
Amperometric determination of promethazine in tablets using an electrochemically reduced graphene oxide modified electrode
Fabiana S.
Felix
a,
Luís M. C.
Ferreira
a,
Fernanda
Vieira
b,
Geraldo M.
Trindade
b,
Viviane S. S. A.
Ferreira
b and
Lúcio
Angnes
*a
aUniversidade de São Paulo, Instituto de Química, São Paulo, 05508-000, Brazil. E-mail: luangnes@iq.usp.br; Tel: +55 11 3091 3828
bNacional Grafite Ltda., Minas Gerais 35550-000, Brazil
First published on 7th November 2014
Abstract
Graphene films were prepared on a glassy carbon electrode for amperometric determination of promethazine hydrochloride in pharmaceutical products. This modified sensor was prepared by chemical oxidation of graphite powder followed by product exfoliation in ultrapure water using an ultrasonic bath. Then, the resultant graphene oxide was electrochemically reduced in 0.10 mol L−1 acetic acid–sodium acetate (pH = 5.0) on a glassy carbon electrode surface. The proposed sensor exhibited reproducible amperometric responses in a wide linear range from 1.99 × 10−6 to 1.03 × 10−3 mol L−1 at +0.78 V (vs. Ag/AgCl). Low detection and quantification limits (1.99 × 10−7 mol L−1 and 6.63 × 10−7 mol L−1, respectively) were achieved. This method was applied to the analyses of commercial tablet samples and all results were in good agreement with those obtained using spectrophotometry and high-performance liquid chromatography.
Introduction
Graphene is composed of a two-dimensional structure with a single atomic layer of sp2-hybridized carbon atoms and has the same thickness as a one carbon atom, with a crystalline hexagonal configuration.1 It was firstly isolated by mechanical exfoliation of graphite and visualized under an optical microscope by Geim et al. in 2004.2This new form of carbon arrangement has attracted enormous attention from different scientific fields due to its exceptional properties, such as high surface area, chemical inertness, optical transmittance, high current density and high electrical and thermal conductivities.3 Because of its ability to promote fast electron transfer, it provides new opportunities to be used as an electrode material. However, the development of strategies for large-scale graphene production with high quality has been challenging since its discovery. Among some, the most reported involves soft chemistry routes focused on the oxidation and exfoliation of graphite followed by a reduction step which are typically used to prepare graphene-related materials such as graphene oxide (GO) and reduced graphene oxide (rGO).1 GO exhibits an excessive number of oxygen-containing functional groups which makes it an electrically insulating material. In this matter, a reduction step is necessary to eliminate some oxygenated functionalities and restore the conjugation to graphene structure.
Among all methods used to produce rGO films from graphene oxide the electrochemical reduction is very simple, fast and low cost, while other methodologies have been using toxic reducing agents or high temperature operating routes.4 Moreover, the use of high negative potentials along the reduction process can overcome the energy barriers for the reduction of oxygen functionalities found on the basal plane and the edge.11 As a consequence, GO can be efficiently reduced and subsequently applied for electrochemical quantification of different analytes.5–10
From the electron and molecular structure perspective, it is a consensus that compounds of the phenothiazine group can be easily oxidized at the graphene film surface. These drugs have a tricyclic aromatic ring with sulfur and nitrogen atoms, and different substituents attached at the 2 and 10 or 3 and 7 positions.12 There are different electrode surfaces which have already been used as detectors for phenothiazine derivative detection, such as the boron-doped diamond,13 the nanodiamond modified with Ag particles,14 gold,15,16 carbon nanotubes,17–19 glassy carbon,20 FTO modified with SiPy+Cl− and CuTsPc film,21 and modified carbon paste electrodes.22,23 To the best of our knowledge, the amperometric detection of promethazine hydrochloride using a rGO modified sensor is reported here for the first time.
Promethazine hydrochloride, (2RS)-N,N-dimethyl-1-(10H-phenothiazin-10-yl)propan-2-amine hydrochloride, is a neuroleptic agent which has sedative and antipsychotic effects.13 In the following sections, the use of the rGO modified glassy carbon electrode as an amperometric sensor for promethazine hydrochloride determination in pharmaceutical products is described. The proposed sensor was prepared just by a simple electrochemical reduction of GO on the electrode surface.
Experimental
Reagents and solutions
Graphite oxide was prepared from natural graphite (powder with 99.80% purity from Nacional Grafite Ltda., Minas Gerais, Brazil) using a modified Hummers method.24,25 The promethazine hydrochloride standard was acquired from Sigma-Aldrich (St. Louis, Mo, USA). All reagents were of analytical grade and used as received. Sulfuric acid, acetic acid, sodium acetate, sodium hydroxide, boric acid, hydrochloric acid, acetonitrile and potassium mono-hydrogen phosphate were purchased from Merck (Darmstadt, Germany). All solutions were prepared with ultrapure water from a Millipore Milli-Q system with resistivity ≥18.2 MΩ cm (Barnstead, Dubuque, IA, USA). A promethazine hydrochloride stock solution (0.10 mol L−1) was prepared by dissolving the solid salt in ultrapure water, which was later stored in a dark flask and under refrigeration. Analyte standard solutions were properly diluted with supporting electrolyte just before measurements. Commercial pharmaceutical products (Fenergan® – tablets, from Sanofi-Aventis Farmacêutica Ltda) from different fabrication lots (09/2013 and 01/2014) were purchased from a local drugstore.The influence of two different supporting electrolytes (Britton–Robinson (B–R) buffer (0.10 mol L−1, pH 2–7) and sulfuric acid solution (0.10 mol L−1)) for promethazine hydrochloride oxidation was evaluated by cyclic voltammetry. Sulfuric acid produced better results and was adopted for the main measurements.
A 0.10 mol L−1 acetic acid–sodium acetate (pH = 5.0) solution was used for the electrochemical reduction of GO film deposited over the glassy carbon surface. This solution was prepared by mixing 0.100 mol L−1 sodium acetate and 0.055 mol L−1 acetic acid.
Instrumentation
Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50). Then, 50 μL of the resulting solution was injected into the chromatograph.
:50). Then, 50 μL of the resulting solution was injected into the chromatograph.
Results and discussion
Production of rGO-GCE
Graphite powder was oxidized in acidic medium according to the modified Hummers method and the formed product (graphite oxide) was then exfoliated in ultrapure water to obtain GO (methodology). Graphite oxide has a layered structure, similar to graphite but it presents oxygen based functional groups on both basal planes and edges which expands the interlayer distance, as well as the atomic-thick layer hydrophilicity. As a consequence, these oxidized layers can be exfoliated in water or polar organic media by ultrasonic bath producing only one or few layers of carbon atoms similar to graphene. Fig. 1 presents scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the obtained GO surface. | ||
| Fig. 1 SEM images of (A) a bare GCE and (B) a GO modified GCE and (C) a TEM image of GO. | ||
Fig. 1 displays the SEM images of GCE (A) with its typical glazed surface and GCE modified with GO (B). It is possible to observe a typical wrinkled structure (which arises from the π–π interaction between graphene sheets) with plenty of corrugations in Fig. 1B. Moreover, this figure is in accordance with previous studies reported in the literature.30 As shown in the TEM image (Fig. 1C), the GO surface has a silk veil-like waved structure with thin and wrinkled sheets, revealing the random overlay of the individual sheets.
Raman spectroscopy is a technique widely used for the characterization of sp2 and sp3 hybridized carbon atoms at carbon-based materials, in a manner to distinguish the order and disorder/defect structures. X-ray diffraction (XRD) is other nondestructive characterization technique used to obtain typical d-spacing (interlayer distance of the (002) peak) measurements. In this study, both techniques were used to confirm material oxidation. Raman spectra (A) and XRD patterns (B) of both graphite (a) and GO (b) are shown in Fig. 2.
 | ||
| Fig. 2 (A) Raman spectra obtained for (a) graphite and (b) GO. (B) XRD patterns of (a) graphite and (b) GO. | ||
Fig. 2A shows a prominent peak at 1586 cm−1 and a weak peak at 1356 cm−1 which corresponds to the G and D bands of raw graphite, respectively (a). The presence of the D band for graphite indicates that there are a considerable number of defective sites within the structure (sp3-hybridized carbons). However, a significant increase in the intensity of the D band and a shift of the G band to 1598 cm−1 were observed for GO (b), indicating that a large number of sp2-hybridized carbons have been converted to sp3-hybridization through chemical oxidation. The relative intensity ratio of D and G bands (ID/IG) is usually considered proportional to the number of defective sites in the material.30 The ID/IG value for GO was of 1.06, it is higher than the one found for raw graphite (∼0.09), which indicates that there are significant edge-plane-like defective sites existing on the material surface due to the oxidation step.25
XRD patterns of raw graphite (a) and GO (b) are shown in Fig. 2B. The diffraction (002) peak of graphite was centered at 2θ = 26.5° (a) with an interlayer d-spacing of 0.336 nm. After chemical oxidation of graphite the (002) peak disappeared, but a new peak at 2θ = 11.6° (b) with a d-spacing of 0.756 nm is observed. This indicates the generation of oxygen based functional groups in the graphite interlayer spaces.30 From these results, GO film was deposited on the GCE surface and then, electrochemically reduced in 0.10 mol L−1 acetic acid–sodium acetate solution (pH = 5.0).
Fig. 3 shows successive scanning for the electrochemical reduction of GO, in a potential range from 0 to −1.5 V (vs. Ag/AgCl), recorded over a scan rate of 50 mV s−1.
 | ||
| Fig. 3 30 successive cyclic voltammograms recorded for the GO reduction process in 0.10 mol L−1 acetic acid–sodium acetate solution (pH = 5.0) using a GCE modified with GO. Scan rate, 50 mV s−1. (Dotted line) 1st scan, and (solid line) 2nd, 10th, 20th and 30th scans. | ||
The cyclic voltammograms of the GCE modified with GO (Fig. 3) show a large cathodic peak at −0.71 V in the first cycle likely due to the reduction of the oxygen based functional groups present at the extremities of GO structures, since reduction of water to hydrogen occurs at a more negative potential (about −1.5 V).10 In the 2nd cycle, the reduction peak decreased considerably and disappeared in the following cycles. No difference could be observed between the 20th and 30th cycles. This voltammetric result demonstrated that the reduction step occurred quickly and irreversibly and GO could be reduced electrochemically on the GCE surface without the use of toxic reagents that would have been required in the chemical reduction of GO.
Voltammetric studies of promethazine at the rGO-GCE
The electrochemical behavior of promethazine hydrochloride was investigated at bare and modified GCE electrodes. Initial studies were carried out with 1.0 × 10−3 mol L−1 of the analyte in 0.10 mol L−1 H2SO4 solution. This electrolyte was chosen based on the fact that many phenothiazine compounds, including promethazine, can easily oxidize in acidic medium.15 The cyclic voltammograms recorded at the bare GCE (a) and rGO-GCE (b) are compared in Fig. 4. For both sensors, it was possible to observe two oxidation waves corresponding to the formation of a cation (at +0.72 V (a) and +0.70 V (b)) and a dication radical (at +0.95 V (a) and +0.90 V (b)) during the forward sweep. | ||
| Fig. 4 Cyclic voltammograms recorded for a bare (a) GCE and rGO-GCE (b) in 0.10 mol L−1 H2SO4 medium (dashed line) and in 1.0 × 10−3 mol L−1 promethazine solution (solid line). Scan rate, 100 mV s−1. The inset shows the probable mechanism of promethazine oxidation. | ||
The inset of Fig. 4 shows the proposed mechanism for the electrochemical reaction of promethazine in acid medium. The first process corresponds to the reversible electrooxidation (reduction at +0.64 V) of the analyte nitrogen atom (situated at the ring) and the second anodic peak is related to the irreversible electrooxidation of the sulfur atom associated with a coupled hydrolysis reaction.
Comparing the oxidation and reduction peaks, the small magnitude of current at the reduction process can be attributed to the fact that the promethazine radical is fairly stable on a voltammetric time scale and at low pH value. For higher scan rates the difference between the cathodic and anodic signals decreases (see Fig. 5). These results are in agreement with those reported by Sackett et al.31 However, rGO-GCE appears to be a more favorable surface for the analyte oxidation and reduction processes, considering the small displacement observed between the anodic and cathodic peaks. The anodic current magnitude at the rGO-GCE was greater than the one at the bare GCE, probably caused by the surface area increase due to the presence of graphene film.
 | ||
| Fig. 5 Cyclic voltammograms obtained with a rGO-GCE in 0.10 mol L−1 H2SO4 for different: promethazine concentrations (A) (a–f, 2.49 to 14.8 × 10−4 mol L−1) and (B) scan rates (0.01 to 0.8 V s−1). Insets: (■) anodic and (●) cathodic currents. | ||
Other voltammetric studies, for a 1.0 × 10−3 mol L−1 promethazine solution (not shown here), involving a 0.1 mol L−1 B–R buffer (in a pH range over 2.0 and 7.0) were also carried out. The results showed that increasing the pH value makes the second peak shift to a less positive potential, until a critical point and above (pH 3) where higher values caused the two oxidation peaks to coalesce. Therefore, a single oxidation peak in the first scan and two new couples in the second scan were observed, implying the formation of new oxidation products, different from the ones formed during the first scan. Moreover, a slight peak current decrease (around 2%) was observed after 11 cycles at pH 2.0. At pH 7.0 this decrease was far more significant (42%) when compared to the initial current value. The reason for this current signal decrease is not obvious but probably at this pH the products generated during the oxidation process remain on the rGO-GCE surface, partially blocking it. Based on these results and considering the good repeatability obtained in 0.10 mol L−1 H2SO4 solution (RSD = 1.82% for 11 scans), this electrolyte was selected for all the experiments performed in the following sections.
Fig. 5A presents cyclic voltammograms corresponding to successive additions of promethazine in 0.10 mol L−1 H2SO4 at the rGO-GCE, with a scan rate of 100 mV s−1. The current signals obtained for each concentration of analyte were very stable and reproducible. A very linear relationship between cathodic and anodic currents and promethazine concentration (from 2.49 to 14.8 × 10−4 mol L−1) was observed (inset of Fig. 5A). Fig. 5B shows the variation of cathodic and anodic currents with a scan rate from 0.01 to 0.8 V s−1, using a 1.0 × 10−3 mol L−1 promethazine solution in 0.10 mol L−1 H2SO4 medium. An increase in the height of the oxidation and reduction peaks occurred and anodic peak potential shifted towards more positive values. The same was not so pronounced for cathodic peak potential. Moreover, the cathodic and anodic current values varied linearly with the square root of the scan rate (inset of Fig. 5B).
Determination of promethazine hydrochloride
The effect of anodic working potential (from +0.70 to +0.80 V, vs. Ag/AgCl, not shown here) was investigated in order to find the most suitable condition for amperometric quantification of promethazine in pharmaceutical products. During the amperometric experiments in 0.10 mol L−1 H2SO4, +0.78 V was selected as the working potential based on the best compromise between sensitivity and reproducibility.Under the optimized conditions (electrolyte: 0.10 mol L−1 H2SO4 and working potential: +0.78 V), a series of experiments were performed using promethazine standard solutions at different concentrations, in order to build the analytical curve. Linearity between oxidation current and analyte concentration was observed over a wide range, from 1.99 × 10−6 to 10.34 × 10−4 mol L−1. Fig. 6A shows a series of amperometric responses obtained in one of these experiments. The dynamic range presented a slope of 6.28 ± 3.67 × 10−2 mA mol−1 L and an intercept of 1.68 × 10−1 ± 1.62 × 10−2 μA, with a correlation coefficient of 0.999. The estimated detection limit was 1.99 × 10−7 mol L−1 (S/N = 3) and the quantification limit was calculated as 6.63 × 10−7 mol L−1.
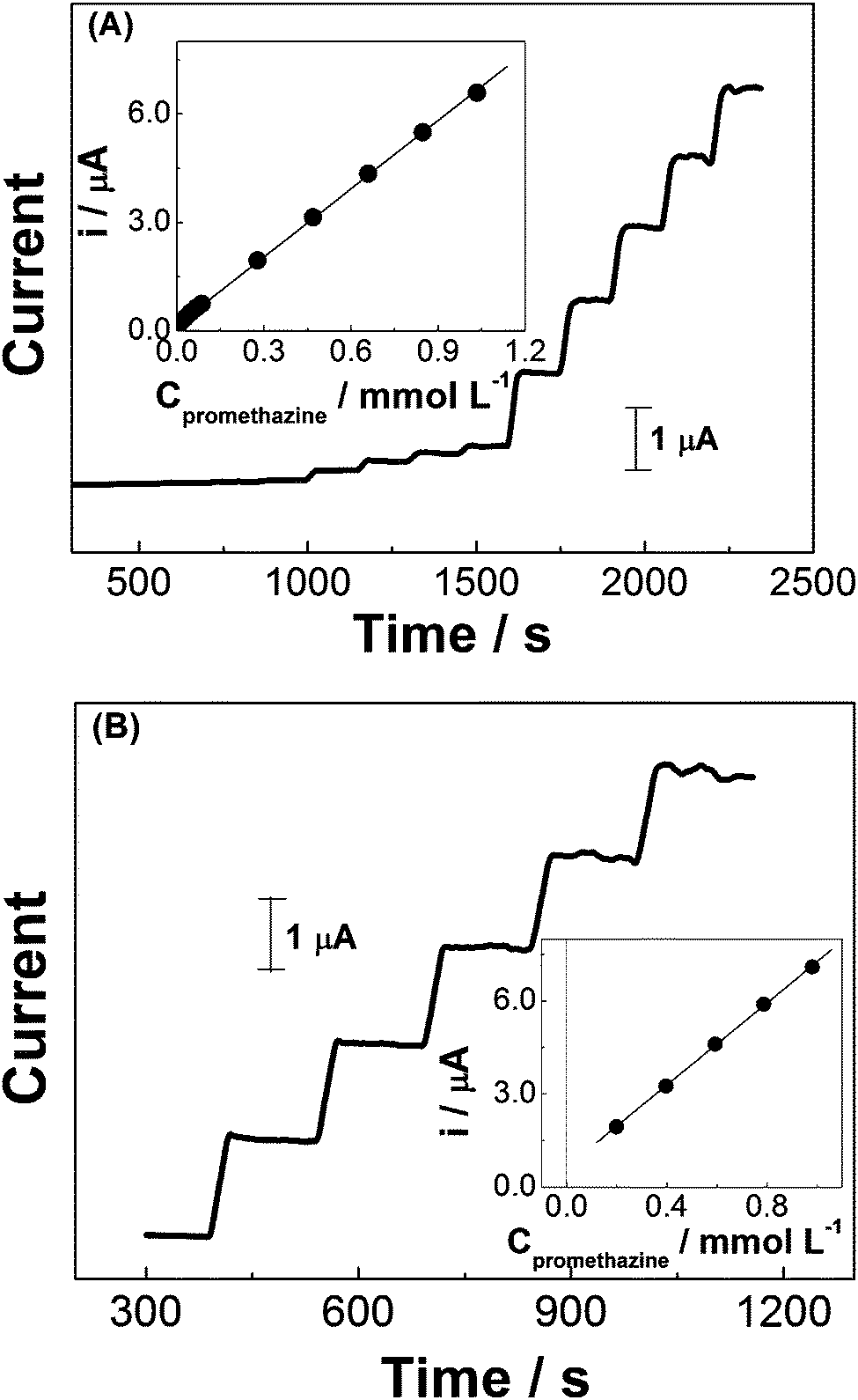 | ||
| Fig. 6 Amperometric response for: (A) successive additions of promethazine standard solution in 0.10 mol L−1 H2SO4 medium (1.99 × 10−6–10.34 × 10−4 mol L−1) and (B) promethazine analysis in commercial tablet sample using successive additions of standard solution (1.99–9.80 × 10−4 mol L−1). E = +0.78 V. | ||
The so prepared rGO-GCE sensor was used for the determination of promethazine hydrochloride in tablet samples which contains also amide, lactose, sugar, hydrated silica, talcum, magnesium stearate, methacrylate copolymer, polyethylene glycol, titanium dioxide and riboflavin. Parallel experiments involving 1 mM of promethazine and 5 mM amide, lactose, glucose or riboflavin showed that these compounds do not cause any interference under the established conditions. Fig. 6B presents the results of successive additions of promethazine standard solution into the electrochemical cell containing a commercial pharmaceutical sample dissolved in 0.10 mol L−1 H2SO4. The inset of this figure depicts a good linear relationship between successive additions of analyte standard solution (from 1.99 to 9.80 × 10−4 mol L−1) and amperometric signals. Values of promethazine hydrochloride concentration in tablet samples were obtained by extrapolating the data of the standard addition plots which correspond to the current variation of the added promethazine.
The results obtained using the proposed method were compared with those from the spectrophotometric and high-performance liquid chromatography procedures.29 Spectrophotometry is the technique recommended by the British Pharmacopoeia for promethazine determination.28Table 1 presents these results together with the corresponding standard deviations calculated from three independent measurements for each sample.
As presented in Table 1, results found by the proposed method are very close to the labeled value (28.2 mg) with a standard deviation of ±1.4%. Moreover, these results were compared favorably with the values obtained for spectrophotometry28 and HPLC.29 In the case of sample 1, the average value obtained by amperometry showed to be the same of that found by spectrophotometry and it was very close to the result obtained by HPLC. A significance test (null hypothesis) was applied to results showed in Table 1, resulting in experimental t-values of 1.00 (for amperometry and spectrophotometry) and 0.33 (for amperometry and HPLC). These values suggest there is no evidence of systematic errors for both situations (amperometry in comparison with spectrophotometric analysis and amperometry in comparison with HPLC analysis), considering 95% of confidence interval and a critical t-value of 12.71.32 The rGO-GCE sensor presented good analytical performance when compared with other carbon sensors modified with different agents for promethazine determination.33,34 Recently, Primo et al. have reported the use of the GCE modified with carbon nanotubes dispersed in DNA and, after 5 min of accumulation at an open circuit potential, they obtained a linear range from 1.0 × 10−7 to 6.0 × 10−6 mol L−1 and a detection limit of 2.3 × 10−8 mol L−1.34
Conclusion
The results obtained in this work demonstrate the potentiality of a GCE modified with an rGO film for the amperometric quantification of promethazine hydrochloride in pharmaceutical products. A low detection limit (1.99 × 10−7 mol L−1) and a wide linear range (from 1.99 × 10−6 to 1.03 × 10−3 mol L−1) with good repeatability were achieved for all amperometric experiments. Moreover, the fabrication process of the proposed sensor includes a simple electrochemical reduction of GO, avoiding the use of an excess of reducing agents that could contaminate the resulting material, which occurs when chemical reduction is chosen. Finally, this amperometric method allows rapid, simple, accurate and precise analysis without any sample pretreatment such as extraction or derivatization, making this methodology very suitable for quality control applications.Acknowledgements
The authors gratefully acknowledge financial support from Brazilian foundations (FAPESP, CAPES and CNPq – Process 306504-2011-1). We thank Mr Marcos Rodrigues Facchini Cerqueira and Justin Claude Kemmegne Mbouguen for suggestions in this paper.References
- L. J. Cote, J. Kim, V. C. Tung, J. Y. Luo, F. Kim and J. X. Huang, Pure Appl. Chem., 2011, 83, 95–110 CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozoy, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- W. Choi, I. Lahiri, R. Seelaboyina and Y. S. Kang, Crit. Rev. Solid State Mater. Sci., 2010, 35, 52–71 CrossRef CAS.
- M. Pumera, A. Ambrosi, A. Bonanni, E. L. K. Chng and H. L. Poh, Trends Anal. Chem., 2010, 29, 954–965 CrossRef CAS PubMed.
- M. A. Raj and S. A. John, Anal. Chim. Acta, 2013, 771, 14–20 CrossRef CAS PubMed.
- Y. H. Tang, R. Huang, C. B. Liu, S. L. Yang, Z. Z. Lu and S. L. Luo, Anal. Methods, 2013, 5, 5508–5514 RSC.
- K. Q. Deng, J. H. Zhou and X. F. Li, Colloids Surf., B, 2013, 101, 183–188 CrossRef CAS PubMed.
- Y. Kong, X. Ren, Z. Huo, G. Wang, Y. Tao and C. Yao, Eur. Food Res. Technol., 2013, 236, 955–961 CrossRef CAS PubMed.
- L. P. Jia and H. S. Wang, J. Electroanal. Chem., 2013, 705, 37–43 CrossRef CAS PubMed.
- J. M. Jian, Y. Y. Liu, Y. L. Zhang, X. S. Guo and Q. Cai, Sensors, 2013, 13, 13063–13075 CrossRef CAS PubMed.
- Y. Matsumoto, H. Tateishi, M. Koinuma, Y. Kamei, C. Ogata, K. Gezuhara, K. Hatakeyama, S. Hayami, T. Taniguchi and A. Funatsu, J. Electroanal. Chem., 2013, 704, 233–241 CrossRef CAS PubMed.
- J. Karpinska, B. Starczewska and H. PuzanowskaTarasiewicz, Anal. Sci., 1996, 12, 161–170 CrossRef CAS.
- F. W. P. Ribeiro, A. S. Cardoso, R. R. Portela, J. E. S. Lima, S. A. S. Machado, P. de Lima, D. de Souza and A. N. Correia, Electroanalysis, 2008, 20, 2031–2039 CrossRef CAS.
- S. Shahrokhian and N. H. Nassab, Electroanalysis, 2013, 25, 417–425 CrossRef CAS.
- D. Daniel and I. G. R. Gutz, J. Pharm. Biomed. Anal., 2005, 37, 281–286 CrossRef CAS PubMed.
- J. W. Li, F. Q. Zhao and B. Z. Zeng, Microchim. Acta, 2007, 157, 27–33 CrossRef CAS.
- X. M. Feng, C. Wang, R. J. Cui, X. Y. Yang and W. H. Hou, J. Solid State Electrochem., 2012, 16, 2691–2698 CrossRef CAS PubMed.
- P. Xiao, W. B. Wu, J. J. Yu and F. Q. Zhao, Int. J. Electrochem. Sci., 2007, 2, 149–157 CrossRef CAS PubMed.
- J. P. Marco, K. B. Borges, C. R. T. Tarley, E. S. Ribeiro and A. C. Pereira, Sens. Actuators, B, 2013, 177, 251–259 CrossRef CAS PubMed.
- Z. S. Yang, J. Zhao, D. P. Zhang and Y. C. Liu, Anal. Sci., 2007, 23, 569–572 CrossRef CAS.
- C. G. de Jesus, C. M. S. Forte, K. Wohnrath, C. A. Pessoa, J. E. D. Soares, S. T. Fujiwara, P. de Lima-Neto and A. N. Correia, Electroanalysis, 2011, 23, 1814–1820 CrossRef.
- T. Alizadeh and M. Akhoundian, Electrochim. Acta, 2010, 55, 5867–5873 CrossRef CAS PubMed.
- M. H. Parvin, M. B. Golivand, M. Najafi and S. M. Shariaty, J. Electroanal. Chem., 2012, 683, 31–36 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- P. H. Deng, Z. F. Xu and Y. F. Kuang, J. Electroanal. Chem., 2013, 707, 7–14 CrossRef CAS PubMed.
- D. W. Zhao, H. Liu, F. Wang, Q. M. Feng and M. Li, Anal. Sci., 2013, 29, 625–630 CrossRef CAS.
- The Stationery Office, British Pharmacopoeia, London, 7th edn, 2013, vol. 3, p. 1014 Search PubMed.
- S. Thumma, S. Q. Zhang and M. A. Repka, Pharmazie, 2008, 62, 562–567 Search PubMed.
- S. J. Li, C. Qian, K. Wang, B. Y. Hua, F. B. Wang, Z. H. Sheng and X. H. Xia, Sens. Actuators, B, 2012, 174, 441–448 CrossRef CAS PubMed.
- P. H. Sackett, J. S. Mayausky, T. Smith, S. Kalus and R. L. Mccreery, J. Med. Chem., 1981, 24, 1342–1347 CrossRef CAS.
- J. N. Miller and J. C. Miller, Statistics for Analytical Chemistry, Prentice Hall, Dorchester, 4th edn, 2000, pp. 52–54 Search PubMed.
- X. Xi, L. Ming and J. Liu, Drug Test. Anal., 2011, 3, 182–186 CrossRef CAS PubMed.
- E. N. Primo, M. B. Oviedo, C. G. Sanchez, M. D. Rubianes and G. A. Rivas, Bioelectrochemistry, 2014, 99, 8–16 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |