
New thermally stable energetic materials: synthesis and characterization of guanylhydrazone substituted furoxan energetic derivatives†
Bo
Wu
a,
Hongwei
Yang
*a,
Qiuhan
Lin
ab,
Zhixin
Wang
a,
Chunxu
Lu
a and
Guangbin
Cheng
*a
aSchool of Chemical Engineering, Nanjing University of Science and Technology, Xiaolingwei 200, Nanjing, P. R. China. E-mail: hyang@mail.njust.edu.cn; gcheng@mail.njust.ed.cn
bSchool of Materials Science & Engineering, Beijing Institute of Technology, 5 South Zhongguancun Street, Beijing, P. R. China
First published on 18th September 2014
Abstract
New guanylhydrazone substituted furoxan energetic derivatives were synthesized via the condensation reactions of 3-methyl-4-furoxancarbaldehyde with aminoguanidine derivatives. The resulting compounds 1–5 were well characterized using IR spectroscopy, multinuclear NMR spectroscopy, differential scanning calorimetry, thermogravimetry analysis as well as elemental analysis. Additionally, the structures of compounds 1, 2 and 5 were confirmed by single crystal X-ray diffraction. Except for the compounds 1 and 2, all the remaining products exhibit good thermal stabilities with decomposition temperatures above 200 °C. The detonation pressure values calculated for these compounds range from 17.0 to 28.3 GPa, and the detonation velocities range from 6906 to 8210 m s−1. These values suggest that the guanylhydrazone substituted furoxan energetic derivatives could be potential candidates for thermally stable energetic materials.
Introduction
In recent years, a new class of energetic compounds containing a large fraction of nitrogen has attracted considerable interest.1 In contrast to conventional energetic materials, the energy of these high nitrogen content energetic compounds is derived from their very high positive heat of formation directly attributed to a large number of inherently energetic N–N and C–N bonds.2 Moreover, their high level of environmental compatibility is another advantage over the traditional energetic materials.3 Nitrogen-rich building blocks such as guanidine,4 furazan,5 1,2,4,5-tetrazine,6 tetrazole,7 or triazole8 are essential units in the design and synthesis high nitrogen energetic materials. Among them, furoxan (1,2,5-oxadiazole-2-oxide) is a N-oxide derivative of furazan. It is also a highly energetic heterocycle used in the field of energetic materials. The combination of N-oxide and “latent” a nitro-group structure could enhance the detonation performances of the energetic furoxan derivatives.9 Additionally, guanidine-containing compounds such as guanidine, aminoguanidine, diaminoguanidine, and triaminoguanidine are important starting materials for the preparation of energetic organic salts. Generally, they pair with the energetic anions to form new energetic salts. These energetic salts oftentimes possess high positive heats of formation and high thermal stabilities. Typical highly thermally stable guanidine-containing energetic salts5c,8d,9a are shown in Scheme 1.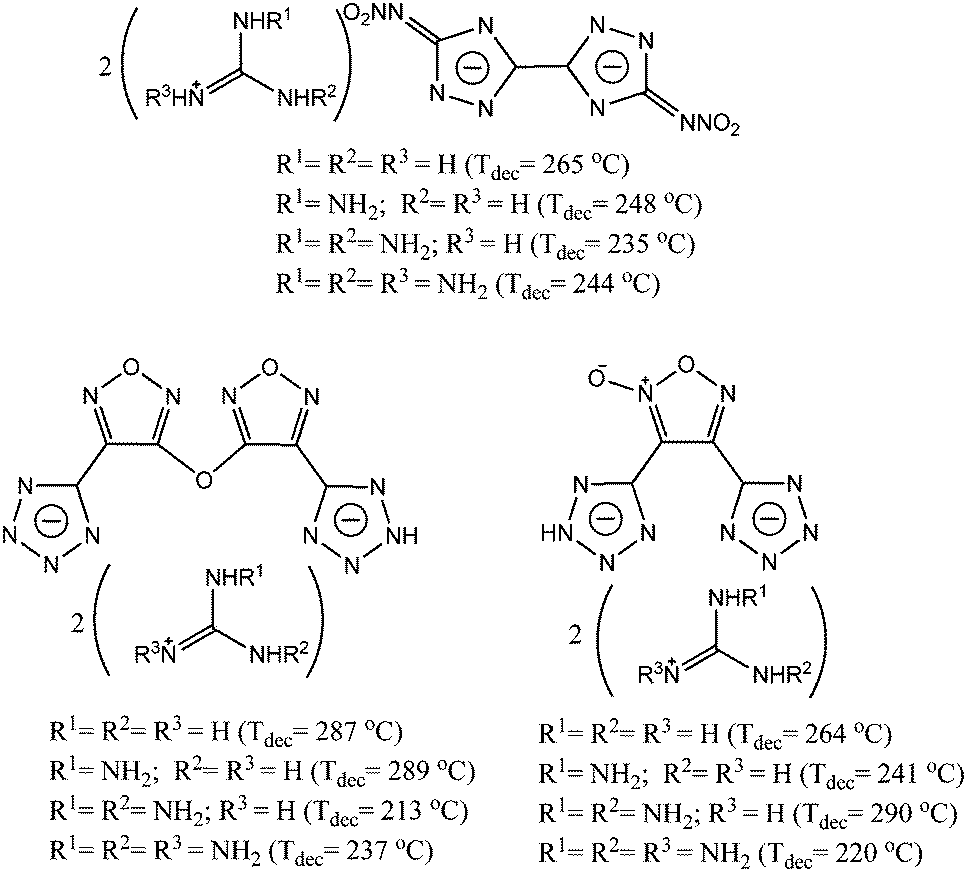 | ||
| Scheme 1 Highly thermally stable energetic salts containing the guanidine moiety. | ||
Guanylhydrazone derivatives have been widely used in medicine field. They can be used as antibacterials, antihypertensives, antitumor agents and so on.10 However, few guanylhydrazones have been reported in the field of energetic materials. In our previous study,9d the condensation reaction of the amino group in 1,5-diaminotetrazole with aldehyde group in 3-methyl-4-furoxancarbaldehyde has been investigated. Herein, the reactions of hydrazine groups in aminoguanidine derivatives with 3-methyl-4-furoxancarbaldehyde are further studied. The resulting new guanylhydrazone substituted furoxan derivatives are characterized for their structural aspects, thermal behaviour and explosive properties using experimental and theoretical methods.
Results and discussion
Synthesis
As shown in Scheme 2, 3-methyl-4-furoxancarbaldehyde was synthesized according to the literature.11 Starting with a 1.2-fold excess of 3-methyl-4-furoxancarbaldehyde, treatment of aminoguanidine salts such as aminoguanidine hydrochloride, aminoguanidium nitrate and aminoguanidinium perchlorate led to mono-guanylhydrazone furoxan derivatives in a high yield (above 90%). After synthesis of these mono-guanylhydrazone furoxan derivatives, the preparation of bisguanylhydrazones and trisguanylhydrazones was also investigated. The desired 1,3-bis-(3-methylfuroxan-4-methyleneamino)guanidine (4) and 1,2,3-tris-(3-methylfuroxan-4-methyleneamino)guanidine (5) were obtained via the reactions of 4-fold excess and 6-fold excess of 3-methyl-4-furoxancarbaldehyde with 1,3-diaminoguanidine monohydrochloride and triaminoguanidine nitrate in the yields of 55% and 85%, respectively.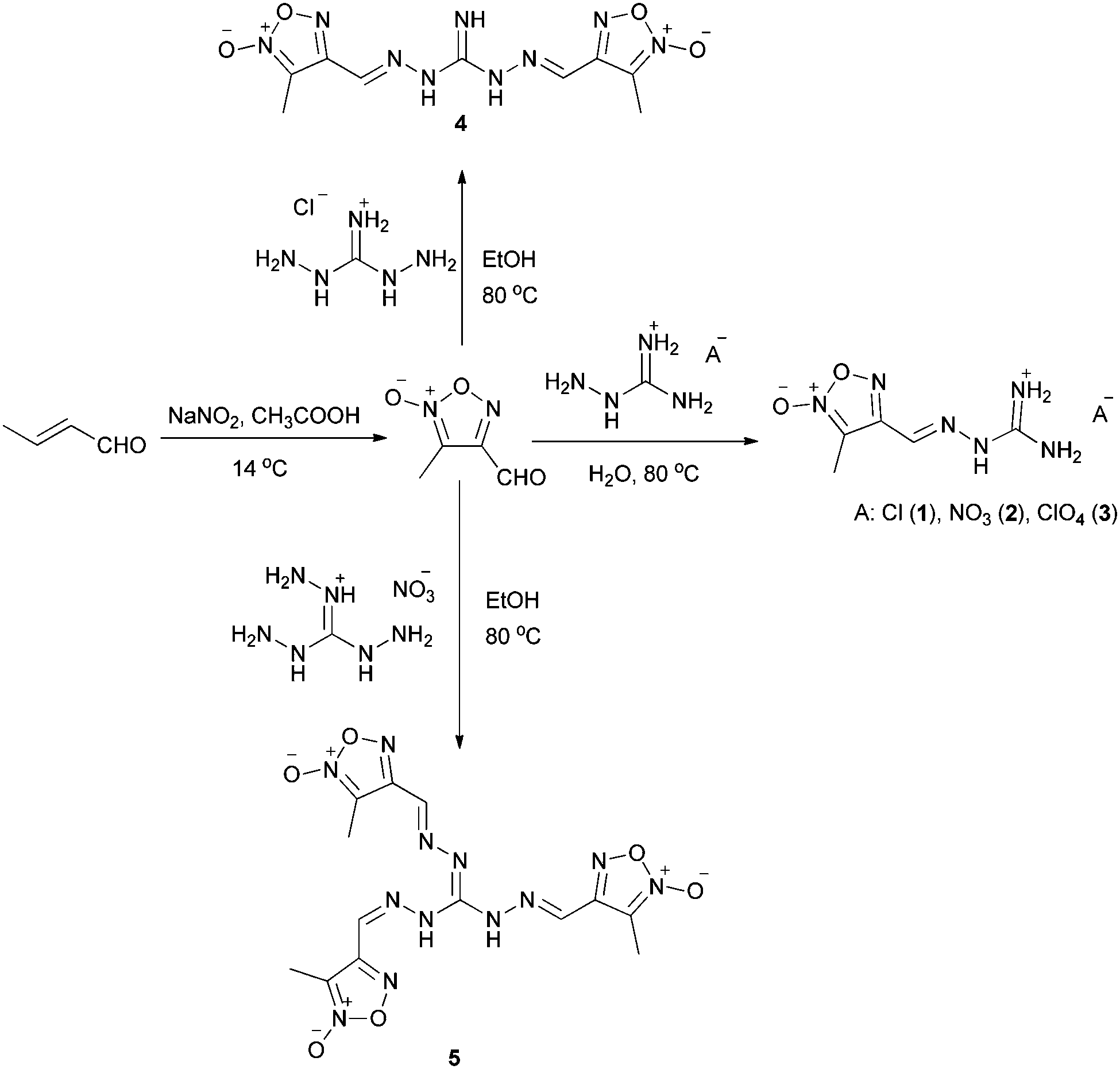 | ||
| Scheme 2 Synthesis of compounds 1–5. | ||
NMR spectroscopy
All compounds were investigated using 1H, 13C spectroscopy. Additionally, the 15N NMR spectra were recorded for compounds 1, 2 and 3. The multinuclear NMR spectra were measured in DMSO-d6 and the chemical shifts are given with respect to DMSO or CH3NO2 as the external standard. The 1H and 13C NMR spectra of all compounds are given in the Electronic Supplementary Information (Fig. S1–S10, ESI†).In the 1H NMR spectra, for all compounds, the proton signals of the methyl group are found at δ = 2.37 to 2.43 ppm. One singlet for the hydrogen atom of –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– can be found at the chemical shifts of 8.29 to 8.66 ppm. The hydrogen signals –NH– for all compounds appear between δ = 11.20 and 12.87 ppm. The carbon atoms in the methylfuroxan moiety occur as four singlets, and the assignments agree with values of similar compounds reported in the literature.9d,11 In compounds 1–3, the carbon atoms of the aminoguanidine-containing moiety are identified at δ = 156.8, 156.4 and 156.4 ppm. The carbon atom in triaminoguanidine structure occurs at δ = 153.9 ppm in compound 5. For compound 4, the carbon atom of diaminoguanidine moiety appears at δ = 154.9 ppm, which is identical to that of the carbon atom bonded with N-oxide nitrogen atom in the furoxan ring. The value agrees with that reported in the literature for similar compounds.4c,5f,6e,9a,c
N– can be found at the chemical shifts of 8.29 to 8.66 ppm. The hydrogen signals –NH– for all compounds appear between δ = 11.20 and 12.87 ppm. The carbon atoms in the methylfuroxan moiety occur as four singlets, and the assignments agree with values of similar compounds reported in the literature.9d,11 In compounds 1–3, the carbon atoms of the aminoguanidine-containing moiety are identified at δ = 156.8, 156.4 and 156.4 ppm. The carbon atom in triaminoguanidine structure occurs at δ = 153.9 ppm in compound 5. For compound 4, the carbon atom of diaminoguanidine moiety appears at δ = 154.9 ppm, which is identical to that of the carbon atom bonded with N-oxide nitrogen atom in the furoxan ring. The value agrees with that reported in the literature for similar compounds.4c,5f,6e,9a,c
The 15N NMR spectra for compounds 1 and 3 show five singlet resonances. And six well-resolved resonances are detected in the 15N NMR of compound 2 (Fig. 1). 15N signal assignments are based on the theoretical and experimental data in the literature.4a,12 Within their similar guanylhydrazone structures, three 15N signals are observed at the high field width ranging from −52.93 to −54.55 ppm. The signals of –NH– group of compounds 1–3 are found at δ = −229.67 (1), −231.45 (2), and −231.78 (3), respectively. The signals of –CN– group bonded with the furoxan ring appear at δ = −52.93 (1), −53.23 (2), and −54.55 (3). Within the furoxan ring, the N-oxide (N1) resonates downfield appearing around −6 ppm while another nitrogen atom appears around −21 ppm due to the electron-withdrawing inductive effect of oxygen atom. In the spectra of 2, signals at lowest field are assigned to nitrate anions appearing at δ = −4.46 ppm.
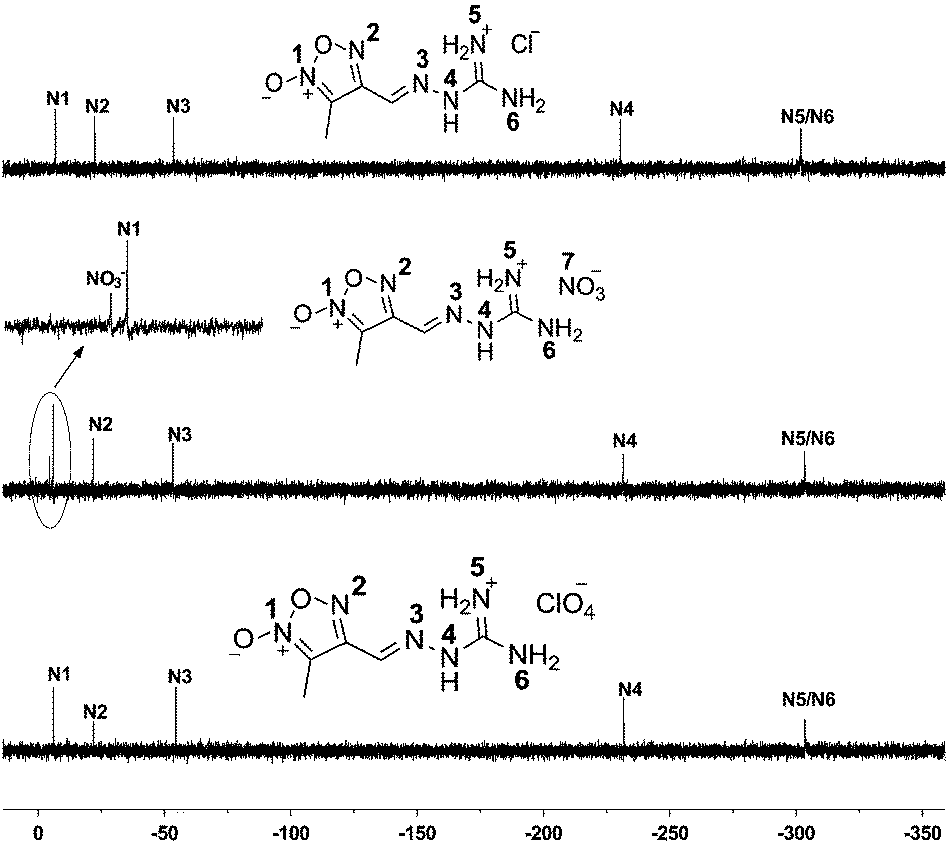 | ||
| Fig. 1 15N NMR spectra of compounds 1–3. | ||
Single-crystal X-ray analysis
Compounds 1, 2 and 5·DMF were characterized by low temperature (173 K) single crystal X-ray structure determination. The crystallographic data are summarized in Table 1.| 1 | 2 | 5·DMF | |
|---|---|---|---|
| Formula | C5H9N6O2Cl | C5H9N7O5 | C16H21N13O7 |
| Formula weight | 220.63 | 247.19 | 507.46 |
| Temperature | 173(2) K | 173(2) K | 173(2) K |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space group | P21 | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| ρ/g cm−3 | 1.526 | 1.587 | 1.475 |
| a/Å | 5.8923(5) | 12.6945(11) | 8.8526(11) |
| b/Å | 11.4413(9) | 7.5903(8) | 11.6239(14) |
| c/Å | 7.6951(7) | 11.6510(11) | 12.4741(15) |
| α/° | 90 | 90 | 109.894(4) |
| β/° | 112.239(3) | 112.837(3) | 101.069(4) |
| γ/° | 90 | 90 | 100.231(4) |
| Goodness-of-fit on F2 | 1.006 | 0.993 | 1.005 |
| Final R indexes [I > 2σ(I)] | R 1 = 0.0507, wR2 = 0.1002 | R 1 = 0.0480, wR2 = 0.1031 | R 1 = 0.0581, wR2 = 0.0641 |
| Final R indexes (all data) | R 1 = 0.0785, wR2 = 0.1137 | R 1 = 0.0921, wR2 = 0.1163 | R 1 = 0.1629, wR2 = 0.0807 |
| CCDC | 996724 | 996725 | 996726 |
3-Methyl-4-furoxancarbaldehyde guanylhydrazone·HCl (1) crystallizes in the monoclinic space group P21 with a cell volume of 480.18(7) Å3. A density of 1.526 g cm−3 was determined from the X-ray crystal structure. As shown in Fig. 2, the molecules are in an E configuration. The furoxan ring and CN bonds are in a plane with a mean deviation of 0.0143 Å resulting from the large π-conjugation system. The protonated guanidine moiety shows a completely planar assembly due to the electron delocalization in the moiety. The dihedral angle between them is 8.4°. The C4–N3 bond length, 1.277(6) Å, shows typical C–N double bond values. Owing to the electron delocalization effect, the C–N bond lengths of the protonated guanidine moiety are longer than that of C–N double bonds (1.277(6) Å) but shorter than that of the C–N single bond (1.460(2) Å). They differ in a range of 1.317(6) Å to 1.337(6) Å, which are close to that of C–N bond within the furoxan ring. In the packing diagram, as shown in Fig. 3, the discrete chloride ion and protonated guanidine moiety are linked into a 3D network by the extensive classical interactions between cations and anions.
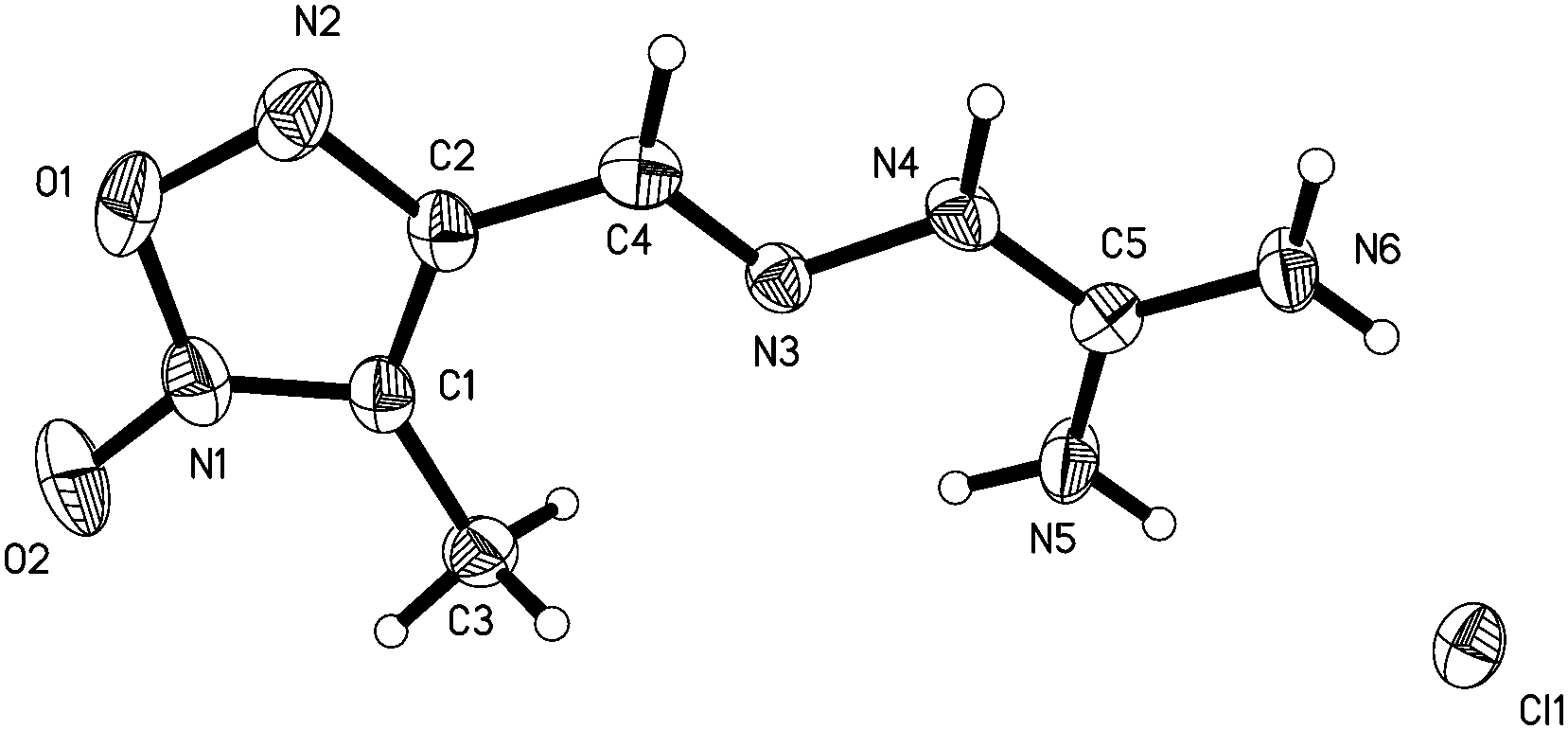 | ||
| Fig. 2 Molecular structure of 1. Thermal ellipsoids are set to 50% probability. Hydrogen atoms are included but are unlabelled for clarity. | ||
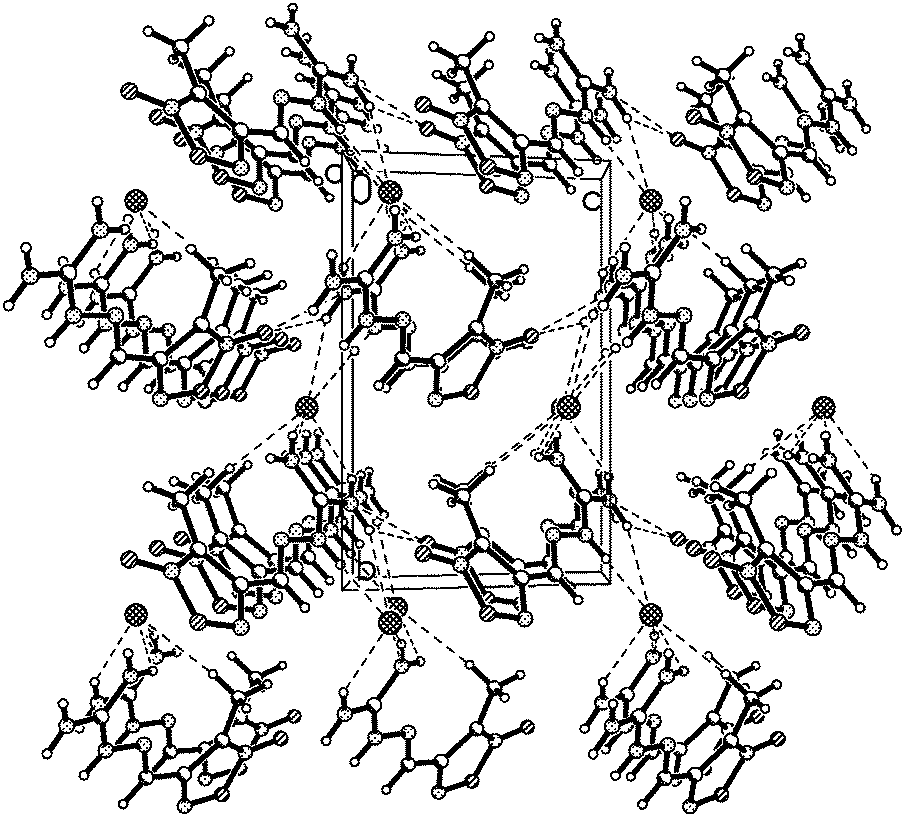 | ||
| Fig. 3 Ball and stick packing diagram of 1 viewed down the a-axis. Unit cell is indicated and dashed lines represent hydrogen bonding. N(4)–H(4A)⋯Cl(1)i 3.048(4) Å; N(5)–H(5A)⋯Cl(1)i 3.196(4) Å; N(6)–H(6B)⋯Cl(1)i 3.495(4) Å; N(5)–H(5B)⋯Cl(1)ii 2.7638(13) Å; N(6)–H(6A)⋯Cl(1)iii 3.303(4) Å. Symmetry code: (i): 1 − x, 1/2 + y, −z; (ii): −1 + x, y, z; (iii): −x, −1/2 + y, 1 − z. | ||
3-Methyl-4-furoxancarbaldehyde guanylhydrazone·HNO3 (2) crystallizes in the monoclinic P21/c with a cell volume of 1034.64(17) Å3 and four molecules in the unit cell. The density is 1.587 g cm−3. The molecular structure is shown in Fig. 4. The molecules are also in an E configuration. The protonated guanidine moiety is planar with a mean deviation of 0.0031 Å. The furoxan ring and CN bonds are in a plane with mean deviation from the ring plane of 0.0399 Å. The dihedral angle between them is 5.0°, which indicates that the planarity of 2 is superior to that of 1. In the packing diagram of 2 (Fig. 5), layers that run parallel to the b axes are formed. The π–π stacking interactions exist between the layers. The perpendicular distance between adjacent sheets is only 3.3 Å, which is even shorter than the interplanar spacing in graphite (3.4 Å).
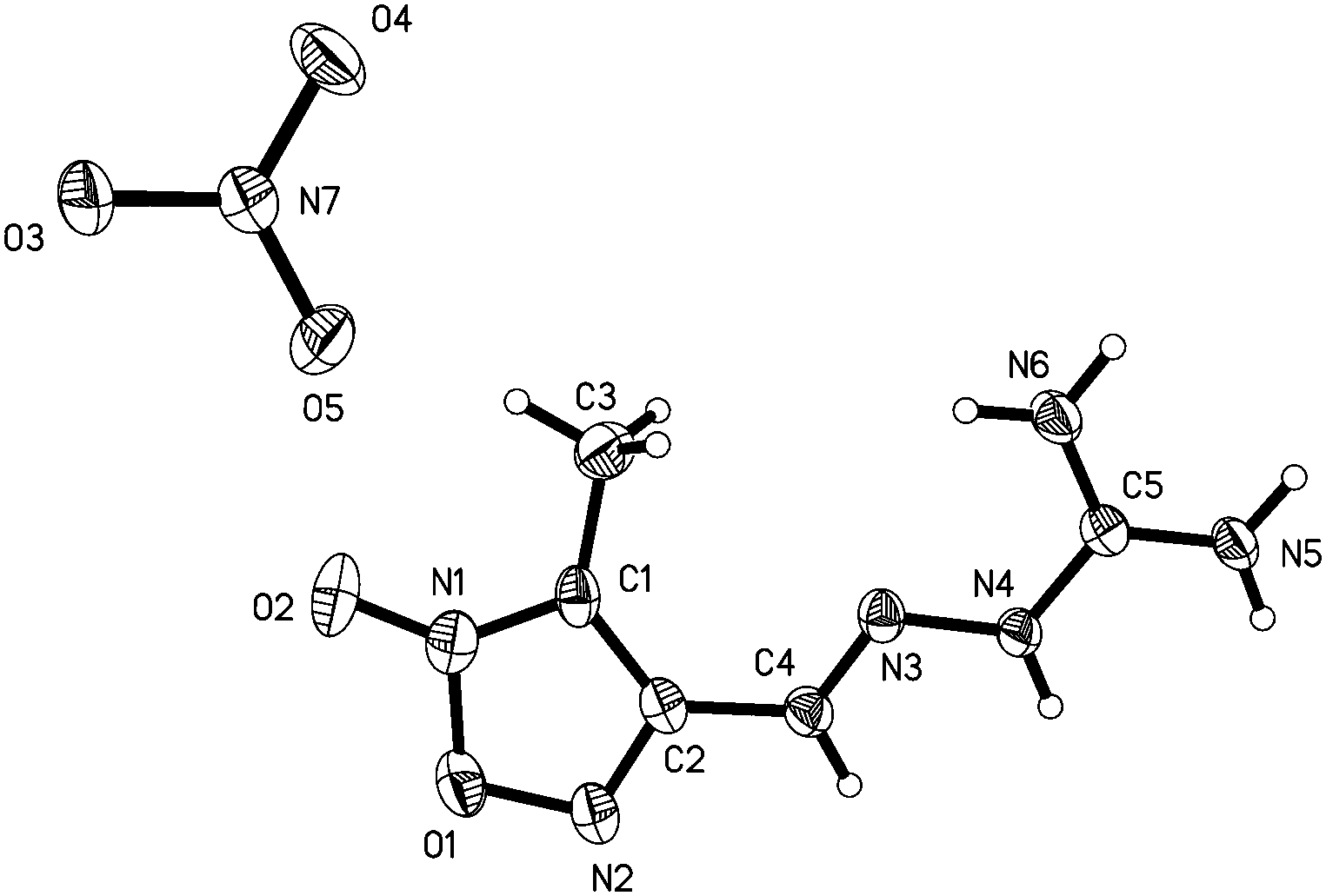 | ||
| Fig. 4 Molecular structure of 2. Thermal ellipsoids are set to 50% probability. Hydrogen atoms are included but are unlabelled for clarity. | ||
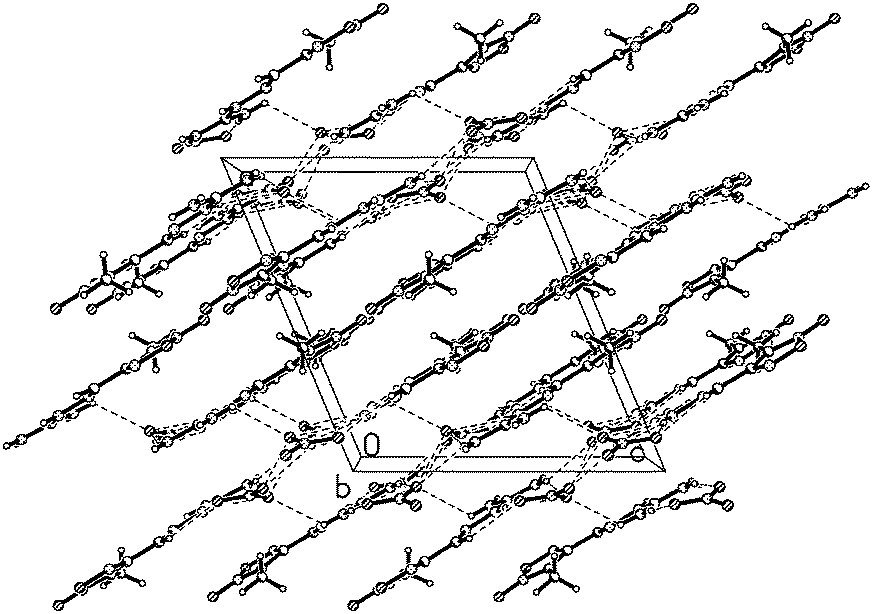 | ||
| Fig. 5 Ball and stick packing diagram of 2 viewed down the b-axis. Unit cell is indicated and dashed lines represent hydrogen bonding. | ||
The structure of compound 5 was confirmed by X-ray crystal structure analysis of 5·DMF. The 5·DMF crystallizes in the triclinic space group, P with a cell volume of 1142.8(2) Å3 and two molecules in the unit cell. A density of 1.475 g cm−3 was determined from the X-ray crystal structure. In contrast to mono-guanylhydrazone furoxan derivatives 1 and 2, the protonated guanidine structure was not observed within the trisguanylhydrazone derivative 5. It can be attributed to a decrease in the imine basicity of the guanidine moiety resulting from the electron-withdrawing inductive effect of the three furoxan rings in the structure of 5. As showed in Fig. 6, the molecules of 5 contain three hydrazone structures. Among them, two hydrazone structures are in an E configuration and another one is in a Z configuration. In the structure of 5, the C1–N9 double bond (1.301(3) Å) of the guanidine moiety is longer than that of the hydrazone moiety (C2–N2 (1.275(3) Å), C6–N6 (1.287(4) Å) and C10–N10 (1.285(3) Å)). But it is shorter than C–N double bond in the furoxan ring whose values differ in the range from 1.315(4) Å to 1.324(4) Å. It indicates that the conjugation was also found in the guanidine structure, which is weaker than that of the furoxan ring. The atoms of the guanidine moiety and three furoxan rings are planar with a mean deviation from the ring plane of 0.0895 Å. The wave-like layer structure can be observed despite the presence of DMF molecules in the crystal, and is shown in Fig. 7. The weak π–π stacking interactions exist between the layers with a centroid-to-centroid distance of 3.623(2) Å.
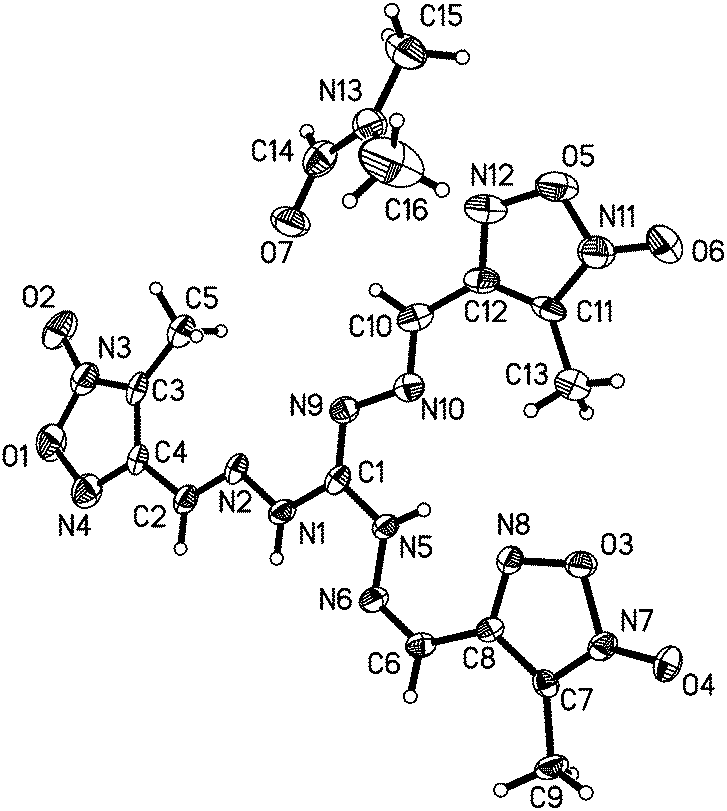 | ||
| Fig. 6 Molecular structure of 5·DMF. Thermal ellipsoids are set to 50% probability. Hydrogen atoms are included but are unlabelled for clarity. | ||
 | ||
| Fig. 7 A view of 5·DMF showing the stacking of layers. | ||
Thermal behavior and sensitivities
The thermal stabilities of compounds 1–5 were determined by differential scanning calorimetry (DSC) and thermogravimetry analysis (TGA) at a heating rate of 10 °C min−1 (ESI,† Fig. S11–S15). For all compounds, the decomposition temperatures are determined by the decomposition onset temperatures. As shown in Table 2, Compounds 1 and 3 undergo a melting process, whereas the other compounds decompose directly. Except for the compounds 1 (Td,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) onset = 199.5 °C) and 2 (Td,onset = 179.0 °C), the decomposition temperature (Td,onset) of all other guanylhydrazone substituted furoxan energetic compounds is higher than 200 °C, thus indicating that these compounds are promising energetic materials that exhibit good thermal stability. Of these, compound 4 has the highest thermal stability, Td,onset = 231.7 °C, which is much higher than that of RDX (205 °C), but lower than that of TNT (295 °C). For all compounds, the exothermic temperature range agrees with that of the weight loss. The least mass loss with the value of 40% can be observed at the exothermic temperature range of compound 1. It would be caused by its lowest nitrogen and oxygen content in all products. A mass loss of 70% can be observed within the decomposing process of compound 2, which is nearly identical to that of compound 4. In all compounds, compound 5 possesses the highest mass loss of 98% within its decomposing process, which indicates that almost all the products of thermal decomposition are gases.
onset = 199.5 °C) and 2 (Td,onset = 179.0 °C), the decomposition temperature (Td,onset) of all other guanylhydrazone substituted furoxan energetic compounds is higher than 200 °C, thus indicating that these compounds are promising energetic materials that exhibit good thermal stability. Of these, compound 4 has the highest thermal stability, Td,onset = 231.7 °C, which is much higher than that of RDX (205 °C), but lower than that of TNT (295 °C). For all compounds, the exothermic temperature range agrees with that of the weight loss. The least mass loss with the value of 40% can be observed at the exothermic temperature range of compound 1. It would be caused by its lowest nitrogen and oxygen content in all products. A mass loss of 70% can be observed within the decomposing process of compound 2, which is nearly identical to that of compound 4. In all compounds, compound 5 possesses the highest mass loss of 98% within its decomposing process, which indicates that almost all the products of thermal decomposition are gases.
| Compd | 1 | 2 | 3 | 4 | 5 | TNT16 | RDX17 |
|---|---|---|---|---|---|---|---|
| a Melting point. b Thermal decomposition temperature under nitrogen gas (determined by the DSC exothermal peak, 10 °C min−1). c Oxygen balance (%) for CaHbNcOd: OB (%) = 1600 × (d − 2a − b/2)/Mw (based on carbon dioxide). d Nitrogen content. e Density from X-ray structure. f Density from theoretical calculation. g Heat of explosion. h Molar enthalpy of formation. i Detonation velocity. j Detonation pressure. k Impact sensitivity. l Electrostatic discharge sensitivity. | |||||||
| T m [°C] | 199.5 | — | 186.6 | — | — | 80.4 | — |
| T d [°C] | 199.5 | 179.0 | 200.0 | 231.7 | 215.3 | 295 | 205 |
| OBc | −90.6 | −61.50 | −47.78 | −100.89 | −99.46 | −73.97 | −21.61 |
| N%d | 38.1 | 39.7 | 29.5 | 40.8 | 38.7 | 18.5 | 37.8 |
| ρ [g cm−3] | 1.526e/1.584f | 1.587e/1.681f | 1.788f | 1.557f | 1.570f | 1.654 | 1.816 |
| Q [J g−1] | 3970 | 5292 | 5336 | 5646 | 5964 | 4271 | — |
| ΔfHmh [kJ mol−1] | 240 | 163 | 160 | 766 | 1214 | −295 | 176.2 |
| D [m s−1] | 6906 | 8210 | 8175 | 7532 | 7528 | 6881 | 8983 |
| P [GPa] | 17.0 | 25.8 | 28.3 | 20.2 | 20.8 | 19.5 | 35.2 |
| ISk [J] | 6.8 | 4.2 | 3.1 | 22 | 18 | 15 | 7.5 |
| ESDl [J] | 0.194 | 0.132 | 0.103 | 0.425 | 0.353 | — | 0.1–0.2 |
Impact sensitivity measurements were performed by using the standard BAM method.13 Additionally, all compounds were tested upon the sensitivity toward electrical discharge using Electric Spark Tester ESD JGY-50 III. As shown in Table 2, the IS values of 1–3 are 6.8 J, 4.2 J and 3.1 J, respectively. It shows that they are more sensitive than RDX (7.4 J). However, the IS values of 4 and 5 are 22 J and 18 J, which are much higher than that of RDX. For all compounds, similar trends are observed in the results of electrostatic discharge sensitivity. Compounds 1–3 are less sensitive to electrostatic discharge, wherein, 3 (103 mJ) is more sensitive than 1 (194 mJ) and 2 (132 mJ). Compounds 4 (425 mJ) and 5 (353 mJ) are significantly less sensitive to electrostatic discharge.
Heats of formation and detonation parameters
The heat of formation is an important parameter to evaluate the performance of these new energetic compounds. The heats of formation were calculated by using the Gaussian 09 suite of programs14 by using the method of isodesmic reactions (ESI,† Scheme S1). The lattice energies of the ionic compounds 1–3 were predicted by using the formula suggested by Jenkins et al.15 As listed in Table 2, the resulting heats of formation of these compounds are in the range of 160 kJ mol−1 (3) to 1214 kJ mol−1 (5). The detonation velocity (D) and detonation pressure (P), which are used to characterize the performance of a high explosive, were calculated by using the EXPLO v6.01 program. For these new compounds, because of the low density of the all compounds, the calculated detonation velocities lie in the range between 6906 and 8210 m s−1, which are lower than that of RDX (8983 m s−1), but higher than that of TNT (6881 m s−1). Among them, compound 2 exhibits the highest detonation velocity (8210 m s−1) which is comparable to that of TATB (8114 m s−1). The detonation pressures range between 17.0 and 28.3 GPa, in which the highest P value of 3 (28.3 GPa) is much higher than that of TNT (19.5 GPa).Conclusions
The reactions of 3-methyl-4-furoxancarbaldehyde with aminoguanidine derivatives generate the previously unknown guanylhydrazone substituted furoxan energetic derivatives. All compounds have been well characterized using IR spectroscopy, multinuclear NMR spectroscopy, differential scanning calorimetry, thermogravimetry analysis and elemental analysis. Single crystal X-ray measurements were accomplished for compounds 1, 2 and 5·DMF and provide insight into structural characteristics as well as inter- and intramolecular interactions. Except for compounds 1 and 2, all the remaining products exhibit good thermal stabilities with decomposition onset temperatures of above 200 °C. All resulting compounds reveal positive heats of formation in the range of 160 kJ mol−1 (3) to 1214 kJ mol−1 (5). The detonation pressure values calculated for these compounds range from 17.0 to 28.3 GPa, and the detonation velocities range from 6906 to 8210 m s−1. The most interesting compound regarding the thermal and energetic properties is 3. It exhibits decomposition temperatures at 200 °C. Its detonation pressure and detonation velocities values are 28.3 GPa and 8175 m s−1, which are significantly higher than that of TNT. In conclusion, the introduction of the aminoguanidine moiety into energetic heterocycles through CN bonds enriches the methodology for synthesizing aminoguanidine-containing energetic materials.
Experimental
Caution: although we experienced no difficulties in handling these energetic materials, small-scale and best safety practices (leather gloves, face shield) are strongly encouraged.General
All chemical reagents and solvents were purchased and were used as supplied without further purification. 1H, 13C, and 15N NMR spectra were recorded on a Bruker Avance III 300 instrument at 25 °C. The chemical shifts are given relative to dimethyl sulfoxide (1H, 13C) or nitromethane (15N) as external standards. Infrared (IR) spectra were recorded on a Perkin-Elmer Spectrum BX FT-IR instrument equipped with an ATR unit at 25 °C. Transmittance values are qualitatively described as “very strong” (vs), “strong” (s), “medium” (m), “weak” (w) and “very weak” (vw). TG and DSC studies were performed at a heating rate of 10 °C min−1 in closed Al containers under a nitrogen flow of 30 mL min−1 using an STD-Q600 instrument. Analyses of C/H/N were performed with a Vario EL III Analyzer. The electrostatic sensitivity test was carried out using an Electric Spark Tester ESD JGY-50 III. The impact sensitivity tests were carried out using a HGZ-1 drop hammer. Test specimens were kept between two hardened anvils and a 2.0 kg drop weight was allowed to fall freely from different heights. Twenty-five tests were conducted for each compound.X-ray crystallography
The data for 1, 2 and 5·DMF were collected using a Bruker three-circle platform diffractometer equipped with a SMART APEX II CCD detector. A Kryo-Flex low-temperature device was used to keep the crystals at a constant 173 K during the data collection. The data collection and the initial unit cell refinement was performed by using APEX2 (v2010.3-0). Data Reduction was performed by using SAINT (v7.68A) and XPREP (v2008/2). Corrections were applied for Lorentz, polarization, and absorption effects by using SADABS (v2008/1). The structure was solved and refined with the aid of the programs in the SHELXTL-plus (v2008/4) system of programs. The full-matrix least-squares refinement on F2 included atomic coordinates and anisotropic thermal parameters for all non-H atoms. The H atoms were included in a riding model. The structure was solved by direct methods with SHELXS-97 and expanded by using the Fourier technique. The non-hydrogen atoms were refined anisotropically. The hydrogen atoms were located and refined.General procedures for the preparation of energetic compounds 1–3
3-Methyl-4-furoxancarbaldehyde11 and aminoguanidium perchlorate4a were synthesized according to the literature.A solution of aminoguanidium salts (5.0 mmol) and 3-methyl-4-furoxancarbaldehyde (0.77 g, 6.0 mmol) in 30 mL ethanol was refluxed for 24 h. Then, the reaction mixture was cooled to room temperature and the precipitate of target product was collected by filtration. The filtrate was separated by column chromatography on silica gel using 10:1 ethyl acetate:methanol as the eluent to give the remaining target product.
N–), 8.03 (s, 4H, –NH2), 2.37 (s, 3H, –CH3); 13C NMR (75 MHz, DMSO): δ (ppm): 156.8, 154.8, 137.8, 113.2, 10.7. IR (KBr): 3421 (s), 3166 (m), 2953 (w), 2840 (m) 1682 (s), 1616 (vs), 1538 (m), 1501 (w), 1460 (s), 1156 (m), 1038 (w), 1013 (w), 804 (w), 678 (w), 608 (m), 516 (w). Elemental analysis: C5H9ClN6O2 (220.62): calcd C 27.22, H 4.11, N 38.09; found C 27.11, H 4.09, N 37.91.
N–), 7.89 (s, 4H, –NH2), 2.37 (s, 3H, –CH3); 13C NMR (75 MHz, DMSO): δ (ppm): 156.4, 154.8, 138.2, 113.2, 10.6. IR (KBr): 3390 (s), 3308 (s), 3212 (s), 3163 (s), 3012 (m), 2903 (w), 1698 (s), 1633 (s), 1608 (vs), 1565 (w), 1503 (w), 1457 (m), 1385 (vs), 1329 (m), 1316 (m), 1302 (m), 1149 (w), 1039 (m), 1011 (w), 924 (w), 798 (w), 702 (w), 599 (m), 512 (w). Elemental analysis: C5H9N7O5 (247.17): calcd C 24.30, H 3.67, N 39.67; found C 23.90, H 3.73, N 39.31.
N–), 7.81 (s, 4H, –NH2), 2.37 (s, 3H, –CH3); 13C NMR (75 MHz, DMSO): δ (ppm): 156.4, 154.8, 138.3, 113.1, 10.6. IR (KBr): 3407 (s), 3316 (s), 3224 (m), 3168 (m), 3006 (w), 2925 (w), 2857 (w), 2361 (m), 2337 (m), 1684 (vs), 1611 (vs), 1540 (m), 1501 (m), 1464 (s), 1402 (m), 1380 (w), 1332 (w), 1310 (w), 1147 (s), 1091 (vs), 1067 (m), 1046 (s), 970 (w), 924 (w), 875 (w), 807 (m), 672 (m), 629 (s), 512 (m). Elemental analysis: C5H9ClN6O6 (284.61): calcd C 21.10, H 3.19, N 29.53; found C 21.19, H 3.14, N 29.44.
1H NMR (300 MHz, DMSO): δ (ppm): 12.63 (br, –NH–), 8.66 (s, 2H, –CHN–), 8.62 (s, 2H, –NH2), 2.43 (s, 6H, –CH3); 13C NMR (75 MHz, DMSO): δ (ppm): 154.9, 140.0, 113.3, 10.7. IR (KBr): 3453 (s), 3210 (s), 2812 (s), 2361 (m), 1669 (vs), 1611 (vs), 1465 (s), 1433 (s), 1378 (s), 1346 (m),1314 (w), 1280 (w), 1189 (w), 1118 (s), 1033 (m), 954 (w), 927 (w), 826 (m), 798 (w), 739 (w), 678 (m), 625 (m), 600 (w), 486 (s). Elemental analysis: C9H11N9O4 (309.24): calcd C 34.96, H 3.59, N 40.76; found C 34.90, H 3.56, N 39.98.
1H NMR (300 MHz, DMSO): δ (ppm): 11.20 (s, 2H, –NH–), 8.38 (s, 3H, –CHN–), 2.41 (s, 9H, –CH3); 13C NMR (75 MHz, DMSO): δ (ppm): 155.6, 153.7, 134.6, 113.4, 10.7 ppm. IR (KBr): 3422 (w), 3330 (m), 2361 (m), 1638 (m), 1604 (vs), 1576 (s), 1544 (s), 1489 (m), 1457 (s), 1409 (m), 1377 (m), 1303 (m), 1284 (w), 1152 (s), 1102 (m), 1033 (m), 963 (w), 929 (w), 869 (w), 820 (m), 728 (vw), 666 (w), 611 (w), 496 (w). C13H14N12O6 (434.33): calcd C 35.95, H 3.25, N 38.70; found C 36.39, H 3.29, N 38.65.
Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK2011696) and the National Natural Science Foundation of China (No. 21376121).Notes and references
- (a) H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377 CrossRef CAS PubMed; (b) D. Chand, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 15383 RSC; (c) D. Fischer, T. M. Klapötke, M. Reymann and J. Stierstorfer, Chem. – Eur. J., 2014, 20, 1 CrossRef.
- (a) A. Hammerl and T. M. Klapötke, Inorg. Chem., 2002, 41, 906 CrossRef CAS PubMed; (b) A. Hammerl, G. Holl, M. Kaiser, T. M. Klapötke and H. Piotrowski, Z. Anorg. Allg. Chem., 2003, 629, 2117 CrossRef CAS; (c) D. E. Chavez, M. A. Hiskey and R. D. Gilardi, Org. Lett., 2004, 6, 2889 CrossRef CAS PubMed; (d) M. H. V. Huynh, M. A. Hiskey, E. L. Hartline, D. P. Montoya and R. Gilardi, Angew. Chem., Int. Ed., 2004, 43, 4924 CrossRef CAS PubMed.
- (a) J. Giles, Nature, 2004, 427, 580 CrossRef CAS PubMed; (b) M. B. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. K. Sikder, B. R. Gandhe and A. S. Rao, J. Hazard. Mater., 2009, 161, 589 CrossRef CAS PubMed; (c) M. Rahm and T. Brinck, Chem. – Eur. J., 2010, 16, 6590 CrossRef CAS PubMed.
- (a) T. M. Klapötke and J. Stierstorfer, Cent. Eur. J. Energ. Mater., 2008, 5, 13 Search PubMed; (b) R. Wang, Y. Guo, Z. Zeng and J. M. Shreeve, Chem. Commun., 2009, 2697 RSC; (c) R. Wang, Y. Guo, Z. Zeng, B. Twamley and J. M. Shreeve, Chem. – Eur. J., 2009, 15, 2625 CrossRef CAS PubMed; (d) Y. Huang, Y. Zhang and J. M. Shreeve, Chem. – Eur. J., 2010, 17, 1538 CrossRef PubMed.
- (a) A. B. Sheremetev and V. O. Kulagina, J. Org. Chem., 1996, 61, 1510 CrossRef CAS; (b) A. B. Sheremetev, S. E. Semenov, V. S. Kuzmin and S. L. Ioffe, Chem. – Eur. J., 1998, 4, 1023 CrossRef CAS; (c) L. Liang, K. Wang, C. Bian, J. Song, L. Ling, F. Zhao and Z. Zhou, J. Mater. Chem., 2012, 22, 21954 RSC; (d) A. B. Sheremetev, N. S. Aleksandrova, N. V. Palysaeva, M. I. Struchkova, V. A. Tartakovsky and K. Y. Suponitsky, Chem. – Eur. J., 2013, 19, 12446 CrossRef CAS PubMed; (e) L. Liang, D. Cao, J. Song, H. Huang, K. Wang, C. Bian, X. Dong and Z. Zhou, J. Mater. Chem. A, 2013, 1, 8857 RSC; (f) J. Zhang and J. M. Shreeve, J. Am. Chem. Soc., 2014, 136, 4437 CrossRef CAS PubMed.
- (a) D. E. Chavez, M. A. Hiskey and R. D. Gilardi, Angew. Chem., Int. Ed., 2000, 39, 1791 CrossRef CAS; (b) M. H. V. Huynh, M. A. Hiskey, J. G. Archuleta, E. L. Roemer and R. Gilardi, Angew. Chem., 2004, 116, 5776 CrossRef; (c) D. E. Chavez, M. A. Hiskey and R. D. Gilardi, Org. Lett., 2004, 6, 2889 CrossRef CAS PubMed; (d) M. H. V. Huynh, M. A. Hiskey, D. E. Chavez, D. L. Naud and R. D. Gilard, J. Am. Chem. Soc., 2005, 127, 12537 CrossRef CAS PubMed; (e) H. Gao, R. Wang, B. Twamley, M. A. Hiskey and J. M. Shreeve, Chem. Commun., 2006, 4007 RSC; (f) Z. Fu, C. He and F. Chen, J. Mater. Chem., 2012, 22, 60 RSC; (g) D. E. Chavez, S. K. Hanson, J. M. Veauthier and D. A. Parrish, Angew. Chem., Int. Ed., 2013, 52, 6876 CrossRef CAS PubMed.
- (a) T. M. Klapötke, P. Mayer, A. Schulz and J. J. Weigand, J. Am. Chem. Soc., 2005, 127, 2032 CrossRef PubMed; (b) T. Abe, G. Tao, Y. Joo, Y. Huang, B. Twamley and J. M. Shreeve, Angew. Chem., Int. Ed., 2008, 47, 7087 CrossRef CAS PubMed; (c) T. M. Klapötke and J. Stierstorfer, J. Am. Chem. Soc., 2008, 131, 1122 CrossRef PubMed; (d) Y. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 564 CrossRef CAS PubMed; (e) T. M. Klapötke, F. A. Martin and J. Stierstorfer, Angew. Chem., Int. Ed., 2011, 50, 4227 CrossRef PubMed; (f) T. M. Klapötke, F. A. Martin and J. Stierstorfer, Chem. – Eur. J., 2012, 18, 1487 CrossRef PubMed; (g) T. M. Klapötke, D. G. Piercey and J. Stierstorfer, Dalton Trans., 2012, 41, 9451 RSC; (h) Q. Zhang, J. Zhang, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2013, 19, 11000 CrossRef CAS PubMed; (i) Q. Lin, Y. Li, C. Qi, W. Liu, Y. Wang and S. Pang, J. Mater. Chem. A, 2013, 1, 6776 RSC; (j) D. Chand, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 15383 RSC.
- (a) H. Xue, Y. Gao, B. Twamley and J. M. Shreeve, Chem. Mater., 2005, 17, 191 CrossRef CAS; (b) R. Wang, H. Gao, C. Ye and J. M. Shreeve, Chem. Mater., 2007, 19, 144 CrossRef CAS; (c) G. Tao, B. Twamley and J. M. Shreeve, J. Mater. Chem., 2009, 19, 5850 RSC; (d) R. Wang, H. Xu, Y. Guo, R. Sa and J. M. Shreeve, J. Am. Chem. Soc., 2010, 132, 11904 CrossRef CAS PubMed; (e) V. Thottempudi, H. Gao and J. M. Shreeve, J. Am. Chem. Soc., 2011, 133, 6464 CrossRef CAS PubMed; (f) V. Thottempudi and J. M. Shreeve, J. Am. Chem. Soc., 2011, 133, 19982 CrossRef CAS PubMed; (g) C. Qi, S. Li, Y. Li, Y. Wang, X. Chen and S. Pang, J. Mater. Chem., 2011, 21, 3221 RSC; (h) V. Thottempudi, F. Forohor, D. A. Parrish and J. M. Shreeve, Angew. Chem., Int. Ed., 2012, 51, 9881 CrossRef CAS PubMed; (i) Q. Lin, Y. Li, Y. Li, Z. Wang, W. Liu, C. Qi and S. Pang, J. Mater. Chem., 2012, 22, 666 RSC; (j) A. A. Dippold and T. M. Klapötke, J. Am. Chem. Soc., 2013, 135, 9931 CrossRef CAS PubMed; (k) Y. Zhang, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2013, 1, 585 RSC; (l) P. Yin, J. Zhang, C. He, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2014, 2, 3200 RSC.
- (a) H. Huang, Z. Zhou, L. Liang, J. Song, K. Wang, D. Cao, C. Bian, W. Sun and M. Xue, Z. Anorg. Allg. Chem., 2012, 638, 392 CrossRef CAS; (b) H. Huang, Z. Zhou, L. Liang, J. Song, K. Wang, D. Cao, W. Sun, C. Bian and M. Xue, Chem. – Asian J., 2012, 7, 707 CrossRef CAS PubMed; (c) L. Liang, K. Wang, C. Bian, L. Ling and Z. Zhou, Chem. – Eur. J., 2013, 19, 14902 CrossRef CAS PubMed; (d) Y. Tang, H. Yang, J. Shen, B. Wu, X. Ju, C. Lu and G. Cheng, Eur. J. Inorg. Chem., 2014, 1231 CrossRef CAS.
- (a) J. R. Doamaral, F. A. French, E. J. Blanz and D. A. French, J. Med. Chem., 1971, 14, 862 CrossRef CAS; (b) A. W. Tai, E. J. Lien, E. C. Moore, C. Yu and J. D. Roberts, J. Med. Chem., 1983, 26, 1326 CrossRef CAS; (c) A. Andreani, M. Rambaldi, A. Locatelli, R. Bossa, A. Fraccari and I. Galatulast, J. Med. Chem., 1992, 35, 4634 CrossRef CAS; (d) A. Andreani, M. Rambaldi, A. Leoni, A. Locatelli, R. Bossa, A. Fraccari, I. Galatulas and G. Salvatore, J. Med. Chem., 1996, 39, 2852 CrossRef CAS PubMed; (e) A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, L. Varoli, M. Rambaldi, G. Farruggia, C. Stefanelli, L. Masotti and M. W. Kunkel, J. Med. Chem., 2006, 49, 7897 CrossRef CAS PubMed; (f) A. Andreani, S. Burnelli, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, L. Varoli, M. Rambaldi, N. Calonghi, C. Cappadone, G. Farruggia, M. Zini, L. Masotti, C. Stefanelli, N. S. Radin and R. H. Shoemaker, J. Med. Chem., 2008, 51, 809 CrossRef CAS PubMed.
- (a) R. Fruttero, B. Ferrarotti, A. serafino, A. D. stilo and A. Gasco, J. Heterocycl. Chem., 1989, 26, 1345 CrossRef CAS; (b) G. Kim, J. Kim, S. Lee, H. Kim and K. Hwang, Bull. Korean Chem. Soc., 2009, 30, 459 CrossRef CAS.
- (a) I. Yavari, R. E. Botto and J. D. Roberts, J. Org. Chem., 1978, 43, 2542 CrossRef CAS; (b) G. A. Olah, A. Burrichter, G. Rasul, M. Hachoumy and G. K. Prakash, J. Am. Chem. Soc., 1997, 119, 12929 CrossRef CAS; (c) P. Cmoch, B. Kamienski, K. Kamien ska-Trela, L. Stefaniak and G. A. Webb, J. Phys. Org. Chem., 2000, 13, 480 CrossRef CAS; (d) R. Marek and A. Lycka, Curr. Org. Chem., 2002, 6, 35 CrossRef CAS; (e) K. L. Anderson, L. H. Merwin, W. S. Wilson and J. C. Facelli, Int. J. Mol. Sci., 2002, 3, 858 CrossRef CAS PubMed; (f) T. L. C. Martins, T. C. Ramalho, J. D. Figueroa-Villar, A. F. C. Flores and C. M. Pereira, Magn. Reson. Chem., 2003, 41, 983 CrossRef CAS; (g) R. Marek, A. Lycka, E. Kolehmainen, E. Sievänen and J. Tousek, Curr. Org. Chem., 2007, 11, 1154 CrossRef CAS.
- Test methods according to the UN Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria, 4th rev.ed.; United Nations Publication, New York, 2003; 13.4.2 Test 3 (a) (ii) BAM Fallhammer, pp. 75–82.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. AlLaham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 09, revision A. 01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H. D. B. Jenkins, D. Tudeal and L. Glasser, Inorg. Chem., 2002, 41, 2364 CrossRef CAS PubMed.
- Y. Zhang, Y. Guo, Y. Joo, D. A. Parrish and J. M. Shreeve, Chem. – Eur. J., 2010, 16, 10778 CrossRef CAS PubMed.
- T. M. Klapötke, D. G. Piercey and J. Stierstorfer, Dalton Trans., 2012, 41, 9451 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 996724–996726. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj01175a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |