
Universal polymer coatings and their representative biomedical applications
Qiang
Wei†
*ab and
Rainer
Haag
ab
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Takustr. 3, 14195, Berlin, Germany. E-mail: wei.weiqiang@gmail.com; wei@is.mpg.de
bHelmholtz Virtual Institute, Multifunctional Biomaterials for Medicine, Kantstraße 55, 14513, Teltow-Seehof, Germany
First published on 27th August 2015
Abstract
Universal polymer coatings have excellent potential for biomedical applications, because of their substrate-independent properties and versatile surface functionalization methods. The goal of this review is to summarize the state-of-art research on universal polymer coatings and their biomedical applications, as well as to present their common features including some general rules for their further development.
 Qiang Wei | Dr Qiang Wei received his Bachelor's and Master's degrees in 2008 and 2011, respectively, from Sichuan University, China. He completed his PhD with Prof. Dr Rainer Haag at Freie Universität Berlin in 2014. His PhD thesis focused on the design of mussel-inspired polymers and the development of multifunctional polymer coatings. He then joined Max-Planck Institute for Intelligent Systems as postdoctoral researcher in the research area of cell and tissue engineering. |
 Rainer Haag | Prof. Dr Rainer Haag obtained his PhD with A. de Meijere at the University of Göttingen in 1995. After postdoctoral work with S. V. Ley, University of Cambridge (UK), and G. M. Whitesides, Harvard University, Cambridge (USA), he completed his habilitation at the University of Freiburg in 2002. He then became associate professor at the University of Dortmund and in 2004 was appointed full Professor of organic and macromolecular chemistry at the Freie Universität Berlin. His main research interests are the mimicry of biological systems by functional dendritic polymers, with particular focus on applications in nanomedicine. |
1. Introduction
Polymer coatings on solid materials play an increasingly important role in physical, chemical, and biomedical sciences.1,2 Thiol and siloxane chemistries are commonly used to modify noble metal and hydroxylated surfaces, respectively.3,4 Besides the widely used self-assembled monolayer (SAM) and chemical surface immobilization that are induced by these and other anchor groups, Langmuir–Blodgett deposition,5 layer-by-layer assembly,6 irradiation,7 and electrostatic or hydrophobic adsorption2 are well established. However, most of these technologies require specific chemical or physical substrate properties, and thus have failed to become universal coatings.Universal coatings are coatings that can modify a wide range of material surfaces and are stable under the applied conditions. Ideally, these coatings are substrate-independent, regardless of the chemical composition and physical characteristics (e.g. topology and stiffness) of the substrates. To develop such coatings, the anchoring interactions between the polymers and various substrates must be well designed. Chemical functionalities for specific covalent binding between the polymeric coating and the surface must be avoided in order to modify different types of substrate, because no anchor can be active on all of the different surface compositions. Although, some irradiation technologies can activate many kinds of surfaces, the efficiency and the density of the active sites are relatively low on some surfaces. Therefore, they should be enhanced by some compensatory methods, e.g., polymerization, to obtain dense surface coatings.8,9 On the other hand, noncovalent interactions, like electrostatic interaction, hydrogen bonding, hydrophobic attraction, and van der Waals interaction, occur on nearly all types of interfaces. Thus, multiple noncovalent interactions can be recognized as the driving forces for constructing polymer coatings on different surface types. Admittedly, most of the noncovalent interactions between interfaces are not strong enough to tether polymer coatings for practical applications. Therefore additional intra-layer interactions, i.e., physical and chemical crosslinking can be used to enhance the stability of the coating.
Crosslinking can either be initiated in situ while anchoring the coating, or in a step-wise fashion after the formation of precast layers (Fig. 1). In the in situ case, one-pot coating is easy and rapid. However, spontaneous crosslinking may cause polymeric modifiers to aggregate, which makes the surface morphology less controllable. In the latter case using precast layers, further crosslinking procedures like heating or irradiation are required. This step, however, must be well designed to avoid decreasing the performance of the coatings.
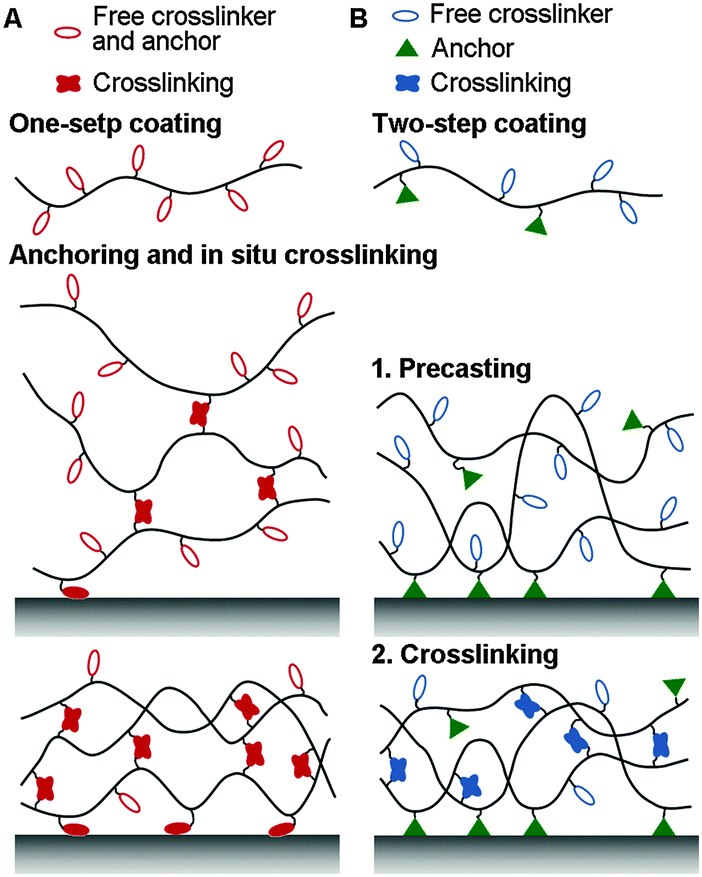 | ||
| Fig. 1 Universal coatings can be stabilized on different kinds of surfaces by interior crosslinking, which can be achieved either (A) by in situ crosslinking together with anchoring or (B) by step-wise crosslinking after the formation of a precast layer. | ||
A secondary functionalization of these universal coatings is normally required to achieve specific surface characteristics. Thus, there must be enough active groups remaining in the coatings for further modification. The most important surface coatings in biomedical applications include bioinert, biospecific, and antibacterial coatings. A bioinert surface on the one hand requires dense and stable coatings to prevent protein adsorption on a molecular level and to further repel cell adhesion.2 These coatings must be hydrophilic and electrically neutral and contain hydrogen bond accepting groups but no hydrogen bond donating groups.10,11 Biospecific surfaces on the other hand, which contain cell recognition motifs, are another approach to modulate cell interaction on the surface of a biomedical device.1 A relatively low density of functionalization is sufficient to trigger cell adhesion. In the case of arginylglycylaspartic acids (RGDs), a minimum density as low as 1 fmol cm−2 was reported for cell spreading and 10 fmol cm−2 for forming focal contacts and stress fibers on a surface.12 Inspired by a cell membrane that contains bioactive carbohydrates and proteins in the bioinert background of a phospholipid bilayer, biospecific molecules can be combined with bioinert coatings to increase the efficacy of biological communication.1,2 As a result, implanted surfaces would only integrate with, for example, endothelial cells, and not with leukocytes and other cells. When constructing such designed coatings, it is important to achieve multifunctional coatings. Moreover, bioinert materials are often combined with antibacterial agents to prevent bacterial adhesion and improve the biocompatibility of the coatings.13 Besides these functional coatings mentioned above, multiple functional surfaces, e.g., infection-resistant, anticoagulated, and self-cleaning surfaces, can be developed by immobilizing different functional molecules14,15 on reactive universal coatings.
In this review, we summarize the characteristics and common features of current universal polymer coating systems, which include surface irradiation, layer-by-layer (LbL) assembly, spin coating, chemical vapor deposition (CVD), laser deposition, blood proteins, mussel-inspired coatings, and plant phenols (Table 1). These systems are categorized by their anchoring interactions, i.e., chemisorption, physisorption, and multiple interactions, to clearly show their similarities and differences. We will therefore draw conclusions on the general rules for developing such coatings in the future. Also, some recent developments in universal coatings in the area of biomedical applications will be described.
| Types | Preparation | Anchoring interactiona | Advantages | Disadvantages |
|---|---|---|---|---|
| a Only the main anchoring interactions for each type of coating are shown. | ||||
| Surface irradiation | Gas phase | Covalent bond | Covalently anchored; highly pure | Specific equipment; potential damage to the substrates; limited by substrate shape |
| Layer-by-layer (LbL) assembly | Solution phase | Electrostatic attraction | Shape independent; dip-coating | Time-consuming; unstable in strong electrolyte conditions |
| Spin coating | Solution phase | Hydrophobic interactions | Well-modulated thickness | Weak anchoring; limited by the shape |
| Chemical vapor deposition (CVD) | Gas phase | Hydrophobic interactions | Highly pure | Specific equipment; weak interactions; limited by substrate shape |
| Laser deposition | Gas or solution phase | Hydrophobic interactions | Comprehensive material source; direct-writing | Specific equipment; limited by substrate scale and shape |
| Blood proteins | Solution phase | Multiple interactions | Shape independent; dip-coating | Degradable |
| Mussel-inspired coatings | Solution phase | Multiple interactions | Shape independent; dip-coating | Surface roughness; dark colour |
| Plant phenols | Solution phase | Multiple interactions | Shape independent; dip-coating; cheap resource | Surface roughness |
2. Irradiative chemisorption
High energy ionizing radiation can directly generate initiation sites by liberating electrons from atoms or molecules near material surfaces. These positively charged initiation sites can immediately react with other molecules to generate functional groups for further surface modification. Different radiation methods, including plasma, ultraviolet (UV), gamma rays, microwaves, lasers, electron beams, etc., have been employed to activate corresponding material surfaces.16Plasma exposure is the most common radiation method. However, plasma, which can easily activate organic surfaces, does not work equally well with inorganic surfaces.8 Thus, plasma polymerization of monomers with vinyl groups has become a general method to functionalize different solid surfaces as an alternative to simple irradiation.8,17,18 As a result, the sparse active sites on organic surfaces can be connected by highly crosslinked polymer films, which results in stable coatings on substrates via polyvalent anchoring. As a result, highly crosslinked polymer films can be stably deposited on substrates via polyvalent anchoring.17 Various chemical surface functionalities, like anhydride-,8 amino-,17 epoxide-,19 and perfluoroalkyl-groups,20 can be achieved by employing different monomers (Fig. 2).
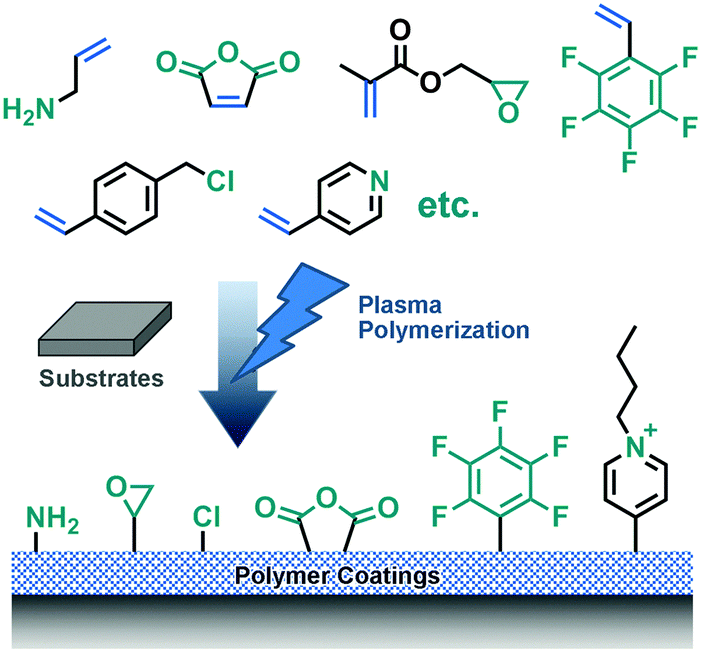 | ||
| Fig. 2 Various functional universal coatings can be achieved by plasma polymerization with different monomers. | ||
Long-term irradiation may change the properties of the functional groups of monomers and degrade newly formed polymers. Pulsed plasma with short on-periods and long off-periods was proven to deposit polymer films with a higher degree of molecular specificity than traditional continuous wave plasma.8 The active sites in the gas phase and at the growing film surface could be generated in a short plasma duty cycle on-period (microseconds), which initiated polymerization during the longer plasma off-period (milliseconds).21
Alternatively, polymeric targets, such as polytetrafluoroethylene (PTFE), polyimide, and polyolefin, have been sputtered to form coatings by radio frequency (RF) magnetron sputtering.22 Powerful magnets cause the emission of volatile fragments from polymeric targets. These fragments take part in the plasma polymerization process and line up on the substrates to form thin films. Since polymeric targets are provided in the solid state, fewer safety precautions are required than for handling the gas of monomers in plasma polymerization.23 To achieve the secondary modification, amino-rich thin films were prepared by sputtering a nylon 6,6 target in a mixture of N2/H2 or N2/Ar. As a result, a high NH2/C ratio in the coatings was achieved.23
The amino groups present in plasma polymerized polyallylamine coatings and RF magnetron sputtered nylon coatings are suitable for immobilization of atom transfer radical polymerization (ATRP) initiators via amide linkages.9,17 Bioinert polymer brushes of poly(oligoethylene glycol methacrylate) or poly(carboxybetaine acrylamide) can be subsequently initiated from the functional surfaces, which has resulted in dramatically decreased protein adsorption on the solid surfaces.9
The plasmachemical functionalization of surfaces with poly(4-vinylpyridine) coatings yielded bactericidal activity towards Staphylococcus aureus (Gram positive) and Klebsiella pneumoniae (Gram negative), after quaternization of the pyridine moieties with bromobutane.24
Patterned functional surfaces were developed by depositing two separate functional nanolayers, including an active bottom layer of poly(glycidyl methacrylate) and a passive release top layer of poly(pentafluorostyrene) on substrates. A selective lift-off of the top layer by a prepatterned adhesive template resulted in the exposure of the underlying active layer.25
Surface irradiation methods are easily controlled methods for film growth on different substrates. In many cases, solvents are not required and coating processes are suitable for large-scale film deposition. Moreover, covalently anchored coatings can remain stable under different solvent conditions. However, irradiation may change the properties of the substrates, especially ultrathin substrate layers, and it can be limited by the shape of the substrates. Additionally, irradiation and deposition require sophisticated equipment, which limits their applications in industry. Thus, physisorbed universal coatings can be considered as alternatives.
3. Physisorption
Typical physisorption of surface coatings includes electrostatic attraction, van der Waals forces, and hydrophobic interaction. Based on these universal interactions, some technologies, including layer-by-layer (LbL) assembly, spin coating, and chemical vapor deposition (CVD), have been developed to obtain some universal coating systems.3.1 Electrostatic attraction
Polyelectrolytes are good candidates to anchor on the substrate surface via electrostatic attraction. However, monolayer brushes of block copolymers, which are immobilized through a polyionic block onto the surface and prevent further adsorption via another flexible block,26 cannot be efficiently adsorbed on uncharged surfaces and are sensitive to salt concentration. Instead, the layer-by-layer (LbL) assembly technique is more universal. It does not significantly depend on the nature, size, and topology of the substrate,27 due to the intra-coating electrostatic interaction. Some LbL assembly systems indeed successfully fabricate multicomponent thin films on a wide range of surfaces by consecutive adsorption of polyanions and polycations. Electrostatic attraction between oppositely charged and flexible polymers has the least steric demand for building all the chemical bonds and stabilizing fuzzy layered LbL assembled multilayers.An alternative electrostatic assembly approach for cationic poly(allylamine hydrochloride) (PAH) and anionic poly(sodium 4-styrenesulfonate) (PSS) has been performed on a variety of material surfaces, including glass, gold, mica, silicon, and other polymers. The properties of the different underlying surfaces were completely converted to the surface properties of the polyelectrolyte coatings.28 The chemically active scaffold can be further utilized to fabricate protein microarrays. Mouse IgG has been immobilized on PAH-capped polyelectrolyte coatings. The rest of the surface was then blocked with bovine serum albumin (BSA). The nearly identical specific signal intensities of anti-mouse IgG with low nonspecific binding can be observed on the tested dissimilar substrates.28
LbL assembly is, however, a time-consuming process, especially for the fabrication of thick films.29 Large dimensional building blocks with fast adsorption kinetics can realize rapid fabrication and have been built with mesoporous silica (MSiO2) nanoparticles with cationic poly(diallyldimethylammonium chloride) (PDDA) to assemble a substrate-independent thick coating with only three coating cycles.30 This coating exhibited both antireflection and antifogging properties, because the rough surface morphology and nanopores in the MSiO2 nanoparticles resulted in superhydrophilic surface performance. A maximum transmittance of 99.9% was achieved in the visible spectral range, under the optimal conditions.
A one-pot electrostatic attraction based coating was achieved by the aggregation of polyallylamine and orthophosphate anions, and can be fabricated within 60 min.31 The orthophosphate anions efficiently crosslinked the coatings by both electrostatic interaction and hydrogen bonding. The presence of amino groups in the coatings led to further functionalization. Biotin immobilized coatings recognized streptavidin.
Electrostatic attraction is a relatively strong non-specific interaction, which has produced a set of universal coatings. However, strong electrolyte solutions, e.g. strong acid or base, can interrupt this attraction and decompose the coatings.32,33
3.2 van der Waals forces and hydrophobic interactions
The relatively weak van der Waals and hydrophobic interactions can also be used to anchor universal coatings, if the intra-coating crosslinking is well designed.A mixture of hydrophilic amine- and epoxy-terminated four-arm polyethylene glycols (PEGs) was spin-coated on a flat substrate. After chemically crosslinking these macromonomers under gentle heating, a hydrogel-like coating with tunable film thicknesses of 4–200 nm was fabricated on a broad variety of solid substrates. Because of its controllable swelling behavior, this coating was able to adsorb a high density of citrate-stabilized gold nanoparticles (AuNPs) from aqueous solution and resulted in PEG/AuNP composite films.34
In a similar approach, hydrophobic benzocyclobutene-functionalized random copolymers of styrene and methyl methacrylate [P(S-r-BCB-r-MMA)] were spin coated on a wide variety of metal, metal oxide, semiconductor, and polymeric surfaces to produce thin films.35 The styrene moieties of the copolymers induced balanced interfacial interactions on the surfaces.36 After heating in the 200–250 °C temperature range, the reactive benzocyclobutene (BCB) moieties underwent crosslinking reactions. The crosslinked films were resistant to solvents and formed a robust coating on the substrates. In another case, a hybrid polymer composed of poly(methylsilsesquioxane) (PMSSQ) blocks and poly(pentafluorophenyl acrylate) (PFPA) blocks was employed to coat different materials.37 The PMSSQ blocks initiated crosslinking after spin coating, while the PFPA blocks enabled various secondary functionalizations of the coatings.
In the case of the crosslinkable PEGs that are mentioned above, the coating mainly interacted with substrates by weak van der Waals forces. Keeping this kind of hydrophilic coating stable in water solution for a long time is a big challenge, because water can shear off the whole coating. Therefore, it is preferable to use hydrophobic coatings like the P(S-r-BCB-r-MMA) and PMSSQ–PFPA cases above. Nonpolar substances tend to aggregate or adsorb on solid surfaces in aqueous solution and repel water molecules. Since water is the most common and secure solvent in our daily life, hydrophobic interactions have successfully generated a set of universal coatings.
Chemical vapor deposition (CVD), which is often used in the semiconductor industry to produce thin films, can also be used to fabricate hydrophobic coatings of poly(p-xylylenes) and derivatives for a wide range of substrates including PTFE (Fig. 3).38 In the CVD polymerization process, diradicals of [2.2]paracyclophane or its derivatives are obtained during vaporization under heating and vacuum conditions. The obtained diradicals are then deposited on the substrate during polymerization. It has been reported that these CVD polymers strongly anchor on substrate surfaces and are insoluble in common organic solvents.39 It is reasonable to speculate that chain transfer in such radical-rich polymerizations may have resulted in chemical crosslinking, which highly stabilized the deposited coatings as well as intermolecular hydrophobic interactions and π-stacking. The copolymers of [2.2]paracyclophane and its functionalized derivatives have generated multifunctionalized CVD coatings, which can be widely used in biomedical applications. Using a vacuum deposition process the limitations caused by solvents and additives in dip coating procedures are overcome,39 so that highly pure coatings can be obtained. However, for production, every CVD step requires expensive equipment, such as high vacuum conditions.
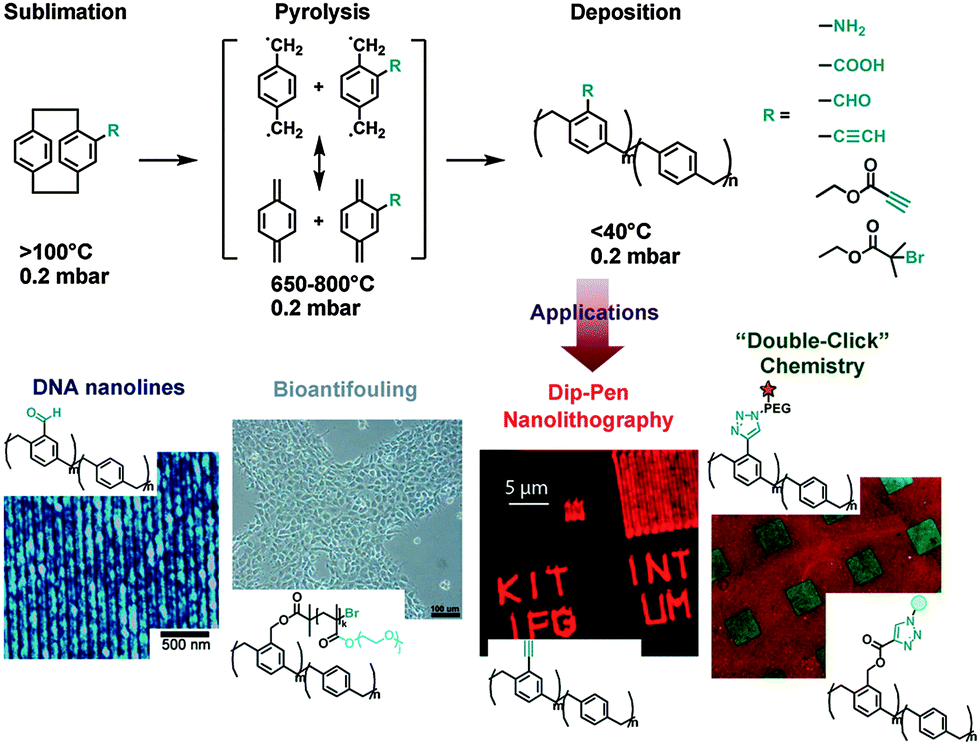 | ||
| Fig. 3 Chemical vapor deposition (CVD) polymerization using various monomers to achieve multifunctional universal coatings. Reprinted from ref. 40–43 with kind permission of Wiley and The American Chemical Society. | ||
Surface active CVD coatings are good platforms for immobilizing biomolecules. The anhydride-rich coating of poly(p-xylylene-2,3-dicarboxylic anhydride) can immobilize amino-terminated biotin ligands which selectively bind to streptavidin. The biotin-conjugated human anti-α5-integrins were then immobilized on streptavidin and specifically interacted with endothelial cells.44 Surfaces that “click” have been developed using alkyne-containing vapor deposited polymer coatings. Polymers with monoalkyne grafted [2.2]paracyclophane have generated excellent adhesion and stability, even at 680 °C and in many organic solvents. On the other hand, enough alkynes were exposed on the surfaces to react with azide-containing biotin-based ligands45 or to support dip-pen nanolithography by “click chemistry”.42 In further developments, a bioorthogonal immobilization of biotins and streptavidins was carried out using a copper-free click reaction on CVD coatings.43 The synthesized [2.2]paracyclophane-4-methyl propiolate, which contained an electron-withdrawing group in close proximity to the alkyne, was identified for the copper-free click reaction with azide groups. Moreover, this [2.2]paracyclophane derivative was compatible with the processing conditions during CVD polymerization without decomposition or side reactions. Using alkynyl moieties in copper-catalyzed “click” reactions, a two-step cascade of bioorthogonal reactions sequentially immobilized different biomolecules on separate areas of the same surface.43 Additionally, aldehyde functionalized CVD coatings could link to 5′ amine modified complementary DNA sequences by forming imine bonds. Thus, poly(4-formyl-p-xylylene-co-p-xylylene) was deposited on different substrates to serve as a “replica” to collect DNA microarrays from microcontact printing.40
Besides the immobilization of biomolecules, initiators for atom transfer radical polymerization (ATRP) can be directly immobilized onto CVD monomers and can be polymerized and deposited on different kinds of substrates, including stainless steel, glass, silicon, poly(dimethylsiloxane), poly(methyl methacrylate), poly(tetrafluoroethylene), and polystyrene.41 This polymeric initiator coating initiated ATRP of oligo(ethylene glycol) methyl ether methacrylate to produce a bioinert polymeric coating as thick as 300 nm. Both protein adsorption and cell adhesion were significantly inhibited on this bioinert coating.
A physical vapour deposition technique, namely laser processing of polymers, also has the potential to modify different material surfaces.46 Lasers can irradiate and vaporize almost every conceivable target material by either photolytic or pyrolytic processes. These materials, including synthetic polymers and natural biopolymers, can be then deposited on substrates.47 By depositing blood proteins48 or mussel inspired polymers,49 the anchoring interactions of the coatings can be enhanced (for details, see Section 4). Interestingly, some of the laser deposition techniques, such as matrix-assisted pulsed laser evaporation and laser guidance approaches, can serve as direct-write techniques to deposit patterns on substrates.50
Overall, versatile physisorption based universal coatings have been developed that on the one hand overcome many problems in irradiated chemisorption and on the other hand suffer from stability problems under some application conditions. The inherently weak anchoring interactions of the physisorbed surface coatings, however, can become thermally unstable. These coatings may also be displaced by other solutes in solution. Therefore, these coatings must be carefully utilized under appropriate conditions, i.e., by avoiding strong electrolyte solutions for electrostatic attraction based coatings, and by avoiding long-term submersion in non-polar solvents or flow environments for hydrophobic interaction based coatings.
4. Bioinspired surface coatings
Nature, due to evolutionary processes, has developed ways to excellently and precisely solve problems from which many artificial systems are suffering. Learning from nature is an endless source of inspiration. In the present section, universal coatings that have been inspired or directly collected from natural biological systems of blood proteins, mussel foot proteins, and plant phenols will be described and discussed. These bioinspired surface coatings bind to substrate surfaces by multiple combined interactions, besides simple chemisorption or physisorption, to enhance the stability of the coatings under different conditions.4.1 Blood proteins as adhesive coatings
It is well known that blood proteins nonspecifically adsorb onto blood contact surfaces within seconds via multiple interactions such as van der Waals forces, ionic or electrostatic attraction, hydrogen bonding, and hydrophobicity.2 An approach involving blood proteins to modify both flat and nonwoven substrates has been reported.51 A set of proteins, including α-lactalbumin, lysozyme, fibrinogen, and soy globulins (glycinin and β-conglycinin), were denatured at their isoelectric point (pI). Under these conditions, a maximum amount of proteins could be adsorbed onto the substrates, because the electrostatic repulsion among the protein molecules was limited.52 Denaturation helped the hydrophobic domains of the proteins to be adsorbed on the substrates with the result that the hydrophilic amino and hydroxyl groups could be exposed on the surface for secondary modification. To stabilize the coatings, the adsorbed protein layers were crosslinked with glutaraldehyde in the presence of sodium borohydride. The ATRP initiator molecules could then be immobilized on the amino and hydroxyl groups, from which poly(2-hydroxyethyl methacrylate) (PHEMA) polymer brushes were grown. By combining fluorinating moieties, these amphiphilic polymer brushes efficiently prevented further nonspecific protein adsorption.53 Although the protein based coatings above were only reported for modifying polyolefin surfaces, it is possible to apply these coatings to a wide range of material surfaces because nonspecific adsorption of proteins is quite general on solid surfaces. In the other case, phase-transited lysozyme was employed to coat a broad range of substrates based on the same concept.54 However, the main problem of this coating system may be long-term stability, since protein layers can be degradable in physiological environments.4.2 Mussel foot proteins as adhesive coatings
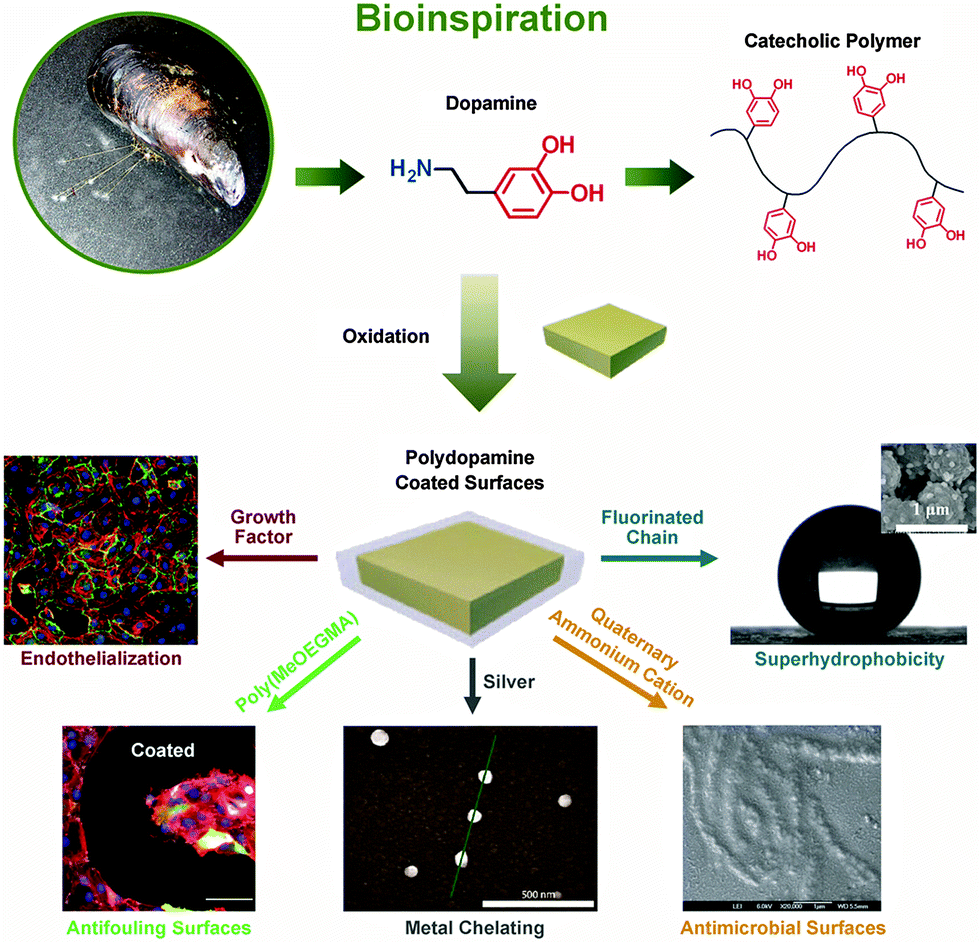 | ||
| Fig. 4 Mussel-inspired polydopamine as universal mltifunctional coatings. Modified from ref. 68–72 with kind permission of Wiley and The American Chemical Society. | ||
Native polydopamine coatings already show low cytotoxicity and can promote the adhesion of osteoblasts73 and endothelial cells,74 because the critical surface tension of polydopamine (39.2 dynes per cm) is in a suitable range for cell adhesion (35–40 dynes per cm).75 Furthermore, a number of secondary modifications can be applied by immobilizing different functional molecules onto polydopamine coatings via residual free amines and catechols.15 Bioinert polymer layers have been created by both “grafting to” and “grafting from” approaches, as well as by LbL assembly on polydopamine coatings to achieve substrate-independent surface modification.69,76,77 Biospecific molecules, such as vascular endothelial growth factor,68 adhesion peptides,78 and glycosaminoglycan,79 have been easily immobilized onto polydopamine coatings with an one-step immersion, and have resulted in specific cell adhesion. The metal chelating ability of the catechol groups in the coatings can cause in situ deposition of silver nanoparticles.70 The silver nanoparticles or the grafted quaternary ammonium groups71 on the coatings have exhibited strong and broad spectrum antimicrobial activities. Moreover, the combination of bioinert layers with antibacterial moieties produced dual fouling resistance and antibacterial properties of the coatings, which significantly improved the antibacterial performance of the surfaces.70,71 The deposited silver nanoparticles on polydopamine coated microparticles resulted in a hierarchical structure similar to the micromorphology of a lotus leaf. These composite particles became extremely water repellent after fluorination.72 Although synthetic polydopamine coatings were only introduced in 2007 by the Messesmith group,56 they have already become one of the most widely applied universal coatings due to their facile procedure and chemical versatility.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) resulted in a colorless coating. Surface-initiated ATRP of 2-hydroxyethyl methacrylate (HEMA) can be performed using this coating.87 A fluorinated dopamine derivative was developed by conjugating a perfluorinated chain to the carboxyl group of DOPA.88 The remaining amine and catechol groups resulted in a structurally rough film with static water contact angles larger than 150° as a superhydrophobic surface.
:2) resulted in a colorless coating. Surface-initiated ATRP of 2-hydroxyethyl methacrylate (HEMA) can be performed using this coating.87 A fluorinated dopamine derivative was developed by conjugating a perfluorinated chain to the carboxyl group of DOPA.88 The remaining amine and catechol groups resulted in a structurally rough film with static water contact angles larger than 150° as a superhydrophobic surface.
Systematic studies on how the grafting amount of catechol groups affects the coatings on different types of surfaces have also been reported.91,92 The thickness and stability of the polymer coatings can be controlled by catechol groups which act as both anchors and crosslinkers. In the case of metal oxide surfaces, although even one catechol group can tether the polymer chain, multiple catechol units are required in the anchor group to prevent oxidative detachment.93,94 In the case of inert polymeric substrates, such as PTFE, polystyrene, and polyolefin, the interaction between the catechol group and these surfaces is relatively weak.65,95 Besides weak anchoring, the other important role of catechol as a crosslinker is to stabilize laterally the coatings on inert substrates.92 Therefore, a relatively large amount of catechol groups is required to achieve universal coatings. For the design of bioinert surface coatings, however, an overrepresentation of catechol groups leads to protein adsorption and cell adhesion. Only a well-balanced amount of catechol groups can supply coatings with both good stability and bioinertness.92
Although catechol is a powerful anchor for surface coating, its effectiveness has been somewhat over praised in some previous publications. Actually, even multiple catechol functionalized polymers have hardly reached a very high surface coverage on inert surfaces.91,92 In some publications, effective coatings on inert surfaces were obtained by polymers that conjugated with a few catechol groups and the success was announced to be fully due to catechol adhesion. We do not doubt these coatings, but we should mention that the hydrophobic effect of the polymer itself has often been ignored, which definitely enhanced the interface interactions besides catechol anchoring. Control experiments should be well designed to explore the further role of catechols in these cases.
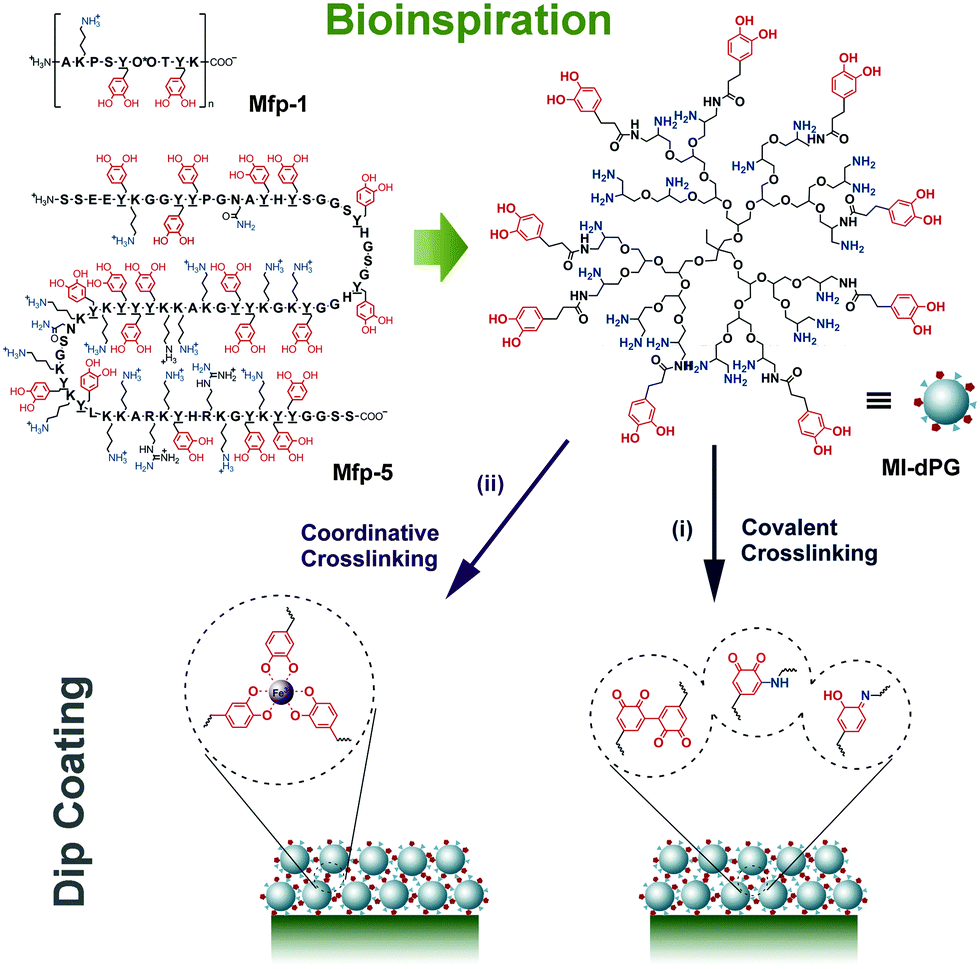 | ||
| Fig. 5 Mussel-inspired dendritic polyglycerol (MI-dPG) that mimicked mfps by the functional groups, molecular weight, and molecular structure resulted in rapid covalent (postulated structure) and coordinative crosslinking for universal surface coatings. Reprinted from ref. 97 with kind permission of Wiley. | ||
Besides catechol induced surface adhesion, mussels limit the auto-oxidation of catechols on the surface of byssal plaques to enhance the adhesion by thiol-rich mfp-6.101 Other hydrophobic amino acid residues, mainly in mfp-3 “slow”, can retard oxidation of catechols by shielding them from the solvent and, more importantly, compensate the adhesion by hydrophobic interactions.102 The adhesion of a mussel byssus, however, is more complicated than a simple catechol-mediated recipe.
4.3 Plant phenols as adhesive coatings
Tea cups are often stained by tea water. Inspired by this phenomenon, a number of phenolic biomolecules that are present in tea, red wine, chocolate, and many other plants have been identified for versatile universal coatings. These biomolecules possess abundant and dense catechol (1,2-dihydroxyphenyl) and gallol (1,2,3-trihydroxyphenyl) functional groups and thus exhibit strong solid–liquid interfacial properties. A plant polyphenol of tannic acid (TA) and a simple phenolic mimic of pyrogallol were deposited from buffered saline (0.6 m NaCl, pH 7.8) to form polydopamine-like films.103 These phenolic films retained most of the advantages of polydopamine films as multifunctional universal coatings, but they were low cost and colorless. In addition, these coatings could scavenge radical and non-radical reactive oxygen species. In subsequent work, a library of about 20 kinds of natural and synthetic phenolic molecules was screened.104 Among them, eight catechol-, gallol-, and resorcinol-rich molecules were identified to form excellent universal coatings. Besides TA and pyrogallol, the other six precursors were epigallocatechin gallate (EGCG), epigallocatechin (EGC), catechin (Ctn), catechol (Ctl), hydroxyhydroquinone (HHQ), and morin (Fig. 6A). Moreover, 5-pyrogallol 2-aminoethane, which contains both the pyrogallol group and a primary amine group, has been shown to form coatings with enhanced stability and coating ability,105 because the presence of the amine group enhances the crosslinking. As the polymerization and deposition of dopamine could be accelerated by an oxidant,30 the laccase-catalyzed polymerization of plant phenols also resulted in a rapid coating formation.106 In fact, oxidation and enzyme catalysis can accelerate the formation of both polydopamine and phenol coatings.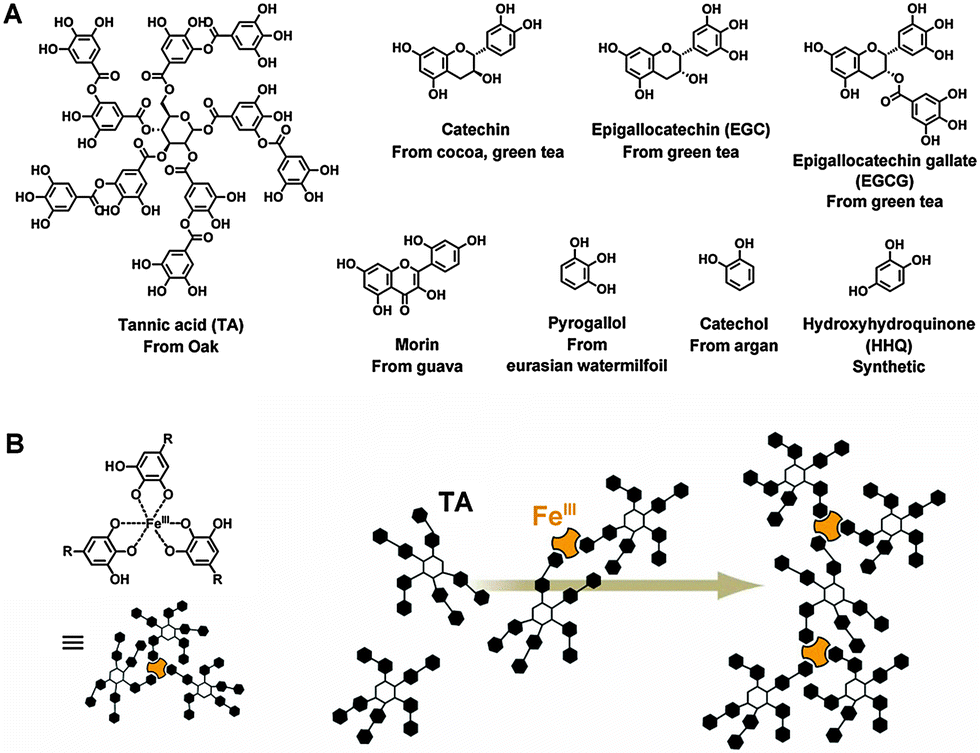 | ||
| Fig. 6 (A) Chemical structures of the natural and synthetic phenols that were identified to form universal coatings.104 (B) Scheme of the assembly of iron-based coordination complexes. Reprinted from ref. 107 with kind permission of AAAS. | ||
Besides polydopamine type crosslinking, another self-assembly process based on polyphenols for surface modification has been explored. Phenolic moieties are weakly acidic and can donate an electron or electron pair to chelate metal ions.107 Thus, polyphenols like TA can be crosslinked by coordination with iron, e.g. Fe(III) (Fig. 6B), then deposited and bound to substrates to form versatile coatings with negligible cytotoxicity.108 This coordinative crosslinking is pH responsive. At low pH, the hydroxyl groups are protonated, which leads to a destabilization of the crosslinking and disassembly of the coatings.109 In the case of coordination between TA and Fe(III), only mono-phenol complexes formed at pH < 2.0, with the result that the coating disassembled. Even at 3 < pH < 6, when bis-complexes existed, the coatings could not be kept stable. Only when tris-complexes formed at pH > 7, did the coatings show good long-term stability. A library of functional TA-metal networks showed that this pH sensitivity was controllable by changing the metal species and feed concentrations.110 Moreover, varying the feed concentration of the lanthanide metals allowed control over the fluorescence intensity of the coatings. Similarly, catecholic polymers have also been reported, which can generate coatings by ion based coordinative crosslinking.97 Therefore, this new type of coating is a potential candidate for biomedical applications.
In summary, all three types of bioinspired universal coating system, i.e., blood proteins, mussel foot proteins, and plant phenols, have been successfully applied on almost all kinds of material surfaces, regardless of the shape of the substrates. Their combined multiple anchoring interactions and the high degree of intra-coating crosslinking resulted in a set of highly stable coatings. However, our natural systems, e.g., mussel byssus, can even adjust the balance of each interaction to reach optimal adhesion on different substrates.55 There is still a long way to go in chemistry and materials science to really mimic natural systems to generate the best universal coating.
5. Conclusion and perspectives
An ancient Chinese proverb says “A single chopstick can be gently broken, a pillar of ten chopsticks firmly holds dough”. Both lateral crosslinking and the polymerization of all monovalent anchorings (one single chopstick) combine the binding forces on the substrate surface together to reach multi-/polyvalent anchoring (a pillar of ten chopsticks). Thus, a coating can be indeed stabilized to reach a universal coating, even if the force of the individual monovalent anchoring is relatively small. Therefore, the common features of the presented universal coatings can be summarized, and the general rules for developing new universal coatings can be proposed that: (1) there must be some interaction between the coating materials and the substrate surfaces, even though the interaction might be relatively weak; (2) lateral crosslinking, either covalent or noncovalent, must be present; (3) the coating should be prepared with the available functions or it can be further functionalized. Stronger interfacial interaction and a higher degree of crosslinking can result in more stable coatings, especially on chemically inert surfaces, such as Teflon.Among the large family of surface modification systems, however, still only a few universal coatings can be successfully used for practical applications. It is necessary to further establish a mechanistic understanding of the stabilization of universal coatings and theoretical guidelines for developing such coatings. Therefore, future research should be focused on exploring the mechanisms of the adsorption of polymers onto different surfaces, quantitatively studying the crosslinking's contribution in stabilizing coatings, and establishing a set of theories to guide the development of universal coatings. Although homogeneous coatings were obtained by all of the approaches mentioned above, most of the coatings were only studied on a lab scale. Quantitative analysis of the uniformity of coatings on a large scale is still lacking, which is also important for practical applications.
Overall, it remains a big challenge to further develop universal coatings to become a real long-term stable tool in our daily life, however, universal polymer coatings have already added a new page to material surface modification.
Acknowledgements
This work was supported by the Helmholtz Virtual Institute on “Multifunctional Biomaterials for Medicine”. We thank Dr Pamela Winchester for proofreading this manuscript.References
- S. R. Meyers and M. W. Grinstaff, Chem. Rev., 2012, 112, 1615–1632 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- S. Onclin, B. J. Ravoo and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2005, 44, 6282–6304 CrossRef CAS PubMed.
- K. Ariga, Y. Yamauchi, T. Mori and J. P. Hill, Adv. Mater., 2013, 25, 6477–6512 CrossRef CAS PubMed.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS PubMed.
- D. M. He, H. Susanto and M. Ulbricht, Prog. Polym. Sci., 2009, 34, 62–98 CrossRef CAS PubMed.
- D. O. H. Teare, W. C. E. Schofield, V. Roucoules and J. P. S. Badyal, Langmuir, 2003, 19, 2398–2403 CrossRef CAS.
- C. Rodriguez-Emmenegger, O. Kylian, M. Houska, E. Brynda, A. Artemenko, J. Kousal, A. B. Alles and H. Biederman, Biomacromolecules, 2011, 12, 1058–1066 CrossRef CAS PubMed.
- R. G. Chapman, E. Ostuni, S. Takayama, R. E. Holmlin, L. Yan and G. M. Whitesides, J. Am. Chem. Soc., 2000, 122, 8303–8304 CrossRef CAS.
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089–1100 CrossRef CAS.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- D. Campoccia, L. Montanaro and C. R. Arciola, Biomaterials, 2013, 34, 8533–8554 CrossRef CAS PubMed.
- Y. L. Liu, K. L. Ai and L. H. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS PubMed.
- B. Yameen, H. U. Khan, W. Knoll, R. Forch and U. Jonas, Macromol. Rapid Commun., 2011, 32, 1735–1740 CrossRef CAS PubMed.
- B. R. Coad, Y. Lu, V. Glattauer and L. Meagher, ACS Appl. Mater. Interfaces, 2012, 4, 2811–2823 CAS.
- C. Tarducci, E. J. Kinmond, J. P. S. Badyal, S. A. Brewer and C. Willis, Chem. Mater., 2000, 12, 1884–1889 CrossRef CAS.
- A. M. Hynes, M. J. Shenton and J. P. S. Badyal, Macromolecules, 1996, 29, 4220–4225 CrossRef CAS.
- M. E. Ryan, A. M. Hynes and J. P. S. Badyal, Chem. Mater., 1996, 8, 37–42 CrossRef CAS.
- H. Biederman, V. Stelmashuk, I. Kholodkov, A. Choukourov and D. Slavinska, Surf. Coat. Technol., 2003, 174, 27–32 CrossRef.
- O. Kylian, J. Hanus, A. Choukourov, J. Kousal, D. Slavinska and H. Biederman, J. Phys. D: Appl. Phys., 2009, 42, 142001 CrossRef.
- W. C. E. Schofield and J. P. S. Badyal, ACS Appl. Mater. Interfaces, 2009, 1, 2763–2767 CAS.
- P. S. Brown, T. J. Wood, W. C. E. Schofield and J. P. S. Badyal, ACS Appl. Mater. Interfaces, 2011, 3, 1204–1209 CAS.
- C. Perrino, S. Lee, S. W. Choi, A. Maruyama and N. D. Spencer, Langmuir, 2008, 24, 8850–8856 CrossRef CAS PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- S. D. Spillman, H. M. McEvoy and B. D. MacCraith, Langmuir, 2009, 25, 1403–1411 CrossRef CAS PubMed.
- B. S. Shim, P. Podsiadlo, D. G. Lilly, A. Agarwal, J. Leet, Z. Tang, S. Ho, P. Ingle, D. Paterson, W. Lu and N. A. Kotov, Nano Lett., 2007, 7, 3266–3273 CrossRef CAS PubMed.
- Q. Wei, F. L. Zhang, J. Li, B. J. Li and C. S. Zhao, Polym. Chem., 2010, 1, 1430–1433 RSC.
- W. A. Marmisollé, J. Irigoyen, D. Gregurec, S. Moya and O. Azzaroni, Adv. Funct. Mater., 2015, 25, 4144–4152 CrossRef PubMed.
- J. Cho and F. Caruso, Macromolecules, 2003, 36, 2845–2851 CrossRef CAS.
- X. Y. Zhu, D. Janczewski, S. S. C. Lee, S. L. M. Teo and G. J. Vancso, ACS Appl. Mater. Interfaces, 2013, 5, 5961–5968 CAS.
- N. Meyerbroker, T. Kriesche and M. Zharnikov, ACS Appl. Mater. Interfaces, 2013, 5, 2641–2649 Search PubMed.
- D. Y. Ryu, K. Shin, E. Drockenmuller, C. J. Hawker and T. P. Russell, Science, 2005, 308, 236–239 CrossRef CAS PubMed.
- P. Mansky, Y. Liu, E. Huang, T. P. Russell and C. J. Hawker, Science, 1997, 275, 1458–1460 CrossRef CAS.
- D. Kessler, N. Metz and P. Theato, Macromol. Symp., 2007, 254, 34–41 CrossRef CAS PubMed.
- K. L. Choy, Prog. Mater. Sci., 2003, 48, 57–170 CrossRef CAS.
- J. Lahann, D. Klee and H. Hocker, Macromol. Rapid Commun., 1998, 19, 441–444 CrossRef CAS.
- S. Thevenet, H. Y. Chen, J. Lahann and F. Stellacci, Adv. Mater., 2007, 19, 4333–4337 CrossRef CAS PubMed.
- X. W. Jiang, H. Y. Chen, G. Galvan, M. Yoshida and J. Lahann, Adv. Funct. Mater., 2008, 18, 27–35 CrossRef CAS PubMed.
- H. Y. Chen, M. Hirtz, X. P. Deng, T. Laue, H. Fuchs and J. Lahann, J. Am. Chem. Soc., 2010, 132, 18023–18025 CrossRef CAS PubMed.
- X. P. Deng, C. Friedmann and J. Lahann, Angew. Chem., Int. Ed., 2011, 50, 6522–6526 CrossRef CAS PubMed.
- J. Lahann, M. Balcells, T. Rodon, J. Lee, I. S. Choi, K. F. Jensen and R. Langer, Langmuir, 2002, 18, 3632–3638 CrossRef CAS.
- H. Nandivada, H. Y. Chen, L. Bondarenko and J. Lahann, Angew. Chem., Int. Ed., 2006, 45, 3360–3363 CrossRef CAS PubMed.
- D. B. Chrisey, A. Pique, R. A. McGill, J. S. Horwitz, B. R. Ringeisen, D. M. Bubb and P. K. Wu, Chem. Rev., 2003, 103, 553–576 CrossRef CAS PubMed.
- K. B. Shepard and R. D. Priestley, Macromol. Chem. Phys., 2013, 214, 862–872 CrossRef CAS PubMed.
- L. Stamatin, R. Cristescu, G. Socol, A. Moldovan, D. Mihalescu, I. Stamatin, I. N. Mihailescu and D. B. Chrisey, Appl. Surf. Sci., 2005, 248, 422–427 CrossRef CAS PubMed.
- R. Cristescu, I. N. Mihailescu, I. Stamatin, A. Doraiswamy, R. J. Narayan, G. Westwood, J. J. Wilker, S. Stafslien, B. Chisholm and D. B. Chrisey, Appl. Surf. Sci., 2009, 255, 5496–5498 CrossRef CAS PubMed.
- R. Modi, H. D. Wu, R. C. Y. Auyeung, C. M. Gilmore and D. B. Chrisey, J. Mater. Res., 2001, 16, 3214–3222 CrossRef CAS.
- K. K. Goli, O. J. Rojas, A. E. Ozcam and J. Genzer, Biomacromolecules, 2012, 13, 1371–1382 CrossRef CAS PubMed.
- M. Wahlgren and T. Arnebrant, Trends Biotechnol., 1991, 9, 201–208 CrossRef CAS.
- K. K. Goli, O. J. Rojas and J. Genzer, Biomacromolecules, 2012, 13, 3769–3779 CrossRef CAS PubMed.
- Z. F. Wu and P. Yang, Adv. Mater. Interfaces, 2015, 2, 1400401 Search PubMed.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. d'Ischia, A. Napolitano, V. Ball, C. T. Chen and M. J. Buehler, Acc. Chem. Res., 2014, 47, 3541–3550 CrossRef PubMed.
- J. Yang, M. A. C. Stuart and M. Kamperman, Chem. Soc. Rev., 2014, 43, 8271–8298 RSC.
- N. F. Della Vecchia, R. Avolio, M. Alfe, M. E. Errico, A. Napolitano and M. d'Ischia, Adv. Funct. Mater., 2013, 23, 1331–1340 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711–4717 CrossRef CAS PubMed.
- R. Rodriguez, M. A. Blesa and A. E. Regazzoni, J. Colloid Interface Sci., 1996, 177, 122–131 CrossRef CAS.
- T. H. Anderson, J. Yu, A. Estrada, M. U. Hammer, J. H. Waite and J. N. Israelachvili, Adv. Funct. Mater., 2010, 20, 4196–4205 CrossRef CAS PubMed.
- S. A. Mian, L. M. Yang, L. C. Saha, E. Ahmed, M. Ajmal and E. Ganz, Langmuir, 2014, 30, 6906–6914 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999–13003 CrossRef CAS PubMed.
- J. Yu, Y. J. Kan, M. Rapp, E. Danner, W. Wei, S. Das, D. R. Miller, Y. F. Chen, J. H. Waite and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15680–15685 CrossRef CAS PubMed.
- Y. R. Li, M. Qin, Y. Li, Y. Cao and W. Wang, Langmuir, 2014, 30, 4358–4366 CrossRef CAS PubMed.
- G. P. Maier, M. V. Rapp, J. H. Waite, J. N. Israelachvili and A. Butler, Science, 2015, 349, 628–632 CrossRef CAS PubMed.
- Y. M. Shin, Y. B. Lee, S. J. Kim, J. K. Kang, J. C. Park, W. Jang and H. Shin, Biomacromolecules, 2012, 13, 2020–2028 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, C. M. Preuss, B. Yameen, O. Pop-Georgievski, M. Bachmann, J. O. Mueller, M. Bruns, A. S. Goldmann, M. Bastmeyer and C. Barner-Kowollik, Adv. Mater., 2013, 25, 6123–6127 CrossRef CAS PubMed.
- T. S. Sileika, H. D. Kim, P. Maniak and P. B. Messersmith, ACS Appl. Mater. Interfaces, 2011, 3, 4602–4610 CAS.
- S. Q. Liu, C. Yang, Y. Huang, X. Ding, Y. Li, W. M. Fan, J. L. Hedrick and Y. Y. Yang, Adv. Mater., 2012, 24, 6484–6489 CrossRef CAS PubMed.
- L. Zhang, J. J. Wu, Y. X. Wang, Y. H. Long, N. Zhao and J. Xu, J. Am. Chem. Soc., 2012, 134, 9879–9881 CrossRef CAS PubMed.
- S. H. Ku, J. Ryu, S. K. Hong, H. Lee and C. B. Park, Biomaterials, 2010, 31, 2535–2541 CrossRef CAS PubMed.
- Z. L. Yang, Q. F. Tu, Y. Zhu, R. F. Luo, X. Li, Y. C. Xie, M. F. Maitz, J. Wang and N. Huang, Adv. Healthcare Mater., 2012, 1, 548–559 CrossRef CAS PubMed.
- C. Ozcan and N. Hasirci, J. Biomater. Sci., Polym. Ed., 2007, 18, 759–773 CrossRef CAS PubMed.
- Q. Wei, B. J. Li, N. Yi, B. H. Su, Z. H. Yin, F. L. Zhang, J. Li and C. S. Zhao, J. Biomed. Mater. Res., Part A, 2011, 96A, 38–45 CrossRef CAS PubMed.
- W. J. Yang, D. Pranantyo, K. G. Neoh, E. T. Kang, S. L. M. Teo and D. Rittschof, Biomacromolecules, 2012, 13, 2769–2780 CrossRef CAS PubMed.
- K. Yang, J. S. Lee, J. Kim, Y. B. Lee, H. Shin, S. H. Um, J. B. Kim, K. I. Park, H. Lee and S. W. Cho, Biomaterials, 2012, 33, 6952–6964 CrossRef CAS PubMed.
- K. Wang and Y. Luo, Biomacromolecules, 2013, 14, 2373–2382 CrossRef CAS PubMed.
- H. L. Wei, J. Ren, B. Han, L. Xu, L. L. Han and L. Y. Jia, Colloids Surf., B, 2013, 110, 22–28 CrossRef CAS PubMed.
- S. Hong, J. Kim, Y. S. Na, J. Park, S. Kim, K. Singha, G. I. Im, D. K. Han, W. J. Kim and H. Lee, Angew. Chem., Int. Ed., 2013, 52, 9187–9191 CrossRef CAS PubMed.
- S. M. Kang, J. Rho, I. S. Choi, P. B. Messersmith and H. Lee, J. Am. Chem. Soc., 2009, 131, 13224–13225 CrossRef CAS PubMed.
- E. Amstad, T. Gillich, I. Bilecka, M. Textor and E. Reimhult, Nano Lett., 2009, 9, 4042–4048 CrossRef CAS PubMed.
- Z. Shafiq, J. X. Cui, L. Pastor-Perez, V. San Miguel, R. A. Gropeanu, C. Serrano and A. del Campo, Angew. Chem., Int. Ed., 2012, 51, 4332–4335 CrossRef CAS PubMed.
- L. Garcia-Fernandez, J. X. Cui, C. Serrano, Z. Shafiq, R. A. Gropeanu, V. San Miguel, J. I. Ramos, M. Wang, G. K. Auernhammer, S. Ritz, A. A. Golriz, R. Berger, M. Wagner and A. del Campo, Adv. Mater., 2013, 25, 529–533 CrossRef CAS PubMed.
- P. Y. Sun, H. X. Lu, X. Yao, X. X. Tu, Z. Zheng and X. L. Wang, J. Mater. Chem., 2012, 22, 10035–10041 RSC.
- M. Kohri, H. Kohma, Y. Shinoda, M. Yamauchi, S. Yagai, T. Kojima, T. Taniguchi and K. Kishikawa, Polym. Chem., 2013, 4, 2696–2702 RSC.
- D. Hong, K. Bae, S. P. Hong, J. H. Park, I. S. Choi and W. K. Cho, Chem. Commun., 2014, 50, 11649–11652 RSC.
- H. Lee, K. D. Lee, K. B. Pyo, S. Y. Park and H. Lee, Langmuir, 2010, 26, 3790–3793 CrossRef CAS PubMed.
- F. Zhang, S. W. Liu, Y. Zhang, Z. G. Chi, J. R. Xu and Y. Wei, J. Mater. Chem., 2012, 22, 17159–17166 RSC.
- Y. K. Gong, L. P. Liu and P. B. Messersmith, Macromol. Biosci., 2012, 12, 979–985 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, R. C. Mutihac, P. L. M. Noeske, F. Paulus, R. Haag and I. Grunwald, Biomacromolecules, 2014, 15, 3061–3071 CrossRef CAS PubMed.
- J. L. Dalsin, L. J. Lin, S. Tosatti, J. Voros, M. Textor and P. B. Messersmith, Langmuir, 2005, 21, 640–646 CrossRef CAS PubMed.
- Q. Wei, S. Krysiak, K. Achazi, T. Becherer, P. L. M. Noeske, F. Paulus, H. Liebe, I. Grunwald, J. Dernedde, A. Hartwig, T. Hugel and R. Haag, Colloids Surf., B, 2014, 122, 684–692 CrossRef CAS PubMed.
- Q. Y. Lu, E. Danner, J. H. Waite, J. N. Israelachvili, H. B. Zeng and D. S. Hwang, J. R. Soc., Interface., 2013, 10, 20120759 CrossRef PubMed.
- J. H. Waite, Results Probl. Cell Differ., 1992, 19, 27–54 CAS.
- Q. Wei, K. Achazi, H. Liebe, A. Schulz, P. L. M. Noeske, I. Grunwald and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 11650–11655 CrossRef CAS PubMed.
- M. J. LaVoie, B. L. Ostaszewski, A. Weihofen, M. G. Schlossmacher and D. J. Selkoe, Nat. Med., 2005, 11, 1214–1221 CrossRef CAS PubMed.
- M. Calderon, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, P. L. M. Noeske, I. Grunwald and R. Haag, Adv. Mater., 2014, 26, 2688–2693 CrossRef CAS PubMed.
- J. Yu, W. Wei, E. Danner, R. K. Ashley, J. N. Israelachvili and J. H. Waite, Nat. Chem. Biol., 2011, 7, 588–590 CrossRef CAS PubMed.
- W. Wei, J. Yu, C. Broomell, J. N. Israelachvili and J. H. Waite, J. Am. Chem. Soc., 2013, 135, 377–383 CrossRef CAS PubMed.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., Int. Ed., 2013, 52, 10766–10770 CrossRef CAS PubMed.
- D. G. Barrett, T. S. Sileika and P. B. Messersmith, Chem. Commun., 2014, 50, 7265–7268 RSC.
- S. Hong, J. Yeom, I. T. Song, S. M. Kang, H. Lee and H. Lee, Adv. Mater. Interfaces, 2014, 1, 1400113 Search PubMed.
- J. R. Jeon, J. H. Kim and Y. S. Chang, J. Mater. Chem. B, 2013, 1, 6501–6509 RSC.
- W. E. Bentley and G. F. Payne, Science, 2013, 341, 136–137 CrossRef CAS PubMed.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. W. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- J. L. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. von Elverfeldt, C. E. Hagemeyer and F. Caruso, Angew. Chem., Int. Ed., 2014, 53, 5546–5551 CrossRef CAS PubMed.
Footnote |
| † Current address: Max-Planck Institute for Intelligent Systems, Stuttgart, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |