
The synthesis, spectroscopic characterization and anticancer activity of new mono and binuclear phosphanegold(I) dithiocarbamate complexes†
Muhammad
Altaf
a,
M.
Monim-ul-Mehboob
a,
Anvarhusein A.
Isab
*a,
Vikram
Dhuna
b,
Gaurav
Bhatia
b,
Kshitija
Dhuna
c and
Saleh
Altuwaijri
d
aDepartment of Chemistry, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia. E-mail: aisab@kfupm.edu.sa
bDepartment of Biotechnology, DAV College, Amritsar-143001, Punjab, India
cDepartment of Molecular Biology and Biochemistry, Guru Nanak Dev University, Amritsar-143005, Punjab, India
dClinical Research Laboratory, SAAD Research Development Center, SAAD Specialist Hospital, Al-Khobar 31952, Saudi Arabia
First published on 24th October 2014
Abstract
A new series of mononuclear [t-Bu3PAuS2CN(C7H7)2] (1), and binuclear [(DPPM)Au2(S2CN(CH3)2)2] (2), [(DPPM)Au2(S2CN(C2H5)2)2] (3) and [(DPPM)Au2(S2CN(C7H7)2)2] (4) [where DPPM = 1,1-bis(diphenylphosphino)methane, S2CN(CH3)2 = dimethyldithiocarbamate, S2CN(C2H5)2 = diethyldithiocarbamate and S2CN(C7H7)2 = dibenzyldithiocarbamate] gold(I) complexes have been prepared by reacting gold(I) precursors and dialkyl–diaryl dithiocarbamate ligands. The complexes were characterized by an analytical technique and spectroscopic methods such as CHNS analysis, FTIR spectroscopy; 1H, 13C and 31P NMR measurements. The molecular structure of the [t-Bu3PAuS2CN(C7H7)2] (1) complex was determined by X-ray diffraction. The gold(I) complexes (2 and 3) were found particularly to be better potent in vitro cytotoxic agents in comparison to cisplatin against HeLa, HCT15 and A549 cancer cell lines. These metal complexes could serve as attractive anticancer agents for the development of novel therapeutic strategies and to treat cervix, lung and colon cancers.
1. Introduction
The triumphant depiction of cisplatin, oxaliplatin and carboplatin as metal-based anticancer drugs is well acknowledged in the field of chemotherapy.1–6 Indeed, such drugs have been used for the treatment of cancer patients worldwide. Cisplatin and its analogues have serious side effects, such as oto-, neuro-, and nephrotoxicity, which decrease their effectiveness in cancer therapy.7–10 As a result, gold(I) and gold(III) complexes have been investigated as non-platinum based anticancer candidates.11–14The study of gold complexes, bearing different functional ligands exhibiting physical, chemical, biological and pharmacological properties, has gained much attention.11–14 The gold(I) complexes have long been studied as anti-arthritic and anti-microbial agents.15–19 For instance, the drugs like Auranofin, Solganol and Myocrisin have frequently been used for the treatment of rheumatoid arthritis.20–24 Interestingly, the extensive cell-based (in vitro) and animal (in vivo) studies have revealed the potent anti-cancer activities of diverse classes of gold(I) and gold(III) complexes with a wide range of ligands against a panel of human cancer cell lines.25–27
An enormous number of bridged di-gold(I) complexes, preferably existing in a linear 2-coordinate configuration like [ClAu(P–P)AuCl] (where P–P is a bisphosphine), are more effective than free ligands and exhibit a broad range of anticancer activities.28,29 This has inspired the synthesis of stable 4-coordinate digold(I) diphosphine complexes.30–32 The effect of structural variation in chelated bis(diphosphine) gold(I) complexes [Au(R2P(CH2)nPR2)]X on their cytotoxicity and activity against P388 leukaemia, B16 melanoma and M5076 reticulum cell sarcoma has been studied.33 J. W. Faamaua et al. reported compounds of general formula [(Ph2P(CH2)nPPh2)(AuS2CNR2)2], n = 1, 2 or 3 and R = Et or c-hexyl.34
Since the first decade of 21st century, a new class of gold complexes with dithiocarbamate ligands has favorably been emerged as anticancer agents. In this regard, Fregona and coworkers synthesized and characterized some novel gold(III) compounds containing N,N-dimethyldithiocarbamate and ethyl sarcosine dithiocarbamate exhibiting a potential chemical and biological profile.35 Dibromo(N,N-dimethyldithiocarbamato)gold(III) also showed a noteworthy inhibition of in vivo MDA-MB-231 breast cancer growth.36 Zhang et al. reported that gold(I)–dithiocarbamato species, namely [Au(ESDT)](2), could hamper the chymotrypsin-like activity of purified 20S proteasome and 26S proteasome in human breast cancer MDA-MB-231 cells, resulting in accumulation of ubiquitinated proteins and proteasome target proteins, and induction of cell death.37 Recently, the modern research has progressively targeted the search for new gold(I) complexes as potential anticancer drugs.38–42
Worldwide, lung and colorectal cancers are frequent causes of cancer-related death regardless of males and females while cervix cancer is still overwhelmingly the reason for cancer deaths in females exclusively. Therefore, there is a dire need for designing new drugs in order to treat such lethal diseases through chemotherapy. In the wake of chrysotherapeutic agents, gold(I) complexes could be developed with a new combination of P and S donor ligands which may have better selectivity and activity with least side effects for cancer treatment.
Gold(I) complexes based on monophosphine–bisphosphine and dithiocarbamate mixed ligands have been addressed. Rationally, we are planning to design the new dithiocarbamato gold(I) with monophosphine–bisphosphine complexes which would potentially lead to the development of new anticancer agents and the treatment of a variety of cancers effectively through chrysotherapy.
In the current study, we present the synthesis of gold(I) complexes of phosphine and dialkyl–diaryldithiocarbamate mixed ligands, their structure analysis by mid-IR spectroscopy and NMR measurements; and molecular structure determination by single crystal X-ray diffraction. Finally, the characterized gold(I) complexes have systematically been examined for in vitro cytotoxic activity against various human cancer cell lines, e.g. A549 (human lung carcinoma), HCT15 (human colon carcinoma), and HeLa (human cervix cancer).
2. Experimental
2.1. Materials and methods
Chemicals and solvents used in the synthesis were of analytical grade and used without further purification. All the reactions were carried under normal ambient conditions. All chemicals were obtained from Sigma-Aldrich St. Louis, Missouri United States and Strem Chemicals, Massachusetts, United States.Elemental analyses were performed on Perkin Elmer Series 11 (CHNS/O), Analyzer 2400. The solid state FTIR spectra of free ligands and their corresponding gold(I) complexes were recorded on a Perkin-Elmer FTIR 180 spectrophotometer or NICOLET 6700 FTIR using KBr pellets over the range 4000–400 cm−1.
1H, 13C and 31P NMR spectra were recorded on a LAMBDA 500 spectrophotometer operating at 500.01, 125.65 and 200.0 MHz respectively; corresponding to a magnetic field of 11.74 T. Tetramethylsilane (TMS) was used as an internal standard for 1H and 13C NMR measurements. Triphenylphosphine (TPP) was used as an external standard for 31P NMR measurements. The 13C NMR spectra were obtained with 1H broadband decoupling. The spectral conditions were: 32k data points, 0.967 s acquisition time, 1.00 s pulse delay and 45° pulse angle.
Structures of the gold(I) precursors and free ligands used in this study are shown in Scheme 1. The 1H, 13C and 31P NMR chemical shifts of metal precursors and free ligands are given in Tables 1S and 2S (see ESI†). Proposed structures of the synthesized complexes (1–4) are given in Scheme 2.
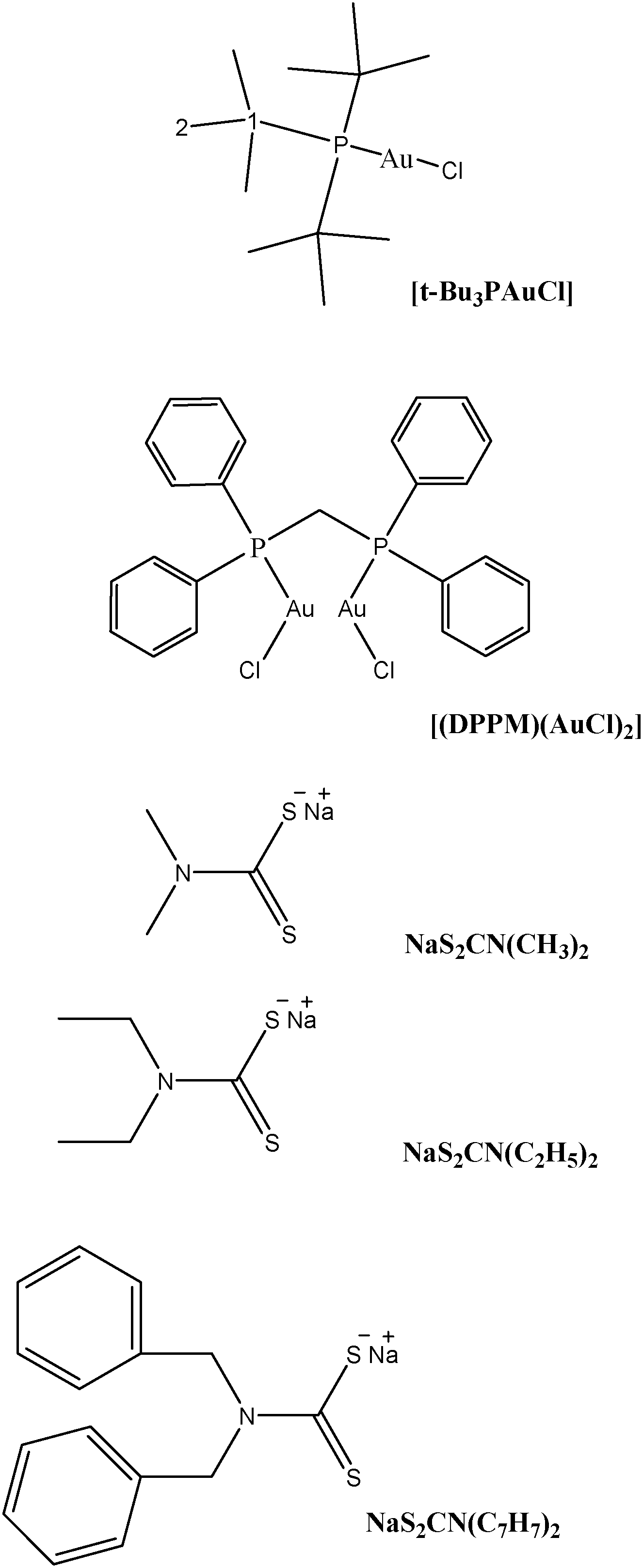 | ||
| Scheme 1 Skeletal structures and condensed formulae of precursors and dithiocarbamate ligands for 1H and 13C NMR data. | ||
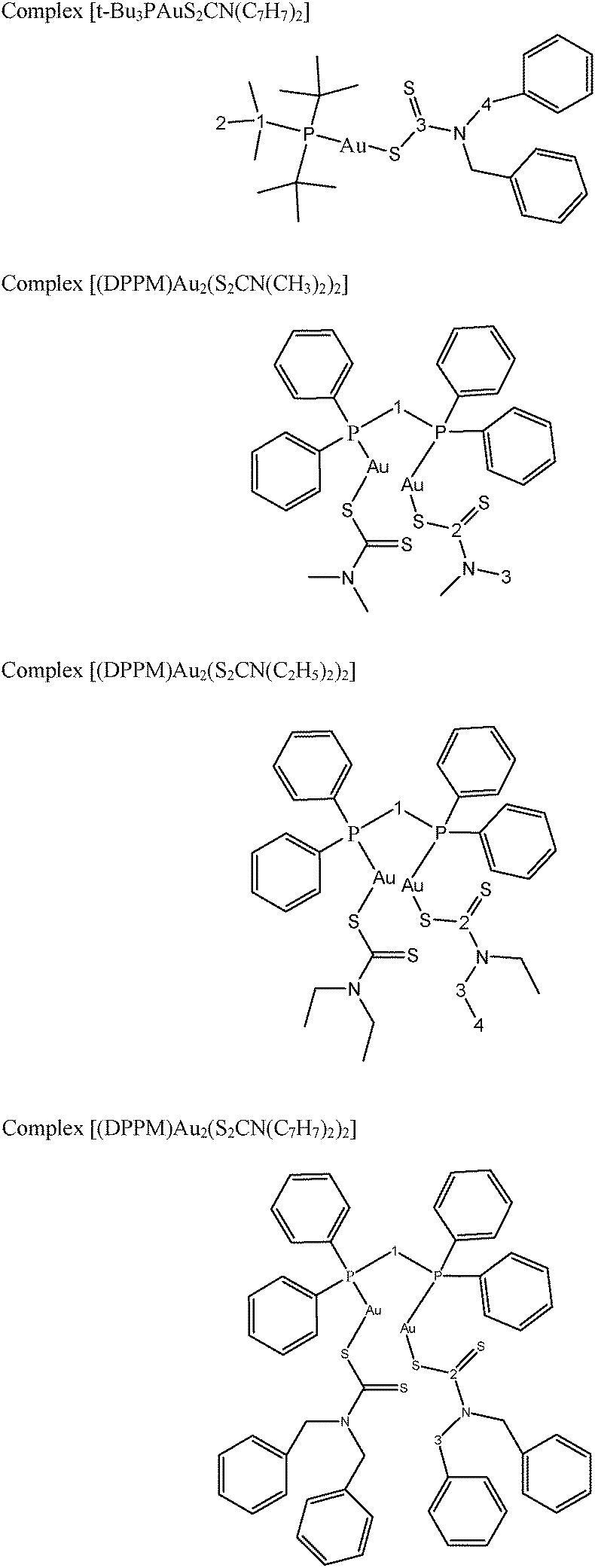 | ||
| Scheme 2 Skeletal structures and condensed formulae of complexes (1–4); representing non-equivalent carbons and protons for 1H and 13C NMR data. | ||
2.2. Synthesis of gold(I) complexes
2.3. Stability test of gold(I) complexes in DMSO-d6
Complexes (1 and 2) were dissolved in DMSO-d6 and analyzed by 1H and 13C {1H} NMR measurements. The extent of decomposition over time was determined by comparing the NMR spectra collected after 1, 6, 12, 24, 48 and 72 h. No significant change in the chemical shifts and the splitting patterns of complexes (1 and 2) was observed in their time dependent 1H NMR spectra.2.4. UV-visible measurements
UV-vis spectroscopy was used to determine the stability of the complexes (1–4) in 1% DMSO solution. Electronic spectra were recorded on freshly prepared solutions and after 72 h of each complex at room temperature. Electronic spectra were obtained for complexes (1–4) using a Lambda 200, Perkin-Elmer UV-vis spectrometer.2.5. X-ray diffraction studies
Pale yellow plate like crystals of compound (1) were obtained by recrystallization of the final product using a mixture of solvents i.e. C2H5OH and H2O in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v ratio under slow evaporation at room temperature. The intensity data were collected at 173 K (−100 °C) on a Stoe Mark II-Image Plate Diffraction System43 equipped with a two-circle goniometer using MoKα graphite mono chromated radiation (λ = 0.71073 Å). The structure was solved by Direct methods with SHELXS-97.44 The refinement and all further calculations were carried out with SHELXL-2013.44 The C-bound H-atoms were included in the calculated positions and treated as riding atoms: C–H = 0.95, 0.99 and 0.98 Å for CH (aromatic), CH2 and CH3, respectively, with Uiso(H) = 1.5Ueq (C-methyl) and = 1.2Ueq (C) for other H-atoms. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using the MULscanABS routine in PLATON.45Fig. 1 was drawn using the programs MERCURY.46 A summary of crystal data and refinement details for gold(I) complex (1) are given in Table 1. Selected bond lengths and bond angles are given in Table 2.
:1 v/v ratio under slow evaporation at room temperature. The intensity data were collected at 173 K (−100 °C) on a Stoe Mark II-Image Plate Diffraction System43 equipped with a two-circle goniometer using MoKα graphite mono chromated radiation (λ = 0.71073 Å). The structure was solved by Direct methods with SHELXS-97.44 The refinement and all further calculations were carried out with SHELXL-2013.44 The C-bound H-atoms were included in the calculated positions and treated as riding atoms: C–H = 0.95, 0.99 and 0.98 Å for CH (aromatic), CH2 and CH3, respectively, with Uiso(H) = 1.5Ueq (C-methyl) and = 1.2Ueq (C) for other H-atoms. The non-H atoms were refined anisotropically, using weighted full-matrix least-squares on F2. A semi-empirical absorption correction was applied using the MULscanABS routine in PLATON.45Fig. 1 was drawn using the programs MERCURY.46 A summary of crystal data and refinement details for gold(I) complex (1) are given in Table 1. Selected bond lengths and bond angles are given in Table 2.
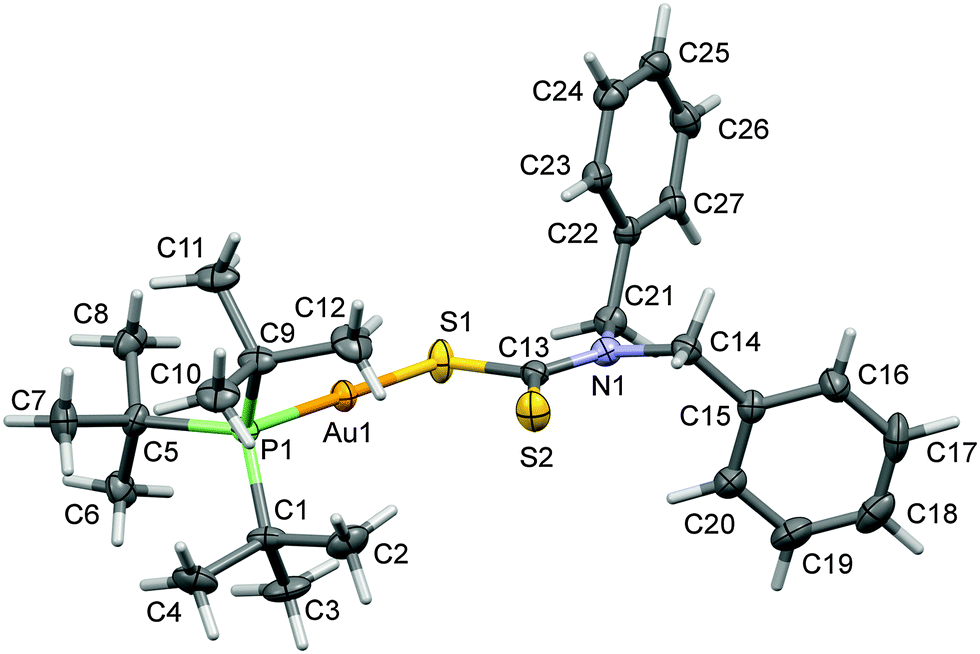 | ||
| Fig. 1 A view of the molecular structure of complex (1) with atom labeling. The displacement ellipsoids are drawn at the 50% probability level. | ||
| Parameters | [t-Bu3PAuS2CN(C7H7)2] |
|---|---|
| Empirical formula | C27H41AuNPS2 |
| Empirical formula weight | 671.66 |
| Crystal size/mm | 0.45 × 0.30 × 0.07 |
| Wavelength/Å | 0.71073 |
| Temperature/K | 173 |
| Crystal symmetry | Orthorhombic |
| Space group | Pbca |
| a/Å | 12.3157 (12) |
| b/Å | 19.6569 (19) |
| c/Å | 22.945 (2) |
| V/Å3 | 6375.6 (4) |
| Z | 8 |
| D c/Mg m−3 | 1.606 |
| μ(Mo-Kα)/mm−1 | 5.52 |
| F(000) | 2688 |
| θ Limits/° | 1.8–26.2 |
| Collected reflections | 17654 |
| Unique reflections | 3454 |
| Observed reflections | 5296 |
| Goodness of fit on F2 | 0.79 |
| R 1[F2 > 2σ(F2)] | 0.028 |
| wR2(F2) | 0.062 |
| Largest diff. peak, hole/e Å−3 | 1.08, −0.79 |
| Bond length (Å) | Bond angles (°) |
|---|---|
| Au1–P1 | 2.2824 (13) |
| Au1–S1 | 2.3365 (13) |
| S1–C13 | 1.749 (5) |
| S2–C13 | 1.701 (5) |
| P1–Au1–S1 | 178.33 (5) |
| C13–S1–Au1 | 100.26 (16) |
| C5–P1–C1 | 110.4 (2) |
| C5–P1–C9 | 110.4 (2) |
| C1–P1–C9 | 109.7 (2) |
| C5–P1–Au1 | 110.40 (16) |
| C1–P1–Au1 | 106.82 (18) |
| C9–P1–Au1 | 109.03 (19) |
2.6. Cell cultures
A549, HeLa and HCT15 human cancer cells were seeded and maintained in triplicate at 4 × 103 cells per well in 100 μL DMEM (Dulbecco's modified Eagle's medium) containing 10% FBS (fetal bovine serum) in a 96-well tissue culture plate and incubated for 72 h at 37 °C, 5% CO2 in air and 90% relative humidity in a CO2 incubator.2.7. MTT assays for anticancer activity of gold(I) complexes (1–4)
100 μL of cisplatin and complexes (0–4) in 50, 25, 12.5 and 6.25 μg mL−1 concentrations, prepared in DMEM, were added to 5000 cancer cells after incubation. The resultant cultures were incubated for 24 h. The medium of wells was discarded. 100 μL DMEM containing MTT (3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium bromide) (5 mg mL−1) was added to the wells and incubated in a CO2 incubator at 37 °C in the dark for 4 h. After incubation, a purple colored formazan (artificial chromogenic dye, a product of the reduction of water insoluble tetrazolium salts e.g., MMT by dehydrogenases and reductases) in the cells is produced and appeared as dark crystals in the bottom of the wells. The medium of culture was discarded from each well carefully to avoid disruption of the monolayer. 100 μL of dimethylsulphoxide (DMSO) was added in each well. The solution was thoroughly mixed in the wells to dissolve the formazan crystals which ultimately results into a purple solution. The absorbance of the 96-well plate was taken at 570 nm with Lab systems Multiskan EX-Enzyme-linked immunosorbent assay (EX-ELISA) reader against a reagent blank. All data presented are mean ± standard deviation.3. Results and discussion
3.1. Chemistry
Addition of dibenzyl dithiocarbamate to tri-tert butylphosphine gold(I) chloride afforded the formation of mononuclear gold(I) crystalline complex (1). Moreover, addition of dimethyl dithiocarbamate, diethyl dithiocarbamate, and dibenzyl dithiocarbamate to [μ-bis(diphenylphosphino)methane]dichlorodigold(I) afforded the formation of three binuclear gold(I) complexes (2–4) respectively in good yields. The mononuclear monophosphine gold(I) complex (1) and binuclear bisphosphine gold(I) complexes (2–4) containing methyl, ethyl and benzyl groups in a dialkyl–diaryl dithiocarbamate ligand have been evaluated to know the steric effects on in vitro cytotoxicity. Complexes (1–4) were completely soluble in polar organic solvents i.e. DMSO and DMF.3.2. Spectroscopic characterization
Dithiocarbamate compounds can be identified via the presence of certain absorbance peaks primarily ν(C–N) and ν(C–S). The region 1480–1550 cm−1 is primarily associated with the S2C–NR2 ‘thioureide’ band in the infrared spectra of dithiocarbamate compounds which defines the carbon–nitrogen bond order between a single bond at 1250–1350 cm−1 and a double bond at 1640–1690 cm−1.47The distinctive thioureide band, ν(C–N) was detected at 1456, 1481, 1486 and 1489 cm−1 in complexes (1–4) respectively. Since these frequency modes lie in between those associated with single C–N and double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, the partial double bond character of the ‘thioureide’ bond was confirmed for all gold(I) complexes.48 The presence of the ‘thioureide’ band between 1545–1430 cm−1 suggest a considerable double bond character in the C⋯N bond vibration of the S2C–NR2 group.49 A strong absorption in this region of the FTIR spectrum results in a strong signal of dithiocarbamato gold(I) complexes.50
N bonds, the partial double bond character of the ‘thioureide’ bond was confirmed for all gold(I) complexes.48 The presence of the ‘thioureide’ band between 1545–1430 cm−1 suggest a considerable double bond character in the C⋯N bond vibration of the S2C–NR2 group.49 A strong absorption in this region of the FTIR spectrum results in a strong signal of dithiocarbamato gold(I) complexes.50
The CS thiocarbonyl stretching splits into two peaks (doublet) with medium intensity at 1022 and 972 cm−1; 1099 and 995 cm−1; 1087 and 982 cm−1; and 1025 and 970 cm−1 for complexes (1–4) respectively. The spectroscopic data suggest monodentate modes of coordination for the dithiocarbamate ligands in complexes (1–4) in analogy of complex [(Ph2P(CH2)2PPh2)(AuS2CNEt2)2].34
In addition to the polar thioureide ion S2CN+R2 band, the common bands for sp3 and sp2 hybridized C–H stretches are observed within 2995–22917 and above 3000 cm−1 respectively which are very much comparable to those of sodium salt of diethyldithiocarbamate. In complexes (1–4), the stretch bands of aromatic (phenyl) and the saturated aliphatic C–H methyl group of coordinated dialkyl–diaryldithiocarbamate correspond above and below 3000 cm−1.51
The 1H NMR chemical shifts of metal precursors [t-Bu3PAuCl], [(DPPM)(AuCl)2] and free dialkyl–diaryldithiocarbamate ligands are given (Table 1S; ESI†). Small upfield and downfield shifts for the mono and bisphosphine coordinated ligands protons have been observed for complexes (1–4); with respect to the chemical shifts of free metal precursors as given in the synthesis part of the experimental section for these complexes. In all four complexes slight downfield and upfield shifts for proton(s) of the coordinated dimethyl dithiocarbamate, diethyl dithiocarbamate and dibenzyldithiocarbamate have also been seen in gold(I) complexes (1–4) respectively in comparison to free dialkyl–diaryldithiocarbamate ligands (see Table 1S; ESI†).
The 13C and 31P NMR chemical shifts of metal precursors [t-Bu3PAuCl], [(DPPM)(AuCl)2] and free dialkyl–diaryldithiocarbamate ligands (Table 2S; ESI†). The 13C NMR spectra of complexes (1–4) showed many resonances as given in the synthesis part of the experimental section for these complexes. There is an up-field chemical shift of CS carbon of coordinated dialkyldithiocarbamate with respect to free dialkyl–diaryldithiocarbamate ligands. The 13C chemical shifts of CS carbon of dimethyl thiocarbamate, diethyl thiocarbamate and dibenzyl thiocarbamate are observed in the range 206–210 ppm. The upfield shifts of CS carbon are additional confirmations for the coordination of dialkyl–diaryl dithiocarbamates ligands in our synthesized complexes (1–4).52
UV-vis spectra of complexes (1–4) were monitored at room temperature for 3 days. The spectra were recorded just after mixing; and after 3 days are illustrated in Fig. 1S (see ESI†). It is observed that the transitions remain relatively unmodified over a period of 3 days. These observations show substantial evidence for the stability of these gold(I) complexes (1–4) under the experimental conditions. However, slight changes in the intensity of characteristic bands were noticed with time; without a significant shift in the absorption peak of spectra.
3.3. Crystal structure of complex [t-Bu3PAuS2CN(C7H7)2] (1)
The X-ray structure of [t-Bu3PAuS2CN(C7H7)2] (1) is shown in Fig. 1. In this structure, gold(I) is coordinated with one P donor atom of tri-tert-butylphosphine and S donor atom of dibenzyldithiocarbamate ligand molecules.The Au–S and Au–P bond distances are 2.3365 (13) and 2.2824 (13) Å respectively. The Au–P and Au–S bond distances are comparable with the [Et3PAu(S2CNEt2)] complex.53 The geometry around the Au(I) metal atom is linear and similar to other analogous Au(I) complexes.54–57 The S–Au–P bond angle is 178.33 (5)° in the molecular structure of [t-Bu3PAuS2CN(C7H7)2] (1) complex which is very close to the angle of 180° for ideal linear geometry. Hence, complex (1) shows a small deviation from ideal linear geometry around the gold(I) atom (Table 2) and confirms the presence of distorted linear geometry in this molecule.
3.4. In vitro cytotoxicity of gold(I) complexes (1–4) in human colon, cervix and lung cancer cells
Human A549 lung cancer cells, human HeLa cervix cancer cells and human HCT15 colon cancer cells have been used to examine the in vitro cytotoxic activity of cisplatin, metal precursor (0) and the synthesized gold(I) complexes (1–4).The concentration (dose) dependent in vitro cytotoxic effect was obtained by the specific increase in concentrations of cisplatin, gold(I) precursor (0) and gold(I) complexes (1–4) against a panel of human cancer cells. The viability of cancer cells vs. concentrations of gold(I) complex is graphically presented in Fig. 2–4. Gold(I) precursor (0) and synthesized complexes (1–4) invariably inhibited the proliferation of all cancer cells in a concentration dependent manner. Generally, the growth inhibition of cancer cells is higher for the synthesized complexes (1–4) in comparison to that of gold(I) precursor (0). Particularly, the degree of anti-proliferation of gold(I) of the synthesized complexes (2 and 3) is significantly greater than those of the synthesized complexes (1 and 4) as illustrated in Fig. 2–4.
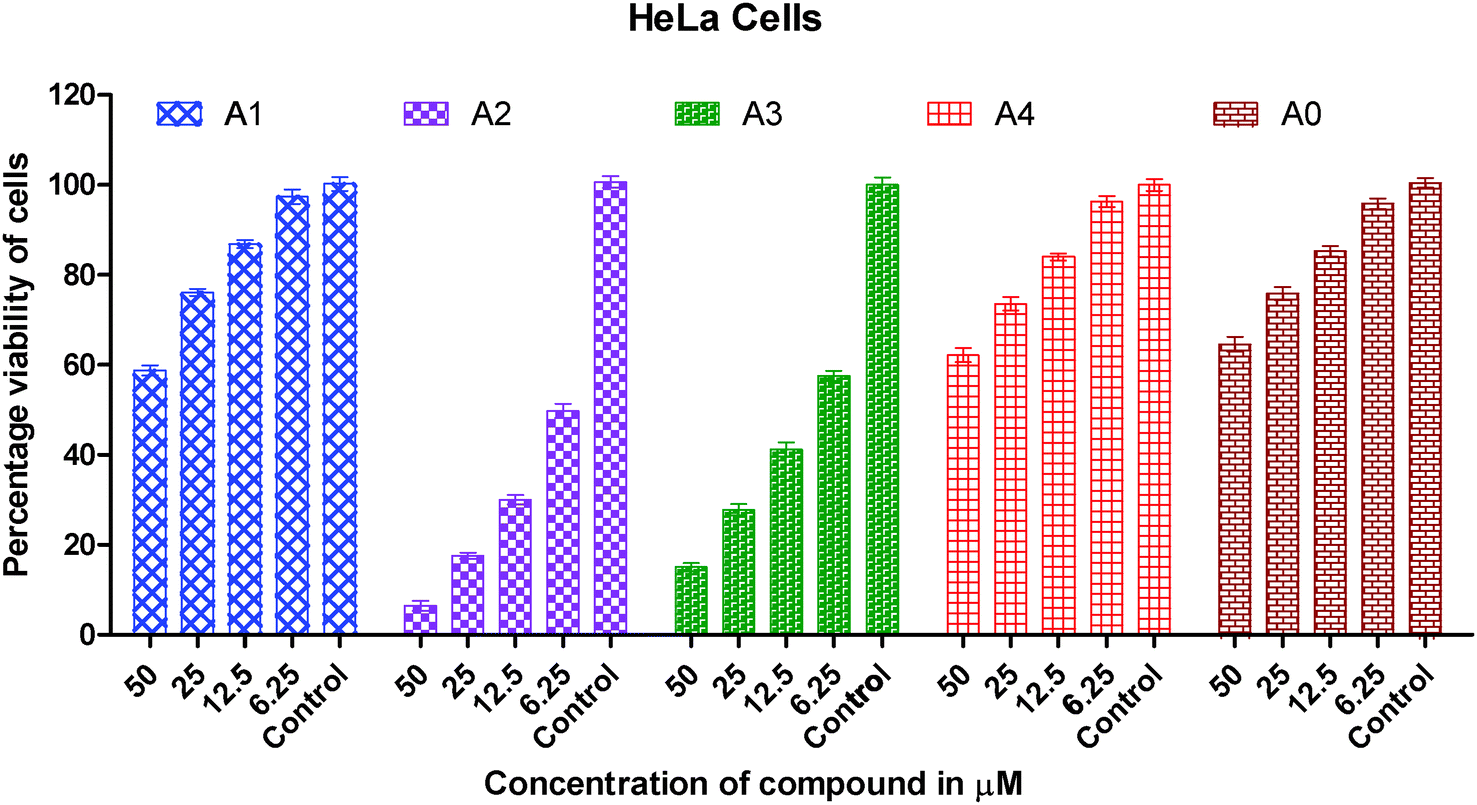 | ||
| Fig. 2 Graph showing the concentration dependent in vitro cytotoxic effect of complexes (0–4) on viability of HeLa cancer cells. | ||
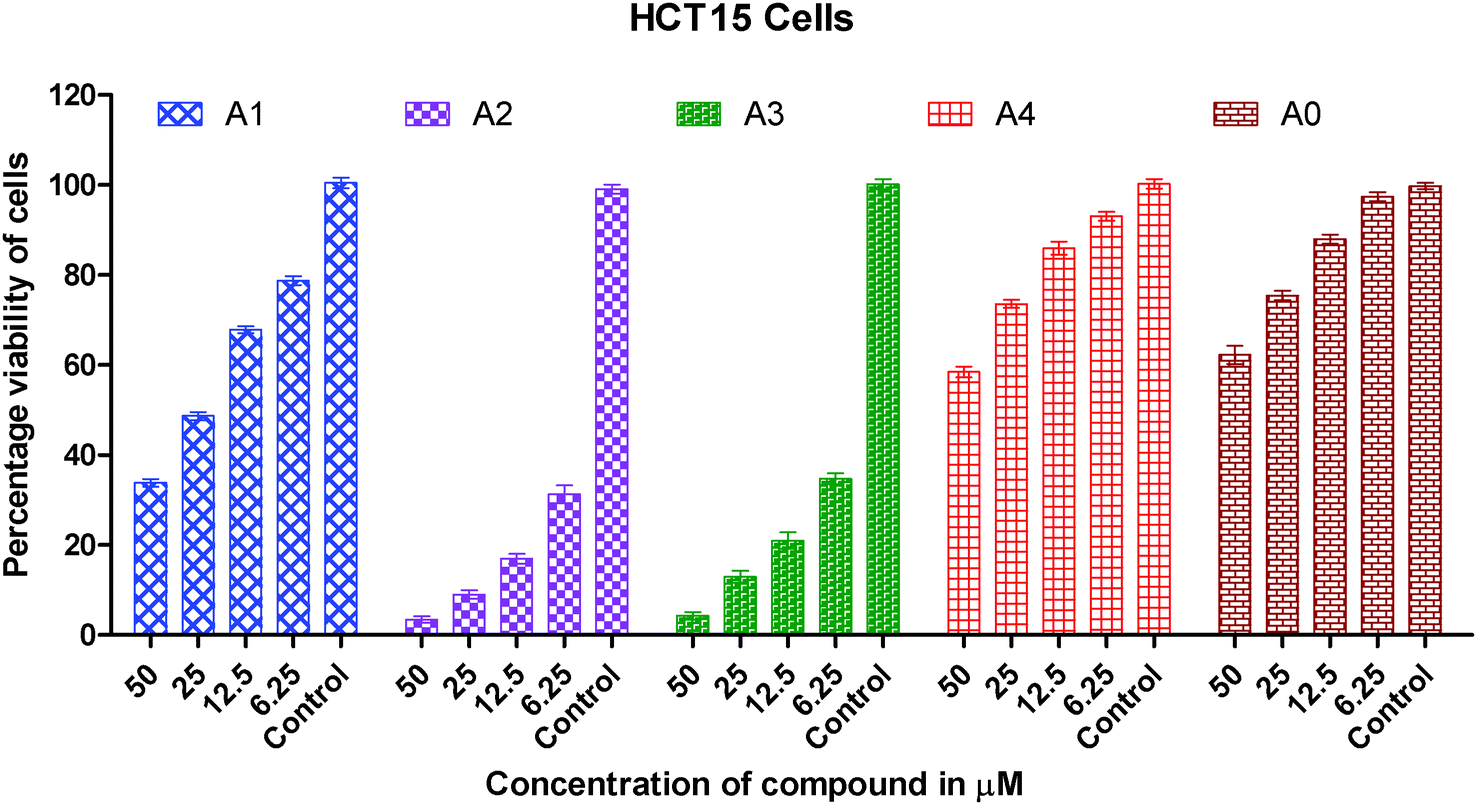 | ||
| Fig. 3 Graph showing the concentration dependent in vitro cytotoxic effect of complexes (0–4) on viability of HCT15 cancer cells. | ||
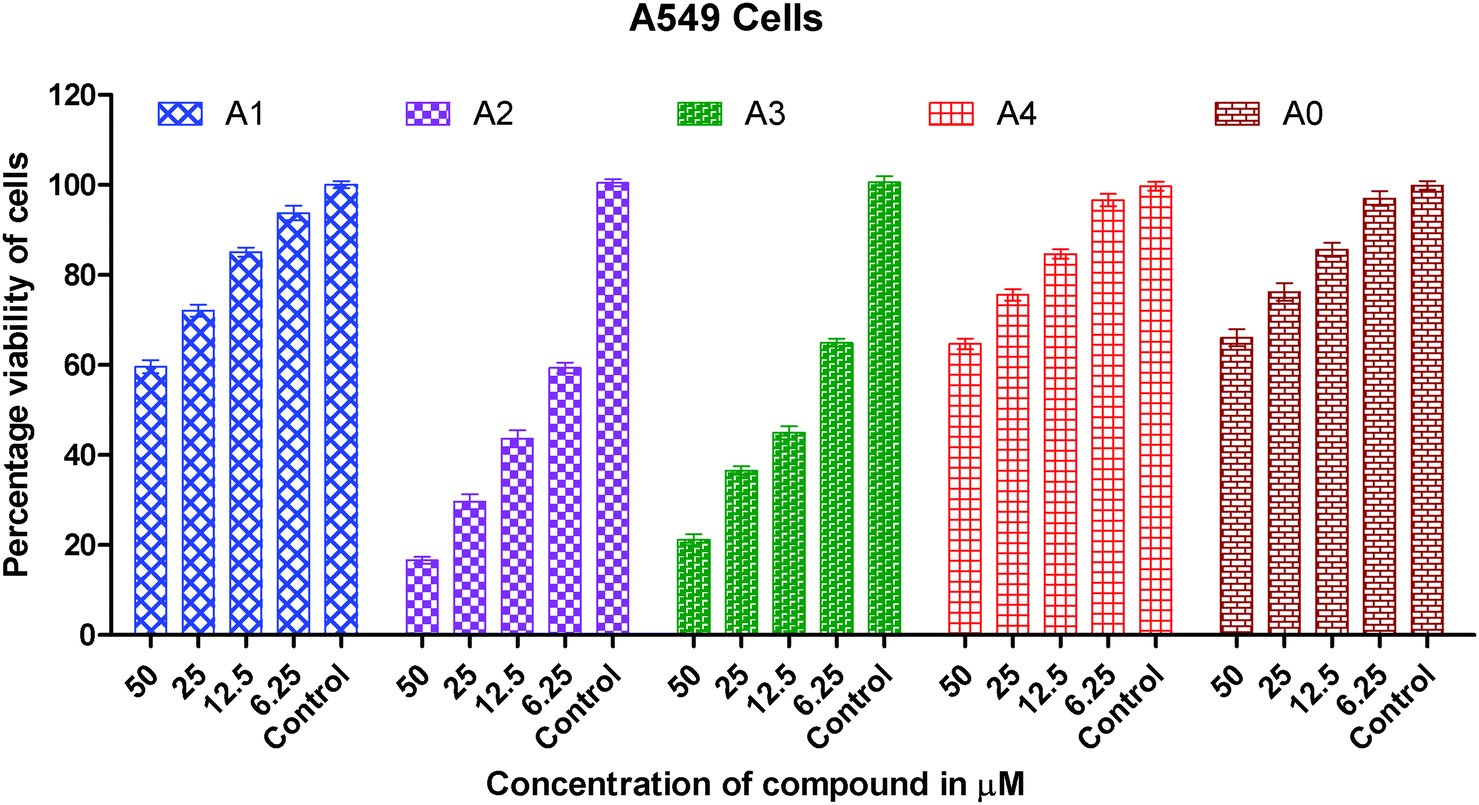 | ||
| Fig. 4 Graph showing the concentration dependent in vitro cytotoxic effect of complexes (0–4) on viability of A549 cancer cells. | ||
The IC50 values for cisplatin, gold(I) precursor (0) and complexes (1–4) against three cancer lines are given in Table 3. The IC50 data for the synthesized gold(I) complexes (1–4) against selected human cancer cell lines i.e. A549, HeLa and HCT15 are in the range of 1.43(0.42) to 133.10(3.62) μM.
| IC50(μM) | |||
|---|---|---|---|
| Complex | A549 | HeLa | HCT15 |
| Cisplatin | 41.67(1.17) | 19.20(1.81) | 29.67(2.35) |
| [(DPPM)(AuCl)2](0) | 136.33(3.17) | 108.73(3.06) | 148.90(3.54) |
| (1) | 96.53(3.25) | 25.90(1.77) | 93.67(2.34) |
| (2) | 5.80(1.93) | 1.43(0.42) | 9.53(1.35) |
| (3) | 9.10(1.61) | 1.63(0.45) | 11.97(1.66) |
| (4) | 105.30(3.68) | 93.87(3.15) | 133.10(3.62) |
It is clearly inferred from the IC50 values against the A549 cell line that in vitro cytotoxicity of complexes (2 and 3) is significantly 15–25 times greater than gold(I) precursor (0) and 5–8 times greater than cisplatin respectively. A similar trend has been observed in the HeLa cell line in which in vitro cytotoxicity of complexes (2 and 3) in terms of IC50 values is improved almost 75 folds than gold(I) precursor; and 12–15 folds than cisplatin respectively. In short, the order of in vitro cytotoxicity is (2) > (3) > cisplatin > (1) > (4) > precursor (0) against A549, HeLa and HCT15 cancer cell lines. It is pertinent to mention that the effectiveness trend of cytotoxicity of complexes (2 and 3) against three cell lines is HeLa > A549 > HCT15. It can be concluded from these studies that complexes (2 and 3) are the most effective cytotoxic agents against the HeLa cancer cell line. Against the HeLa cell line cytotoxicity of complexes (2 and 3) is better than the equivalent Au(PEt3)Cl, as the following IC50(μM) value against the same line show: Au(PEt3)Cl: 1.7(0.06) μM.58
As far as in vitro cytotoxicity against A549, HeLa and HCT15 cell lines is concerned, two out of four synthesized complexes (2 and 3) show much better anticancer activity than classical and well known anticancer drug cisplatin. The much better inhibition of growth of cancer cells by synthesized complexes than gold(I) precursor complex can be attributed to dithiocarbamate as labile co-ligands bonded with central gold(I) ions in synthesized complexes (1–4) by replacing chloride ions in these mononuclear and binuclear complexes.
As we know in drug design and discovery; selectivity and inhibition of target biomolecules is very important. In this regard our results are fruitful and very encouraging for further exploration of anticancer activity of gold(I) complexes. In short the IC50 values of gold(I) complexes (2 and 3) having dialkyldithiocarbamate ligands show much better cytotoxicity than gold(I) complexes (1 and 4) having the diaryldithiocarbamate ligand. The lower cytotoxic activity of gold(I) complexes (1 and 4) is due to the bulky size of the dithiocarbamate ligand, this fact is well understood. The steric hindrance of the bulky ligand makes the approach of gold(I) ions difficult towards biomolecules in these complexes. Overall the anticancer activity of synthesized complexes against A549, HeLa and HCT15 human cancer cell lines is interesting and in the μM range as found in previous anticancer studies of gold complexes.59–62
4. Conclusions
In this study, the anticancer properties of four thiolate Au(I) derivatives with phosphine ligands against three human cancer cell lines, HCT15, HeLa and A549 cell lines have been evaluated. Two complexes exhibit very strong cytotoxic effects in vitro against HCT15, HeLa and A549 cell lines especially complexes with the dimethyldithiocarbamate and diethyldithiocarbamate ligands which showed excellent cytotoxic activity against all tumorous cell lines. These gold(I) complexes are interesting examples of a group of Au(I) thiolate compounds, which contain the S–Au–P arrangement and have also attracted interest as potential anticancer agents. Structural changes like that of the dithiocarbamate ligand are useful to increase the activity of the compounds, reaching the maximum value for complex (2). From this accumulated experience, these dinuclear complexes seem to be the most promising compounds.Gold(I) complexes (1–4) illustrates the inhibitory effect on the growth of all cancer cells in the concentration dependent mode. The screening of the cytotoxic activity based on IC50 data against the HCT15 (human colon cancer cells), HeLa (human cervical cancer cells) and A549 (human lung carcinoma cells) line based IC50 data shows that the compounds (2 and 3) are highly effective, particularly against HeLa and HCT15 cell lines. It shows an ability to circumvent the cellular resistance to cisplatin. When this ability is compared to the equivalent amount of cisplatin and gold(I) precursor complexes, we observe a much better behavior of the binuclear gold(I) complexes (2 and 3). The higher anticancer activity of gold(I) complexes than cisplatin is very encouraging and could be very useful impetus for anticancer drug discovery.
Acknowledgements
The author(s) would like to acknowledge the financial support provided by King Abdulaziz City for Science and Technology (KACST) through the Science & Technology Unit at King Fahd University of Petroleum and Minerals (KFUPM) for this research work under project No. 11-MED1670-04 as part of the National Science, Technology and Innovation Plan (NSTIP).References
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698 CrossRef CAS.
- N. Cutillas, G. S. Yellol, C. de Haro, C. Vicente, V. Rodriguez and J. Ruiz, Coord. Chem. Rev., 2013, 257, 2784 CrossRef CAS PubMed.
- Ž. D. Bugarčić, J. Bogojeski, B. Petrović, S. Hochreuther and R. Van Eldik, Dalton Trans., 2012, 41, 12329 RSC.
- C. Vetter, C. Wagner, J. Schmidt and D. Steinborn, Inorg. Chim. Acta, 2006, 359, 4326 CrossRef CAS PubMed.
- A. Casini and L. Messori, Curr. Top. Med. Chem., 2011, 11, 2647 CrossRef CAS.
- E. Márta Nagy, L. Ronconi, C. Nardon and D. Fregona, Mini-Rev. Med. Chem., 2012, 12, 1216 CrossRef.
- S. Ahmad, A. A. Isab and S. Ali, Transition Met. Chem., 2006, 31, 1003 CrossRef CAS PubMed.
- S. R. McWhinney, R. M. Goldberg and H. L. McLeod, Mol. Cancer Ther., 2009, 8, 10 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755 RSC.
- X. Yao, K. Panichpisal, N. Kurtzman and K. Nugent, Am. J. Med. Sci., 2007, 334, 115 CrossRef PubMed.
- S. S. Al-Jaroudi, M. Monim-ul-Mehboob, M. Altaf, M. Fettouhi, M. I. M. Wazeer, S. Altuwaijri and A. A. Isab, New J. Chem., 2014, 38, 3199 RSC.
- S. M. Janković, A. Djeković, Ž. D. Bugarčić, S. V. Janković, G. Lukić, M. Folic and D. Čanović, BioMetals, 2012, 25, 919 CrossRef PubMed.
- S. S. Al-Jaroudi, M. I. M. Wazeer, A. A. Isab and S. Altuwaijri, Polyhedron, 2013, 50, 434 CrossRef CAS PubMed.
- R. B. Bostancioglu, K. Isik, H. Genc, K. Benkli and A. T. Koparal, J. Med. Chem., 2012, 27, 458 CAS.
- O. Crespo, V. V. Brusko, M. C. Giameno, M. L. Tornil, A. Laguna and N. G. Zabirov, Eur. J. Inorg. Chem., 2004, 423 CrossRef CAS.
- K. Nomiya, R. Noghuchi and M. Oda, Inorg. Chim. Acta, 2000, 298, 24 CrossRef CAS.
- H.-Q. Liu, T.-C. Cheung, S.-M. Peng and C.-M. Che, J. Chem. Soc., Chem. Commun., 1995, 1787 RSC.
- C. J. O'Connor and E. Sinn, Inorg. Chem., 1978, 17, 2067 CrossRef.
- M. A. Cinellu, G. Minghetti, M. V. Pinna, S. Stoccoro, A. Zucca and M. Manassero, J. Chem. Soc., Dalton Trans., 1998, 1735 RSC.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089 CrossRef CAS PubMed.
- R. Noghuchi, A. Hara, A. Sugie and K. Nomiya, Inorg. Chem. Commun., 2006, 9, 355 CrossRef PubMed.
- K. Nomiya, R. Noghuchi, K. Ohsawa, K. Tsuda and M. Oda, J. Inorg. Biochem., 2000, 78, 363 CrossRef CAS.
- B. P. Howe, Met.-Based Drugs, 1997, 4, 273 CrossRef CAS PubMed.
- V. J. Ctalano and A. O. Etogo, J. Organomet. Chem., 2005, 690, 6041 CrossRef PubMed.
- J. C. Lima and L. Rodríguez, J. Med. Chem., 2011, 11, 921 CAS.
- C.-M. Che and R. W.-Y. Sun, Chem. Commun., 2011, 47, 9554 RSC.
- P. Calami, A. Carotti, T. Guerri, L. Messori, E. Mini, P. Orioli and G. P. Speroni, J. Inorg. Biochem., 1997, 66, 103 CrossRef.
- R. K. Johnson, C. K. Mirabelli, L. F. Faucette, F. L. McCabe, B. M. Sutton, D. L. Bryan, G. R. Girard and D. T. Hill, Proc. Am. Assoc. Cancer Res., 1985, 26, 254 Search PubMed.
- C. K. Mirabelli, L. F. Faucette, F. L. McCabe, B. M. Sutton, D. L. Bryan, G. R. Girard, D. T. Hill, J. O. Bartus, S. T. Crooke and R. K. Johnson, J. Med. Chem., 1987, 30, 2181 CrossRef CAS.
- S. J. Berners-Price, M. A. Mazid and P. J. Sadler, J. Chem. Soc., Dalton Trans., 1984, 969 RSC.
- S. J. Berners-Price and P. J. Sadler, Inorg. Chem., 1986, 25, 3822 CrossRef CAS.
- D. T. Hill and G. R. Girard, US Pat., 4755611, 1988 Search PubMed.
- G. F. Rush, D. W. Albers, P. Meunies, K. Leffler and P. F. Smith, Toxicologist, 1987, 7, 59 Search PubMed.
- J. W. Faamaua and E. R. T. Tiekinka, J. Coord. Chem., 1994, 31(2), 93 CrossRef.
- L. Ronconi, L. Giovagnini, C. Marzano, F. Bettio, R. Graziani, G. Pilloni and D. Fregona, Inorg. Chem., 2005, 44, 1867 CrossRef CAS PubMed.
- V. Milacic, D. Chen, L. Ronconi, K. R. Landis-Piwowar, D. Fregona and Q. P. Dou, Cancer Res., 2006, 66, 10478 CrossRef CAS PubMed.
- X. Zhang, M. Frezza, V. Milacic, L. Ronconi, Y. Fan, C. Bi, D. Fregona and Q. P. Dou, J. Cell. Biochem., 2010, 109, 162 CAS.
- S. Ahmad, A. A. Isab, S. Ali and A. R. Al-Arfaj, Polyhedron, 2006, 25, 1633 CrossRef CAS PubMed.
- D. V. Partyka, T. J. Robilotto, M. Zeller, A. D. Hunter and T. G. Gray, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14293 CrossRef CAS PubMed.
- Y. Wang, Q.-Y. He, C.-M. Che and J.-F. Chiu, Proteomics, 2006, 6, 131 CrossRef CAS PubMed.
- Y. Shi, W. Chu, Y. Wang, S. Wang, J. Du, J. Zhang, S. Li, G. Zhou, X. Qin and C. Zhang, Inorg. Chem. Commun., 2013, 30, 178 CrossRef CAS PubMed.
- M. Monim-ul-Mehboob, M. Altaf, M. Fettouhi, A. A. Isab, M. I. M. Wazeer, M. N. Shaikh and S. Altuwaijri, Polyhedron, 2013, 61, 225 CrossRef CAS PubMed.
- Stoe & Cie, X-Area & X-RED32, GmbH, Darmstadt, Germany, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- A. J. Odola and J. A. O. Woods, J. Chem. Pharm. Res., 2011, 3, 865 CAS.
- F. Jian, Z. Wang, Z. Bai, X. You, H. Fun, K. Chinnakali and L. A. Razak, Polyhedron, 1999, 18, 3401 CrossRef CAS.
- A. Jayaraju, M. M. Ahamad, R. M. Rao and J. Sreeramulu, Pharma Chem., 2012, 4, 1191 CAS.
- J. Chatt, L. A. Duncanson and L. M. Venanzi, Nature, 1956, 177, 1042 CrossRef CAS.
- C. J. Pouchert, Aldrich Library of FT-IR Spectra, Aldrich Chemical Company, Milwaukee, 2nd edn, 1997, vol. 1 Search PubMed.
- M. Altaf, M. Monim-ul-Mehboob, A. A. A. Seliman, A. A. Isab, V. Dhuna, G. Bhatia and K. Dhuna, J. Organomet. Chem., 2014, 765, 68 CrossRef CAS PubMed.
- S. Y. Ho and E. R. T. Tiekink, Z. Kristallogr., 2005, 220, 342 CAS.
- I. Sänger, H.-W. Lerner, T. Sinke and M. Bolte, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m708 Search PubMed.
- P. Lu, T. C. Boorman, A. M. Z. Slawin and I. Larrosa, J. Am. Chem. Soc., 2010, 132, 5580 CrossRef CAS PubMed.
- R. E. Marsh, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 893 Search PubMed.
- H. Schmidbaur, B. Brachthiuser, O. Steigelmann and H. Beruda, Chem. Ber., 1992, 125, 2705 CrossRef CAS.
- E. Barreiro, J. S. Casas, M. D. Couce, A. Sánchez, A. Sánchez-Gonzalez, J. Sordo and E. M. Vázquez-López, J. Inorg. Biochem., 2014, 138, 89 CrossRef CAS PubMed.
- E. Barreiro, J. S. Casas, M. D. Couce, A. Sánchez, J. Sordo and E. M. Vázquez-López, J. Inorg. Biochem., 2014, 131, 68 CrossRef CAS PubMed.
- R. Kivekäs, E. Colacio, J. Ruiz, J. D. López-González and P. León, Inorg. Chim. Acta, 1989, 159, 103 CrossRef.
- L. Ortego, F. Cardoso, S. Martins, M. F. Fillat, A. Laguna, M. Meireles, M. D. Villacampa and M. C. Gimeno, J. Inorg. Biochem., 2014, 130, 32 CrossRef CAS PubMed.
- I. Ott, T. Koch, H. Shorafa, Z. Bai, D. Poeckel, D. Steinhilber and R. Gust, Org. Biomol. Chem., 2005, 3, 2282 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 994019 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj00747f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2015 |