
High activity and durability of novel perovskite electrocatalysts for water oxidation†
Wei
Zhou
*ab,
Mingwen
Zhao
c,
Fengli
Liang
b,
Sean C.
Smith
d and
Zhonghua
Zhu
*b
aState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemistry & Chemical Engineering, Nanjing Tech University, No. 5 Xin Mofan Road, Nanjing 210009, P.R. China. E-mail: zhouwei1982@njtech.edu.cn
bSchool of Chemical Engineering, The University of Queensland, St. Lucia, Queensland 4072, Australia. E-mail: z.zhu@uq.edu.au
cSchool of Physics and State Key Laboratory of Crystal Materials, Shandong University, Jinan, Shandong 250100, People's Republic of China
dIntegrated Materials Design Centre, School of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia
First published on 26th June 2015
Abstract
Development of highly active and cost-effective electrocatalysts is central to the large-scale electrolysis of water for renewable energy generation. Perovskite oxides are a group of promising candidates to lower the oxygen evolution reaction (OER) barriers in water splitting and further improvement of their activity and durability is an important objective. Here we report scandium and niobium cation (Sc3+ and Nb5+) doped strontium cobaltite perovskites (SrScxNbyCo1−x−yO3−δ) as a family of highly active and durable electrocatalysts for the OER in alkaline solution. These perovskites not only manifest up to a factor of 50 increase of the intrinsic activity compared to the gold-standard OER electrocatalysts (such as IrO2 and RuO2) and a factor of 5.8 enhancement to the perovskite-Ba0.5Sr0.5Co0.8Fe0.2O3−δ at an overpotential of 0.35 V, but also, more importantly, show excellent durability in alkaline solutions under operation.
Conceptual insightsElectrochemical splitting of water into molecular oxygen (O2), protons, and electrons could provide a way to store the electricity generated from sustainable but intermittent energy sources, such as wind and solar power, as fuels. The water oxidation is also an essential process for metal–air batteries, and regenerative fuel cells. A major challenge is that efficient catalysts for water electrolysis are expensive and contain rare noble metals, so cost effective approaches will require the discovery of efficient electrocatalysts that contain only Earth-abundant elements. Although perovskite oxides represent a promising group of candidates to lower the oxygen evolution reaction (OER) barriers in water splitting, further improvement of their activity and durability is imperative. Herein we report doping of strontium cobaltite perovskites with scandium and niobium cations (Sc3+ and Nb5+) to generate a family of highly active and durable electrocatalysts (SrScxNbyCo1−x−yO3−δ) for the OER in alkaline solution. The intrinsic OER activity of these perovskites is enhanced by up to a factor of 5.8 over that of Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) at an overpotential of 0.35 V. The higher OER activity is correlated with the higher affinity for surface hydroxide adsorption and more abundant electron states near the Fermi level of the developed perovskites compared with BSCF. |
The oxygen evolution reaction (OER) is an essential but sluggish process for many energy storage applications, such as electrolyzers, solar-driven water-splitting devices, metal–air batteries, and regenerative fuel cells.1–5 A variety of transition metal oxides have been developed to mitigate the high overpotential loss in the OER process, including rutile oxides (e.g. RuO2 and IrO2),6,7 spinel oxides (e.g. NiCo2O4),8 and perovskite oxides (e.g. LaNiO3 (LN) and LaxSr1−xCoyFe1−yO3−δ).9–11 Low abundance, high cost and instability in alkaline solution are all issues that inhibit practical utilization of the precious-metal electrocatalysts such as RuO2 and IrO2. In contrast, the low cost and high efficiency of perovskites clearly indicate an important direction for OER electrocatalyst development.12–18
Conventional perovskite-type OER electrocatalysts have the general formula ABO3 with the A-site occupied by alkaline earth and/or rare earth metal(s) and the B-site occupied by first-row transition metal(s) with 3d electrons. The active centers for the OER are transition metal couples with high redox ability at the B-site such as Co3+/4+and Fe3+/4+. Based on a density functional theory (DFT) study, Man et al. theoretically predicted the cobaltite perovskite SrCoO3 as exhibiting near-optimal OER activity.19 However, the preparation of fully oxidized SrCoO3 perovskite is difficult. Instead, the doped SrCoO3 perovskites with oxygen vacancies, such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) and SrCo0.8Fe0.2O3−δ (SCF), are more facile to prepare and show up to one order of magnitude in enhancement of activity towards water oxidation compared with IrO2 and RuO2 at an overpotential of 0.4 V.6,16 However, the BSCF and SCF perovskites readily lose the perovksite structure to become amorphous as the OER proceeds.20,21 To this end, the development of perovskites with both high activity and durability is imperative in order to enable substantial market penetration in the relevant applications.
Here, we report a new series of perovskite-type OER electrocatalysts using scandium (Sc3+) and niobium (Nb5+) to partially replace cobalt in SrCoO3 to form SrScxNbyCo1−x−yO3−δ perovskites. The doping of Sc3+ and Nb5+ retains the perovskite-type structure and keeps a similar electronic configuration to the SrCoO3 perovskite host structure. These SrScxNbyCo1−x−yO3−δ perovskites exhibit substantive improvements in both catalytic activity and durability for OER in alkaline solution.
We synthesized the SrScxNbyCo1−x−yO3−δ perovskites through a conventional ceramic process,22i.e. solid state reaction. Among all Sc and Nb co-doped samples, only SrSc0.025Nb0.025Co0.95O3−δ (SSNC1) and SrSc0.175Nb0.025Co0.8O3−δ (SSNC2) show single phase with optimized synthetic parameters (Fig. S1, ESI†). The samples with other compositions show additional phase(s) depending on the synthetic temperature. The formation of a brownmillerite phase was found on all other SrScxNbyCo1−x−yO3−δ perovskites (except SSNC1 and SSNC2) at 1250 °C. This is due to the significantly different reaction activity of Sc2O3 and Nb2O5 causing the phase separation in SrScxNbyCo1−x−yO3−δ perovskites23 (Fig. S2, ESI†). Enhancing the synthesis temperature to 1300 °C can diminish this difference to some extent but leads to the separation of CoO as a new phase (Fig. S3, ESI†). The formation of additional phase(s) is detrimental to the OER activity (as shown in next paragraphs), hence SSNC1 and SSNC2 are selected to demonstrate the optimal performance determined to date. Due to the higher calcination temperature, the as-synthesized SSNC1 and SSNC2 show much larger particle size as compared to those of BSCF, SCF, LN, La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) and La0.6Sr0.4CoO3−δ (LSC) (Fig. S4, ESI†).
We collected the OER currents of perovskites using a thin-film rotating-disk electrode with well-defined oxygen transport in ultra-high purity O2-saturated 0.1 M KOH solution at room temperature. Carbon black was used to provide a conductive medium to facilitate the utilization of the perovskite electrocatalysts. The OER activity of the carbon additive is negligible in the potential regime studied here (Fig. S5, ESI†). The electrochemical behavior of the electrocatalysts is shown in Fig. 1a. Linear sweep voltammogram (LSV) measurements revealed a sharp increase in current at ∼1.55 V for SSNC1 and SSNC2, which corresponds to catalytic water oxidation. Based on enlarged LSV profiles (inset in Fig. 1a), the co-doping of Sc and Nb shifts the onset voltage more negatively for the SSNC1 and SSNC2 perovskites compared with those of BSCF, SCF, RuO2 and LN. The onset potential of RuO2 is about 1.45 V vs. RHE, which is in line with those reported in the literature.6,24,25 The OER behavior of SrCoO3−δ (δ ≈ 0.5) in a brownmillerite structure (not a perovskite structure) is tested (Fig. S6, ESI†). As expected it shows much lower OER activity as compared with SSNC1, SSNC2 and BSCF (Fig. S7, ESI†).
 | ||
| Fig. 1 (a) iR-corrected polarization curves of SSNC1, SSNC2, BSCF, SCF, RuO2 and LN electrocatalysts in 0.1 M KOH at a scan rate of 10 mV s−1. The inset shows the enlarged picture to compare the onset potential of OER for the electrocatalysts. (b) The Tafel plots of OER on SSNC1, SSNC2, BSCF, SCF, RuO2 and LN electrocatalysts. (c) The comparison of intrinsic OER activity of SSNC1-8, BSCF, BSCF1, BSCF2, SCF, LSCF, LSC, LN, RuO2 and IrO2 electrocatalysts at 1.58 V vs. RHE (overpotential η = 0.35 V). SSNC3: SrSc0.025Nb0.075Co0.9O3−δ; SSNC4: SrSc0.15Nb0.05Co0.8O3−δ; SSNC5: SrSc0.125Nb0.075Co0.8O3−δ; SSNC6: SrSc0.05Nb0.05Co0.9O3−δ; SSNC7: SrSc0.1Nb0.05Co0.85O3−δ; SSNC8: SrSc0.1Nb0.1Co0.8O3−δ; LSCF: La0.6Sr0.4Co0.2Fe0.8O3−δ; LSC: La0.6Sr0.4CoO3−δ. BSCF was synthesized by the EDTA-citrate method calcinated at 900 °C for 5 hours with a specific surface area of 0.53 m2 g−1; BSCF1 was synthesized by the EDTA-citrate method calcinated at 1000 °C for 5 hours with a specific surface area of 0.33 m2 g−1; BSCF2 was synthesized by solid state reaction calcinated at 1050 °C for 5 hours with a specific surface area of 0.15 m2 g−1. | ||
The redox couple peaks of transition metals (Co, Fe, Ni and Ru) were observed in cyclic voltammogram (CV) measurements for SSNC1, BSCF, SCF, RuO2 and LN electrocatalysts (Fig. S8, ESI†). The redox couples varied with the doping strategy. The sequence of voltage for the redox couple occurring is SSNC1 < BSCF ≈ SCF < RuO2 < LaNiO3. Rasiyah et al.26 and Maiyalagan et al.27 have shown a relationship between the onset potential for OER and the potential of the redox couples. They observed the lower potential of redox couples related to the lower onset potential of OER, which is also the case in our study. The shift of the redox couple peak in SSNC1 suggests that the co-doping of Sc and Nb affects the properties of Co.
Fig. 1b shows the Tafel plots for the electrocatalysts. In the lower potential range (<1.6 V), the Tafel slopes of 55–60 mV dec−1 were observed for SSNC1 and SSNC2, which are close to that for LN (∼55 mV dec−1) and lower than those for RuO2 (∼80 mV dec−1), SCF (∼90 mV dec−1) and BSCF (∼75 mV dec−1). In the higher over potential range (>1.6 V), the Tafel slopes of SSNC1 and SSNC2 changed to ∼70 mV dec−1, while the slopes increased to 110, 145, 150 and 110 mV dec−1 for BSCF, SCF, RuO2 and LN respectively.
The intrinsic activity normalized to the surface area is used to evaluate the performance of the electrocatalysts as reported in the literature.12,27 Selection of an appropriate method to measure the surface area of electrocatalysts is of importance to calculate their intrinsic OER activity. The surface area of the electrocatalysts can be measured geometrically or electrochemically. The electrochemical surface area (ECSA) should ideally represent the material surface available for electrochemical processes, which can be obtained by measuring the double-layer capacitance in a suitably small voltage range. This method has been recommended as a tool for benchmarking oxide electrocatalysts.28 However, the use of the ECSA to quantify the surface activity of oxide materials is not well established,29 because the dependence of capacitance upon potential in oxides is not well known and that faradaic reactions might be present even within a small potential window. A recent study reported that the pseudocapacitance is related to the oxygen stoichiometry of the perovskite oxides.30 This leads to even more difficulty to compare the ECSA between different perovskite oxides because the oxygen stoichiometry of perovskites could vary in a large range based on their chemical composition and crystallite structure. In some recent studies, surface areas based on imaging the particle size distribution directly using an electron microscope, or obtained via gas adsorption, are more accurate.12,27,31,32 As a result, the intrinsic activity of the oxides was normalized to the specific surface area determined from Brunauer, Emmett, and Teller (BET) measurement in the present study. The as-measured OER activity of the thin-film electrocatalyst is capacity-corrected by taking an average of forward and backward (positive and negative-going) scans. The capacity-corrected OER current is then ohmically corrected with the measured ionic resistance to yield the final electrode OER activity (Fig. S9, ESI†). All the samples were measured at least three times to obtain the data corrected with the standard deviations (Fig. S10, ESI†). The phase purity of all the samples was studied by X-ray diffraction (XRD) and the specific surface area of the samples was determined from nitrogen BET area measurements (Table S1, ESI†). Fig. 1c shows the intrinsic OER activity of SSNC1 and SSNC2 compared with some other electrocatalysts measured at an overpotential of 0.35 V. The intrinsic OER activities of IrO2, RuO2 and LN measured by us are comparable to those found in previous studies.6,9 The intrinsic OER activities of BSCF is about 10 times as large as that of IrO2 at an overpotential of 0.35 V, which is in line with that reported by Y. Shao-Horn's group.12 SSNC1 and SSNC2 show up to a factor of 50 increase of the intrinsic activity compared to the gold-standard OER electrocatalysts IrO2 and RuO2 and a factor of 5.8 enhancement to the perovskite-BSCF at an overpotential of 0.35 V.12 We synthesized BSCF electrocatalysts through different methods and at various temperatures to evaluate a possible effect under different synthetic conditions. As shown in Fig. 1c, the intrinsic activity is similar for all the BSCF perovskites synthesized in this study, with no obvious correlation with the particle size of the sample and synthetic methods.
It has been reported that the amount of hydroxide species formed on the surface of the metal oxide electrocatalysts correlates with their OER activity in alkaline solution.10,33 The least energetically favorable (rate-determining) step of OER is surface OH− oxidation based on the previous studies of OER kinetics on perovskites. Therefore, the chemical properties of the oxide surfaces (e.g., hydroxide species) have a significant impact on the OER catalysis. The OER rate is governed by the concentration of hydroxide species that participate in the formation of the O–O bond in hydroperoxide.33 Here, we analyzed the oxygen species on the surface of electrocatalysts by X-ray photoelectron spectroscopy (XPS). The XPS spectra of the O1s levels are shown in Fig. 2a. The O1s XPS spectra of both samples present two features at about 529 and 531 eV. The lower binding energy peak can be ascribed to the lattice oxygen species (O2−) and the higher one can be assigned to less electron-rich oxygen species. By deconvoluting the broad peak at higher binding energy (530–534 eV), three peaks can be obtained: the one with lower BE (∼530.5 eV) is assigned to oxygen in the form of O22−/O− on the surface; the next (∼531.5 eV) is ascribed to the hydroxyl groups (–OH) or the surface-adsorbed oxygen (O2); and the third peak (∼533 eV) is due to molecular water or carbonates adsorbed on the surface.34 Table S2 (ESI†) lists the relative concentrations of the different kinds of oxygen species which are estimated from the relative area of these subpeaks. As can be seen, the relative concentration of hydroxide species is the highest for SSNC1 and SSNC2.
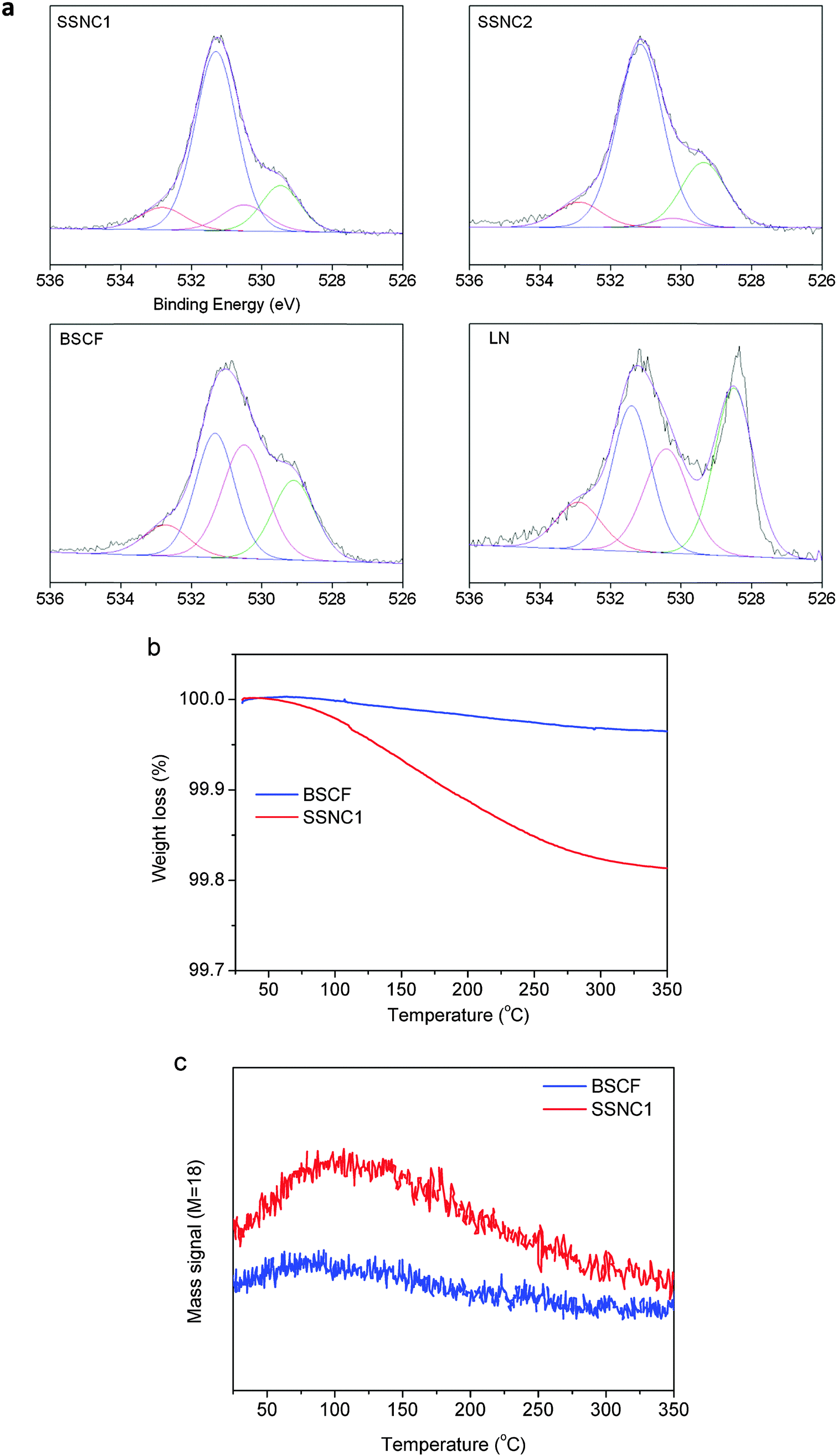 | ||
| Fig. 2 (a) XPS spectra of O1s species on the surface of SSNC1, SSNC2, BSCF and LN. (b) Weight losses for SSNC1 and BSCF from 25 to 350 °C. (c) Desorption of water from SSNC1 and BSCF measured by mass spectroscopy (M = 18) from 25 to 350 °C. | ||
As XPS can only provide semi-quantitative information on the surface oxygen species, we performed thermogravimetry-mass spectrometry (TG-MS) analysis to provide more accurate measurement on the OH amount in SSNC1 and BSCF (Fig. 2b). Based on the MS results (Fig. 2c), the weight loss below 250 °C is due to water desorption. Since the sample subjected to the TG-MS test was pretreated to remove the adsorbed water under vacuum conditions at 200 °C, most of the desorbed water (detected by TG-MS for mass = 18) originates from hydroxide species rather than surface adsorbed molecular water. This is also well supported by the fact that most of the water desorbed from SSNC1 is between 100 and 250 °C. The thermogravimetric analysis (TGA) results show that the amount of water released from SSNC1 is around 5 times larger than that from BSCF. Given a similar ratio of intrinsic activities to those measured herein, it is likely that the higher affinity for surface hydroxide adsorption of the SSNC1 perovskite compared to BSCF is correlated with its higher OER activity.
To aid in understanding the unprecedentedly high activity of Sc and Nb co-doped SrCoO3 perovskite (SSNC), the partial electronic density of states (PDOS) projected onto the active Co atoms of SSNC and BSCF were calculated. As we can see, there are abundant electron states near the Fermi level for both cases, which facilitate the activity of the surface (Fig. 3a and b). For SSNC, the peak of the PDOS of dxz/dyz orbitals is closer to the Fermi level than that of BSCF. This may be one reason why SSNC is more active than BSCF, since the dxz/dyz orbitals dominate the OER activity of the surface. The SrCoO3 perovskite was theoretically predicted to have the highest OER activity:19 there is abundant electronic density of states of dxz/dyz orbitals at the Fermi level and the peak of the PDOS of dxz/dyz orbitals is close to the Fermi level (Fig. 3c). These features are largely preserved in the PDOS profile of SSNC. For BSCF, however, the degeneracy of the PDOS of dxz/dyz orbitals is lifted and the peaks of the PDOS are away from the Fermi level. This suggests that the co-doping of Sc and Nb largely maintains electronic configuration of SrCoO3, such that SSNC achieves very high OER activity. We also evaluated the over-potential of SSNC and BSCF theoretically19 using data from first-principles calculations (Table S3, ESI†). The overpotential difference (ΔOEP) between SSNC and BSCF is about 77 mV. Assuming that the reaction rate obeys an Arrhenius relation with respect to ΔOEP and that similar pre-exponential factors apply in each case, i.e., rate ∝ exp(ΔOEP/kBT), where kB is the Boltzmann constant, the reaction rate of SSNC is estimated to be about 19 times higher than that of BSCF at room temperature. Given the complicated experimental conditions and the simplifications of the theoretical treatment and the structural model, this represents a reasonable supportive correlation between modeling and the experimentally observed trends in activity.
 | ||
| Fig. 3 The electron density of states (PDOS) projected onto the atomic orbitals of the active Co atom on the upmost surface of (a) SSNC, (b) BSCF, and (c) SC. The contributions of the five d orbitals are marked by the lines in different colours. The PDOS of the dxz and dyz orbitals are degenerated for (a) and (c) due to the symmetry of the surfaces. The energy at the Fermi level was set to zero. | ||
Another important challenge for OER electrocatalysts is to increase the durability. Very interestingly, SSNC1 and SSNC2 are very stable under OER potentials. No significant changes were detected in the repeated CV scans for SSNC1 or chronoamperometry measurement for SSNC2 (Fig. 4a and b). The steps in the chronoamperometry test (Fig. 4b) are related to the release of oxygen gas bubbles built up during the OER, as also reported in ref. 24 and 35. Since no obvious degradation of the electrocatalyst was observed during the long term stability test, the carbon corrosion is negligible. It has been reported that the instability of BSCF and SCF stems from the degeneration of their near-surface regions to an amorphous structure caused by the leaching of the Ba and Sr cations from the perovskite lattice.20,21 The stability of the SSNC2 during OER was further supported by high resolution transmission electron microscopy (HRTEM) images. No obvious change in the surfaces of SSNC2 was observed after OER measurements, while TEM imaging showed that the surfaces of SSNC2 after chronoamperometry testing are comparable to that of the as-synthesized SSNC2 (Fig. 4c and d).
 | ||
Fig. 4 (a) The 1st and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000th CV scans of SSNC1 electrocatalysts. The CV scans were performed at 100 mV s−1. (b) Chronoamperometric response of the SSNC2 electrocatalyst kept at 0.7 V vs. Ag|AgCl (3 M NaCl) in 0.1 M KOH solution for 17 hours. The HRTEM image of as-synthesized (c) SSNC2 perovskite and (d) the SSNC2 electrocatalyst after chronoamperometry test for 17 hours. 000th CV scans of SSNC1 electrocatalysts. The CV scans were performed at 100 mV s−1. (b) Chronoamperometric response of the SSNC2 electrocatalyst kept at 0.7 V vs. Ag|AgCl (3 M NaCl) in 0.1 M KOH solution for 17 hours. The HRTEM image of as-synthesized (c) SSNC2 perovskite and (d) the SSNC2 electrocatalyst after chronoamperometry test for 17 hours. | ||
In conclusion, we have demonstrated a new family of perovskites, Sc- and Nb co-doped SrCoO3 with high performance and stability for the OER. The intrinsic OER activity of these perovskites is enhanced by up to a factor of 5.8 over that of the perovskite Ba0.5Sr0.5Co0.8Fe0.2O3−δ at an overpotential of 0.35 V. Moreover, these perovskites also display excellent durability in alkaline solutions during operation. The results presented here encourage further study of perovskite-type electrocatalysts for OER to enable the development of efficient water oxidation in water splitting devices, rechargeable metal–air batteries, regenerative fuel cells and other rechargeable air-based energy storage devices. Because of the lower specific surface area of the SSNC perovskite oxides, the mass activity of the electrocatalysts can be further improved by enlarging their effective surface area or involving active elements into the lattice of the perovskites.36,37
Conflict of interest
There is no competing financial interest in this work.Contributions
W.Z. and Z.H.Z. conceived the project and designed the experiments. W.Z. and F.L.L. performed synthesis and characterization. M.W.Z. did the first principles modelling. W.Z., S.C.S. and Z.H.Z. analyzed the data and co-wrote the manuscript. All authors discussed the results and worked on the manuscript.Acknowledgements
This research was funded by an Australian Research Council (ARC) Discovery Project (DP130102151). The Australian Microscopy & Microanalysis Research Facility is also acknowledged for providing characterization facilities. W. Zhou thanks the Program for Jiangsu Specially-Appointed Professors. We would like to thank Hassan Tahini who designed the back cover graphic for this work.References
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- R. F. Service, Science, 2009, 324, 1257 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. P. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- Y. M. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1980, 111, 125 CrossRef CAS.
- C. R. Davidson, G. Kissel and S. Srinivasan, J. Electroanal. Chem., 1982, 132, 129 CrossRef CAS.
- W. Zhou and J. Sunarso, J. Phys. Chem. Lett., 2013, 4, 2982 CrossRef CAS.
- J. O. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS PubMed.
- Y. Matsumoto, S. Yamada, T. Nishida and E. Sato, J. Electrochem. Soc., 1980, 127, 2360 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- A. Vojvodic and J. K. Nørskov, Science, 2011, 334, 1355 CrossRef CAS PubMed.
- Y. L. Zhao, L. Xu, L. Q. Mai, C. H. Han, Q. Y. An, X. Xu, X. Liu and Q. J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19569 CrossRef CAS PubMed.
- Z. Chen, A. P. Yu, D. Higgins, H. Li, H. J. Wang and Z. W. Chen, Nano Lett., 2012, 12, 1946 CrossRef CAS PubMed.
- J.-J. Xu, D. Xu, Z.-L. Wang, H.-G. Wang, L.-L. Zhang and X.-B. Zhang, Angew. Chem., Int. Ed., 2013, 52, 3887 CrossRef CAS PubMed.
- Y. L. Zhu, W. Zhou, Y. B. Chen, C. Su, M. O. Tadé and Z. P. Shao, Angew. Chem., Int. Ed., 2015, 54, 3897 CrossRef CAS PubMed.
- M. Bursell, M. Pirjamali and Y. Kiros, Electrochim. Acta, 2002, 47, 1651 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nøskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159 CrossRef CAS PubMed.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264 CrossRef CAS.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8626 Search PubMed.
- W. Zhou, J. Sunarso, M. W. Zhao, F. L. Liang, T. Klande and A. Feldhoff, Angew. Chem., Int. Ed., 2013, 52, 14036 CrossRef CAS PubMed.
- P. W. Barnes, M. W. Lufaso and P. M. Woodward, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 384 Search PubMed.
- R. C. Liu, F. L. Liang, W. Zhou, Y. S. Yang and Z. H. Zhu, Nano Energy, 2015, 12, 115 CrossRef CAS PubMed.
- F. L. Liang, Y. Yu, W. Zhou, X. Y. Xu and Z. H. Zhu, J. Mater. Chem. A, 2015, 3, 634 CAS.
- P. Rasiyah and A. C. C. Tseung, J. Electrochem. Soc., 1984, 131, 803 CrossRef CAS PubMed.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- S. Trasatti and O. Petrii, Pure Appl. Chem., 1991, 63, 711 CrossRef CAS.
- J. T. Mefford, W. G. Hardin, S. Dai, K. P. Johnston and K. J. Stevenson, Nat. Mater., 2014, 13, 726 CrossRef CAS PubMed.
- V. Augustyn and A. Manthiram, ChemPlusChem, 2015, 80, 422 CrossRef CAS PubMed.
- N. Colligan, V. Augustyn and A. Manthiram, J. Phys. Chem. C, 2015, 119, 2335 CAS.
- K.-N. Jung, J.-H. Jung, W. B. Im, S. Yoon, K.-H. Shin and J.-W. Lee, ACS Appl. Mater. Interfaces, 2013, 5, 9902 CAS.
- Y. G. Wang, J. W. Ren, Y. Q. Wang, F. Y. Zhang, X. H. Liu, Y. Guo and G. Z. Lu, J. Phys. Chem. C, 2008, 112, 15293 CAS.
- M. R. Gao, W. C. Sheng, Z. B. Zhuang, Q. R. Fang, S. Gu, J. Jiang and Y. S. Yan, J. Am. Chem. Soc., 2014, 136, 7077 CrossRef CAS PubMed.
- Y. S. Yang, W. Zhou, R. C. Liu, M. R. Li, T. E. Rufford and Z. H. Zhu, ChemElectroChem, 2014, 2, 200 CrossRef PubMed.
- Y. L. Zhu, W. Zhou, Y. B. Chen, J. Yu, X. M. Xu, C. Su, M. O. Tadé and Z. P. Shao, Chem. Mater., 2015, 27, 3048 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and associated references are available. See DOI: 10.1039/c5mh00096c |
| This journal is © The Royal Society of Chemistry 2015 |