-Ethyl-2,3,4,5-tetramethylimidazolium hydrogen carbonate [C2C1m3im][HCO3] (9a)
1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 1.31 (t, 3J(H,H) = 7.3 Hz, 3H; CH2CH3), 2.23 (s, 3H; CH3C4/5), 2.24 (s, 3H; CH3C4/5), 2.70 (s, 3H, C2CH3), 3.71 (s, 3H; NCH3), 4.15 ppm (q, 3J(H,H) = 7.3 Hz, 2H; CH2CH3); 13C NMR (75 MHz, CDCl3, 25 °C, TMS): δ = 8.50 (CH3C4/5), 8.75 (CH3C4/5), 10.23 (C2CH3), 14.93 (CH2CH3), 32.15 (NCH3), 40.55 (NCH2), 124.69 (C4/5), 126.08 (C4/5), 142.07 (C2), 160.27 ppm (HCO3); IR (ATR): δ = 740, 1372, 1626, 2928, 3390 cm−1; MW (g mol−1): 214.26; yield: 47%, transparent crystals, Tm (°): 37.1-Ethyl-2-isopropyl-3,4,5-trimethylimidazolium hydrogen carbonate [C2Ci3m3im][HCO3] (9b)
1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 1.36 (t, 3J(H,H) = 7.4 Hz, 3H; CH2CH3), 1.51 (t, 3J(H,H) = 7.1 Hz, 6H; CH(CH3)2), 2.27 (s, 6H; CH3C4/5), 3.58–3.72 (m, 1H; CH(CH3)2), 3.83 (s, 3H; NCH3), 4.24 ppm (q, 3J(H,H) = 7.4 Hz, 2H; CH2CH3); 13C NMR (75 MHz, CDCl3, 25 °C, TMS): δ = 8.69 (CH3C4/5), 8.86 (CH3C4/5), 15.73 (CH2CH3), 19.30 (CH(CH3)2), 25.16 (CH(CH3)2), 32.98 (NCH3), 40.87 (NCH2), 125.15 (C4/5), 127.15 (C4/5), 147.43 (C2), 160.41 ppm (HCO3); IR (ATR): δ = 1334, 1385, 1522, 1649, 3388 cm−1; MW (g mol−1): 242.32; yield: 89%, brown oil.1-Ethyl-3,4,5-trimethylimidazolium chloride [C2m3im][Cl] (18c)
1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 1.55 (t, J3(H,H) = 7.3 Hz, 3H; CH2CH3), 2.26 (s, 3H; CH3C4/5), 2.27 (s, 3H; CH3C4/5), 3.95 (s, 3H; NCH3), 4.24 (q, J3(H,H) = 7.3 Hz, 2H; CH2CH3) 10.74 ppm (s, 1H; C2H); 13C NMR (75 MHz, CDCl3, 25 °C, TMS): δ = 8.47 (CH3C4/5), 8.55 (CH3C4/5), 15.42 (CH2CH3), 33.85 (NCH3), 42.45 (NCH2), 125.98 (C4/5), 127.08 (C4/5), 136.27 ppm (C2); IR (ATR): δ = 1200, 1252, 1457, 1572, 1637, 2342, 2362, 2953, 3390 cm−1; MW (g mol−1): 174.67; elemental analysis calcd (%) for C8H15ClN2: C 55.01, H 8.66, N 16.04; found: C 54.32, H 8.75, N 15.79; yield: 81%, opaque powder, Tm (°): 54.1-Ethyl-2,3,4,5-tetramethylimidazolium chloride [C2C1m3im][Cl] (18a)
1H NMR (300 MHz, CDCl3, 25 °C, TMS): δ = 1.40 (d, 3J(H,H) = 7.4 Hz, 3H; CH2CH3), 2.26 (s, 6H; CH3C4/5), 2.94 (s, 3H; C2CH3), 3.87 (s, 3H; NCH3), 4.23 ppm (q, J3(H,H) = 7.4 Hz, 2H; CH2CH3); 13C NMR (75 MHz, CDCl3, 25 °C, TMS): δ = 8.70 (CH3C4/5), 8.99 (CH3C4/5), 11.18 (C2CH3), 15.12 (CH2CH3), 32.89 (NCH2), 41.07 (NCH3), 124.69 (C4/5), 126.16 (C4/5), 142.45 ppm (C2); IR (ATR): δ = 1091, 1346, 1446, 1649, 2978, 3393 cm−1; MW (g mol−1): 188.70; yield: 99%, white powder, Tm (°): 59.Procedure for the synthesis of 1-ethyl-2-isopropyl-3,4,5-trimethylimidazolium bis(trifluorosulfonyl)amide [C2Ci3m3im] [NTf2]
An excess (1.1 equiv., for full conversion) of hydrogen bis(trifluorosulfonyl)imide (HNTf2) was added to a vigorously stirred aqueous solution (cont. 20% CH3CN) of freshly prepared [C2Ci3m3im][HCO3], taking into account the weight of completely converted imidazole. The acid was added dropwise to prevent acid evaporation induced by the exothermic reaction. During addition, gas (CO2) evolution was visible. The mixture was allowed to stir for 30 minutes at room temperature after which the solvents were evaporated. Slow crystallisation of the liquor occurred in a sealed flask at room temperature. Spectral data for the compound were in agreement with the data found in the experimental part of ref. 17.Procedure for the synthesis of 1-ethyl-2-isopropyl-3,4,5-trimethylimidazolium dicyanamide [C2Ci3m3im][ N(CN)2]
To an aqueous solution (cont. 20% CH3CN) of freshly prepared [C2Ci3m3im][HCO3] (taking into account the weight of completely converted imidazole) was added an excess (1.1 equiv., for full conversion) of NH4N(CN)2. The reaction was stirred at room temperature for 24 h. After metathesis, the mixture was evaporated, consecutively dissolved in methanol and again evaporated. The excess ammonium dicyanamide could be precipitated from a CHCl3 solution of the evaporated residue at −18 °C. Spectral data for the compound were in agreement with the data found in the experimental part of ref. 17.Single crystal X-ray diffraction
For the structures of compounds 9c, 18c and 18a, X-ray intensity data were collected on a Agilent Supernova Dual Source (Cu at zero) diffractometer equipped with an Atlas CCD detector using CuKα radiation (λ = 1.54178 Å) and ω scans. The images were interpreted and integrated using the program CrysAlisPro (CrysAlisPro, Agilent Technologies, version 1.171.36.28). Using Olex2,31 the structure was solved by direct methods using the ShelXS structure solution program and refined by full-matrix least-squares on F2 using the ShelXL program package.32 Non-hydrogen atoms were anisotropically refined and the hydrogen atoms in the riding mode and isotropic temperature factors were fixed at 1.2 times U (eq.) of the parent atoms (1.5 times for methyl groups and the hydroxyl group), with the C/O–H distances free to refine. All hydrogen atoms could be unambiguously located from a difference Fourier electron density map. CCDC 967637–967639.Crystal data for compound 9
C9H16N2O3, M = 200.24, triclinic, space group P (no. 2), a = 6.9516(3) Å, b = 8.1294(3) Å, c = 9.6234(4) Å, α = 74.586(4)°, β = 80.157(4)°, γ = 83.657(3)°, V = 515.35(4) Å3, Z = 2, T = 293(2) K, ρcalc = 1.290 g cm−3, μ(Cu-Kα) = 0.806 mm−1, F(000) = 216, 8198 reflections measured, 2005 unique (Rint = 0.0187) which were used in all calculations. The final R1 was 0.0387 (I > 2σ(I)) and wR2 was 0.1125 (all data).
Crystal data for compound 18c
C8H15ClN2, M = 174.67, monoclinic, space group P21/c (no. 14), a = 6.77997(17) Å, b = 14.4971(4) Å, c = 9.8785(3) Å, β = 100.572(3)°, V = 954.48(5) Å3, Z = 4, T = 293(2) K, ρcalc = 1.215 g cm−3, μ(Cu-Kα) = 3.066 mm−1, F(000) = 376, 4198 reflections measured, 1807 unique (Rint = 0.0261) which were used in all calculations. The final R1 was 0.0366 (I > 2σ(I)) and wR2 was 0.1064 (all data).Crystal data for compound 18a
C9H17ClN2, M = 188.70, trigonal, space group R (no. 148), a = b = 19.8873(6) Å, c = 14.4048(4) Å, V = 4933.9(4) Å3, Z = 18, T = 100(2) K, ρcalc = 1.143 g cm−3, μ(Cu-Kα) = 2.702 mm−1, F(000) = 1836, 10529 reflections measured, 2230 unique (Rint = 0.0529) which were used in all calculations. The final R1 was 0.0399 (I > 2σ(I)) and wR2 was 0.0981 (all data). Disordered solvent peaks (probably from acetone or diethyl ether) were omitted from the refinement model using the Squeeze procedure in Platon.33
Acknowledgements
The authors gratefully acknowledge the financial support from the Agentschap voor Innovatie door Wetenschap en Technologie (IWT) (SBO project “Materials processing in ionic liquids”, MAPIL) and KVH thanks the Hercules Foundation (project AUGE/11/029 “3D-SPACE: 3D Structural Platform Aiming for Chemical Excellence”) for funding.Notes and references
- B. Albert and M. Jansen, ZAAC, 1995, 621, 1735 CrossRef CAS.
- S. Mori, K. Ida and M. Ue, US Pat., 4,892,944, 1990 Search PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS PubMed.
- U. Romano, F. Rivetti and N. Di Muzio, US Pat., 4,318,862, EniChem Anic S.p.A., Italy, 1978 Search PubMed.
- J. D. Holbrey, W. M. Reichert, I. Tkatchenko, E. Bouajila, O. Walter, I. Tommasi and R. D. Rogers, Chem. Commun., 2003, 28 RSC.
- M. Besnard, M. I. Cabaco, F. V. Chavez, N. Pinaud, P. J. Sebastiao, J. A. P. Coutinho and Y. Danten, Chem. Commun., 2012, 48, 1245 RSC.
- G. Gurau, H. Rodriguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Angew. Chem., Int. Ed., 2011, 50, 12024 CrossRef CAS PubMed.
- Y. Zhang, Z. Wu, S. Chen, P. Yu and Y. Luo, Ind. Eng. Chem. Res., 2013, 52, 6069 CrossRef CAS.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112 RSC.
- M. Smiglak, J. D. Holbrey, S. T. Griffin, W. M. Reichert, R. P. Swatloski, A. R. Katritzky, H. F. Yang, D. Z. Zhang, K. Kirichenko and R. D. Rogers, Green Chem., 2007, 9, 90 RSC.
- M. Fèvre, P. Coupillaud, J. Vignolle and D. Taton, J. Org. Chem., 2012, 77, 10135 CrossRef PubMed.
- I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2006, 47, 6453 CrossRef CAS PubMed.
- P. U. Naik, L. Petitjean, K. Refes, M. Picquet and L. Plasseraud, Adv. Synth. Catal., 2009, 351, 1753 CrossRef CAS.
- I. Tommasi and F. Sorrentino, Tetrahedron Lett., 2005, 46, 2141 CrossRef CAS PubMed.
- N. J. Bridges, C. C. Hines, M. Smiglak and R. D. Rogers, Chem. – Eur. J., 2007, 13, 5207 CrossRef CAS PubMed.
- G. Degen and K. Ebel, WO Pat., 2006/027069, BASF, 2006 Search PubMed.
- C. Maton, N. De Vos, B. I. Roman, E. Vanecht, N. R. Brooks, K. Binnemans, S. Schaltin, J. Fransaer and C. V. Stevens, ChemPhysChem, 2012, 13, 3146 CrossRef CAS PubMed.
- J. D. Holbrey, R. D. Rogers, S. S. Shukla and C. D. Wilfred, Green Chem., 2010, 12, 407 RSC.
- J.-C. Hsu, Y.-H. Yen and Y.-H. Chu, Tetrahedron Lett., 2004, 45, 4673 CrossRef CAS PubMed.
- R. Lo and B. Ganguly, New J. Chem., 2012, 36, 2549 RSC.
- A. K. L. Yuen, A. F. Masters and T. Maschmeyer, Catal. Today, 2013, 200, 9 CrossRef CAS PubMed.
- M. J. Earle and K. R. Seddon, WO Pat., 2001/077081, 2001 Search PubMed.
- S. Pirketta, Acta Chem. Scand., Ser. A, 1986, 40, 207 Search PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- Y. A. Borisov, E. E. Arcia, S. L. Mielke, B. C. Garrett and T. H. Dunning, J. Phys. Chem. A, 2001, 105, 7724 CrossRef CAS.
- W. J. Xiao, X. X. Wang, Q. Chen, T. H. Wu, Y. Wu, L. Z. Dai and C. S. Song, Chem. Lett., 2010, 39, 1112 CrossRef CAS.
- Z. Q. Zheng, T. H. Wu, R. W. Zheng, Y. Wu and X. P. Zhou, Catal. Commun., 2007, 8, 39 CrossRef CAS PubMed.
- Z. Q. Zheng, J. Wang, T. H. Wu and X. P. Zhou, Adv. Synth. Catal., 2007, 349, 1095 CrossRef CAS.
- P. Tundo and M. Selva, Green Chem., 2005, 7, 464 RSC.
- B. V. Lotsch, J. Senker, W. Kockelmann and W. Schnick, J. Solid State Chem., 2003, 176, 180 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and crystallographic images. CCDC 967637–967639. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01301h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |