
Highly enantioselective Michael reaction employing cycloheptanone and cyclooctanone as nucleophiles†
Ai-Bao
Xia
a,
Long
Zhao
a,
Tao
Wang
a,
Yan-Peng
Zhang
a,
Ai-Guo
Zhong
b,
Dan-Qian
Xu
*a and
Zhen-Yuan
Xu
*a
aCatalytic Hydrogenation Research Centre, State Key Laboratory Breeding Base of Green Chemistry-Synthesis Technology, Zhejiang University of Technology, Hangzhou, 310014, China. E-mail: chrc@zjut.edu.cn; greenchem@zjut.edu.cn
bDepartment of Pharmaceutical and Chemical Engineering, Taizhou College, Linhai Zhejiang, 317000, China
First published on 24th October 2014
Abstract
An organocatalytic Michael reaction of cycloheptanone and cyclooctanone with nitrodienes and nitroolefins catalyzed by a hydroquinine-based primary amine catalyst has been accomplished. The corresponding Michael adducts were obtained in good yields (up to 88%) with good to excellent diastereoselectivities (up to >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and enantioselectivities (up to >99% ee). The absolute configuration of the Michael product was assigned by TDDFT simulation of the ECD spectrum. And the Michael products can be readily converted into analogues of cycloalkano[b] fused pyrrolidines.
:1) and enantioselectivities (up to >99% ee). The absolute configuration of the Michael product was assigned by TDDFT simulation of the ECD spectrum. And the Michael products can be readily converted into analogues of cycloalkano[b] fused pyrrolidines.
Introduction
Over the past decade, chemists have witnessed a rapid development in organocatalysis, a powerful and attractive field involving the synthesis of chiral building blocks, natural products, and biologically active compounds using inexpensive and environmentally benign organocatalysts under mild conditions. Numerous elegant enantiocontrol organocatalytic methodologies have been developed.1 The Michael reaction has been widely used to build valuable carbon–carbon bonds in modern organic chemistry.2 Accordingly, the asymmetric organocatalytic Michael reaction has attracted considerable attention, and significant progress has been widely made over the past 10 years with a diverse combination of Michael donors and acceptors.3 Specifically, the asymmetric organocatalytic Michael addition of carbonyl compounds to nitroalkenes has stimulated extensive interest because the chiral adducts (γ-nitrocarbonyl compounds) serve as key precursors of various key complex organic targets.4Therefore, much effort has been directed to determine new transformations to prepare these powerful intermediates.5 To date, poor enantioselectivity has been achieved in most cases when cycloheptanone or cyclooctanone serves as the Michael donor.6 Recently, Wang reported one example of secondary amine catalysed asymmetric Michael addition of cycloheptanone with nitroolefin, giving excellent enantioselectivity.6e However, numerous biologically active compounds, both naturally occurring and artificial, contain complex seven- and eight-membered carbocycles in their core structures, such as cycloalkano[b]fused pyrrolidines (Fig. 1)7 and α-methylene butyrolactones.8 Consequently, the development of an efficient organocatalytic method for the Michael reaction of cycloheptanone and cyclooctanone with electrophiles with high enantioselectivity remains challenging and indispensable.
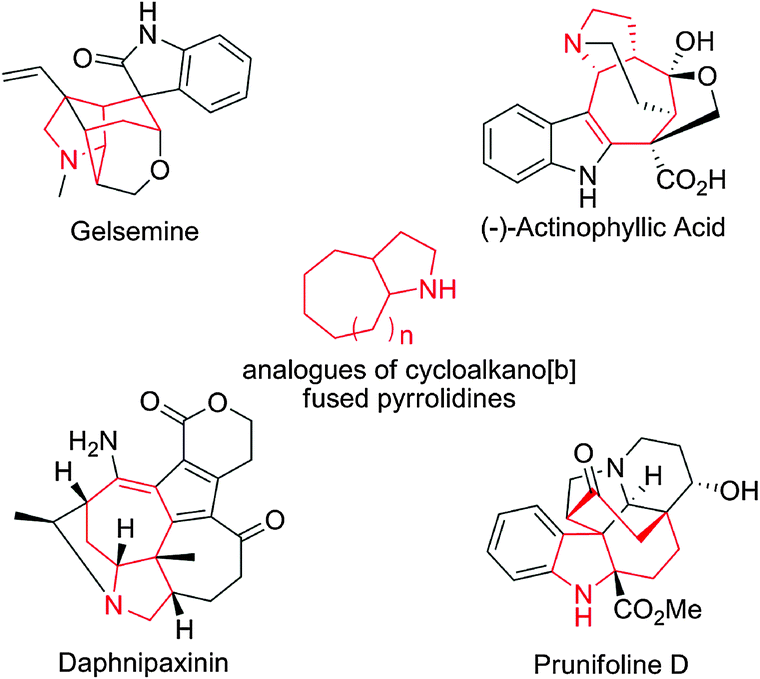 | ||
| Fig. 1 Examples of biologically active compounds containing seven- and eight-membered carbocycles. | ||
Results and discussion
We initiated our study using catalyst 3 for the Michael reaction of cycloheptanone 1a with nitrodiene 2a (Scheme 1).9Table 1 shows that the secondary amine catalysts L-prolinol 3a, L-proline 3b, Jørgensen-Hayashi catalyst 3c, and MacMillan catalyst 3d were almost completely inactive (entries 1 to 4) for the Michael transformation. The multifunctional Xu catalysts 3e and 3f led to moderate catalytic activity (10% and 52% yield) and good enantioselectivity (70% ee and 67% ee) (entries 5 and 6). The chiral primary amine catalyst 3g and the chiral primary amine-thiourea bifunctional catalyst 3h could not promote the reaction (entries 7 and 8). By contrast, higher selectivity was obtained using the Cinchona alkaloid-based primary amine catalysts 3i, 3j, 3k, 3l, and 3m,10 which promoted the formation of 5a with high diastereoselectivity (9:1 dr to 29:1 dr) and enantioselectivity (67% ee to 88% ee), although low yields (11% to 19%) were obtained (entries 9 to 13). The loadings of catalysts 3i, 3k, and 3m were then verified, and the results showed that the yield increased with increasing catalyst loading. The yield and enantioselectivity increased from 19% and 86% ee to 73% and 89% ee, respectively, when the loading amount of catalyst 3m increased from 20 mol% to 50 mol% (entries 13 to 18). Additionally, increasing the reaction temperature led to higher yield but lower selectivity (entry 19 vs. entry 17). Notably, xylene and PhCO2H were found to be the comparatively suitable solvent and additive, respectively, among a series of organic solvents and acids (see the ESI†).11 Thus, an efficient catalyst 3m/PhCO2H/xylene system was developed for the highly enantioselective Michael reaction.
 | ||
| Scheme 1 The catalysts used in this study. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | T (°C) | Yieldd (%) | dre (anti:syn) |
eee (%) (anti) |
| a Unless otherwise stated, the reaction was conducted by stirring in xylene (0.5 ml) using 1a (0.5 mmol) and 2a (0.13 mmol) with 20 mol% catalyst 3 and 20 mol% PhCO2H 4a at room temperature. b 30 mol% catalyst 3 and 30 mol% PhCO2H 4a were used. c 50 mol% catalyst 3 and 50 mol% PhCO2H 4a were used. d Isolated yield. e Determined by HPLC analysis on a Chiralcel AS-H. f The opposite configuration. | |||||
| 1 | 3a | 25 | 5< | n.d. | n.d. |
| 2 | 3b | 25 | Trace | n.d. | n.d. |
| 3 | 3c | 25 | Trace | n.d. | n.d. |
| 4 | 3d | 25 | Trace | n.d. | n.d. |
| 5 | 3e | 25 | 10 | 4:1 |
70 |
| 6 | 3f | 25 | 52 | 3:1 |
67 |
| 7 | 3g | 25 | Trace | n.d. | n.d. |
| 8 | 3h | 25 | Trace | n.d. | n.d. |
| 9 | 3i | 25 | 16 | 24:1 |
85f |
| 10 | 3j | 25 | 12 | 9:1 |
69f |
| 11 | 3k | 25 | 11 | 29:1 |
88 |
| 12 | 3l | 25 | 12 | 9:1 |
67 |
| 13 | 3m | 25 | 19 | 13:1 |
86 |
| 14b | 3i | 25 | 23 | 8:1 |
83f |
| 15b | 3k | 25 | 25 | 8:1 |
89 |
| 16b | 3m | 25 | 37 | 11:1 |
88 |
| 17c | 3k | 25 | 48 | 6:1 |
90 |
| 18c | 3m | 25 | 73 | 7:1 |
89 |
| 19c | 3k | 50 | 70 | 4:1 |
83 |

The scope of the reaction with respect to nitrodiene was explored using the established optimized reaction conditions (Table 2). With cycloheptanone 1a, aryl and alkyl substituents were well tolerated on nitrodiene reactants and provided the respective Michael product 5a–l with moderate to high yields (60% to 88%), diastereoselectivities (2:1 to 9:1 dr), and enantioselectivities (78% to 91% ee) (entries 1 to 12). Apparently, electron-donating and electron-withdrawing substituents on the aromatic ring of the nitrodienes had limited effect on yield and selectivity (entries 1 to 9). Accordingly, the nitrodiene scope was further explored using cyclooctanone 1b as the substrate (entries 13 to 18). In this case, various nitrodiene derivatives with different substitution patterns on the aromatic ring all provided the expected products 5m to 5q with excellent levels of diastereoselectivity (23:1 to 99:1 dr) and enantioselectivity (91% to 94% ee) (entries 13 to 17). Notably, alkyl-substituted nitrodiene could also be used as a reaction partner, and the product 5r was obtained in 83% yield with 99:1 dr and 99% ee (entry 18). Compared with the previous results with 1a, the use of 1b as the substrate generally led to enhanced diastereoselectivities and enantioselectivities.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | n/1 | R1 | Product | Yieldc (%) | drd (anti:syn) |
eed (%) (anti) |
| a Unless otherwise stated, the reaction was conducted by stirring in xylene (0.5 ml) using 1 (0.5 mmol) and 2 (0.13 mmol) with 30 mol% catalyst 3m and 30 mol% PhCO2H 4a at room temperature. In the case of racemic samples, 50 mol% pyrrolidine and 50 mol% PhCO2H 4a were used. b 50 mol% catalyst 3m and 50 mol% PhCO2H 4a were used. c Isolated yield. d Determined by HPLC analysis. | ||||||
| 1 | 1/1a | C6H5 | 5a | 73 | 7:1 |
89 |
| 2 | 1/1a | 4-MeC6H4 | 5b | 72 | 8:1 |
90 |
| 3 | 1/1a | 4-MeOC6H4 | 5c | 67 | 7:1 |
87 |
| 4 | 1/1a | 4-FC6H4 | 5d | 80 | 7:1 |
89 |
| 5 | 1/1a | 4-ClC6H4 | 5e | 75 | 6:1 |
90 |
| 6 | 1/1a | 4-BrC6H4 | 5f | 62 | 6:1 |
88 |
| 7 | 1/1a | 3-FC6H4 | 5g | 78 | 7:1 |
86 |
| 8 | 1/1a | 3-ClC6H4 | 5h | 75 | 6:1 |
91 |
| 9 | 1/1a | 3-BrC6H4 | 5i | 60 | 6:1 |
86 |
| 10 | 1/1a | Pr | 5j | 84 | 8:1 |
80 |
| 11 | 1/1a | iPr | 5k | 78 | 9:1 |
83 |
| 12 | 1/1a | CO2Et | 5l | 88 | 2:1 |
78 |
| 13 | 2/1b | C6H5 | 5m | 45 | 23:1 |
92 |
| 14 | 2/1b | 4-MeC6H4 | 5n | 41 | 47:1 |
93 |
| 15 | 2/1b | 4-FC6H4 | 5o | 23 | 99:1 |
91 |
| 16 | 2/1b | 4-ClC6H4 | 5p | 37 | 99:1 |
92 |
| 17 | 2/1b | 4-BrC6H4 | 5q | 23 | 26:1 |
94 |
| 18 | 2/1b | iPr | 5r | 83 | 99:1 |
99 |

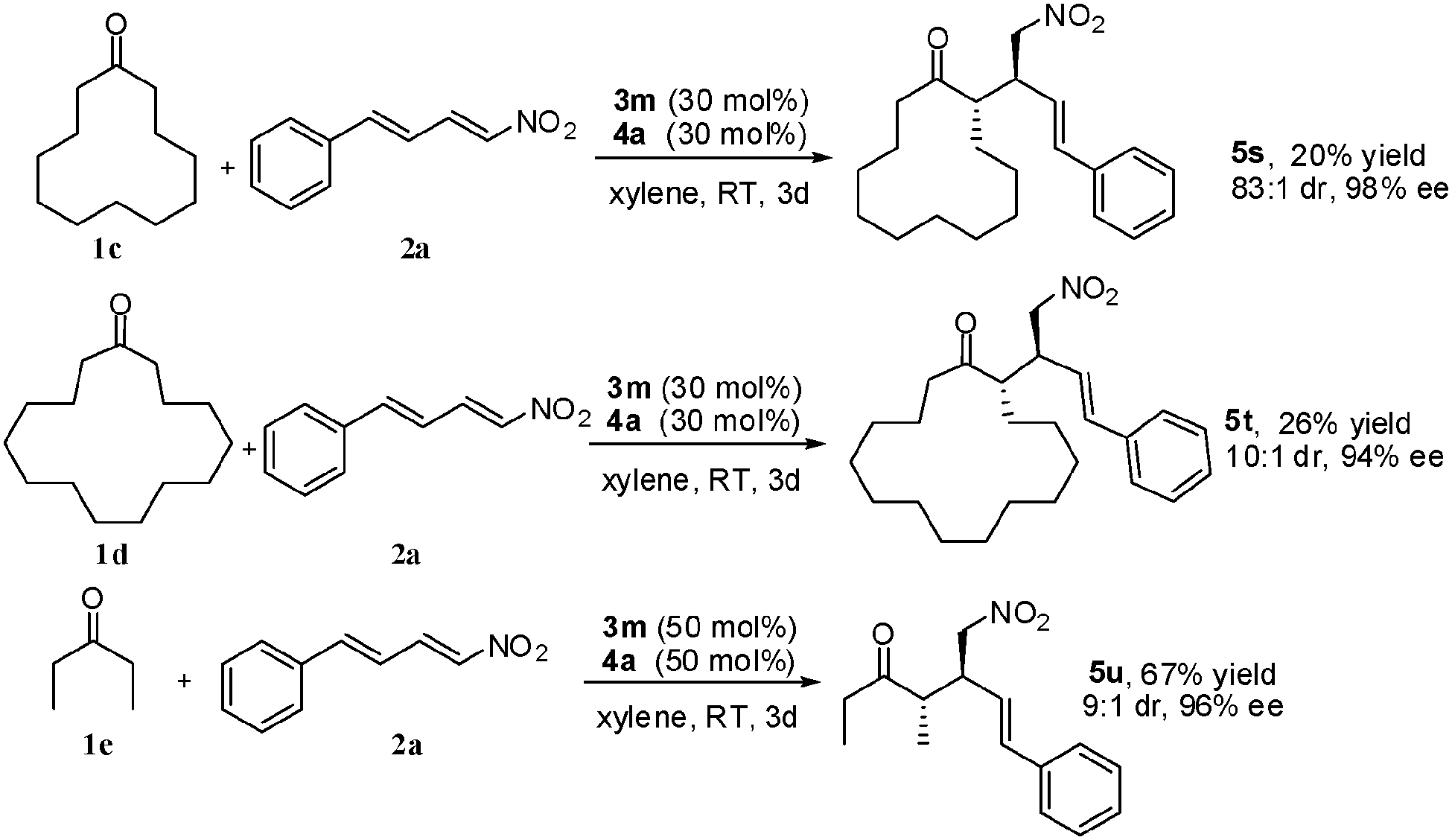
The macrocyclic ketones 1c and 1d could also readily participate in the Michael transformation as nucleophiles, which resulted in the additional products 5s and 5t with high ee values of 98% and 94% and dr values of 83:1 and 10:1, respectively. Acyclic ketone 1e also served as a suitable carbon nucleophile for the Michael reaction, and afforded the corresponding adduct 5u with good results in terms of yield (67%), diastereoselectivity (9:1 dr), and enantioselectivity (96% ee).
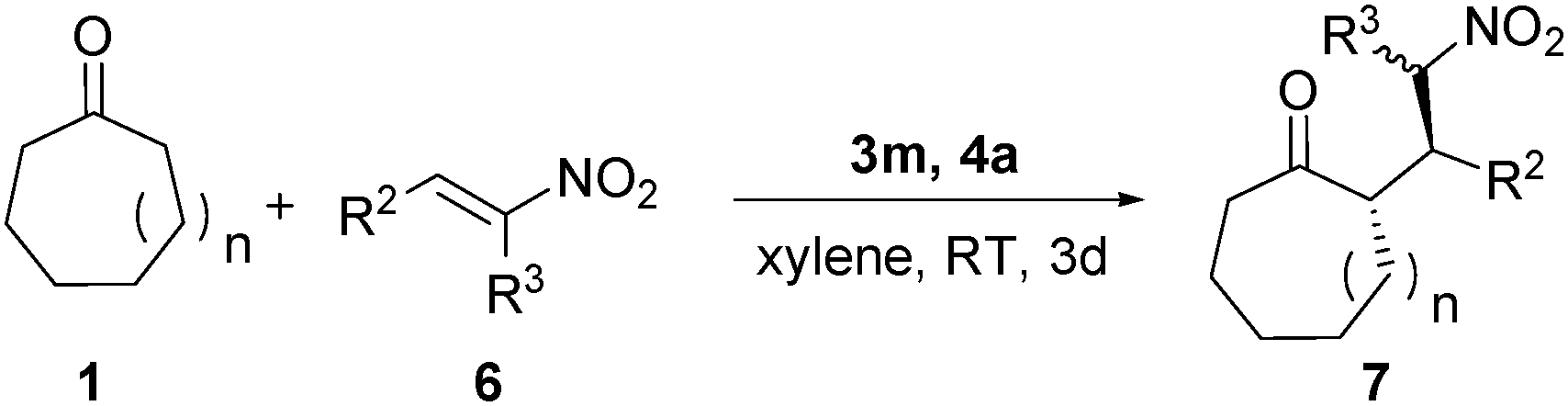
With the above success, the reaction scope was further extended to nitroolefins. As illustrated in Table 3, the reaction of cycloheptanone 1a or cyclooctanone 1b with nitroolefins were performed under the above optimal reaction conditions. The reactions proceeded smoothly in moderate to high yields (up to 84% yield), high to excellent diastereoselectivities (up to >100:1 dr) and good to excellent enantioselectivities (up to >99 ee) (entries 1 to 15). It is noteworthy that reactions between 1b and nitroolefins gave better results than those nitrodienes that were used as the nucleophiles (Table 3, entries 9 to 15 vs.Table 2, entries 13 to 18), and reactions between 1a and nitroolefins obtained better diastereoselectivities than those between 1a and nitrodienes (Table 3, entries 1 to 8 vs.Table 2, entries 1 to 9). The reactions of β-disubstituted nitroalkenes including β-methyl nitroalkene (Table 3, entries 16 and 17) reacted successfully to give the corresponding products 7p and 7q in good to high diastereoselectivities with high to excellent enantioselectivities (Scheme 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | n/1 | R2 | R3 | Product | Yieldc (%) | drd (anti:syn) |
eed (%) (anti) |
| a Unless otherwise stated, the reaction was conducted by stirring in xylene (0.5 ml) using 1 (0.5 mmol) and 6 (0.13 mmol) with 30 mol% catalyst 3m and 30 mol% PhCO2H 4a at room temperature. In the case of racemic samples, 50 mol% pyrrolidine and 50 mol% PhCO2H 4a were used. b 50 mol% catalyst 3m and 50 mol% PhCO2H 4a were used. c Isolated yield. d Determined by HPLC analysis. | |||||||
| 1 | 1/1a | C6H5 | H | 7a | 70 | 19:1 |
89 |
| 2 | 1/1a | 4-MeC6H4 | H | 7b | 69 | 30:1 |
84 |
| 3 | 1/1a | 4-MeOC6H4 | H | 7c | 84 | 20:1 |
90 |
| 4 | 1/1a | 4-FC6H4 | H | 7d | 76 | 38:1 |
92 |
| 5 | 1/1a | 4-ClC6H4 | H | 7e | 79 | 24:1 |
87 |
| 6 | 1/1a | 4-BrC6H4 | H | 7f | 79 | 38:1 |
83 |
| 7 | 1/1a | 3-MeOC6H4 | H | 7g | 75 | 22:1 |
81 |
| 8 | 1/1a | 3-BrC6H4 | H | 7h | 50 | 57:1 |
86 |
| 9 | 2/1b | C6H5 | H | 7i | 67 | 86:1 |
99 |
| 10 | 2/1b | 4-MeC6H4 | H | 7j | 60 | 59:1 |
95 |
| 11 | 2/1b | 4-MeOC6H4 | H | 7k | 57 | >100:1 |
99 |
| 12 | 2/1b | 4-FC6H4 | H | 7l | 39 | 43:1 |
99 |
| 13 | 2/1b | 4-BrC6H4 | H | 7m | 54 | 26:1 |
99 |
| 14 | 2/1b | 3-ClC6H4 | H | 7n | 58 | >100:1 |
>99 |
| 15 | 2/1b | 3-BrC6H4 | H | 7o | 50 | 16:1 |
98 |
| 16 | 1/1a | C6H5 | Me | 7p | 34 | 3:1 |
93 |
| 17 | 2/1b | C6H5 | Me | 7q | 38 | 8:1 |
99 |
 | ||
| Scheme 2 Further investigation of the substrate scope. | ||
The absolute configuration (AC) of the Michael product 5a was determined to be S, S by comparing the experimental CD spectrum with the results of time-dependent density functional theory (TDDFT) calculations of electronic circular dichroism (ECD) spectra.11,12 As shown in Fig. 2 (Fig. S6, ESI†), in the selected data in the 200–390 nm UV region, the experimental CD spectrum is consistent with the calculated data of 1-SS. Then, a transition state model was proposed (Scheme 3). Nitrodiene 2a was activated well through the hydrogen-bonding interaction between the protonated bridgehead nitrogen atom of 3m and the nitro group of 2a. Therefore, the enamine formed from 3m and 1a attacked the activated 2a from the Si face to afford the major stereoisomer of Michael adduct 5a with the configuration of (S, S).
 | ||
| Fig. 2 Experimental (dotted trace) and calculated ECD spectra (full trace) of the Michael product 5a. | ||
 | ||
| Scheme 3 Proposed transition state for the reaction. | ||
The enantioselective Michael reaction can be performed successfully on the gram-scale to obtain 2.09 g of 5a (73% yield) with the same diastereoselectivity and enantioselectivity under modified conditions (Scheme 4). The synthetic utility of this Michael reaction was also demonstrated in the synthesis of chiral cycloalkano[b]fused pyrrolidine 9a (Scheme 4).13 The transformation involves the Zn/HCl-mediated reductive cyclization of adduct 5a to obtain imine 8a in 95% yield. A reduction of imine 8a with NaBH4 followed by Bn protection resulted in 9a with good overall yield and without racemization.
 | ||
| Scheme 4 Enantioselective gram-scale synthesis of 5a, and derivatization of Michael adduct 5a to cycloalkano[b]fused pyrrolidine 9a.11 | ||
Conclusions
In summary, an organocatalytic enantioselective Michael reaction of nucleophiles, cycloheptanone or cyclooctanone, with nitrodienes and nitroolefins catalyzed by hydroquinine-based primary amine catalysts has been established. The corresponding adducts were obtained in good yields (up to 88%) with good to excellent diastereoselectivities (up to >100:1 dr) and good to excellent enantioselectivities (up to >99% ee). The product can be readily converted into analogs of cycloalkano[b] fused pyrrolidines, which further enhances the utility of this transformation for the synthesis of potentially valuable chiral molecules.
Experimental section
General information
The 1H NMR and 13C NMR spectra were recorded in deuterated solvents and are reported in ppm relative to tetramethylsilane. GC-MS experiments were performed on a GC system with a mass selective detector. HRMS data were measured using a TOF mass spectrometer. Column chromatography and flash chromatography experiments were performed on silica gel (200–300 mesh) eluting with ethyl ether and petroleum ether. TLC experiments were carried out on glass-backed silica plates. In each case, the enantiomeric ratio was determined on a chiral column in comparison with authentic racemates by chiral HPLC. Chemicals were used without purification as commercially available. Nitrodienes14 and organocatalysts 3d,153e–3f,163h,173i–3l,183m19 were synthesized according to literature.Typical experimental procedure for the Michael reaction
Xylene (0.5 mL) was added to a mixture of cycloheptanone 1a or cyclooctanone 1b (0.5 mmol) with nitrodienes 2 or nitroolefins 6 (0.126 mmol) in the presence of a 30 mol% or 50 mol% catalyst 3m and 30 mol% or 50 mol% PhCO2H 4a at room temperature with vigorous stirring. The reaction conversion was monitored by GC-MS. After three days, the reaction mixture was extracted with DCM, washed with water, dried and concentrated. The residue was purified by flash chromatography on silica gel (ether/petroleum ether = 1:4 to 1:2 as an eluent) to give the slightly white solid of the Michael addition products 5a–5i, 5m–5q, 7a–7b, 7e–7f, 7i–7k, 7n–7q, the slightly yellow liquid products 5j–5l, 5r, 7g and colorless oil 7c, 7l. The enantiomeric excesses (% ee) were determined by HPLC analysis using chiral stationary phases.
Acknowledgements
This work was supported by the NSFC (21202149), the Zhejiang Natural Science Foundation (Y4110348), the Foundation of Zhejiang Education Committee (Y201225109), the Foundation of Zhejiang Key Course of Chemical Engineering and Technology, and the Foundation of Zhejiang Key Laboratory of Green Pesticides and Cleaner Production Technology.Notes and references
- For selected books and reviews on organocatalysis, see: (a) Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, ed. A. Berkessel and H. Gröger, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) B. List and J. W. Yang, Science, 2006, 313, 1584 CrossRef CAS PubMed; (d) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. C. Vo, Drug Discovery Today, 2007, 12, 8 CrossRef CAS PubMed; (e) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (f) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS PubMed; (g) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed; (h) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (i) E. Marqués-López, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC; (j) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605 CrossRef CAS PubMed; (k) J. Alemán and S. Cabrera, Chem. Soc. Rev., 2013, 42, 774 RSC.
- Cinchona Alkaloids in Synthesis and Catalysis, ed. C. E. Song and P. D. Magnus, Wiley-VCH, Weinheim, 2009 Search PubMed.
- For selected reviews, see: (a) R. Ballini, G. Bosica, D. Fiorini, A. Palmieri and M. Petrini, Chem. Rev., 2005, 105, 933 CrossRef CAS PubMed; (b) D. Almasi, D. A. Alonso and C. Najera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS PubMed; (c) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS; (d) J. L. Vicario, D. Badia and L. Carrillo, Synthesis, 2007, 2065 CrossRef CAS PubMed; (e) B. Westermann, M. Ayaz and S. S. van Berkel, Angew. Chem., Int. Ed., 2010, 49, 846 CrossRef CAS PubMed; (f) J. Wang, P. Li, P. Y. Choy, A. S. C. Chan and F. Y. Kwong, ChemCatChem, 2012, 4, 917 CrossRef CAS; (g) C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988 RSC.
- For selected reviews, see: (a) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS; (b) S. Sulzer-Mosse and A. Alexakis, Chem. Commun., 2007, 3123 RSC; (c) D. Roca-Lopez, D. Sadaba, I. Delso, R. P. Herrera, T. Tejero and P. Merino, Tetrahedron: Asymmetry, 2010, 21, 2561 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (b) Y. Zhang and W. Wang, Catal. Sci. Technol., 2012, 2, 42 RSC.
- (a) P. Kotrusz, S. Toma, H.-G. Schmalz and A. Adler, Eur. J. Org. Chem., 2004, 1577 CrossRef CAS; (b) J. Wang, H. Li, B. Lou, L. Zu, H. Guo and W. Wang, Chem. – Eur. J., 2006, 12, 4321 CrossRef CAS PubMed; (c) L. Gu, Y. Wu, Y. Zhang and G. Zhao, J. Mol. Catal. A: Chem., 2007, 263, 186 CrossRef CAS PubMed; (d) D.-Q. Xu, H.-D. Yue, S.-P. Luo, A.-B. Xia, S. Zhang and Z.-Y. Xu, Org. Biomol. Chem., 2008, 6, 2054 RSC; (e) D.-Z. Xu, S. Shi and Y. Wang, Eur. J. Org. Chem., 2009, 4848 CrossRef CAS; (f) C. Wang, C. Yu, C. Liu and Y. Peng, Tetrahedron Lett., 2009, 50, 2363 CrossRef CAS PubMed; (g) S. Belot, A. Quintard, N. Krause and A. Alexakis, Adv. Synth. Catal., 2010, 352, 667 CrossRef CAS; (h) A. Lu, R. Wu, Y. Wang, Z. Zhou, G. Wu, J. Fang and C. Tang, Eur. J. Org. Chem., 2011, 122 CrossRef CAS; (i) Y.-M. Chuan, L.-Y. Yin, Y.-M. Zhang and Y.-G. Peng, Eur. J. Org. Chem., 2011, 578 CrossRef CAS; (j) M. Tsakos, M. Trifonidou and C. G. Kokotos, Tetrahedron, 2012, 68, 8630 CrossRef CAS PubMed; (k) V. Gauchot, J. Gravel and A. R. Schmitzer, Eur. J. Org. Chem., 2012, 6280 CrossRef CAS.
- For seven-membered carbocycle example, see: (a) K. E. O. Ylijoki and J. M. Stryke, Chem. Rev., 2012, 113, 2244 CrossRef PubMed; (b) C. L. Martin, L. E. Overman and J. M. Rohde, J. Am. Chem. Soc., 2008, 130, 7568 CrossRef CAS PubMed; (c) T. B. Dunn, J. M. Ellis, C. C. Kofink, J. R. Manning and L. E. Overman, Org. Lett., 2009, 11, 5658 CrossRef CAS PubMed; (d) C. L. Martin, L. E. Overman and J. M. Rohde, J. Am. Chem. Soc., 2010, 132, 4894 CrossRef CAS PubMed; (e) D. S. Belov, E. R. Lukyanenko, A. V. Kurkin and M. A. Yurovskaya, Tetrahedron, 2011, 67, 9214 CrossRef CAS PubMed; (f) D. S. Belov, E. R. Lukyanenko, A. V. Kurkin and M. A. Yurovskaya, J. Org. Chem., 2012, 77, 10125 CrossRef CAS PubMed , for eight-membered carbocycle example, see: ; (g) G. Mehta and V. Singh, Chem. Rev., 1999, 99, 881 CrossRef CAS PubMed; (h) W. G. Earley, J. E. Jacobsen, A. Madin, G. P. Meier, C. J. O'Donnell, T. Oh, D. W. Old, L. E. Overman and M. J. Sharp, J. Am. Chem. Soc., 2005, 127, 18046 CrossRef CAS PubMed; (i) H. Zhou, H.-P. He, N.-C. Kong, Y.-H. Wang, X.-D. Liu and X.-J. Hao, Helv. Chim. Acta, 2006, 89, 515 CrossRef CAS; (j) K.-H. Lim, O. Hiraku, K. Komiyama, T. Koyano, M. Hayashi and T.-S. Kam, J. Nat. Prod., 2007, 70, 1302 CrossRef CAS PubMed; (k) K.-H. Lim and T.-S. Kam, Phytochemistry, 2008, 69, 558 CrossRef CAS PubMed.
- (a) J. A. Marshall and N. Cohen, J. Org. Chem., 1965, 30, 3475 CrossRef CAS; (b) J. W. Patterson and J. E. Mcmurry, Chem. Commun., 1971, 488 RSC; (c) S. Krihnamurthy and H. C. Brown, J. Am. Chem. Soc., 1976, 98, 3394 Search PubMed; (d) T. F. Murray, E. G. Samsel, V. Varma and J. R. Norton, J. Am. Chem. Soc., 1981, 103, 7520 CrossRef CAS; (e) M. Ando, H. Kusaka, H. Ohara, K. Takase, H. Yamaoka and Y. Yanagit, J. Org. Chem., 1989, 54, 1952 CrossRef CAS; (f) Y. Higuchi, F. Shimoma and M. Ando, J. Nat. Prod., 2003, 66, 810 CrossRef CAS PubMed.
- For selected review, see: (a) R. Ballini, N. Araújo, M. V. Gil, E. Román and J. A. Serrano, Chem. Rev., 2013, 113, 3493 CrossRef CAS PubMed ; For selected examples of Michael reaction to nitrodienes, see: ; (b) B. Tan, P. J. Chua, Y. Li and G. Zhong, Org. Lett., 2008, 10, 2437 CrossRef CAS PubMed; (c) S. Belot, A. Massaro, A. Tenti, A. Mordini and A. Alexakis, Org. Lett., 2008, 10, 4557 CrossRef CAS PubMed; (d) H. Ma, K. Liu, F.-G. Zhang, C.-L. Zhu, J. Nie and J.-A. Ma, J. Org. Chem., 2010, 75, 1402 CrossRef CAS PubMed; (e) Z.-B. Li, S.-P. Luo, Y. Guo, A.-B. Xia and D.-Q. Xu, Org. Biomol. Chem., 2010, 8, 2505 RSC.
- For selected examples, see: (a) S. H. McCooey and S. J. Connon, Org. Lett., 2007, 9, 599 CrossRef CAS PubMed; (b) J.-W. Xie, W. Chen, R. Li, M. Zeng, W. Du, L. Yue, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Angew. Chem., Int. Ed., 2007, 46, 389 CrossRef CAS PubMed.
- For more details, please see the ESI†.
- For selected reviews, see: (a) X.-C. Li, D. Ferreira and Y. Ding, Curr. Org. Chem., 2010, 14, 1678 CrossRef CAS PubMed ; For selected examples of the use of this technique to assign the absolute configurations of chiral organic molecules, see: ; (b) F. Furche, R. Ahlrichs, C. Wachsmann, E. Weber, A. Sobanski, F. Vogtle and S. Grimme, J. Am. Chem. Soc., 2000, 122, 1717 CrossRef CAS; (c) C. Diedrich and S. Grimme, J. Phys. Chem. A, 2003, 107, 2524 CrossRef CAS.
- (a) I. H. Sanchez, M. I. Larraza, I. Rojas, F. Kuri Brena, H. J. Flores and K. Jankowski, Heterocycles, 1985, 23, 3033 CrossRef CAS; (b) W. Glassco, J. Suchocki, C. George, B. R. Martin and E. L. May, J. Med. Chem., 1993, 36, 3381 CrossRef CAS; (c) T. J. Dickerson, T. Lovell, M. M. Meijler, L. Noodleman and K. D. Janda, J. Org. Chem., 2004, 69, 6603 CrossRef CAS PubMed; (d) N. Ruiz, E. Reyes, J. L. Vicario, D. BadÍa, L. Carrillo and U. Uria, Chem. – Eur. J., 2008, 14, 9357 CrossRef CAS PubMed; (e) S. V. Pansare, R. Lingampally and R. L. Kirby, Org. Lett., 2010, 12, 556 CrossRef CAS PubMed; (f) P. Chauhana and S. S. Chimnia, Adv. Synth. Catal., 2011, 353, 3203 CrossRef.
- S. Belot, A. Massaro, A. Tenti, A. Mordini and A. Alexakis, Org. Lett., 2008, 10, 4557 CrossRef CAS PubMed.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- D. Q. Xu, A. B. Xia, S. P. Luo, J. Tang, S. Zhang, J. R. Jiang and Z. Y. Xu, Angew. Chem., Int. Ed., 2009, 48, 3821 CrossRef CAS PubMed.
- X.-J. Zhang, S.-P. Liu, J.-H. Lao, G.-J. Du, M. Yan and A. S. C. Chan, Tetrahedron: Asymmetry, 2009, 20, 1451 CrossRef CAS PubMed.
- B. Vakulya, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967 CrossRef CAS PubMed.
- S. H. McCooey and S. J. Connon, Org. Lett., 2007, 9, 599 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, NMR spectra and HPLC spectra. See DOI: 10.1039/c4nj01206b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |