
The near infra red (NIR) chiroptical properties of nickel dithiolene complexes†
Yann
Le Gal
,
Antoine
Vacher
,
Vincent
Dorcet
,
Marc
Fourmigué
,
Jeanne
Crassous
and
Dominique
Lorcy
*
Institut des Sciences Chimiques de Rennes, UMR 6226 CNRS-Université de Rennes 1, Campus de Beaulieu, Bât 10A, 35042 Rennes cedex, France. E-mail: dominique.lorcy@univ-rennes1.fr
First published on 12th September 2014
Abstract
Enantiomerically pure dianionic, monoanionic and neutral dithiolene complexes formulated as [Ni((R,R)-CHMePh-thiazdt)2]−2,−1,0 and [Ni((S,S)-CHMePh-thiazdt)2]−2,−1,0 (CHMePh-thiazdt: N-(1-phenylethyl)-1,3-thiazoline-2-thione-4,5-dithiolate) have been synthesized from the enantiopure N-(1-phenylethyl)-1,3-thiazoline-2-thione precursors. The electrochemical and spectro-electrochemical investigations carried out on these complexes show that the CHMePh-thiazdt ligands act as electron rich ligands and that the complexes are strong near infrared (NIR) absorbers in the range of 800–1100 nm for the neutral species and 1050–1500 nm for the reduced radical anion species. Circular dichroism (CD) thin layer spectro-electrochemical experiments carried out on the neutral (R,R) enantiomer, [Ni((R,R)-CHMePh-thiazdt)2], revealed a redox switching of CD-active bands, not only in the UV-vis but also in the NIR region.
Introduction
Nickel bis(dithiolene) complexes exhibit versatile and unique properties, associated with different redox states, for potential applications in optics and electronics.1 For instance, these dithiolene complexes are strong near infrared (NIR) absorbers in the range of 700–1400 nm, and this absorption range can be tuned not only by the electron withdrawing or releasing ability of the ligand but also by controlling the degree of oxidation of the complex.2 The introduction of chirality into such metal dithiolene complexes has also recently emerged,3 either to generate NIR chiro-optical effects4 or for the possible observation, in conducting dithiolene salts,5 of electrical magneto-chiral anisotropy,6 as recently reported in fully organic conductors.7 Interestingly,4 the chirality of cholesterol substituents was transferred to the NIR response of the complexes only in a gel supramolecular organisation, but was absent in the diluted molecular complexes. It is therefore an important issue to be able to introduce chirality in the close vicinity of NIR-absorbing square-planar dithiolene complexes. In that respect, two strategies have been essentially reported to prepare such chiral salts: either the chirality is introduced by the counter-ion associated with the anionic metal dithiolene complex5,8 or a chiral substituent is covalently linked to the dithiolene ligand.3Our current interest in metal dithiolene complexes involving the N-alkyl-1,3-thiazoline-2-thione-4,5-dithiolate (R-thiazdt) ligand9–12 prompted us to investigate the synthesis of chiral ligands on this attractive platform. Indeed, the thiazoline core offers the possibility of numerous structural modifications which can be brought about through the substituent on the nitrogen atom. Therefore, we decided to investigate the synthesis of a thiazoline core bearing a chiral substituent and to use this chiral precursor in the preparation of homoleptic dithiolene complexes of the general formula [Ni(R*-thiazdt)2] (Chart 1), with a chiral center very close to the two metallacycles. The enantiopure (R- or S-) and racemic, commercially available 1-phenyl-ethylamine was chosen here as a first proof of concept. Herein, we report the synthesis of the enantiopure nickel dithiolene complexes together with spectroscopic and electrochemical investigations in solution. The X-ray crystal structures, magnetic properties as well as the chiroptical properties will be also presented, demonstrating for the first time a circular dichroism effect in the NIR region in diluted solution.
 | ||
| Chart 1 Examples of reported chiral dithiolene complexes3,4 and the desired [Ni(R*-thiazdt)2] complex derived from 1-phenylethylamine. | ||
Results and discussion
Syntheses
The dithiolene complexes were prepared either from the enantiopure or the racemic mixture of 1-phenylethylamine according to the chemical route described in Scheme 1. The (R)-, (S)- and (R,S)-3-(1-phenylethyl)-1,3-thiazoline-2-thione 1 were prepared according to a modified procedure of Sandström.13 Addition of carbon disulfide and triethylamine to a suspension of 1-phenylethylamine leads to the dithiocarbamate salt which reacts with chloroacetaldehyde. Cyclization and dehydration in the presence of sulfuric acid affords the 1,3-thiazoline-2-thione (R)-, (S)- and rac-1 in 55% yield.14 In order to prepare the organic dithiolate protected form of the ligand, the N(-1-phenylethyl)-4,5-bis(cyanoethylthio)-1,3-thiazoline-2-thione 3 where the two thiolates are protected by cyanoethyl groups, we choose a two-step approach. The first step consists in the formation of the dithiolate which is trapped with ZnCl2 as described in Scheme 1.9b | ||
| Scheme 1 Synthetic procedure of (R,R) and (S,S)-4. | ||
Subsequent reaction of this Zn salt 2 with 3-bromopropionitrile affords the organic dithiolate protected form 3. Even if this route is longer than the direct formation of 3 from 1, it allows an easier isolation of 3, thanks to the precipitation in the first step of the Zn salt 2. It is worth mentioning that at room temperature the 1H NMR spectra of (R)-, (S)- and rac-3 exhibit two broad unresolved signals, one for the C–H at 7.12 ppm (Fig. 1) and the other one at 2.22 ppm for the CH2 of one cyanoethyl chain. This is reminiscent of what was observed for other N(-1-phenylethyl)-thiazoline-2-thione derivatives where broad signals were attributed to the presence of two rotamers designated as the rotamer syn and anti, depending on the orientation of the smallest substituent (C–H) in the plane of the thiazole ring (Chart 2).13,15 A temperature-dependent 1H NMR study in CDCl3 was carried out on (S)-3 from ambient temperature to 223 K. Upon lowering the temperature, the broad signal observed at 7.12 ppm, for the C–H, becomes a quadruplet at 233 K indicating that at this temperature one of the rotamers predominates strongly. Similar results have been observed in previous studies related to other N-(1-phenylethyl)-1,3-thiazoline-2-thiones, substituted in the 4 and 5 positions, where it was demonstrated that these compounds exist predominantly in the anti form.13
 | ||
| Chart 2 | ||
 | ||
| Fig. 1 1H NMR spectra of the phenyl and the benzylic protons of (S)-3 (top) and the methyl and the cyanoethyl groups (bottom) at various temperatures. | ||
Single crystals of good quality for X-ray diffraction studies were obtained by slow diffusion of diethyl ether into a concentrated solution of (S)-3 in dichloromethane. Fig. 2 shows the two crystallographically independent molecules, for the structure solved in the orthorhombic system, space group P212121. Both crystallographically independent molecules (S)-3 exhibit a planar thiazole skeleton with the C*–H of the chiral substituent oriented towards the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S thione, that is the anti rotamer (Chart 2). The C–H bonds lie in the plane of the thiazole ring with short C–H⋯S distances (2.5662(1) and 2.577(1) Å). Within this anti conformer, as observed in the NMR 1H study, the phenyl group exerts a strong shielding effect on the closest cyanoethyl group.
S thione, that is the anti rotamer (Chart 2). The C–H bonds lie in the plane of the thiazole ring with short C–H⋯S distances (2.5662(1) and 2.577(1) Å). Within this anti conformer, as observed in the NMR 1H study, the phenyl group exerts a strong shielding effect on the closest cyanoethyl group.
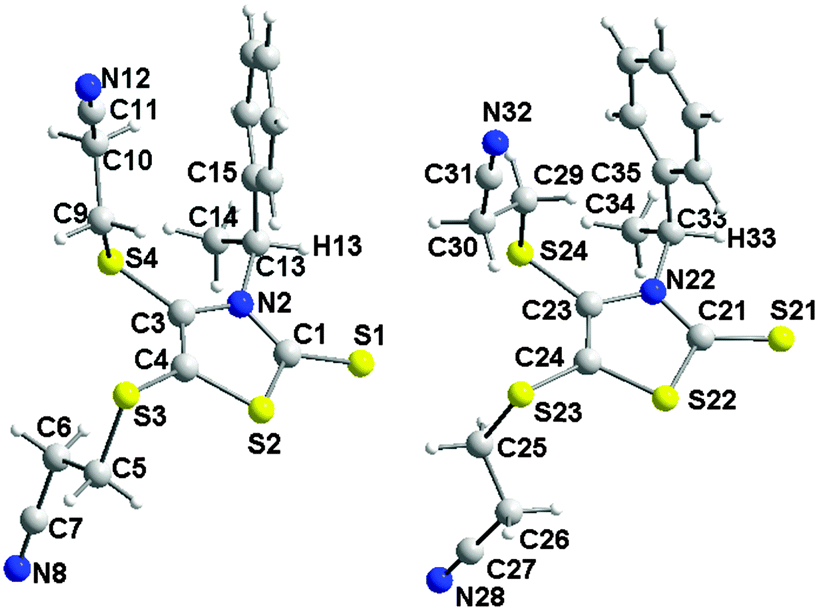 | ||
| Fig. 2 Molecular structures of the two crystallographically independent molecules (S)-3. | ||
Deprotection of the dithiolate precursor 3 was performed in basic medium using NaOMe,16 followed by addition of NiCl2 and Et4NBr, to afford the dianionic dithiolene complexes 4 as a dark red crystalline material (Scheme 1). The reaction was conducted on the enantiopure (R)-3 and (S)-3 affording respectively the dianionic complexes (R,R)-4 and (S,S)-4. Attempts to analyze the dianionic complexes using H1 NMR spectroscopy were unsuccessful due to the fact that these complexes, as their methyl analogues [NEt4][Ni(Me-thiazdt)2],9b are easily oxidized in solution upon exposure to traces of air to the paramagnetic radical anion species. This was confirmed by the investigation of their redox properties by cyclic voltammetry (CV). These studies were carried out in CH3CN using [Bu4N][PF6] as the supporting electrolyte and the redox potentials of (R,R)-4 and (S,S)-4 are collected in Table 1 together with those of [NEt4][Ni(Me-thiazdt)2] and [NBu4]2[Ni(dmit)2]17 for comparison. The Ni complexes 4 exhibit two reversible oxidation waves at E1 = −0.33 V and E2 = +0.25 V vs. SCE, corresponding to the oxidation of the dianionic species into the radical anion and then to the neutral species (Fig. 3). A third irreversible redox process is also observed at Epa3 = +0.86 V vs. SCE (Epa = anodic peak potential) associated with the oxidation of the neutral complex into the radical cation. Comparison with the Ni dithiolene complexes belonging to the same family, [NEt4][Ni(Me-thiazdt)2], and investigated under the same conditions, shows no difference in the redox potentials, indicating that the presence of the –C*HMePh substituent on the thiazole ring has no significant effect. In contrast, comparison with the well-known Ni(dmit)2 derivative shows a cathodic shift of about 150 mV for the potential of the −2/−1 redox process for 4, confirming the electron rich character of the thiazole dithiolene ligands.
| E 1 | E 2 | E pa 3 | |
|---|---|---|---|
| a Not fully reversible process. b E pa/Epc: anodic and cathodic peak potentials. c E values have been determined in CH2Cl2. d DL-bordt stands for bornylenedithiolate. | |||
| 4 | −0.33 | +0.25 | 0.86a |
| [Ni(Me-thiazdt)2]9b | −0.29 | +0.20/0.01b | |
| [Ni(DL-bordt)2]c,d | −1.20 | −0.43 | +1.00a |
| [Ni(dmit)2]17 | −0.175 | +0.34/0.228b | |
 | ||
| Fig. 3 Cyclic voltammogram of (R,R)-4 in CH3CN with 0.1 M Bu4NPF6 as supporting electrolyte at a scan rate of 100 mV s−1. Pt working electrode. | ||
In order to generate the radical anion species, (R,R)-4 and (S,S)-4 were reacted with one equivalent of [Cp2Fe][PF6]. The resulting anionic complexes (R,R)-5 and (S,S)-5, [NEt4][Ni((R)-CHMePh-thiazdt)2] were then crystallized in an acetonitrile–toluene solution, and crystals of the enantiopure (R,R)-5 suitable for an X-ray diffraction study were obtained.
Structural properties
Complex (R,R)-5 crystallizes in the monoclinic system, space group C2 with both Et4N+ cation and [Ni((R)-CHMePh-thiazdt)2]−˙ radical anion located in the general position in the unit cell. The molecular structure represented in Fig. 4 shows a slightly distorted square planar geometry around the Ni atom and the formation of the trans-isomer. Concerning the thiazole skeleton, the C–H groups of the chiral substituents are oriented towards the thione bonds leading to the anti/anti rotamer (Chart 3) as observed for the organic precursor 3 (Chart 2). | ||
| Fig. 4 Molecular structure of the radical anion [Ni((R)-CHMePh-thiazdt)2]˙− in [NEt4][Ni((R)-1-PhEt-thiazdt)2] 5 showing the atom labelling. | ||
 | ||
| Chart 3 | ||
In the solid state, the enantiopure complexes are isolated from each other by the Et4N+ counterion in the ac plane while several short lateral S⋯S intermolecular contacts can be observed along the b axis (Fig. 5). Actually, steric hindrance due to the presence of the bulky N-1-phenylethyl substituent on the nitrogen impedes shorter lateral contacts in the bc plane. The temperature dependence of the magnetic susceptibility for the enantiopure monoanionic radical (R,R)-5 shows a weak Curie-type behavior with a magnetic susceptibility accounting for only 3.8% S = 1/2 species, demonstrating that the radical anions in (R,R)-5 are interacting antiferromagnetically within the chains running along b, to such an extent that the remaining susceptibility in the investigated temperature range (5–300 K) is negligible.
 | ||
| Fig. 5 Top: projection view along the ac plane of the unit cell of the monoanionic radical (R,R)-5 the NEt4 groups have been omitted for clarity. Bottom: view of the shortest S⋯S contacts in the chains of radical anions running along b. | ||
Oxidation of the radical anion species, (R,R)-5 and (S,S)-5, with one more equivalent of [Cp2Fe][PF6] leads to the neutral species [Ni(CHMePh-thiazdt)2] (R,R)-6 and (S,S)-6. These neutral diamagnetic species have been investigated by 1H NMR at room temperature where the signals associated with the benzylic protons are well resolved, compared to those observed for the protected ligands (R)-3 and (S)-3. This indicates the presence of only one rotamer at room temperature, most probably because of the steric hindrance generated by the metallacycles.
UV-vis-NIR spectroelectrochemical investigations
UV-vis-NIR absorption spectra of freshly prepared solutions of the neutral dithiolene species in dichloromethane exhibit absorption bands in the UV-vis region between 250 and 450 nm and also in the near-IR region with a strong absorption band at 1013 nm (ε = 28 454 M−1 cm−1). The gradual reduction of this neutral Ni complex to the monoanionic species induces the growth of a lower energy band in the near-IR region, at 1260 nm, concomitantly with the disappearance of the band at 1013 nm, as shown in Fig. 6. These values are in accordance with those obtained on the Ni(Et-thiazdt)2 analogue, where the neutral and monoanionic species exhibit indeed absorption bands at comparable wavelengths, i.e. 1030 nm (in toluene) and 1219 nm (in CH2Cl2), respectively.18 These low-energy absorption bands are characteristic of square-planar nickel dithiolene complexes and are attributed to a π–π* transition of the HOMO–LUMO character within these neutral species.19 | ||
| Fig. 6 UV-vis-NIR monitoring of the electrochemical reduction of [Ni(CHMePh-thiazdt)2] (R,R)-6 from 0.25 V to −0.2 V in CH2Cl2 with 0.1 M Bu4NPF6 as the supporting electrolyte. | ||
As mentioned in the Introduction, the chiral character of these complexes opens perspectives toward the elaboration of electrochemically driven chiroptical switches.20,21 Accordingly, the optical activity and circular dichroism of the dithiolene complexes, as well as some organic chiral precursors were investigated, in the UV-vis range as well as in the NIR range when possible. The optical activity of two organic precursors, the non-substituted thiazole 1 and the organic protected dithiolate form 3, as well as the radical anion complex 5 are reported in Table 2, as specific and molar optical rotations at three different wavelengths, (436, 546 and 578 nm), in the visible range. Note that both enantiomers of 5, (R,R)-5 and (S,S)-5, exhibit practically no optical rotation (|α| < 0.002) at 365 nm and between 546 nm and 589 nm but show a weak optical activity at 436 nm (|α| < 0.048).
Circular dichroism (CD) spectra in the UV-visible range (Fig. 7) were recorded in CH2Cl2 at room temperature for the neutral complexes (R,R)- and (S,S)-6, together with those of starting thiazoline (R)- and (S)-1 for comparison. These CD spectra of both enantiomers, for 1 and for 6, exhibit mirror-image relationships. The spectrum for the (R)-1 enantiomer exhibits a weak negative band around 340 nm, a positive band at 320 nm and at shorter wavelengths a positive band at 270 nm. This spectrum is similar to the one reported for this compound by Sandström et al.13 The CD spectrum of the (S)-1 enantiomer is the mirror-image of the (R)-1 spectrum. Besides some similarities in the short wavelengths such as a positive band at around 270 nm for (R,R)-6, the CD spectra of the neutral complexes (R,R)- and (S,S)-6 exhibit two weak CD-active bands above 400 nm ((R,R)-6: Δε = −7.22 M−1 cm−1 at 403 nm and Δε = −1.9 M−1 cm−1 at 564 nm).
 | ||
| Fig. 7 CD spectra in CH2Cl2 (inserts: UV-vis absorption spectra) of thiazoline 1 (top) and of neutral complexes 6 (bottom). | ||
In order to investigate the CD spectra of the other redox species such as the dianion in 4 and the radical anion in 5, we set up a thin-layer spectro-electrochemical experiment on the CD spectrometer, starting with a solution of the neutral (R,R)-6 enantiomer (Fig. 8). In the UV-vis region, the CD spectra of the electrogenerated radical anion (R,R)-5 and the dianion (R,R)-4 show some similarities, a positive band at 280 nm, a broad negative band ranging from 300 nm to 455 nm band with two maxima at 320 nm and 430 nm and a broad positive band from 455 nm to 660 nm. Most of these CD-active bands undergo changes in their intensities upon reduction accompanied by slight bathochromic shifts.
 | ||
| Fig. 8 CD spectra monitored electrochemically in CH2Cl2 at 293 K of the neutral (R,R)-6, radical anion (R,R)-5 and dianion (R,R)-4 complexes (insert: CD spectra from 900 to 1030 nm). | ||
The CD spectra of the neutral and the radical anion species have been also investigated in the NIR region (insert Fig. 8), where both compounds are known to absorb (see Fig. 6). Remarkably, the neutral species exhibits a negative CD band centered around 1000 nm, related to its 1013 nm absorption. This CD active band in the NIR disappears upon reduction to the radical anion, while no clear CD signal can be observed for the radical anion species at lower energies, due to spectrophotometer limitations in the wavelength range. Note also that the UV-vis-NIR CD spectra monitored electrochemically exhibit a very good reversibility, as the CD spectrum of the neutral complex (R,R)-6 could be recovered from the dianionic and monoanionic complexes, upon oxidation.
Conclusions
In conclusion, we have described here the synthesis of a novel chiral Ni dithiolene complex where the chirality was provided by the dithiolene ligand, in the close proximity of the complex metallacycles. Electrochemical and spectro-electrochemical experiments showed that these Ni complexes exhibit four redox states from the dianion, radical anion, neutral to the radical cation with different UV-visible-near infrared (UV-vis-NIR) spectra. The spectro-electrochemical circular dichroism (CD) spectroscopy studies revealed changes in several CD active bands when reducing the neutral enantiopure complex (R,R)-6 to the radical anion (R,R)-5 and dianion (R,R)-4. Interestingly, the appearance of a CD-active band in the NIR region is observed in the neutral Ni–dithiolene complex upon oxidation of the radical anion. These results suggest that chiral Ni–dithiolene complexes may be potentially used as redox chiroptical switches both in the UV-vis and NIR regions.21Experimental section
General
All air-sensitive reactions were carried out under an argon atmosphere. Melting points were measured on a Kofler hot-stage apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker AV300III spectrometer and chemical shifts are quoted in parts per million (ppm) referenced to tetramethylsilane. Optical rotation of the enantiopure compounds was determined using a Perkin-Elmer 341 polarimeter. The circular dichroism (CD) spectra were recorded on a Jasco spectropolarimeter J-815. Mass spectra were recorded using a Thermo-fisher Q-Exactive instrument by the Centre Régional de Mesures Physiques de l'Ouest, Rennes. Elemental analyses were performed at the Centre Régional de Mesures Physiques de l'Ouest, Rennes. Tetrahydrofuran was distilled from sodium–benzophenone. Column chromatography was performed using silica gel Merck 60 (70-260 mesh). The spectroelectrochemical setup was performed in 0.2 M CH2Cl2-[NBu4][PF6]. A Cary 5000 spectrophotometer was employed to record the UV-vis-NIR spectra. Cyclic voltammetry were carried out on a 10−3 M solution of the complex in CH2Cl2, containing 0.1 M nBu4NPF6 as the supporting electrolyte. Voltammograms were recorded at 0.1 V s−1 on a platinum disk electrode (A = 1 mm2). The potentials were measured versus the saturated calomel electrode.Syntheses
(R)-1: yield 54%. 1H NMR (300 MHz, CDCl3) δ 3.35 (t, 3H, CH3, J = 6.9 Hz), 6.53 (m, 2H, CH,*CH), 6.88 (d, 1H, CH, J = 4.8 Hz), 7.33 (m, 5H, Ar). 13C NMR (75 MHz, CDCl3) δ 18.4, 56.2, 111.2, 126.8, 128.2, 128.4, 128.8, 138.9, 186.0; HRMS (ESI) calcd for C11H11NNaS2+: 244.02306. Found: 244.0230.
(S)-1: yield 55%. 1H NMR (300 MHz, CDCl3) δ 3.35 (t, 3H, CH3, J = 6.9 Hz), 6.53 (d, 1H, CH, J = 4.8 Hz), 6.59 (q, 1H, CH, J = 6.9 Hz), 6.88 (d, 1H, CH, J = 4.8 Hz), 7.33 (m, 5H, Ar). 13C NMR (75 MHz, CDCl3) δ 18.4, 56.2, 111.7, 126.9, 128.2, 128.4, 128.8, 138.9, 186.9; Anal. calcd for C11H11NS2: C, 59.68; H 5.01; N, 6.33; S, 28.97. Found: C, 59.81; H, 5.01; N, 6.35; S, 28.80; HRMS (ESI) calcd for C11H11NNaS2+: 244.02306. Found: 244.0229.
(R/S)-1: yield 55%. 1H NMR (300 MHz, CDCl3) δ 3.35 (t, 3H, CH3, J = 6.9 Hz), 6.53 (m, 2H, CH,*CH), 6.88 (d, 1H, CH, J = 4.8 Hz), 7.33 (m, 5H, Ar). 13C NMR (75 MHz, CDCl3) δ 18.42, 56.2, 111.2, 126.8, 128.2, 128.4, 128.8, 138.9, 186.0; Anal calcd for C11H11NS2: C, 59.68; H, 5.01; N, 6.33; S, 28.97. Found: C, 59.41; H, 5.03; N, 6.11; S, 28.86; HRMS (ESI) calcd for C11H11NNaS2+: 244.02306. Found: 244.0231.
(R)-3: yield 41%, Mp = 108 °C. 1H NMR (300 MHz, (CDCl3)2 1.97 (d, 3H, CH3, J = 7.2 Hz), 2.19 (broad s, 4H, CH2CN), 2.70 (t, 2H, SCH2, J = 6.9 Hz), 3.09 (t, 2H, SCH2, J = 6.9 Hz), 7.05 (broad signal, 1H, CH), 7.36 (m, 5H, Ar)). 13C NMR (75 MHz, (CDCl3) δ 16.4, 17.4, 18.6, 31.3, 31.4, 57.1, 117.0, 117.2, 126.5, 127.7, 127.8, 128.6, 135.7, 138.7, 188.6). UV-vis (CH2Cl2) λ (nm) (ε [L mol−1 cm−1]) 318 (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 586); Anal. calcd for C17H17N3S4: C, 52.14; H, 4.38; N, 10.73; S, 32.75. Found: C, 52.00; H, 4.42; N, 10.70; S, 32.55. HRMS (ESI) calcd for C17H17N3NaS4+: 414.0203. Found: 414.0201.
586); Anal. calcd for C17H17N3S4: C, 52.14; H, 4.38; N, 10.73; S, 32.75. Found: C, 52.00; H, 4.42; N, 10.70; S, 32.55. HRMS (ESI) calcd for C17H17N3NaS4+: 414.0203. Found: 414.0201.
(S)-3: yield 40%, Mp = 109 °C. 1H NMR (300 MHz, (CDCl3)2 1.99 (d, 3H, CH3, J = 7.2 Hz), 2.20 (broad s, 4H, CH2CN), 2.72 (t, 2H, SCH2, J = 6.9 Hz), 3.11 (t, 2H, SCH2, J = 6.9 Hz), 7.12 (broad signal, 1H, CH), 7.38 (m, 5H, Ar)). 13C NMR (75 MHz, (CDCl3) δ 16.3, 17.3, 18.5, 31.2, 31.3, 57.0, 117.0, 117.2, 126.5, 126.9, 127.6, 128.5, 135.6, 138.5, 188.4); UV-vis (CH2Cl2) λ (nm) (ε [L mol−1 cm−1]) 318 (14918); Anal. calcd for C17H17N3S4: C, 52.14; H, 4.38; N, 10.73; S, 32.75. Found: C, 51.54; H, 4.46; N, 10.64; S, 32.59. HRMS (ESI) calcd for C17H17N3NaS4 [M+Na]+: 414.0203. Found: 414.0203.
(R,S)-3: yield 38%, Mp = 108 °C. 1H NMR (300 MHz, (CDCl3)2 1.97 (d, 3H, CH3, J = 7.2 Hz), 2.19 (broad s, 4H, CH2CN), 2.71 (t, 2H, SCH2, J = 6.9 Hz), 3.08 (t, 2H, SCH2, J = 6.9 Hz), 7.09 (broad signal, 1H, CH), 7.37 (m, 5H, Ar)). 13C NMR (75 MHz, (CDCl3) δ 16.5, 17.5, 18.6, 31.4, 31.6, 57.2, 117.0, 117.1, 126.5, 127.1, 127.8, 128.8, 135.9, 138.8, 188.8). UV-vis (CH2Cl2) λ (nm) (ε [L mol−1 cm−1]) 318 (15227); Anal. calcd for C17H17N3S4: C, 52.14; H, 4.38; N, 10.73; S, 32.75. Found: C, 52.03; H, 4.38; N, 10.73; S, 32.81; HRMS (ESI) calcd for C17H17N3NaS4+: 414.0203. Found: 414.0201.
(S,S)-5. Mp > 250 °C; Anal. calcd for C30H38N3S8Ni: C, 47.67; H, 5.07; N, 5.56. Found: C, 47.63; H, 5.07; N, 5.56. HRMS (ESI) calcd for A−(C22H18N2S8Ni): 623.85947. Found: 623.8596; calcd for the ionization cluster [2A−,Et4N+]−: 1377.87797. Found: 1377.8788. UV-vis (CH2Cl2) λ (nm) (ε [L mol−1 cm−1]) 324 (16442), 366 (18846), 432 (6256), 1252 (13558). [α]25436 = −1677, [Φ]25436 = −12400.
(R,R)-5. Mp > 250 °C; Anal. calcd for C30H38N3S8Ni: C, 47.67; H, 5.07; N, 5.56; S, 33.94. Found: C, 47.50; H, 5.02; N, 5.34; S, 34.36. HRMS (ESI) calcd for A−(C22H18N2S8Ni): 623.85947. Found: 623.8601; calcd for the ionization cluster [2A−,Et4N+]−: 1377.87797. Found: 1377.8798. UV-vis (CH2Cl2) λ (nm) (ε [L mol−1 cm−1]) 323 (15220), 366 (18693), 432 (5720), 1250 (12870). [α]25436 = +1500, [Φ]25436 = +11300.
875), 390 (14462), 432 (5720), 1013 (28454).
| Compound | (S)-3 | (R,R)-5 |
|---|---|---|
| Formula | 2(C17H17N3S4) | C30H38N3NiS8 |
| FW (g mol−1) | 783.15 | 755.88 |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | P212121 | C2 |
| a (Å) | 8.5565(3) | 16.624(4) |
| b (Å) | 11.6085(3) | 7.8685(14) |
| c (Å) | 37.5518(10) | 28.101(5) |
| α (°) | 90 | 90 |
| β (°) | 90 | 102.325(7) |
| γ (°) | 90 | 90 |
| V (Å3) | 3729.95(19) | 3591.2(12) |
| T (K) | 150(2) | 293(2) |
| Z | 4 | 4 |
| D calc (g cm−3) | 1.395 | 1.398 |
| μ (mm−1) | 0.513 | 1.031 |
| Total refls | 33125 |
15925 |
| Uniq. refls (Rint) | 8535 (0.0551) | 7860 (0.0828) |
| Unique refls (I > 2σ(I)) | 7759 | 4396 |
| R 1, wR2 | 0.0365, 0.0756 | 0.0635, 0.1283 |
| R 1, wR2 (all data) | 0.042, 0.078 | 0.1250, 0.1560 |
| GoF | 1.026 | 0.996 |
Acknowledgements
Financial support was obtained from ANR (France) under contract no. 12-BS07-0032-01. CD spectra were acquired on the spectroscopy facility from Biosit (Université de Rennes 1) and X-ray data from the CDIFX facility (ISCR, Université of Rennes1).References
- (a) R. Kato, Bull. Chem. Soc. Jpn., 2014, 87, 355–374 CrossRef CAS; (b) R. Kato, Chem. Rev., 2004, 104, 5319–5346 CrossRef CAS PubMed; (c) B. Garreau-de Bonneval, K. I. Moineau-Chane Ching, F. Alary, T.-T. Bui and L. Valade, Coord. Chem. Rev., 2010, 254, 1457–1467 CrossRef CAS PubMed; (d) M. L. Kirk, R. L. McNaughton and M. E. Helton, Dithiolene Chemistry: Syntheses, Properties and Applications, Prog. Inorg. Chem., 2004, ch. 3, vol. 52, p. 111 Search PubMed.
- (a) U. T. Mueller-Westerhoff, B. Vance and D. I. Yoon, Tetrahedron, 1991, 47, 909–932 CrossRef CAS; (b) B. S. Lim, D. V. Fomitchev and R. H. Holm, Inorg. Chem., 2001, 40, 4257–4262 CrossRef CAS PubMed; (c) R. K. Szilagyi, B. S. Lim, T. Glaser, R. H. Holm, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 9158–9169 CrossRef CAS PubMed; (d) W. Freyer, J. Prakt. Chem., 1986, 328, 253–260 CrossRef CAS.
- (a) H. Kisch, B. Eisen, R. Dinnebier, K. Shankland, W. I. F. David and F. Knoch, Chem. – Eur. J., 2001, 7, 738–748 CrossRef CAS; (b) R. Perochon, C. Poriel, O. Jeannin, L. Piekara-Sady and M. Fourmigué, Eur. J. Inorg. Chem., 2009, 5413–5421 CrossRef CAS.
- S. Debnath, J. F. Bergamini, F. Artzner, C. Mériadec, F. Camerel and M. Fourmigué, Chem. Commun., 2012, 48, 2283–2285 RSC.
- J. Lieffrig, O. Jeannin, P. Auban-Senzier and M. Fourmigué, Inorg. Chem., 2012, 51, 7144–7152 CrossRef CAS PubMed.
- (a) V. Krstic, S. Roth, M. Burghard, K. Kern and G. L. J. A. Rikken, J. Chem. Phys., 2002, 117, 11315–11319 CrossRef CAS PubMed; (b) V. Krstic and G. L. J. A. Rikken, Chem. Phys. Lett., 2002, 364, 51 CrossRef CAS.
- (a) F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2014, 5 DOI:10.1038/ncomms4757; (b) F. Pop, S. Laroussi, T. Cauchy, C. J. Gomez-Garcia, J. D. Wallis and N. Avarvari, Chirality, 2013, 25, 466–474 CrossRef CAS PubMed; (c) F. Pop, P. Auban-Senzier, A. Frackowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS PubMed.
- Q. Ye, T. Akutagawa, T. Endo, S. I. Noro, T. Nakamura and R. G. Xiong, Inorg. Chem., 2010, 49, 8591–8600 CrossRef CAS PubMed.
- (a) S. Eid, M. Guerro and D. Lorcy, Tetrahedron Lett., 2006, 47, 8333–8336 CrossRef CAS PubMed; (b) S. Eid, M. Fourmigué, T. Roisnel and D. Lorcy, Inorg. Chem., 2007, 46, 10647–10654 CrossRef CAS PubMed; (c) A. Filatre-Furcate, N. Bellec, O. Jeannin, P. Auban-Senzier, M. Fourmigué, A. Vacher and D. Lorcy, Inorg. Chem., 2014, 53, 8681–8690 CrossRef CAS PubMed.
- (a) N. Tenn, N. Bellec, O. Jeannin, L. Piekara-Sady, P. Auban-Senzier, J. Íñiguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2009, 131, 16961–16967 CrossRef CAS PubMed; (b) G. Yzambart, N. Bellec, N. Ghassan, O. Jeannin, T. Roisnel, M. Fourmigué, P. Auban-Senzier, J. Íñiguez, E. Canadell and D. Lorcy, J. Am. Chem. Soc., 2012, 134, 17138–17148 CrossRef CAS PubMed; (c) Y. Le Gal, T. Roisnel, P. Auban-Senzier, T. Guizouarn and D. Lorcy, Inorg. Chem., 2014, 53, 8755–8761 CrossRef CAS PubMed.
- S. Eid, T. Roisnel and D. Lorcy, J. Organomet. Chem., 2008, 693, 2755–2760 CrossRef CAS PubMed.
- T. Bsaibess, M. Guerro, Y. Le Gal, D. Sarraf, N. Bellec, M. Fourmigué, F. Barrière, V. Dorcet, T. Guizouarn, T. Roisnel and D. Lorcy, Inorg. Chem., 2013, 52, 2162–2173 CrossRef CAS PubMed.
- J. Roschester, U. Berg, M. Pierrot and J. Sandström, J. Am. Chem. Soc., 1987, 109, 492–507 CrossRef CAS.
- (a) N. Bellec, D. Lorcy, A. Robert, R. Carlier, A. Tallec, C. Rimbaud, L. Ouahab, R. Clérac and P. Delhaes, Adv. Mater., 1997, 9, 1052–1056 CrossRef CAS; (b) N. Bellec, D. Guérin, D. Lorcy, A. Robert, R. Carlier and A. Tallec, Acta Chem. Scand., 1999, 53/10, 861–867 CrossRef PubMed; (c) N. Bellec, D. Lorcy, K. Boubekeur, R. Carlier, A. Tallec, S. Los, W. Pukacki, M. Trybula, L. Piekara-Sady and A. Robert, Chem. Mater., 1999, 11, 3147–3153 CrossRef CAS; (d) D. Guérin, R. Carlier and D. Lorcy, J. Org. Chem., 2000, 65, 6069–6072 CrossRef PubMed.
- C. Roussel, A. Liden, M. Chanon, J. Metzger and J. Sandström, J. Am. Chem. Soc., 1976, 98, 2847–2852 CrossRef CAS.
- (a) N. Svenstrup, K. M. Rasmussen, T. K. Hansen and J. Becher, Synthesis, 1994, 809–812 CrossRef CAS PubMed; (b) N. Svenstrup and J. Becher, Synthesis, 1995, 215–235 CrossRef CAS PubMed.
- R.-M. Olk, A. Röhr, J. Sieler, K. Köhler, R. Kirmse, W. Dietzsch, E. Hoyer and B. Olk, Z. Anorg. Allg. Chem., 1989, 577, 206–216 CrossRef CAS.
- M. C. Aragoni, M. Arca, F. A. Devillanova, F. Isaia, V. Lippolis, A. Mancini, L. Pala, A. M. Z. Slawin and J. D. Woollins, Inorg. Chem., 2005, 44, 9610–9612 CrossRef CAS PubMed.
- U. T. Mueller-Westerhoff and B. Vance, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, UK, 1987 Search PubMed.
- Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, 2nd edn, 2011 Search PubMed.
- J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
Footnote |
| † CCDC 1015857 and 1015858. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01244e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |