
Solvent-free, [Et3NH][HSO4] catalyzed facile synthesis of hydrazone derivatives†
Mehtab
Parveen
*a,
Shaista
Azaz
a,
Ali Mohammed
Malla
a,
Faheem
Ahmad
a,
Pedro Sidonio
Pereira da Silva
b and
Manuela
Ramos Silva
b
aDivision of Organic Synthesis, Aligarh Muslim University, Aligarh, 202002, India. E-mail: mehtab.organic2009@gmail.com; Tel: +91-9897179498
bCEMDRX Physics Department, University of Coimbra, 3004-516 Coimbra, Portugal
First published on 5th November 2014
Abstract
In the present study, a library of hydrazone analogues 2(a–j) and 4(a–e) were synthesized, which were typically accessed via a solvent-free facile nucleophilic addition between hydrazine hydrate and appropriately substituted aromatic aldehydes 1(a–j) and 3-formylchromones 3(a–e). The molecular structure of compound (2f) was well supported by single crystal X-ray crystallographic analysis and also verified by DFT calculations. This new synthetic, eco-friendly, sustainable protocol resulted in a remarkable improvement in the synthetic efficiency (90–98% yield), high purity, using [Et3NH][HSO4] as a catalyst and an environmentally benign solvent eliminating the need for a volatile organic solvent and additional catalyst. This ionic liquid is air and water stable and easy to prepare from cheap amine and acid. The present methodology is a green protocol offering several advantages such as, excellent yield of products, minimizing production of chemical wastes, shorter reaction profile, mild reaction conditions, simple operational procedure, easy preparation of catalyst and its recyclability up to five cycles without any appreciable loss in catalytic activity. The optimization conditions carried out in the present study revealed that 20 mol% of ionic liquid catalyst under solvent-free condition at 120 °C are the best conditions for the synthesis of hydrazone derivatives in excellent yields.
1. Introduction
At present time, ionic liquids have attracted substantial research interest in the context of green synthesis due to their adjustable physical and chemical properties.1 They have been introduced as an alternative green reaction media due to their unique features such as the advantages of optimization of compound characteristics through a broad selection of anion and cation combinations, non-volatility, low flammability, low vapor pressure, high thermal and chemical stability, good solvating ability, ease of recyclability and controlled miscibility.2–11 In the last decade, ionic liquids have been used as environmentally benign solvents or catalysts,12 fuel cells,13 thermal fluids,14 sensors,15 batteries,16 capacitors,17 plasticizers,18 lubricants19 and extractants.20,21 The first involvement of ionic liquids in 2003 for first industrial process by BASF (BASIL10 process) opened new avenues for the application of ionic liquids in new chemical processes.22Although ionic liquids have been used as alternative reaction media and catalyst,12 their high cost, difficulty in separation and toxicity confine their applicability. Therefore, there is a need for exploring the cheap and easily available ionic liquids in organic synthesis. Noda et al.23 reported the preparation and application of the Bronsted acid–base ionic liquids from imidazole and bis(trifluoromethanesulfonyl)amide, similarly Gao et al.24 prepared new ionic liquids by neutralization of 1,1,3,3-tetramethylguanidine with different acids. Bronsted acid ionic liquids (BAILs) are of special significance as they possess simultaneously the proton acidity and the characteristic properties of an ionic liquid.25 These ionic liquids have been proved to be very efficient catalysts as well as solvents for many organic transformations.26–37
It is pertinent to mention that C–N bond is of significant importance as it opens new avenues for the introduction of nitrogen in organic molecules. Despite significant advancement in this field, the construction of the C–N bond is still a major challenge for organic chemists, due to the involvement of harsh reaction conditions or the use of expensive catalysts.38,39 In this regard, hydrazone and their derivatives constitute an important class of compounds in organic chemistry due to their promising biological activities.40 It has been documented in the literature that hydrazone derivatives exhibit a wide spectrum of biological properties such as anti-inflammatory,41 analgesic,42 antipyretic43 as well as chelating properties towards various metal ions.44 Savini et al. have reported anticancer, anti-HIV, and antimicrobial activity45 of heterocyclic hydrazones, furthermore the latter have also been found useful as anti-malaria drugs.46 The synthetic efforts for this class of compounds are very well studied and generally entail the reaction of carbonyl compounds with hydrazine hydrate in organic solvents.47
The catalyst promoted organic synthesis has become one of the hot areas in organic synthesis in the last few decades. At present, a wide range of methods for synthesizing hydrazones in the presence of catalysts are available viz. ZnCl2,48 TiCl2,49 K-10 clay,50 MgSO4-PPTL,51 Mg (ClO4)252 and also SiO2–NaHSO4,53 PSSA.54 However, most of these methods suffer from certain shortcomings including prolonged reaction times, unsatisfactory yields, high temperature, use of organic solvents and expensive non-reusable catalysts.38,39
Thus, it is a challenge to develop alternative greener, milder, cheap and efficient methodologies for the construction of C–N bonds. In this regard, ionic liquids (ILs) offer promising efficiency over other catalyzed reactions.55 Few ionic liquids have been reported for the synthesis of hydrazone derivatives such as [Bmim] BF4, [Bmim] PF6, [Bmim] CH3COO, however these imidazolium-based ionic liquids are of high cost as compared to simple ammonium ionic liquids.56
In continuation of our previous work on the progress of novel synthetic methodologies for organic transformations,57 herein we report for the first time the development of an efficient Bronsted acid ionic liquid (BAIL) [Et3NH][HSO4] promoted synthesis of hydrazone derivatives. In comparison with the current methods of hydrazone formation, our approach displays specific advantages: (i) it proceeds faster and gives excellent yields (90–98%); (ii) it requires an inexpensive catalyst; (iii) it is applicable to a broader substrate scope (electron-rich and electron-deficient).
2. Results and discussions
2.1. Characterization of the catalyst [Et3NH][HSO4]
The catalyst [Et3NH][HSO4] used in the study has been characterized on the basis of 1H NMR and 13C NMR spectral analysis. 1H NMR spectrum displayed a triplet at around δ 1.20 integrating for nine protons has been assigned to methyl group (3 × CH3) protons. Similarly a multiplet resonating at δ 3.15 corresponding to six protons has been attributed to methylene (3 × CH2) protons. A sharp singlet at around δ 8.85 for one proton has been assigned to –NH (D2O-exchangeable) proton (Fig. 1). 13C NMR spectrum showed a pair of resonance signals at around δ 10.23 and 52.01 assigned to methyl (CH3) and methylene (CH2) carbons, respectively (Fig. 2).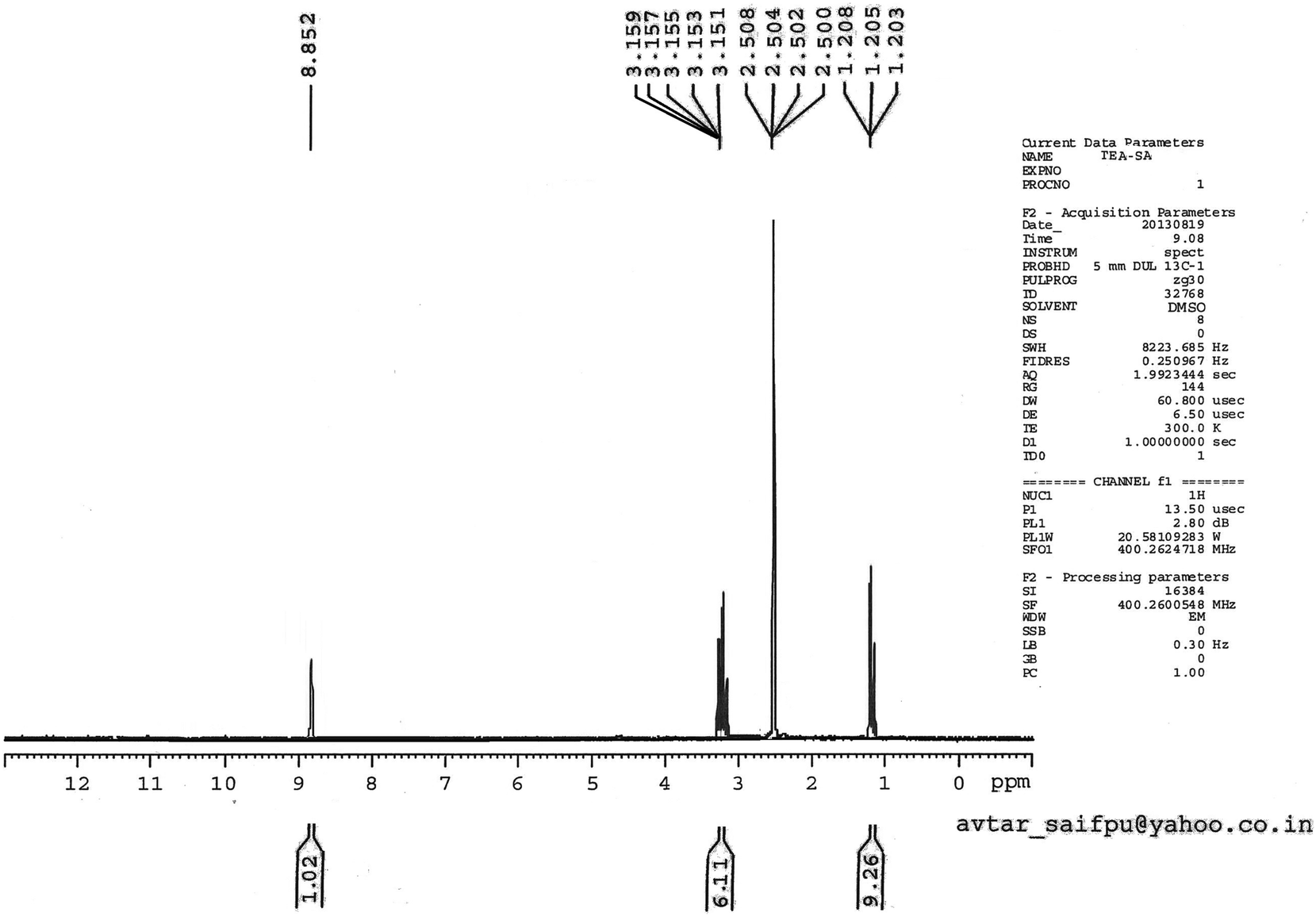 | ||
| Fig. 1 1H NMR of [Et3NH][HSO4]. | ||
 | ||
| Fig. 2 13C NMR of [Et3NH][HSO4]. | ||
2.2. Chemistry
The synthetic pathways of a series of hydrazone derivatives 2(a–j) and 4(a–e) are shown in Schemes 1 and 2, respectively. Herein, each series was typically accessed via a nucleophilic addition between hydrazine hydrate and appropriately substituted aromatic aldehydes 1(a–j) and 3-formylchromones 3(a–e) to yield target hydrazone derivatives. All the compounds were obtained in excellent yields (90–98%) with high purity. | ||
| Scheme 1 Synthetic pathway for the synthesis of hydrazone derivatives of substituted aromatic aldehydes 2(a–j). | ||
 | ||
| Scheme 2 Synthetic pathway for the synthesis of hydrazone derivatives of substituted 3-formylchromone derivatives 4(a–e). | ||
The structural elucidation of the synthesized compounds 2(a–j) and 4(a–e) was established on the basis of elemental analysis, IR, 1H NMR, 13C NMR and mass spectral analysis. The analytical results for C, H and N were within ±0.4% of the theoretical values. IR spectrum of all the synthesized compounds, showing the absence of absorption signal for carbonyl moiety, authenticates the reaction at carbonyl group. Moreover, all the synthesized compounds exhibited a characteristic peak at around (1585–1595), assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group. Other diagnostic peaks for functional groups such as OH, NO2, COγ-pyrone are discussed in the Experimental section. In the 1H NMR spectra, each compound displayed a sharp singlet at around δ 8.65–8.94 ascribed to the –CHN proton. Similarly sharp singlets resonating at around δ 7.23, 7.21, 7.29, 7.24, 7.23 each integrating for two protons, has been attributed to H-2 and H-2′ protons of γ-pyrone ring of chromones 4a, 4b, 4c, 4d and 4e, respectively. 13C NMR spectra, showed a series of signals resonating at around δ 107.50–140.15 which have been assigned to aromatic carbons, peaks resonating at around δ 112.9–116.4 corresponds to –CN moiety and the signals at δ 178.50, 178.42, 178.49, 177.42 and 179.50 have been attributed to carbonyl group (COγ-pyrone) of compounds 4a, 4b, 4c, 4d, and 4e, respectively. The mass spectral analysis of the synthesized compounds was also in good conformity with the proposed structures.
N group. Other diagnostic peaks for functional groups such as OH, NO2, COγ-pyrone are discussed in the Experimental section. In the 1H NMR spectra, each compound displayed a sharp singlet at around δ 8.65–8.94 ascribed to the –CHN proton. Similarly sharp singlets resonating at around δ 7.23, 7.21, 7.29, 7.24, 7.23 each integrating for two protons, has been attributed to H-2 and H-2′ protons of γ-pyrone ring of chromones 4a, 4b, 4c, 4d and 4e, respectively. 13C NMR spectra, showed a series of signals resonating at around δ 107.50–140.15 which have been assigned to aromatic carbons, peaks resonating at around δ 112.9–116.4 corresponds to –CN moiety and the signals at δ 178.50, 178.42, 178.49, 177.42 and 179.50 have been attributed to carbonyl group (COγ-pyrone) of compounds 4a, 4b, 4c, 4d, and 4e, respectively. The mass spectral analysis of the synthesized compounds was also in good conformity with the proposed structures.
The configuration around CN was authenticated by single crystal X-ray crystallographic analysis of compound (2f), where both CN were found to have E,E-geometry (Fig. 3). Among the three possible geometrical isomers (E,E/Z,Z/E,Z), (E,E)-isomers were obtained as the exclusive product (Fig. 3), which has been well established further on the basis of density functional theory (DFT) calculations (Fig. 4). This E,E-selectivity can be interpreted as a way to minimize steric interactions among various substituents. To compare the relative stability of the three possible isomers E,E, Z,Z and E,Z, we have performed the calculation of the vacuum single-point energies of the optimized geometries (Fig. 5) to obtain the energy differences. It was found that the E,E-isomer is stabilized by 5.68 and 9.24 kcal mol−1 more than the E,Z and Z,Z-isomers, respectively (Table 1). This difference in energy is the reason that during the crystallization process, the E,E-isomer gets exclusively crystallized out. The rotations about single bonds (intramolecular torsions) are worth 1–3 kcal mol−1 but can be as high as 10 kcal mol−1 due to steric factors or restricted rotations so this can elucidate the calculated energy differences.58 The single crystal X-ray diffraction analysis of compound (2f) was found to be in good agreement with the previous report.59
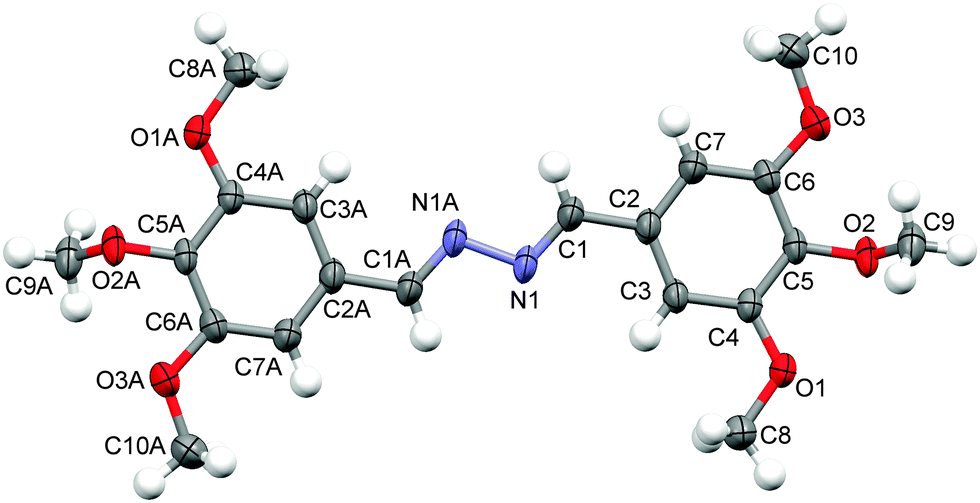 | ||
| Fig. 3 X-ray molecular structure of compound (2f). Displacement ellipsoids are plotted at the 50% probability level. | ||
 | ||
| Fig. 4 Comparison of the molecular conformation of compound (2f), as established from the X-ray study (red) with the optimized geometry (blue). | ||
 | ||
| Fig. 5 Optimized structures of different isomers of compound (2f) (a) E,E-isomer (b) Z,Z-isomer and (c) E,Z-isomer. | ||
| Isomers | Energya (hartree) | Energy (kcal mol−1) | E (X) − E(E,E)b (kcal mol−1) |
|---|---|---|---|
| a 1 hartree = 627.47237 kcal mol−1. b Energy difference between different isomers and E,E-isomer. | |||
| E,E-Isomer | −1337.65585 | −839342.0864438645 | — |
| E,Z-Isomer | −1337.64679 | −839336.4015441923 | 5.68 |
| Z,Z-Isomer | −1337.64112 | −839332.8437758544 | 9.24 |
In our present work, a series of hydrazone derivatives of substituted aromatic aldehydes 2(a–j) and 3-formylchromones 4(a–e) were synthesized by nucleophilic addition of hydrazine hydrate to substituted aromatic aldehydes and 3-formylchromones under reflux in ethanol in the absence of catalyst. The reaction took a prolonged time period (6–8 hours) for completion with a moderate yield (60–72%) of the products (Table 2).
| Product | Structure | Reaction in absence of catalyst | Reaction in presence of catalyst | M.P. (°C) | ||
|---|---|---|---|---|---|---|
| Time (h) | Yield (%) | Time (min) | Yield (%) | |||
| 2a |

|
6.0 | 69 | 30 | 98 | 213–214 (215)59 |
| 2b |

|
7.5 | 72 | 42 | 96 | 75–78 |
| 2c |

|
6.0 | 70 | 37 | 94 | 198 (196–197)60 |
| 2d |

|
7.0 | 70 | 40 | 98 | 296 (297–298)60 |
| 2e |

|
6.5 | 68 | 35 | 90 | 191–193 (193)61 |
| 2f |

|
6.0 | 72 | 30 | 97 | 194 (192–194)58 |
| 2g |

|
8.0 | 65 | 45 | 92 | 211–213 |
| 2h |
|
7.5 | 67 | 42 | 94 | 220(216–218)62 |
| 2i |

|
8.0 | 63 | 35 | 97 | 205–206 |
| 2j |

|
7.0 | 60 | 40 | 96 | >300 (308)63 |
| 4a |

|
6.5 | 70 | 35 | 98 | 290–292 |
| 4b |

|
6.0 | 68 | 30 | 96 | 294 |
| 4c |

|
7.0 | 64 | 40 | 98 | 293–295 |
| 4d |

|
6.5 | 66 | 37 | 94 | 297–299 |
| 4e |

|
8.0 | 62 | 45 | 92 | >300 |
In order to develop an eco-friendly approach for the synthesis of biologically active hydrazone derivatives, we explored the efficacy of [Et3NH][HSO4] by carrying out the reaction of substituted aromatic aldehydes and 3-formylchromones with hydrazine hydrate in (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) molar ratio. In our experiments we investigated the optimum reaction conditions regarding the choice of solvent, temperature of reaction and amount of catalyst on a model reaction using p-dimethylaminobenzaldehyde (1a) and hydrazine hydrate to establish best reaction conditions.
:1) molar ratio. In our experiments we investigated the optimum reaction conditions regarding the choice of solvent, temperature of reaction and amount of catalyst on a model reaction using p-dimethylaminobenzaldehyde (1a) and hydrazine hydrate to establish best reaction conditions.
To achieve the optimum concentration of catalyst, the model reaction was investigated for different concentrations 5, 10, 15, 20 and 25 mol% (Table 3, entries 1–5) of [Et3NH][HSO4] at 120 °C under solvent-free condition. It is obvious from (Table 3, entry 4) that 20 mol% of the catalyst is satisfactory to gain the optimum yield in the shortest reaction time. Using less than 20 mol% of catalyst, moderate yields of the product (76–89%) were obtained with extended reaction times, while with an excess mol% of catalyst (25 mol%) there was no further increase in the yield of the product, probably due to the saturation of the catalytic sites of the catalyst.
In order to study the solvent effect, the model reaction was carried out in different solvent systems. The model reaction was first investigated in MeOH and EtOH (Table 4, entries 1 and 2) the reaction took a longer time (6–8 h) with moderate yields of 64% and 60%, respectively, whereas in water (Table 4, entry 3), the product was obtained in better yield (68%) after refluxing for 5 h. In CH2Cl2 and DMF, moderate yields of the product were obtained after a stretched reaction periods (Table 4, entries 5–6), whereas in acetic acid, the reaction period was reduced to 4 h (Table 4, entry 4) and there was an enhancement in the yield by 5% in comparison to CH2Cl2, probably due to an electromeric effect offered by acetic acid activating the carbonyl group of the reactants, decreasing the activation energy, thereby rendering it more reactive towards nucleophilic attack. Furthermore, when the model reaction was carried out under solvent-free condition, there was a noteworthy increase in the yield of the product in a shorter time period (Table 4, entry 7). In view of the above results, it was concluded that solvent-free is the best reaction condition for the synthesis of present hydrazone derivatives in excellent yields.
| Entry | Solvent | Temp. (°C) | Timeb (h) | Yieldc (%) |
|---|---|---|---|---|
| a Reaction conditions: p-dimethylamino benzaldehyde (1a, 2 mmol), hydrazine hydrate (1 mmol), different solvents (20 mL, entries 1–6, refluxing temperature), solvent free (entries 7–11, temperature 25–140 °C), catalyst (20 mol%). b Reaction progress monitored by TLC (entries 1–9, h). c Isolated yield of products. d Reaction progress monitored by TLC (entries 10–11, min). | ||||
| 1 | MeOH | Reflux | 6 | 64 |
| 2 | EtOH | Reflux | 8 | 60 |
| 3 | Water | Reflux | 5 | 68 |
| 4 | CH3COOH | Reflux | 4 | 76 |
| 5 | CH2Cl2 | Reflux | 10 | 71 |
| 6 | DMF | Reflux | 12 | 68 |
| 7 | Solvent free | Room temp. | 2.5 | 84 |
| 8 | Solvent free | 60 | 2.0 | 89 |
| 9 | Solvent free | 80 | 1.5 | 92 |
| 10 | Solvent free | 120 | 30d | 98 |
| 11 | Solvent free | 140 | 30d | 98 |
To optimize the reaction temperature, the model reaction was carried out at different temperatures under solvent free condition (Table 4, entries 7–11). It was observed that the increase in temperature from 25 °C to 120 °C, has a significant effect on the reaction in terms of the yield and reaction time. The yield of the product increased from 84–98% during the course of reaction (Table 4, entries 7–11). However, no increase in the yield of product was observed when the reaction temperature was raised from 120 °C to 140 °C (Table 4, entry 11). Keeping in view the above optimize conditions, reactions were carried out at 120 °C in the presence of 20 mol% of [Et3NH][HSO4] under solvent-free conditions.
Using these optimized reaction conditions discussed above, the efficacy of this approach was explored for the synthesis of hydrazone derivatives (Schemes 1 and 2) and the results obtained are presented in Table 2.
In Scheme 3, a plausible mechanistic pathway is proposed to illustrate the synthesis of hydrazone derivatives catalyzed by [Et3NH][HSO4]. The initial step involves the protonation of formyl group (–CHO) of differently substituted aromatic aldehydes and 3-formylchromones (I) by protic ionic liquid catalyst [Et3NH][HSO4] to form intermediate (II), which facilitates the nucleophilic attack of hydrazine hydrate to promote the formation of C–N bond to yield intermediate (III). The subsequent elimination of water molecule from intermediate (III) enhanced by catalyst [Et3NH][HSO4] eventually yield compound (IV) followed by regeneration of the catalyst. The repetition of catalytic loop for compound (IV) with another molecule (I) finally ends up with target hydrazone products 2(a–j) and 4(a–e).
 | ||
| Scheme 3 Plausible mechanistic pathway for the synthesis of target hydrazone derivatives 2(a–j) and 4(a–e). | ||
A comparative study of a variety of other Bronsted acid ionic liquid catalysts was conducted to investigate the superiority of [Et3NH][HSO4]. It is obvious from (Table 5) that the catalytic activity was strongly affected by the anionic part of the ionic liquids. In case of [HSO4] anion, higher yields were obtained (Table 5, entries 1–3). However, when [H2PO4] and [CH3COO] anions were probed for their efficiency, lower yields (81–90%) were obtained as compared to [HSO4] anion (92–98%), probably due to the weaker acidity of the phosphate and acetate anions than [HSO4]. These results suggest that [Et3NH][HSO4] is the best ionic liquid catalyst for the synthesis of present hydrazone derivatives.
| Entry | Catalyst | Timeb (min) | Yieldc (%) |
|---|---|---|---|
| a Reaction conditions: p-dimethylamino benzaldehyde (1a, 2 mmol), hydrazine hydrate (1 mmol), solvent free, 120 °C, different catalysts (20 mol%). b Reaction progress monitored by TLC. c Isolated yield of products. | |||
| 1 | [Et3NH][HSO4] | 30 | 98 |
| 2 | [Me3NH][HSO4] | 45 | 92 |
| 3 | [Et2NH2][HSO4] | 40 | 96 |
| 4 | [Et3NH][H2PO4] | 48 | 90 |
| 5 | [Me3NH][H2PO4] | 52 | 86 |
| 6 | [Et2NH2][H2PO4] | 46 | 89 |
| 7 | [Et3NH][CH3COO] | 57 | 84 |
| 8 | [Me3NH][CH3COO] | 62 | 81 |
The reusability of the catalyst was also explored for the selected model reaction. The catalyst was reused five times and the results demonstrate that the catalyst can be reused without a significant reduction in the yield (Table 6). After the completion of the reaction, cold water was added to the reaction mixture, and the products were isolated by filtration. The ionic liquid was recovered from the filtrate by removing the water under reduced pressure.
2.3. Results of density functional theory (DFT) calculations
In order to gain some insight into the influence of the intermolecular interactions on the molecular geometry, we have performed a DFT calculation of the equilibrium geometry of the free molecule starting from the experimental X-ray geometry. The DFT calculations closely reproduce the solid-state geometry of the molecule (2f). The corroboration between the experimental and calculated bond lengths and angles was found to be very good, with the differences being smaller than 0.02 Å and 1.65°, respectively, but the calculated torsion angle of one of the methoxy group deviates appreciably by 24.7° from the experimental value (Table S1, ESI,†Fig. 4).Overall, our data suggests that the supramolecular aggregation has some significance in the stabilization of the observed geometry of compound (2f), in spite of all the interactions being weak.
3. Conclusions
The present protocol reports the convenient and eco-friendly approach for the synthesis of hydrazone derivatives 2(a–j) and 4(a–e) in excellent yields (90–98%) by employing [Et3NH][HSO4] as a catalyst. This solvent-free, green synthetic procedure eliminates the use of toxic solvents and thus makes it attractive one in organic synthesis. The notable features of this protocol are shorter reaction time, high purity, mild reaction conditions, operational simplicity, cleaner reaction profile, enhanced reaction rates and easy workup. The ionic liquid [Et3NH][HSO4] used in the present study exhibits stability towards air and water and is easy to prepare from cheap amine and acid. The recyclability study of the catalyst demonstrates that it can be used up to five cycles without any appreciable loss in catalytic activity.4. Experimental section
4.1. Materials and general methods
Chemicals were purchased from Merck and Sigma-Aldrich as ‘synthesis grade’ and used without further purification. Elemental analysis (C, H, N) was conducted using Carlo Erba analyzer model 1108. Melting points were determined on a Kofler apparatus and are uncorrected. The IR spectra were recorded with a Shimadzu IR-408 Perkin-Elmer1800 instrument (FTIR), and the values are given in cm−1. 1H NMR and 13C NMR spectra were run in DMSO-d6 on a Bruker Avance-II 400 MHz instrument with TMS as an internal standard and J values measured in Hertz. Chemical shifts are reported in ppm (δ) relative to TMS. Mass spectra were recorded on a JEOL D-300 mass spectrometer. Thin layer chromatography (TLC) glass plates (20 × 5 cm) were coated with silica gel G (Merck) and exposed to iodine vapor to check the homogeneity as well as the progress of the reaction.4.2. Synthesis of ionic liquids
The simple ammonium ionic liquids of general type [amine][HSO4] were synthesized by the known standard literature methods31 in the following way.The following ionic liquids were synthesized by the same procedure.31
4.3. General procedure for the synthesis of hydrazone derivatives
To a mixture of an aromatic aldehyde 2(a–j) or 3-formylchromone 4(a–e) (0.02 mol) and hydrazine hydrate (0.01 mol), 20 mol% of [Et3NH][HSO4] was added and the reaction mixture was heated on an oil bath at 120 °C for (20–30 min) with stirring. During the reaction process, the reaction mixture spontaneously solidified. After completion of the reaction as evident from thin layer chromatography (TLC), the reaction mixture was allowed to cool at room temperature. Water was added and the reaction mixture was further stirred for 5 min. The solid obtained was removed by filtration, washed with appropriate solvents and then recrystallized from methanol. The water was removed from filtrate under reduced pressure to recover [Et3NH][HSO4], which was then reused in subsequent cycles.4.4. Spectral characterization
Caromatic), 1594 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 3.10 (s, 6H, 4 × CH3), 6.76 (dd, 2H, C-3 and C-5), 6.78 (dd, 2H, C-3′ and C-5′), 7.20 (dd, 2H, C-2 and C-6), 7.21 (dd, 2H, C-2′ and C-6′), 8.68 (s, 1H, –CHNazine), 8.65 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 42.10 (–CH3). 111.12 (C-3 and C-5), 111.13 (C-3′ and C-5′), 112.54 (CN), 122.50 (C-1), 122.52 (C-1′), 133.21 (C-2 and C-6), 133.24 (C-2′ and C-6′), 152.41 (C-4), 152.43 (C-4′). MS (EI): (m/z) 294.18 [M+˙].
Caromatic),1583 (CN), 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.93 (dd, 2H, H-2′ and H-6′), 7.95 (dd, 2H, H-2 and H-5), 8.31 (dd, 2H, H-3′ and H-5′), 8.32 (dd, 2H, H-3 and H-5), 8.67 (s, 1H, –CHNazine), 8.70 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 114.54 (CN), 115.11 (C-3 and C-5), 115.14 (C-3′ and C-5′), 130.75 (C-2 and C-6), 130.78 (C-2′ and C-6′), 131.20 (C-1), 131.21 (C-1′), 164.23 (C-4), 164.24 (C-4′). MS (EI): (m/z) 244.08 [M+˙].
Caromatic), 1334, 1510 (NO2), 1586 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.72 (m, 1H, C-5), 7.74 (m, 1H,C-5′), 8.10 (d, 1H, C-4), 8.13 (d, 1H, C-4′), 8.62 (s, 1H, C-2), 8.63 (s, 1H, C-2′), 8.69 (s, 1H, –CHNazine), 8.73 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 114.44 (CN), 120.32 (C-2), 120.35 (C-2′), 125.45 (C-4), 125.47 (C-4′), 129.56 (C-5), 129.59 (C-5′), 132.12 (C-1), 132.15 (C-1′), 134.97 (C-6), 134.98 (C-6′), 150.10 (C-3), 150.11 (C-3′). MS (EI): (m/z) 298.07 [M+˙].
Caromatic), 1338, 1512 (NO2), 1585 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.92 (dd, 2H, C-2 and C-6), 7.94 (dd, 2H, C-2′ and C-6′), 8.12 (dd, 2H, C-3 and C-5), 8.16 (dd, 2H, C-3′ and C-5′), 8.79 (s, 1H, –CHNazine), 8.82 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 114.13 (CN), 125.68 (C-2 and C-4), 125.69 (C-2′ and C-4′), 140.15 (C-1), 140.18 (C-1′), 150.01 (C-6), 150.02 (C-6′). MS (EI): (m/z) 298.07 [M+˙].
Caromatic), 1591 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 3.79 (s, 12H, 4 × CH3), 6.98 (d, 1H, C-5), 6.99 (d, 1H, C-5′), 7.42 (d, 1H, C-6), 7.43 (d, 1H, C-6′), 7.69 (s, 1H, C-2), 7.70 (s, 1H, C-2′), 8.85 (s, 1H, –CHNazine), 8.89 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 57.65 (–CH3), 108.89 (C-5), 108.90 (C-5′), 111.56 (C-2), 111.57 (C-2′), 112.80 (CN), 123.23 (C-6), 123.25 (C-6′), 127.87 (C-1), 127.89 (C-1′), 148.78 (C-3), 148.79 (C-3′), 152.66 (C-4), 152.68 (C-4′). MS (EI): (m/z) 328.14 [M+˙].
Caromatic), 1595 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 3.89 (s, 18H, 6 × CH3), 7.18 (dd, 2H, C-2 and C-6), 7.20 (dd, 2H, C-2′ and C-6′), 8.90 (s, 1H, –CHNazine), 8.92 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 58.45 (–OCH3), 106.11 (C-2 and C-6), 106.13 (C-2′ and C-6′), 112.9 (CN), 131.20 (C-1), 131.22 (C-1′), 142.32 (C-6), 142.34 (C-6′), 152.32 (C-3 and C-5), 152.35 (C-3′ and C-5′). MS (EI): (m/z) 388.16 [M+˙].
Caromatic), 1617 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 3.73 (s, 6H, 2 × CH3), 5.76 (s, 1H, –OH), 6.87 (d, 1H, C-5), 6.89 (d, 1H, C-5′), 7.42 (s, 1H, C-2), 7.44 (s, 1H, C-2′), 7.45 (d, 1H, C-6), 7.46 (d, 1H, C-6′), 8.84 (s, 1H, –CHNazine), 8.87 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 56.12 (–OCH3), 114.02 (C-2), 114.04 (C-2′), 113.44 (CN), 118.97 (C-5), 118.99 (C-5′), 123.42 (C-6), 123.44 (C-6′), 129.12 (C-1), 129.13 (C-1′), 150.12 (C-3), 150.14 (C-3′), 152.43 (C-4), 152.47 (C-4′). MS (EI): (m/z) 300.11 [M+˙].
Caromatic), 1588 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 5.40 (s, 2H, –OH), 7.01 (m, 1H, C-5), 7.02 (m, 1H, C-5′), 7.08 (d, 1H, C-3), 7.09 (d, 1H, C-3′), 7.59 (m, 1H, C-4), 7.60 (m, 1H, C-4′), 7.88 (d, 1H, C-6), 7.89 (d, 1H, C-6′), 8.91 (s, 1H, CHNazine). 8.94 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 115.08 (CN), 117.67 (C-3), 117.69 (C-3′), 118.23 (C-1), 118.25 (C-1′), 125.50 (C-5), 125.51 (C-5′), 132.21 (C-6), 132.23 (C-6′), 134.56 (C-4), 134.70 (C-4′), 162.34 (C-2). 162.35 (C-2′). MS (EI): (m/z) 240.09 [M+˙].
Caromatic), 1592 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 5.42 (s, 6H, –OH), 6.92 (s, 2H, C-2 and C-6), 6.93 (s, 2H, C-2′ and C-6′), 8.67 (s, 1H, –CHNazine), 8.69 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 107.94 (C-2 and C-6), 107.96 (C-2′ and C-6′), 114.21 (CN), 132.98 (C-1), 132.99 (C-1′), 137.33 (C-4), 137.35 (C-4′). 147.01 (C-3 and C-5), 147.02 (C-3′ and C-5′). MS (EI): (m/z) 304.07 [M+˙].
Caromatic), 1586 (CN). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 5.45 (s, 2H, –OH), 7.53 (m, 1H, C-6), 7.55 (m, 1H, C-6′), 7.62 (m, 1H, C-5), 7.63 (m, 1H, C-5′), 7.69 (d, 1H, C-3), 7.70 (d, 1H, C-3′), 7.90 (d, 1H, C-4), 7.93 (d, 1H, C-4′), 7.95 (m, 1H, C-7), 7.96 (m, 1H, C-7′), 8.40 (d, 1H, C-8), 8.42 (d, 1H, C-8′), 8.72 (s, 1H, –CHNazine), 8.75 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 107.50 (C-1), 107.52 (C-1′) 110.89 (C-3), 110.90 (C-3′), 115.22 (CN), 115.56 (C-8), 115.57 (C-8′), 123.90 (C-6), 123.91 (C-6′), 124.96 (C-9), 124.92 (C-9′), 127.01 (C-4), 127.03 (C-4′), 127.22 (C-5), 127.24 (C-5′), 128.21 (C-7), 128.23 (C-7′), 141.11 (C-10), 141.12 (C-10′), 158.02 (C-2), 158.03 (C-2′). MS (EI): m/z: 340.12 [M+˙].
Caromatic), 1594 (CN), 1612 (CCγ-pyrone), 1652 (COγ-pyrone). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.23 (s, 2Hγ-pyronering, H-2 and H-2′), 7.42 (m, 4H, H-6, H-6′, H-7 and H-7′), 7.81 (dd, 2H, H-5 and H-5′), 7.82 (dd, 2H, H-8 and H-8′), 8.74 (s, 1H, –CHNazine), 8.78 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 110.74 (C-3), 110.79 (C-3′), 115.15 (CN), 121.26 (C-8), 121.27 (C-8′), 123.90 (C-4a), 123.97 (C-4a′), 124.23 (C-6), 124.25 (C-6′), 127.85 (C-5), 127.82 (C-5′), 133.50 (C-7), 133.59 (C-7′), 151.62 (C-8a), 151.65 (C-8a′), 163.23 (C-2′), 163.25 (C-2), 178.50 (C-4), 178.55 (C-4′). MS (EI): (m/z) 344.08 [M+˙].
Caromatic), 1592 (CN), 1604 (CCγ-pyrone), 1660 (COγ-pyrone). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.21 (s, 2Hγ-pyronering, H-2 and H-2′), 7.30 (dd, 2H, H-7 and H-7′), 7.32 (dd, 2H, H-8 and H-8′), 7.78 (s, 2H, H-5 and H-5′), 8.81 (s, 1H, –CHNazine), 8.83 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 110.71 (C-3), 110.73 (C-3′), 116.4 (CN), 121.12 (C-5), 121.20 (C-8), 121.15 (C-5′), 122.25 (C-8′), 124.50 (C-4a), 124.56 (C-4a′), 128.50 (C-7), 128.46 (C-7′), 135.23 (C-6), 135.21 (C-6′), 152.80 (C-8a), 152.82 (C-8a′), 162.95 (C-2), 162.99 (C-2′), 178.42 (C-4), 178.47 (C-4′). MS (EI): (m/z) 380.06 [M+˙].
Caromatic), 1588 (CN), 1610 (CCγ-pyrone), 1656 (COγ-pyrone). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 1.83 (s, 6H, 2 × CH3), 7.29 (s, 2Hγ-pyronering, H-2 and H-2′), 7.31 (dd, 2H, H-7 and H-7′), 7.35 (dd, 2H, H-8 and H-8′), 7.75 (s, 2H, H-5 and H-5′), 8.92 (s, 1H, –CHNazine), 8.94 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 29.02 (–CH3), 110.72 (C-3), 110.78 (C-3′), 115.13 (CN), 121.24 (C-8), 121.22 (C-8′), 123.91 (C-4a), 123.95 (C-4a′), 129.15 (C-5), 129.19 (C-5′), 133.52 (C-7), 133.51 (C-7′), 135.12 (C-6), 135.10 (C-6′), 151.63 (C-8a), 151.65 (C-8a′). 163.21 (C-2), 163.23 (C-2′), 178.49 (C-4), 178.55 (C-4′). MS (EI) m/z: 372.11 [M+˙].
Caromatic), 1590 (CN), 1607 (CCγ-pyrone,), 1650 (COγ-pyrone). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.24 (s, 2Hγ-pyronering, H-2′ and H-2), 7.31 (dd, 2H, H-8 and H-8′), 7.40 (dd, 2H, H-7 and H-7′), 7.77 (s, 2H, H-5 and H-5′), 8.66 (s, 1H, –CHNazine), 8.69 (s, 1H, –CHNazine). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 110.12 (C-3), 110.13 (C-3′), 115.89 (CN), 120.98 (C-8), 120.99 (C-8′), 123.54 (C-4a), 123.59 (C-4a′), 124.17 (C-5), 124.15 (C-5′), 128.35 (C-6), 128.38 (C-6′), 133.72 (C-7), 133.76 (C-7′), 151.67 (C-8a), 151.68 (C-8a′), 162.21 (C-2), 162.26 (C-2′), 177.42 (C-4), 177.48 (C-4′). MS (EI): m/z 501.90 [M+˙].
Caromatic), 1589 (CN), 1602 (CCγ-pyrone), 1656 (COγ-pyrone). 1H NMR (400 MHz, DMSO-d6, δ, ppm): 7.23 (s, 2Hγ-pyrone ring, H-2′ and H-2), 7.39 (m, 4H, H-6, H-6′, H-7 and H-7′), 7.80 (dd, 2H, H-8 and H-8′), 7.90 (dd, 2H, H-5 and H-5′), 8.68 (s, 1H, –CHNazine), 8.71 (s, 1H, –CHNazine), 9.02 (brs, 4H, D2O exchangeable). 13C NMR (100 MHz, DMSO-d6, δ, ppm): 112.40 (C-3), 112.42 (C-3′), 115.21 (CN), 121.45 (C-8), 121.49 (C-8′), 123.42 (C-4a), 123.44 (C-4a′), 124.21 (C-6), 124.23 (C-6′), 125.63 (C-5), 125.65 (C-5′), 133.43 (C-7), 133.45 (C-7′), 152.10 (C-8a), 152.12 (C-8a′), 166.80 (C-2), 166.82 (C-2′), 179.50 (C-4), 179.53 (C-4′). MS (EI): m/z 374.10 [M+˙].
4.5. X-ray analysis and DFT studies of compound (2f)
A crystal of compound (2f) with a block habit and having approximate dimensions of 0.55 mm × 0.33 mm × 0.31 mm was glued on a glass fibre and mounted on a Bruker Apex II diffractometer. The diffraction data was collected at room temperature 293(2) K using graphite monochromated Mo Kα (λ = 0.71073 Å). Data reduction was performed with APEX II.65 Lorentz and polarization corrections were applied. Absorption correction was applied using SADABS.66 The crystallographic structure was solved by direct methods (SHELXS-97).67 Refinements were carried out with SHELXL-97 package. All refinements were made by full-matrix least-squares on F2, with anisotropic displacement parameters for all non-hydrogen atoms. All the hydrogen atoms were located in a difference Fourier synthesis, placed at calculated positions and then, included in the structure factor calculation in a riding model using SHELXL-97 defaults. The final least-squares cycle was based on 2300 observed reflections [I > 2σ(I)], 130 variable parameters, converged with R = 0.0396 and wR = 0.1091. Pertinent crystallographic data for compound (2f) is summarized in Table S2 (ESI†), while as selected structural parameters has been presented in Table S1 (ESI†).The geometry optimization of the compound (2f) was performed using the PC GAMESS/Firefly QC package,68 which is partially based on the GAMESS (US) source code,69 starting from the experimental X-ray geometry (E,E-isomer). The calculation was performed within density functional theory (DFT) using B3LYP (Becke three-parameter Lee–Yang–Parr) for exchange and correlation, which combines the hybrid exchange functional of Becke70,71 with the correlation functional of Lee, Yang and Parr.72 The calculation was performed with an extended 6-311G(d,p) basis set. Tight conditions for convergence of both the self-consistent field cycles and the maximum density and energy gradient variations were imposed (10-5 atomic units). At the end of this geometry optimization, we conducted a Hessian calculation to guarantee that the final structure corresponds to a true minimum, using the same level of theory as in the geometry optimization. The geometries of the Z,Z and Z,E isomers were also optimized with the same level of theory and obtained from the optimized geometry of the E,E-isomer, performing the necessary rotations of bond/torsion angles using the UCSF Chimera software package version 1.8.73 For the optimized geometries of the E,E, Z,Z and E,Z-isomers, we performed single-point energy calculations with the conditions mentioned above (DFT: B3LYP functional and 6–311G(d,p) basis set).
Acknowledgements
S. Azaz thanks the Chairman, Department of Chemistry, AMU, Aligarh, for providing the necessary research facility, University Sophisticated Instrumentation Facility (USIF), AMU, Aligarh and Sophisticated Analytical Instrumentation Facility (SAIF), Chandigarh, is credited for spectral analysis. P. S. P. Silva acknowledges the support by Fundação para a Ciência e a Tecnologia, under the scholarship SFRH/BPD/84173/2012. UGC is also gratefully acknowledged for research fellowship to S. Azaz, A. Mohammed and F. Ahmad.References
- V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS PubMed.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed.
- M. Kosmulski, J. Gustafsson and J. B. Rosenholm, Thermochim. Acta, 2004, 412, 47–53 CrossRef CAS PubMed.
- H. Matsumoto, M. Yanagida, K. Tanimoto, K. Nomura, Y. Kitagawa and Y. Miyazaki, Chem. Lett., 2000, 29, 922–923 CrossRef.
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772–3789 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2003, p. 41 Search PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- Q. Yao, Org. Lett., 2002, 4, 2197–2199 CrossRef CAS PubMed.
- H. M. Zerth, N. M. Leonard and R. S. Mohan, Org. Lett., 2003, 5, 55–57 CrossRef CAS PubMed.
- A. Kumar and S. S. Pawar, J. Org. Chem., 2004, 69, 1419–1420 CrossRef CAS PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- R. F. de Souza, J. C. Padilha, R. S. Goncalves and J. Dupont, Electrochem. Commun., 2003, 5, 728–731 CrossRef CAS.
- M. E. V. Valkenburg, R. L. Vaughn, M. Williams and J. S. Wilkes, Thermochim. Acta, 2005, 425, 181–188 CrossRef PubMed.
- D. S. Silvester, Analyst, 2011, 136, 4871–4882 RSC.
- V. Chakrapani, F. Rusli, M. A. Filler and P. A. Kohl, J. Phys. Chem. C, 2011, 115, 22048–22053 CAS.
- T. Sato, G. Masuda and K. Takagi, Electrochim. Acta, 2004, 49, 3603–3611 CrossRef CAS PubMed.
- M. P. Scott, C. S. Brazel, M. G. Benton, J. W. Mays, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2002, 1370–1371 RSC.
- F. Zhou, Y. Liang and W. Liu, Chem. Soc. Rev., 2009, 38, 2590–2599 RSC.
- J. Q. Zhua, J. Chen, C. Y. Li and W. Y. Fei, Sep. Purif. Technol., 2007, 56, 237–240 CrossRef PubMed.
- B. Tang, W. Bi, M. Tian and K. H. Row, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 904, 1–21 CrossRef CAS PubMed.
- M. Volland, V. Seitz, M. Maase, M. Flores, R. Papp, K. Massonne, V. Stegmann, K. Halaritter, R. Noe, M. Bartsch, W. Siegel, M. Becker and O. Huttenloch, Chem. Eng. News, 2003, 81, 9 Search PubMed.
- A. Noda, M. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- H. Gao, B. Han, J. Li, T. Jiang, Z. Liu, W. Wu, Y. Chang and J. Zhang, Synth. Commun., 2004, 34, 3083–3089 CrossRef CAS PubMed.
- R. Sugimura, K. Qiao, D. Tomida and C. Yokoyama, Catal. Commun., 2007, 8, 770–772 CrossRef CAS PubMed.
- S. T. Handy and X. Zhan, Org. Lett., 2001, 3, 2233–2236 CrossRef PubMed.
- H. Olivier-Bourbigou and L. Magna, J. Mol. Catal. A: Chem., 2002, 182, 419–437 CrossRef.
- N. S. Suryawanshi, P. Jain, M. Singhal and I. Khan, J. Appl. Chem., 2012, 1, 18–23 CAS.
- Z. Zhou and X. Deng, J. Mol. Catal. A: Chem., 2013, 367, 99–102 CrossRef CAS PubMed.
- A. Rajendran, S. Ramu and C. Karthikeyan, Int. J. Recent Sci. Res., 2011, 2, 36–39 Search PubMed.
- C. Wang, L. Guo, H. Li, Y. Wang, J. Weng and L. Wu, Green Chem., 2006, 8, 603–607 RSC.
- H. Jiang, C. Wang, H. Li and Y. Wang, Green Chem., 2006, 8, 1076–1079 RSC.
- P. A. Ganeshpure, G. George and J. Das, J. Mol. Catal. A: Chem., 2008, 279, 182–186 CrossRef CAS PubMed.
- C. Wang, W. Zhao, H. Li and L. Guo, Green Chem., 2009, 11, 843–847 RSC.
- Z. Zhou and X. Deng, J. Mol. Catal. A: Chem., 2013, 367, 99–102 CrossRef CAS PubMed.
- E. T. Kermani, H. Khabazzadeh and T. Jazinizadeh, J. Heterocycl. Chem., 2011, 48, 1192–1196 CrossRef CAS.
- J. Weng, C. Wang, H. Li and Y. Wang, Green Chem., 2006, 8, 96–99 RSC.
- E. M. Beccalli, G. Broggini, A. Fasana and M. Rigamonti, J. Organomet. Chem., 2011, 696, 277–295 CrossRef CAS PubMed.
- A. Abdukader, Q. Xue, A. Lin, M. Zhang, Y. Cheng and C. Zhu, Tetrahedron Lett., 2013, 54, 5898–5900 CrossRef CAS PubMed.
- M. Xue, M. Xu, W. Lu, J. Huang, H. Li, Y. Xu, X. Liu and Z. Zhao, J. Enzyme Inhib. Med. Chem., 2012, 1–6 Search PubMed.
- S. M. Sondhi, M. Dinodia and A. Kumar, Bioorg. Med. Chem., 2006, 14, 4657–4663 CrossRef CAS PubMed.
- A. C. Cunha, J. L. M. Tributino, A. L. P. Miranda, C. A. M. Fraga and E. J. Barreiro, Farmaco, 2002, 57, 999 CrossRef CAS.
- S. K. Sridhar and A. Ramesh, Biol. Pharm. Bull., 2001, 24, 1149–1152 CAS.
- M. R. Maurya, S. Agarwal, M. Abid, A. Azam, C. Bader, M. Ebel and D. Rehder, Dalton Trans., 2006, 937–947 RSC.
- L. Savini, P. Massarelli, V. Travagli, C. Pellerano, E. Novellino, S. Cosentino and M. B. Pisano, Eur. J. Med. Chem., 2004, 39, 113–122 CrossRef CAS PubMed.
- P. Melnyk, V. Leroux, C. Sergheraert and P. Grellier, Bioorg. Med. Chem. Lett., 2006, 16, 31–35 CrossRef CAS PubMed.
- (a) B. K. Banik, K. J. Barakat, W. R. Wagle, M. S. Manhas and A. K. Bose, J. Org. Chem., 1999, 64, 5746–5753 CrossRef CAS; (b) S. Gadhwal, M. Baruah and J. S. Sandhu, Synlett, 1999, 1573–1574 CrossRef CAS PubMed.
- J. H. Billman and K. M. Tai, J. Org. Chem., 1958, 23, 535–539 CrossRef CAS.
- H. Weingart, J. P. Chupp and W. A. White, J. Org. Chem., 1967, 32, 213–214 CrossRef.
- (a) R. S. Vaas, J. Dudas and R. S. Varma, Tetrahedron Lett., 1999, 40, 4951–4954 CrossRef; (b) S. M. Landge, V. Atanassova, M. Thimmaih and B. Torok, Tetrahedron Lett., 2007, 48, 5161–5164 CrossRef CAS PubMed.
- B. P. Branchaud, J. Org. Chem., 1983, 48, 3531–3538 CrossRef CAS.
- A. K. Chakraborti, S. Bhagat and S. Rudrawar, Tetrahedron Lett., 2004, 45, 7641–7644 CrossRef CAS PubMed.
- A. Bazgir, J. Chem. Res., 2006, 2006, 1–2 CrossRef.
- V. Polshettiwar and R. S. Varma, Tetrahedron Lett., 2007, 48, 5649–5652 CrossRef CAS PubMed.
- C. E. Song, M. Y. Yoon and D. S. Choi, Bull. Korean Chem. Soc., 2005, 26, 1321–1330 CrossRef CAS.
- H. Wang, L. Sun, X. Li, J. Duan and W. Pei, Synth. Commun., 2011, 41, 3223–3227 CrossRef CAS.
- M. Parveen, A. M. Malla, A. Ali, M. Alam and M. F. Mustafa, Chem. Res. Chin. Univ., 2014, 30, 55–62 CrossRef CAS.
- J. Starbuck, R. Docherty, M. H. Charlton and D. Buttar, J. Chem. Soc., Perkin Trans. 2, 1999, 677–692 RSC.
- B. Zhao, Y. Q. Feng, J. W. Guo and J. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o2413–o2414 CAS.
- H. Loghmani-Khouzami, M. M. Sadeghi, J. Safari, M. S. Bdorrezaie and M. Jafapisheh, J. Chem. Res., 2001, 2001, 80–81 CrossRef.
- R. B. Elkan and M. G. Ralph, J. Am. Chem. Soc., 1945, 67, 13–17 CrossRef.
- S. N. Shah and N. K. Chudgar, Molecules, 2000, 5, 657–664 CrossRef CAS PubMed.
- T. D. Smith, et al. , J. Chem. Soc., Dalton Trans., 1982, 363–372 RSC.
- M. S. S. Artur, L. M. S. Vera, R. M. Claramunt, D. S. María, M. B. Ferraro, F. Reviriego, I. Alkorta and J. Elguero, Magn. Reson. Chem., 2013, 51, 530–540 CrossRef PubMed.
- Bruker APEX2 and SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2003 Search PubMed.
- G. M. Sheldrick, SADABS, University of Gottingen, Germany, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. A. Granovsky, PC GAMESS/Firefly version 7.1.G, http://www http://classic.chem.msu.su/gran/gamess/index.html, 2009.
- M. W. Schmidt, K. K. Baldrige, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. J. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Sue, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1026311. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01666a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |