
Facile one-pot method synthesis CNT–GeO2 nanocomposite for high performance Li ion battery anode material
Fangfang
Jia
a,
Lingxiao
Song
b,
Wei
Wei
*b,
Peng
Qu
b and
Maotian
Xu
b
aCollege of Life Science, Shangqiu Normal University, Wenhua Road No. 298, Shangqiu, 476000, P. R. China
bSchool of Chemistry and Chemical Engineering, Shangqiu Normal University, Wenhua Road No. 298, Shangqiu, 476000, P. R. China. E-mail: weiweizzuli@163.com
First published on 22nd October 2014
Abstract
For the first time, we synthesized a carbon nanotubes–GeO2 nanocomposite via a facile one-pot solution method. The as-prepared carbon nanotubes–GeO2 nanocomposite appears as webs of curved nanotubes and nanoparticles, forming strong intertwined entanglements with a three-dimensional (3D) network structure. Such a hybrid structure can not only greatly improve the electron conductivity of the nanocomposite, but also effectively buffer the volume change of GeO2 during the charge–discharge process. The carbon nanotubes–GeO2 nanocomposite retains 813.7 mA h g−1 even after 500 cycles at the current rate of 0.2 A g−1 as an electrode material for Li ion batteries (LIBs) and shows high rate capability at variable current rates.
1 Introduction
Since the 1990s, LIBs have played an important role in rechargeable batteries. Now, Li ion battery research is focused on their durability, energy density, power density, cost and intrinsic safety. In fact, electrode material is a determining factor of the battery performance. Although graphite performs well as an anode for commercial LIBs, its theoretical capacity (372 mA h g−1) is too low to satisfy the persistent, increasing market demand. For the purpose of improving the energy density of LIBs, scientists have made great efforts to explore alternative anode materials with higher capacity. To increase the capacity, Li alloys including Si,1 Sn,2–4 Sb,5,6 Pb,7 In,8 Zn,9 and Ge10–24 have been considered as possible anodes for LIB owing to their low operating voltage, high volume energy density, and high theoretical capacity. Among these, Ge has emerged as an attractive anode for Li ion batteries due to its many fascinating properties, such as high capacity, a low working potential of about 0 to 0.4 V vs. Li, and high Li diffusivity at room temperature (400 times higher than the well-studied Si). Such characteristics offer Ge great advantages for high-power LIBs. However, similar to other Li alloy anodes, the fully lithiated Li4.4Ge undergoes a large volume expansion, which leads to pulverization and capacity fading in the bulk electrodes. To minimize such volume-strain during charge and discharge, many methods have been proved to be effective and these methods can be divided into two categories. The first method is to prepare various Ge nanostructures, such as nanowires,13,14 nanotubes,15 porous structures16,17 and thin films.18 The second is to prepare Ge composite materials, including carbon,19,20 graphene21,22 and so on. Among these composite materials, carbon nanotubes (CNTs), which are considered to be a powerful nanomaterial to functionalize other materials aiming at improving their electrical conductivity and mechanical property,25–27 have been widely used as a high-efficiency material to composite with other electrode materials.28,29 Although CNTs have successfully improved the Li storage performance of many electrode materials, GeO2 has not been the focus of much research.GeO2 has great potential application as an anode material in next-generation LIBs, due to its high theoretical reversible capacity (1125 mA h g−1 based on 4.25 mol Li per mol Ge), low operating voltage (<1.5 V) and higher thermal stability compared with Ge.30 However, similar to the SnO2 anode, at the end of the Li insertion process, the GeO2 particle size was enlarged by 127% in diameter, corresponding to a large volume expansion. The huge volume variation causes the pulverization and electrical detachment of active material, thus lead to a fast capacity fading. Recently, Cho31 and Yan32 demonstrated the promising electrochemical performance of GeO2 anodes via preparation of their carbon composite materials.
In this work, for the first time, to the best of our knowledge, we have developed a one step solution route to the in situ chemical synthesis of a CNT–GeO2 nanocomposite by using CNTs and Ge4+ solution. In the as-prepared nanocomposite, GeO2 nanoparticles and CNTs formed an efficient 3D conductive network structure. The CNTs, which have excellent mechanical strength, flexibility and electronic conductivity, can not only effectively buffer the large volume change of GeO2 during cycling, but also improve the conductivity of the composites. Therefore, such a hybrid structure is an ideal electrode material for LIBs. As anode material for LIBs, the CNT–GeO2 exhibits a remarkably high reversible charge capacity (1086.0 mA h g−1), excellent cycling stability (remains 813.7 mA h g−1 even after 500 cycles) and excellent rate capability.
2 Experimental
Preparation of materials
CNTs were pretreated according to the method reported elsewhere.28,29 In a typical synthesis of the CNT–GeO2 nanocomposite, 40 ml GeCl4 solution (0.05 M) was first mixed with 80 mg of the as-prepared CNTs and the mixture was heated in an oil bath to 110 °C under stirring. Next, 10 ml NaBH4 (0.25 g) solution was added dropwise, and the mixture was refluxed at 110 °C for 10 h. After cooling naturally, the obtained black mixture was collected by centrifugation. The mixture was first dried at 60 °C in air and then annealed at 400 °C in vacuum for 2 h. In order to make a comparison, pure GeO2 nanoparticles were prepared with the same process in the absence of CNTs.Characterizations
The morphologies of the samples were studied by a field-emission scanning electron microscope (Hitachi S-4800, 5 KV). The X-ray diffraction (XRD) spectra of the samples was recorded by a Rigaku Dmax 2200 X-ray diffractometer with Cu Kα radiation (λ = 1.5416 Å). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) investigations were carried out by a JEOL JEM-2100F microscope with an accelerating voltage of 20 KV. The as-prepared samples were dispersed in ethanol and dropped onto a carbon film supported on a copper grid. TGA was performed in an oxygen atmosphere using a Pyris Diamond TG/DTA (PerkinElemer Inc., U.S.A.). The samples were heated from 50 °C to 1000 °C at 10 °C min−1.Electrochemical performance
The electrochemical reactions of samples with lithium were investigated using a simple two-electrode cell. The working electrode consists of 80 wt% CNT–GeO2, 10 wt% Super P carbon as the conducting agent, 10 wt% polyvinylidene fluoride (PVDF) as binder, and Cu foil as substrate (current collector). N-Methyl pyrrolidinone (NMP) slurry consisting of the above mixture was uniformly coated on a copper disk of 14 mm in diameter. The disk electrodes were dried overnight at 60 °C followed by compression at 1.0 × 106 Pa. The 2016 type coin cells were assembled in an Ar-filled glove box using polypropylene (PP) micro-porous film as the separator, a solution of 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as the electrolyte and metallic lithium foil as the counter electrode. The electrochemical tests were performed on CT2001A Land battery testing systems (Jinnuo Electronics Co. Ltd., China). The cells were cycled at various currents and a constant temperature of 25 °C in the voltage range of 0.005–1.5 V.
:1, v/v) as the electrolyte and metallic lithium foil as the counter electrode. The electrochemical tests were performed on CT2001A Land battery testing systems (Jinnuo Electronics Co. Ltd., China). The cells were cycled at various currents and a constant temperature of 25 °C in the voltage range of 0.005–1.5 V.
3 Results and discussion
The process of preparing the CNT–GeO2 nanocomposite is briefly illustrated by Scheme 1. As mentioned in the Experimental section, the CNTs were pretreated before use. The pretreatment is known to create oxygen-containing surface functional groups on the chemically inert surface of the CNTs, thus facilitating good dispersion of the CNTs, uniform distribution of absorbed cations and, finally, uniform coatings.33 When thr Ge4+ solution was mixed with CNTs, Ge4+ bonded selectively with the oxygenated groups by electrostatic force. After NaBH4 solution was added, the Ge4+ cations were reduced to metallic Ge and aggregated to Ge nanoparticles on the surface of the CNTs. Since the reaction was carried out at 110 °C, the as-formed Ge nanoparticles were slowly oxidized by the dissolved oxygen to GeO2 nanoparticles. Thus, the CNT–GeO2 nanocomposite was formed. | ||
| Scheme 1 Scheme illustrating the synthesis of the CNT–GeO2 nanocomposite. | ||
Fig. 1 shows the XRD patterns of CNTs, GeO2 nanoparticles and CNT–GeO2 nanocomposite. From Fig. 1 it can be observed that the XRD patterns of the CNT–GeO2 nanocomposite, which match well with the hexagonal phase (JCPDS no. 36-1463) GeO2, have no distinct difference from pure GeO2 nanoparticles except the peak intensity is relatively weak. In the nanocomposite, no obvious CNT-related peaks can be seen, because the most obvious characteristic diffraction peak ((002) plane) located at around 27° (as shown in Fig. 1) overlapped with the characteristic diffraction peak of GeO2 ((101) plane).
 | ||
| Fig. 1 XRD patterns of the CNT–GeO2 nanocomposite and bare CNTs. | ||
As shown in Fig. 2a, CNTs with a lengths up to the micrometer scale were partly embedded in GeO2 nanoparticles. The CNT–GeO2 nanocomposite appears as a web of curved nanotubes and nanoparticles, forming strong intertwined entanglements with a three-dimensional (3D) network structure. Fig. 2b is the enlarged image of Fig. 2a, from which we can observe that: (1) the diameter of the CNT is 20–50 nm; (2) the GeO2 nanoparticles have a diameter at around 40 nm; (3) the GeO2 nanoparticles and CNTs interweave together, thus forming an excellent conductivity network. Such a 3D conductive network morphology coupled with the good flexibility and mechanical strength of the CNTs can facilitate fast ion/electron transportation, which is beneficial for the high-rate capability and cycling stability of the composite. Fig. 2c depicts the morphology of the as-prepared pure GeO2 nanoparticles; we can observe that the GeO2 nanoparticles have a diameter of ∼100 nm, which is much larger than those in the nanocomposite. This result also implies that the CNTs in the solution can prevent effectively the growth of GeO2 nanocrystals.
 | ||
| Fig. 2 SEM images of the CNT–GeO2 nanocomposite and GeO2 nanoparticles. (a) Low and (b) high magnification SEM images of the CNT–GeO2 nanocomposite, (c) SEM image of GeO2 nanoparticles. | ||
Fig. 3 shows TEM images of the CNT–GeO2 sample. Low magnification TEM imaging (Fig. 3a) revealed clumps of small GeO2 nanoparticles coating the surfaces of the carbon nanotubes, while the CNTs tangled together. High magnification imaging (Fig. 3b) revealed that the crystallite sizes of most of GeO2 crystals were 20–40 nm in diameter. It is well known that nano-sized alloying of anode materials benefits the electrochemical performance. The high resolution TEM (HRTEM) imaging of GeO2 nanoparticles is shown in Fig. 3c. The interplanar spacing of 0.35 nm matches the (101) plane of hexagonal structured GeO2.
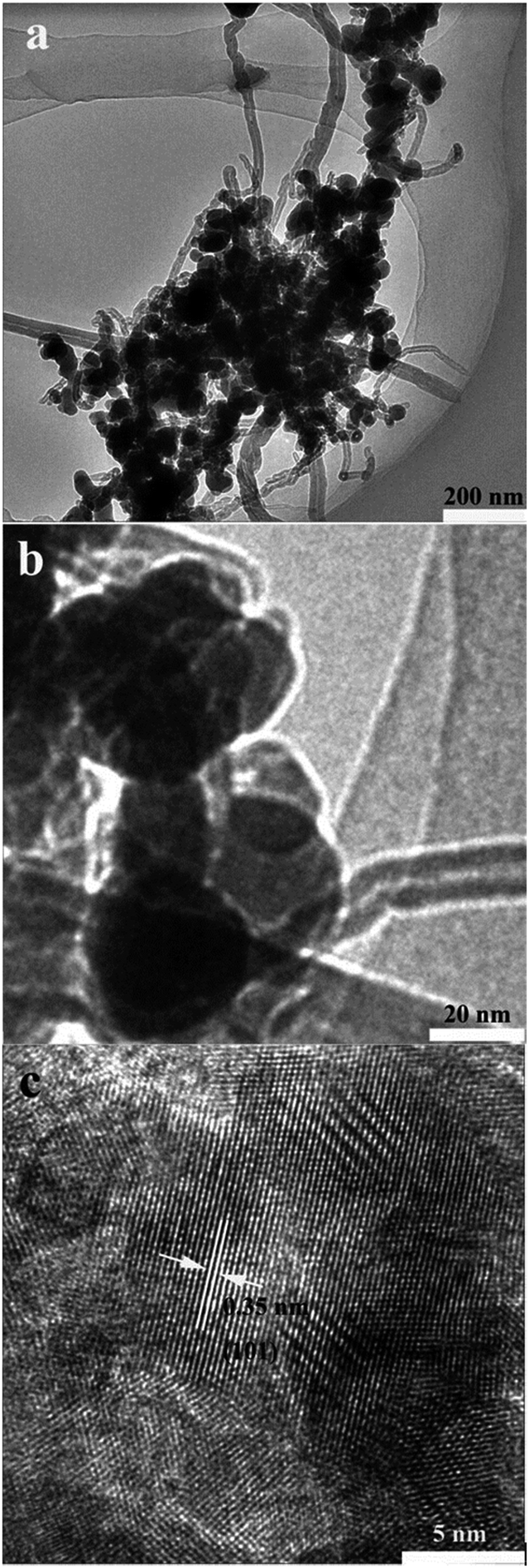 | ||
| Fig. 3 TEM images of CNT–GeO2 nanocomposites. (a) Low magnified image, (b) high magnified image and (c) high resolution TEM image. | ||
The specific surface area and pore structure of the bare CNTs and CNT–GeO2 nanocomposite were characterized by liquid-nitrogen adsorption–desorption tests. As shown in Fig. 4a, the bare CNTs show a type IV isotherm with H1-type hysteresis loop, which is generated by regularly shaped mesopores.34 As shown in Fig. 4b, the CNT–GeO2 nanocomposite shows a type IV isotherm with a typical H3-type hysteresis loop, indicating the characteristics of mesoporous materials.35 According to IUPAC nomenclature, these curves are characteristic of the different processes between adsorption into and desorption from the mesopores.36 The result reconfirmed that most of the pores in both of the bare CNTs and CNT–GeO2 nanocomposite were mesoporous. The BET surface area of the bare CNTs is 158.8 m2 g−1. The surface area of the nanocomposite was significantly reduced after the incorporation of GeO2 particles, namely from 158.8 to 55.7 m2 g−1. In fact, based on the morphology and TGA analysis in the later section, it is revealed that the GeO2 nanoparticles were not only on the surface of CNTs, but also wrapped up a large part of CNTs due to the high weight ratio of GeO2, which decreases greatly the measured surface area. Nevertheless, the BET value is still much larger than the as-synthesized pure GeO2 nanoparticles (24.3 m2 g−1). The relatively large surface area provided by the CNT–GeO2 nanocomposite would be beneficial for easy intercalation/extraction of Li ions and electrolyte molecules.
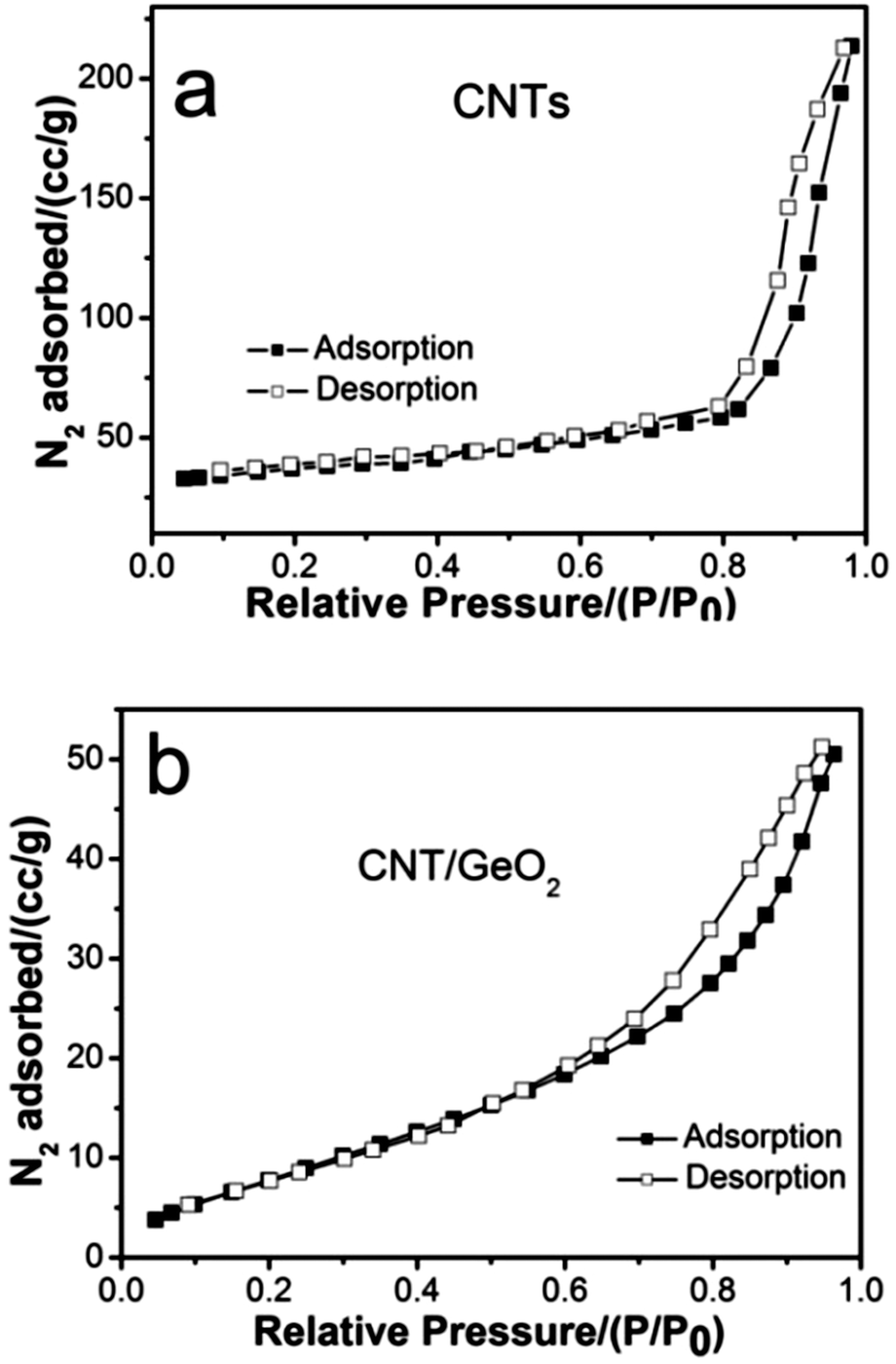 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms obtained at 77 K of (a) CNTs and (b) the CNT–GeO2 nanocomposite. | ||
As shown in Fig. 5a, the weight ratio of CNTs in the nanocomposite was firstly estimated by using energy-dispersive X-ray spectroscopy (EDS) analysis. The EDS spectrum of the CNT–GeO2 nanocomposite clearly showed the existence of germanium, oxygen, and carbon. The weight ratio of 34.3% for carbon indicated the CNTs in the nanocomposite were ∼34.3%. To determine more accurately the amount of CNTs, TGA was carried out in air. The sample was heated from 50 to 1000 °C at a rate of 10 °C min−1. Fig. 5b shows the TGA curve for the CNT–GeO2 nanocomposite along with that of CNT powders. It can be seen that the CNT–GeO2 nanocomposite starts to decompose slowly in air at temperatures above 100 °C, with the CNTs finally burning out at about 700 °C. Since the bare GeO2 powder remains stable in this temperature range, any weight change corresponds to the oxidation of CNTs. Therefore, the change in weight before and after the oxidation of CNTs can be translated into the amount of GeO2 in the CNT–GeO2 nanocomposite. With this method, the approximate amount of GeO2 can be estimated by first subtracting the residual amount of impurities contained in the as-purchased CNTs. Therefore, the final amount of GeO2 is approximately 67.8%, and correspondingly, the weight ratio of CNTs is 32.2%. This value is very close to the EDS analysis.
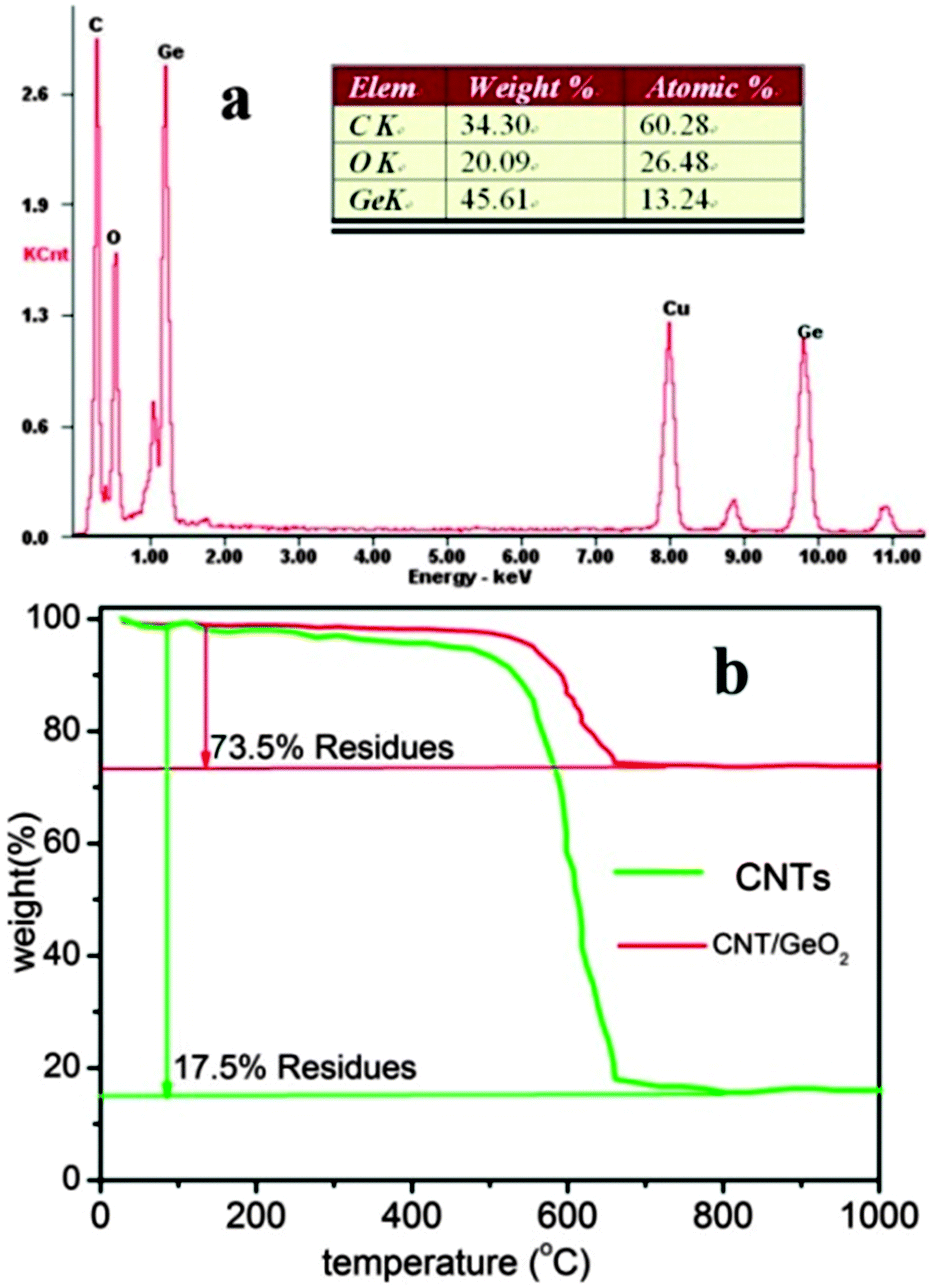 | ||
| Fig. 5 (a) EDS analysis of the CNT–GeO2 nanocomposite and (b) TGA curves of bare CNTs and the CNT–GeO2 nanocomposite. | ||
The initial discharge–charge capacities of the composite are 2131.4 and 1081.6 mA h g−1, respectively, corresponding to an initial coulombic efficiency of 50.7%, as shown in Fig. 7a. The large irreversible initial capacity can be attributed to irreversible insertion of lithium in the CNTs (as shown in Fig. 6) and to the formation of a massive solid electrolyte interface (SEI) due to the high surface area.3 In addition, irreversibility also results from irreversible formation of Li2O before transformation into LixGe alloys during the first reduction.2 Indeed, as shown in Fig. 7a, the plateau at 0.55–0.25 V in the discharge curve is attributed to the reduction of GeO2 into Ge and Li2O.37 Lithium-rich LixGe alloys such as Li7Ge2, Li15Ge4, Li11Ge6, Li9Ge4, and Li22Ge5 are generally formed at lower potentials between 0.01 and 0.25 V, as shown in Fig. 7a.2,38,39Fig. 7b shows the cycling performance of the CNT–GeO2 nanocomposite electrode in the potential range of 5 mV–1.5 V at a current of 0.2 A g−1. As expected, the electrode exhibited excellent capacity retention up to 500 cycles. The discharge–charge capacities of the electrodes after 500 cycles were 844.8 and 813.7 mA h g−1, respectively. The reversible charge capacity retention reaches up to 75.2%. In order to make a comparison, the as-prepared pure GeO2 nanoparticles mixed with 32.2 wt% carbon black were also tested. As shown in Fig. 8a, when cycled only at 0.2 A g−1 for 50 cycles, the discharge–charge capacities of the mixture decreased to 383.5 and 376.5 mA h g−1, respectively, corresponding to a reversible charge capacity retention of only 51.2%. We attribute the superior cycling stability of the CNT–GeO2 nanocomposite to its special nanocomposite structure. The rate capability of CNT–GeO2 is evaluated to investigate further the effect of CNT incorporation on the electrochemical performance of GeO2 (Fig. 8b). The samples were charged and discharged at various current densities. The initial reversible charge capacity of the composite is 1060.0 mA h g−1 at 0.2 A g−1; it decreased to 904.5 mA h g−1 after 10 cycles. The charge capacities reach up to 793.4, 630.1, 459.6 and 209.3 mA h g−1 at 0.5 A g−1, 1 A g−1, 2 A g−1 and 5 A g−1, respectively. When the current rate was gradually decreased to 2 A g−1, 1 A g−1 and 0.2 A g−1 again, the electrode could deliver reversible charge capacities to 451.3, 599.7, 851.3 mA h g−1, respectively. By comparison it can be found that the nanocomposite electrode recovers almost fully its charge capacity upon decreasing the current density. This result also confirms that our CNT–GeO2 nanocomposite is tolerant to a variable charge–discharge current, which is a desirable characteristic required for high power application. Although the electrochemical performance of our CNT–GeO2 is slightly poorer than the recently reported results,31,32 it is comparable to many of the previously reported CNT composites.3,40,41 To improve further the Li storage performance of the CNT–GeO2 composite, the preparation smaller GeO2 (e.g. 2–5 nm) nanoparticles that are more uniformly coated on CNTs may be a possible method.
 | ||
| Fig. 6 The first two cycle charge–discharge profiles of the bare CNTs electrode. The initial discharge capacity of the bare CNTs reaches up to 1940 mA h g−1, while the initial reversible charge capacity is only 703 mA h g−1. | ||
 | ||
| Fig. 7 (a) Initial charge–discharge curves of the CNT–GeO2 nanocomposite; (b) cycling performance of the nanocomposite at 0.2 A g−1. | ||
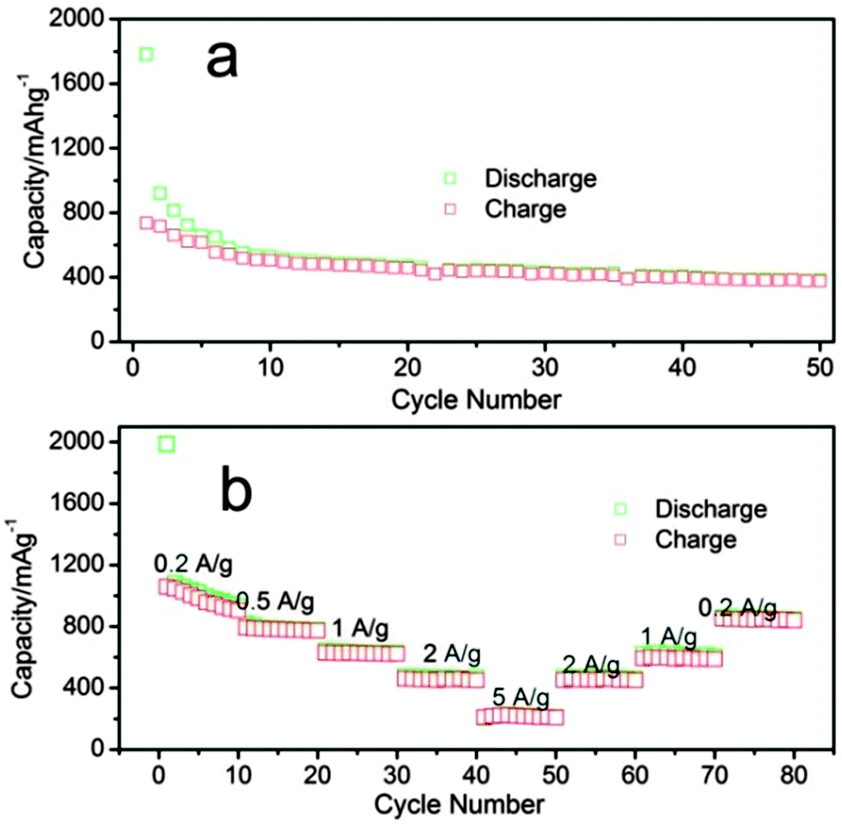 | ||
| Fig. 8 (a) Cycling performance of the carbon black–GeO2 mixture, and (b) rate capability of the CNT–GeO2 nanocomposite. | ||
The electrochemical test results clearly demonstrate that the CNTs play an important role in improving the electrochemical performance of GeO2. It is believed that the enhanced electrochemical performance originates from three factors: first, the enhanced cycling stability can be attributed to the in situ incorporated CNTs that not only effectively buffer the volume changes during the lithiation–delithiation reactions but also restrain the agglomeration of GeO2 nanoparticles upon long-term cycling. Fig. 9 compares the electrode surface morphologies of the CNT–GeO2 nanocomposite and the carbon black–GeO2 mixture. It is obvious that the mixed electrode showed cracks after only 50 cycles (Fig. 9a), while the composite electrode did not show obvious cracks even after 500 cycles (Fig. 9b). This result confirms that the use of the CNTs is an effective way to enhance the cycling performance of GeO2.42 Second, the highly conductive CNTs supply 3D electronically conducting networks for the GeO2 nanoparticles, thus improving greatly the rate capability of the nanocomposite. Third, the small GeO2 nanoparticles (∼40 nm) coupled with the mesoporous structure are beneficial for better electrolyte wetting of the CNT–GeO2 nanocomposite and the rapid Li-ion transport across the electrode/electrolyte interface, which is beneficial for both cycling performance and rate capability.
 | ||
| Fig. 9 SEM images of (a) the carbon black–GeO2 mixture electrode after 50 cycles, and (b) the CNT–GeO2 nanocomposite electrode after 500 cycles. | ||
4 Conclusions
In summary, we have developed one-step solution route to in situ chemically synthesise a carbon nanotubes–GeO2 nanocomposite for the first time. As an anode material for Li-ion batteries, the nanocomposite presented superior cycling stability; even after 500 cycles, a reversible charge capacity of 813.7 mA h g−1 can be obtained, corresponding to a capacity retention up to 75.2%. The nanocomposite also shows excellent rate capability; at current rates of 0.5 A g−1, 1 A g−1, 2 A g−1 and 5 A g−1, the charge capacity reached 793.4, 630.1, 459.6 and 209.3 mA h g−1, respectively. Such good electrochemical performance of the nanocomposite was ascribed to the incorporation of carbon nanotubes, which have high electronic conductivity and possesses good flexibility.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (20145085 & 21271125/B010601).Notes and references
- H. Wu, G. Zheng, N. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904 CrossRef CAS PubMed.
- I. A. Courtney and J. R. Dahn, J. Electrochem. Soc., 1997, 144, 2045 CrossRef CAS PubMed.
- Z. Wen, Q. Wang, Q. Zhang and J. Li, Adv. Funct. Mater., 2007, 17, 2772 CrossRef CAS.
- J. Huang, L. Zhong, C. Wang, J. P. Sullivan, W. Xu, L. Q. Zhang, S. X. Mao, N. S. Hudak, X. H. Liu, A. Subramanian, H. Fan, L. Qi, A. Kushima and J. Li, Science, 2010, 330, 1515 CrossRef CAS PubMed.
- H. Bryngelsson, J. Eskhult, L. Nyholm, M. Herranen, O. Alm and K. Edström, Chem. Mater., 2007, 19, 1170 CrossRef CAS.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255 CrossRef CAS PubMed.
- C. M. Park, J. H. Kim, H. Kim and H. J. Sohn, Chem. Soc. Rev., 2010, 39, 3115 RSC.
- D. W. Kim, I. S. Hwang, S. J. Kwon, H. Y. Kang, K. S. Park, Y. J. Choi, K. J. Choi and J. G. Park, Nano Lett., 2007, 7, 3041 CrossRef CAS PubMed.
- Y. Hwa, J. H. Sung, B. Wang, C. M. Park and H. J. Sohn, J. Mater. Chem., 2012, 22, 12767 RSC.
- X. H. Liu, S. Huang, S. T. Picraux, J. Li, T. Zhu and J. Y. Huang, Nano Lett., 2011, 11, 3991 CrossRef CAS PubMed.
- D. D. Vaughn II and R. E. Schaak, Chem. Soc. Rev., 2013, 42, 2861 RSC.
- J. Graetz, C. C. Ahn, R. Yazami and B. Fultz, J. Electrochem. Soc., 2004, 151, A698 CrossRef CAS PubMed.
- M. H. Seo, M. Park, K. T. Lee, K. Kim, J. Kim and J. Cho, Energy Environ. Sci., 2011, 4, 425 CAS.
- L. P. Tan, Z. Lu, H. T. Tan, J. Zhu, X. Rui, Q. Yan and H. H. Hng, J. Power Sources, 2012, 206, 253 CrossRef CAS PubMed.
- M. H. Park, Y. H. Cho, K. Kim, J. Kim, M. Liu and J. Cho, Angew. Chem., Int. Ed., 2011, 50, 9647 CrossRef CAS PubMed.
- M. H. Park, K. Kim, J. Kim and J. Cho, Adv. Mater., 2010, 22, 415 CrossRef CAS PubMed.
- L. C. Yang, Q. S. Gao, L. Li, Y. Tang and Y. P. Wu, Electrochem. Commun., 2010, 12, 418 CrossRef CAS PubMed.
- N. G. Rudawski, B. L. Darby, B. R. Yates, K. S. Jones, R. G. Elliman and A. A. Volinsky, Appl. Phys. Lett., 2012, 100, 083111 CrossRef PubMed.
- G. Cui, L. Gu, L. J. Zhi, N. Kaskhedikar, P. A. van Aken, K. Müllen and J. Maier, Adv. Mater., 2008, 20, 3079 CrossRef CAS.
- G. Jo, I. Choi, H. Ahn and M. J. Park, Chem. Commun., 2012, 48, 3987 RSC.
- D. J. Xue, S. Xin, Y. Yan, K. C. Jiang, Y. X. Yin, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 2512 CrossRef CAS PubMed.
- J. G. Ren, Q. H. Wu, H. Tang, G. Hong, W. Zhang and S. T. Lee, J. Mater. Chem. A, 2013, 1, 1821 CAS.
- J. S. Pena, I. Sandu, O. Joubert, F. S. Pascual, C. O. Arean and T. Brousse, Electrochem. Solid-State Lett., 2004, 7, A278 CrossRef CAS PubMed.
- Y. Kim, H. Hwang, K. Lawler, S. W. Martin and J. Cho, Electrochim. Acta, 2008, 53, 5058 CrossRef CAS PubMed.
- M. P. Rossi, H. Ye, Y. Gogotsi, S. Babu, P. Ndungu and J. C. Bradley, Nano Lett., 2004, 4, 989 CrossRef CAS.
- P. Cherukuri, S. M. Bachilo, S. H. Litovsky and R. B. Weisman, J. Am. Chem. Soc., 2004, 126, 15638 CrossRef CAS PubMed.
- Z. Wu, Z. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner, A. F. Hebard and A. G. Rinzler, Science, 2004, 305, 1273 CrossRef CAS PubMed.
- H. Fang, S. Zhang, X. Wu, W. Liu, B. Wen, Z. Du and T. Jiang, J. Power Sources, 2013, 235, 95 CrossRef CAS PubMed.
- H. Fang, S. Zhang, T. Jiang, R. Lin and Y. Lin, Electrochim. Acta, 2014, 125, 427 CrossRef CAS PubMed.
- W. Wei and L. Guo, Part. Part. Syst. Charact., 2013, 30, 658 CrossRef CAS.
- K. H. Seng, M. Park, Z. P. Guo, H. K. Liu and J. Cho, Nano Lett., 2013, 13, 1230 CrossRef CAS PubMed.
- Y. Chen, C. Yan and O. G. Schmidt, Adv. Energy Mater., 2013, 3, 1269 CrossRef CAS.
- J. Lu, Carbon, 2007, 45, 1599 CrossRef CAS PubMed.
- G. Giambastiani, S. Cicchi, A. Giannasi, L. Luconi, A. Rossin, F. Mercuri, C. Bianchini, A. Brandi, M. Melucci, G. Ghini, P. Stagnaro, L. Conzatti, E. Passaglia, M. Zoppi, T. Montini and P. Fornasiero, Chem. Mater., 2011, 23, 1923 CrossRef CAS.
- B. Liu and H. C. Zeng, Chem. Mater., 2008, 20, 2711 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscow, R. A. Pierotti, T. Rouquerol and T. Siemienewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- H. A. Wilhelm, C. Marino, A. Darwiche, L. Monconduit and B. Lestriez, Electrochem. Commun., 2012, 24, 89 CrossRef CAS PubMed.
- L. Baggetto and P. H. L. Notten, J. Electrochem. Soc., 2009, 156, A169 CrossRef CAS PubMed.
- A. S. Jing Li, R. J. Sanderson, T. D. Hatchard, R. A. Dunlap and J. R. Dahn, J. Electrochem. Soc., 2009, 156, A283 CrossRef PubMed.
- B. Zhang, Q. B. Zheng, Z. D. Huang, S. W. Oh and J. K. Kim, Carbon, 2011, 49, 4524 CrossRef CAS PubMed.
- L. Noerochim, J. Z. Wang, S. L. Chou and D. Wexler, Carbon, 2012, 50, 1289 CrossRef CAS PubMed.
- C. Li, W. Wei, S. Fang, H. Wang, Y. Zhang, Y. Gui and R. Chen, J. Power Sources, 2010, 195, 2939 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |