
Transient supramolecular reconfiguration of peptide nanostructures using ultrasound†
Charalampos G.
Pappas
ad,
Tapiwa
Mutasa
b,
Pim W. J. M.
Frederix
a,
Scott
Fleming
a,
Shuo
Bai
ad,
Sisir
Debnath
a,
Sharon M.
Kelly
c,
Anthony
Gachagan
b and
Rein V.
Ulijn
*ad
aWestCHEM, Department of Pure and Applied Chemistry, Thomas Graham Building, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: rein.ulijn@asrc.cuny.edu
bCentre for Ultrasonic Engineering, Department of Electronic & Electrical Engineering, University of Strathclyde, 204 George Street, Glasgow G1 1XW, UK
cInstitute of Molecular, Cell and Systems Biology College of Medical, Veterinary and Life Sciences, University of Glasgow, G128QQ, UK
dAdvanced Science Research Center (ASRC), City University of New York, 85 St Nicholas Terrace, New York NY10031, USA
First published on 3rd December 2014
Abstract
Ultrasound, i.e. high frequency oscillating pressure waves, is commonly used to overcome kinetic barriers associated with dissolution, assembly and gelation. We demonstrate that ultrasound energy may also be used to achieve transient reorganization of supramolecular nanostructures, which revert back to the original state when sound is switched off. Aromatic peptide amphiphiles, Fmoc-FL and -YL were used to study the transient acoustic response. These systems showed temporary supramolecular transitions that were sequence dependent. The changes observed were due to an altered balance between H-bonding and π-stacking, giving rise in changes in chiral organisation of peptide building blocks. Transient reconfiguration was visualized by TEM and changes in supramolecular interactions characterized by fluorescence, FT-IR and CD. Remarkably, significant differences are observed when compared to thermal heating, which relates to the oscillating and directional characteristics of ultrasound when delivering heat to a system.
Conceptual insightsFunctional nanoscale systems are becoming more complex and dynamic. Most laboratory-based self-assembled systems operate towards equilibrium. Dynamic self-assembly systems are reliant on energy input. Significant effort is devoted to the development of transient supramolecular systems, which show kinetics and life times that are dependent on energy input. The use of (ultra-) sound, i.e. oscillating pressure waves of varying frequency, provides substantial and largely unexplored opportunities to provide mechanical energy to direct transient nanostructure formation. The use of sound waves to transiently control supramolecular assembly is conceptually novel and may have broad applications in controlling and directing molecular self-assembly. |
Molecular self-assembly plays a key role in biological systems and also provides a versatile approach for materials fabrication.1–3 Molecular self-assembly can be controlled using a variety of stimuli including chemical4,5 and mechanical triggers.6,7 By definition, self-assembly systems operate in the direction that lowers their free energy. Non-equilibrium, transient nanostructures, that only exist away from thermodynamic equilibrium are increasingly of interest. Such systems require energy input to maintain a certain assembled state, which relaxes back to the un-assembled state when energy supply is stopped.8–10 (Ultra-) sound, i.e. oscillating pressure waves of varying frequency provides substantial and largely unexplored opportunities to provide mechanical energy to direct transient nanostructure formation.
The ability of pressure waves to help overcome energy barriers is well known e.g. to dissolve molecules or disperse particles by disrupting (non-specific) intermolecular interactions. For example, using low frequencies (manual shaking at approximately 2 Hz) may result in reversible sol–gel transitions.11 Audible sound frequencies (<1000 Hz) have been used to trigger anisotropy changes (fibre alignment).12 Ultrasound (>20 kHz) may also be used to control self-assembly and gelation processes, resulting in dramatic changes in nanoscale morphology and material properties.13–16 Recently, reversible sol–gel transitions using sonication of supramolecular metallopolymers was reported.17
Peptides are versatile building blocks in molecular self-assembly18–21 with recent reports revealing that ultrasound energy may be used to trigger supramolecular transitions, resulting from changes in supramolecular interactions. Mostly these studies used organic solvents,22–27 with some reports using aqueous media.28,29 Yokoi et al. reported the dynamic re-assembly of a 16 amino-acid residue peptide in water using sonication. They demonstrated that upon sonication the peptide nanostructure broke up into smaller fragments that recombine into nanofibers when sonication is switched off.30 Very recently, non-equilibrium dynamic simulations were used to investigate the effect of ultrasonic cavitation on amyloid fibre nucleation and disruption.31
Clearly, ultrasound may be used to influence nanostructure assembly and dis-assembly mechanisms. In here, we investigate whether sound waves may be used to trigger temporary supramolecular reconfiguration, and consequently change nanostructure morphology, which will dissipate when sound is switched off. We use aromatic peptide amphiphiles as model systems.32,33 These systems assemble as a result of a combination of aromatic stacking interactions and H-bonding, the balance between these two components has a dramatic impact on the nanoscale morphology observed. H-bonding and aromatic stacking have different thermal dependencies and therefore it seems reasonable that ultrasound as a directional means to supply heat may influence their reorganization.
The objectives of this study are therefore (i) to demonstrate the ability of ultrasound to achieve transient changes in materials properties of aromatic peptide amphiphiles (schematically shown in Scheme 1a); (ii) to investigate the relative impact on aromatic stacking and H-bonding interactions, by studying two closely related aromatic dipeptide amphiphiles with different electron densities; (iii) to compare acoustic response with thermal heating in transient supramolecular reorganisation.
 | ||
| Scheme 1 (a) Energy diagram of supramolecular transitions (b) chemical structures. | ||
We used a purpose built ultrasonic setup (it should be noted that most previous reports relied on commercial sonication baths or probes that may not allow for regulation of frequency and amplitude). In our system, an ultrasonic transducer provides high-energy frequencies (80 kHz) of precisely defined wavelength and amplitude. As shown in Fig. S1† the acoustic pressure was measured to be in a range of 0–120 kPa for 0–100 mV amplitude values. In this work, acoustic pressure of 120 kPa was used.
Two aromatic dipeptide amphiphiles with different sequences (fluorenyl-methoxycarbonyl-phenylalanine-leucine, Fmoc-FL and the equivalent tyrosine derivative, Fmoc-YL, Scheme 1b) were used. These are well suited for the current study because Fmoc-FL/-YL are clearly similar in structure, but have different electron density on the aromatic side chain residue (phenyl/phenol) giving rise to an altered relative contribution of aromatic and hydrogen bonding type interactions,34 with the phenyl/phenol substitution giving rise to more effective aromatic stacking. The resulting changes in supramolecular interactions upon thermal heating were investigated using temperature dependent 1H NMR Spectroscopy. NMR in the aromatic region shows increase in the multiplicity for Fmoc-YL upon thermal heating with peaks becoming sharper, indicating increased disorder at 338 K. By contrast, for Fmoc-FL the peaks become broader upon increasing temperature, suggesting increased order and stronger supramolecular interactions (Fig. S2†). Thus, aromatic stacking interactions increase upon heating for Fmoc-FL resulting in retention of gel state, in line with previous observations for other aromatic dipeptide amphiphiles, Fmoc-FG35 and Fmoc-LG.36 By contrast, heating disrupts aromatic interactions for Fmoc-YL, resulting in gel dissolution. These observations show that upon altering the electron density of the aromatic side chain residue (in this case, replacing phenyl by phenol in F and Y) the relative contribution of aromatic and hydrogen bonding type interactions, and therefore the thermal effects on molecular self-assembly can be regulated.34 We concluded that the Fmoc-YL/FL systems provides a suitable model system to investigate acoustic heating to control peptide assembly.
For the ultrasound experiments, 20 mmol of Fmoc-YL was dissolved in 1 ml sodium phosphate buffer (100 mM) pH 8 and heated up to 363 K for two minutes until fully dissolved. The sample was then left to cool to room temperature for 20 minutes. A translucent gel was obtained as evidenced by vial inversion. The Fmoc-dipeptide was then exposed to ultrasound during five minutes. Gradually, the gel converted to a solution and when the ultrasound was switched off, the gel reformed after 10 minutes. We noticed that during ultrasonication the temperature of water in the glass beaker rose to 320 K, which is considerably below the melting temperature of the gel (338 K), as discussed in more detail below. For Fmoc-FL 20 mmol was dissolved in 1 ml sodium phosphate buffer (100 mM) pH 6. An opaque gel phase material was observed after 10 minutes in this pH value, while a viscous suspension was observed at pH 8. In this case, ultrasound exposure during five minutes did not reveal observable macroscopic difference as the Fmoc-dipeptide was maintained in the gel phase. Therefore, macroscopically the effect of ultrasound is qualitatively comparable to that of heating for both peptides.
In order to acquire insights into the supramolecular transitions induced by ultrasound we firstly used fluorescence spectroscopy, focusing on the hydrophobic-stacking interactions among the fluorenyl moieties.37–40 Specifically, for Fmoc-YL in the (pre-sonication) gel state, two characteristic peaks were observed, a narrow band at 331 nm corresponds to the monomeric state and a broad red shifted band at 420–450 nm, corresponding to the aggregated state.37 After exposure to ultrasound (five minutes) and immediate transfer to the fluorimeter (<5 s) a 7 nm blue shift is observed for the main peak at 331 nm. This result demonstrates that there is a disruption of the stacking interactions during sonication. After 10 minutes in the absence of sonication, the emission intensity at 331 nm reverts back to the initial value (Fig. S3a†), suggesting stacking interactions among the aromatics are reformed (Fig. 1a). In the case of Fmoc-FL a peak appears at 348 nm, with a second peak covering the region from 415–450 nm. After ultrasound exposure during five minutes, a red shift is observed and a significant quenching for the emission for the peak at 348 nm, providing an indication that sound waves promote formation of extended stacking interactions among the fluorenyl moieties, that can be interpreted as a more ordered supramolecular structure. When the ultrasound was switched off, the emission increased (Fig. S3c†) back to the initial level (Fig. 1b). Fig. 1c highlights the fluorescence response of the main peak upon application of ultrasound, with Fmoc-YL showing increased emission (unquenching), and Fmoc-FL showing enhanced quenching. The effects are reversed as sound is switched off.
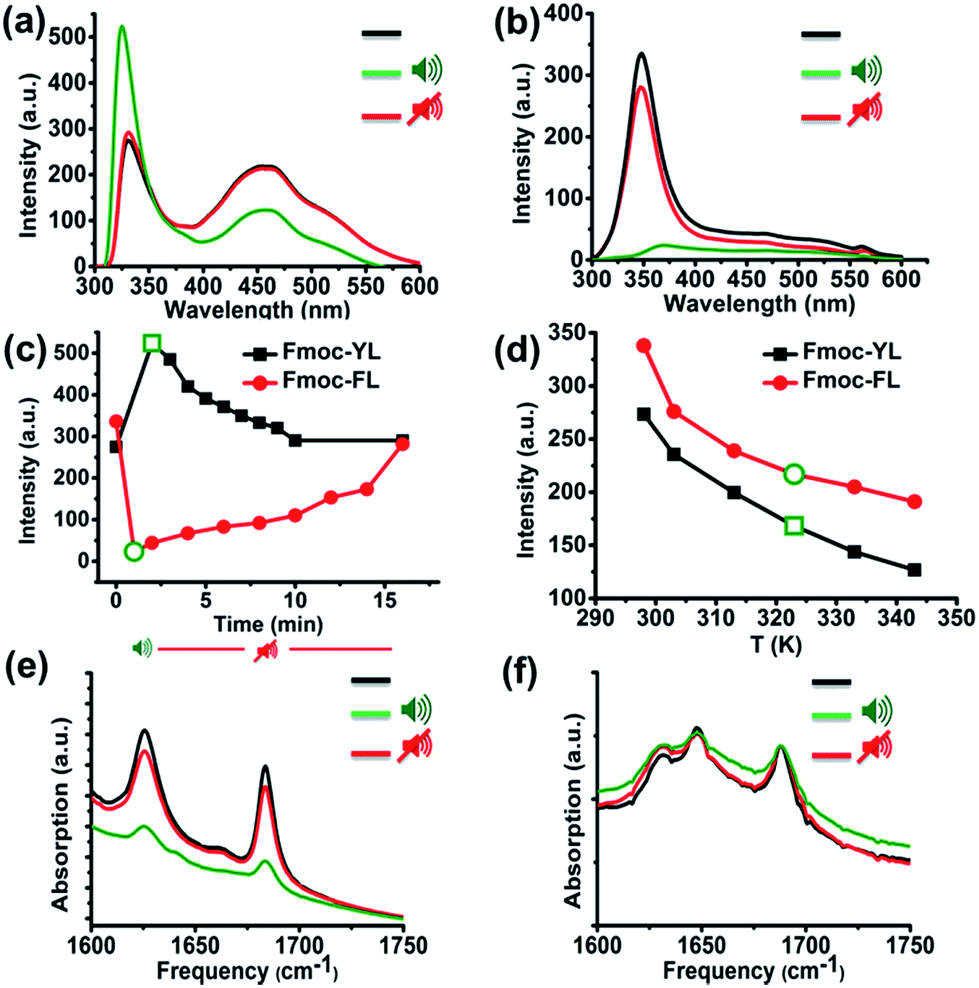 | ||
| Fig. 1 (a) and (b) Fluorescence emission spectra (excitation at 280 nm) of 20 mM Fmoc-YL in phosphate buffer pH 8 and 20 mM of Fmoc-FL in phosphate buffer pH 6 before, after 5 minutes ultrasound exposure and when the sound is switched off. (c) Plot of the fluorescence intensity of on/off ultrasound experiments (323–331 nm for Fmoc-YL) and (348–360 nm for Fmoc-FL). Green symbols indicate values immediately after sound exposure. (d) Plot of fluorescence intensity for Fmoc-YL and Fmoc-FL respectively upon thermal heating from 298–350 K. Green symbol represent the temperature observed upon sonication, for comparison. (e), (f) FT-IR of the Fmoc-YL in D2O pH 8 and Fmoc-FL in phosphate buffer pH 6 before, after 5 minutes ultrasound exposure and when the sound is switched off. | ||
Naturally, exposure to ultrasound energy results in heating of an exposed sample. To establish whether the observed effects can also be obtained by thermal heating, we compared the results with those obtained by thermal heating to the same temperature. Short exposure times of five minutes gave rise to heating of the sample from 298 to 320 K. While ultrasound induced a significant emission enhancement for the main peak at 331 nm, temperature dependent fluorescence experiments revealed quenching at the same temperature (320 K) and no blue-shifted structure was observed for Fmoc-YL. Temperature dependent fluorescence experiments for Fmoc-FL revealed that the emission at 348 nm also decreased but to a smaller extent (Fig. 1d). Clearly, the observed effects on supramolecular structures for ultrasound are different from those achieved using thermal heating.
Next, we investigated ultrasound-induced changes in hydrogen bonding type interactions via the amide groups of the backbone. Analysis by Fourier transform infrared (FT-IR) in D2O of 20 mM of Fmoc-YL pH 8 prior to ultrasonication, revealed two characteristic peaks in the amide I region. The peak at 1624 cm−1 shows formation of H-bonded type interactions via amide while 1682 cm−1 corresponds to the stacked carbamate.41 After ultrasound exposure during five minutes, both of the peaks transiently showed a reduction in intensity, indicating that ultrasound energy disrupts the H-bonds. When the ultrasound was switched off, there is reappearance of the hydrogen bonding and the intensity of the β-sheet like peak was close to the initial values (Fig. 1e). FT-IR spectra of 20 mM of Fmoc-FL in D2O pH 6 prior to sonication revealed 3 characteristic peaks. Peaks at 1624 cm−1 and 1682 cm−1 as previously observed for Fmoc-YL, but also one peak at 1648 cm−1 suggesting that there is also substantial unstructured peptide in these structures. Notably, sound irradiation for five minutes showed a minor reduction in intensity (Fig. 1f) suggesting that ultrasound reduced the H-bonding intensity for both systems.
Having established the relative importance of the aromatic interactions (enhanced in Fmoc-FL, reduced in Fmoc-YL) and hydrogen bonding (reduced in both) we used Circular Dichroism spectroscopy (CD) to investigate the ultrasound effect on the chiral organization of the supramolecular self-assembly. On self-assembly of aromatic dipeptide amphiphiles, CD signals derive from the supramolecular chirality, induced by the chiral organization of chromophores rather than the molecular chirality of the chromophores themselves.37 For Fmoc-YL, a negative signal was observed for the Fmoc peak (303 nm). After ultrasound exposure for five minutes no CD signal was observed as previously observed for micellar aggregates.42 The chiral organization was restored when the ultrasound was switched off (Fig. 2a). For Fmoc-FL initially a weak negative peak was observed at 307 nm. After 5 minutes ultrasound exposure a strong negative peak was noticed, indicating that supramolecular chirality can be induced by ultrasound. The temporary enhanced CD signal of the supramolecular organization was reduced when the sound was switched off (Fig. 2b). Temperature dependent CD experiments for both of the aromatic dipeptide amphiphiles show that similar disruption or enhancement of the chirality can be achieved thermally, but at significantly higher temperatures (Fig. S4 and S5†).
 | ||
| Fig. 2 (a) and (b) Circular dichroism spectra of 20 mM Fmoc-YL in phosphate buffer pH 8 and 20 mM of Fmoc-FL in phosphate buffer pH 6 before, after 5 minutes ultrasound exposure and when the sound is switched off. | ||
The ultrasound induced transient supramolecular reconfiguration was further investigated by TEM (Transmission Electron Microscopy). As reported previously Fmoc-YL assembled into fibers (Fig. 3a).37 Upon ultrasound exposure during five minutes the fibrillar network was temporarily disrupted, showing the formation of spherical aggregates of approximately 300 nm in size (Fig. 3b). For the ultrasonic experiments three drops of the solution was drop casted immediately onto the carbon-coated copper grid and allowed to dry for few minutes at RT before imaging. When the ultrasound was switched off, the Fmoc-YL reassembled into a fibrillar network (Fig. 3c). A second ultrasonication cycle revealed the same transient supramolecular reconfiguration (Fig. S6a and b†). The results appear to be in agreement with a reconfiguration schematically shown in Scheme 1: ultrasound exposure results in disruption of the H-bonded chiral nanofiber structure to form achiral spherical aggregates.
 | ||
| Fig. 3 (a)–(c) TEM images of Fmoc-YL prior to, and following 5 minutes ultrasound exposure and when the ultrasound is off and (d)–(h) TEM images of Fmoc-FL prior to, and following five minutes ultrasound exposure and when the ultrasound is off. Scale bar 200 nm. | ||
In the case of Fmoc-FL initially tapes were observed (Fig. 3d). After 5 minutes of ultrasound irradiation the Fmoc-dipeptide adopted twisted fibrillar architecture (Fig. 3e). When the ultrasound was switched off the initial organization state was reformed (Fig. 3f). The same temporary supramolecular reconfiguration may be achieved by a second ultrasonication cycle (Fig. S6c and d†). In this case the dominance of the directional aromatic stacking interactions retains a fibre morphology, with the reduced H-bonding upon sonication favouring a more twisted orientation.
Conclusions
In summary, we have demonstrated that in aqueous buffer solution ultrasound energy may have a temporary effect on supramolecular self-assembly of aromatic dipeptide amphiphiles showing supramolecular reconfiguration from tapes to coiled fibers (Fmoc-FL) and straight fibers to spherical aggregates (Fmoc-YL) respectively, reverting back to the initial organization state as the sound is switched off. In the case of Fmoc-YL ultrasound energy disrupts the fibrillar network, giving rise to the formation of spherical aggregates, where the Fmoc-dipeptide adopted a less ordered organized structure accompanied with a reduction of fluorenyl order and H-bonding. For Fmoc-FL, sound waves enhance the stacking interactions among the fluorophores, while having a less effect on hydrogen bonding. The work shows that ultrasound energy may be used to temporarily modulate the interplay between hydrophobic/H-bonding interactions. These insights provide a step towards a rational use of ultrasound to fuel transient, energy dissipating supramolecular systems.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/EMERgE/ERC Grant Agreement no. (258775). CGP would like to thank Linn Products Ltd for funding.Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- G. Fichman and E. Gazit, Acta Biomater., 2014, 10, 1671–1682 CrossRef CAS PubMed.
- P. Jonkheijm, P. van der Schoot, A. P. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed.
- G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437–442 CrossRef CAS PubMed.
- J. M. Carnall, C. A. Waudby, A. M. Belenguer, M. C. Stuart, J. J. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed.
- S. Bai, C. Pappas, S. Debnath, P. W. Frederix, J. Leckie, S. Fleming and R. V. Ulijn, ACS Nano, 2014, 8, 7005–7013 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, K. N. Kowlgi, G. J. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed.
- S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789–16792 CrossRef CAS PubMed.
- S. C. Warren, O. Guney-Alta and B. A. Grzybowski, J. Phys. Chem. Lett., 2012, 3, 2103–2111 CrossRef CAS.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- A. Tsuda, Y. Nagamine, R. Watanabe, Y. Nagatani, N. Ishii and T. Aida, Nat. Chem., 2010, 2, 977–983 CrossRef CAS PubMed.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- G. Cravotto, E. C. Gaudino and P. Cintas, Chem. Soc. Rev., 2013, 42, 7521–7534 RSC.
- T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127, 9324–9325 CrossRef CAS PubMed.
- S. Zhang, S. Yang, J. Lan, Y. Tang, Y. Xue and J. You, J. Am. Chem. Soc., 2009, 131, 1689–1691 CrossRef CAS PubMed.
- D. W. Balkenende, S. Coulibaly, S. Balog, Y. C. Simon, G. L. Fiore and C. Weder, J. Am. Chem. Soc., 2014, 136, 10493–10498 CrossRef CAS PubMed.
- L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2008, 41, 1674–1684 CrossRef CAS PubMed.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- D. Bardelang, F. Camerel, J. C. Margeson, D. M. Leek, M. Schmutz, M. B. Zaman, K. Yu, D. V. Soldatov, R. Ziessel, C. I. Ratcliffe and J. A. Ripmeester, J. Am. Chem. Soc., 2008, 130, 3313–3315 CrossRef CAS PubMed.
- Y. Wang, C. Zhan, H. Fu, X. Li, X. Sheng, Y. Zhao, D. Xiao, Y. Ma, J. S. Ma and J. Yao, Langmuir, 2008, 24, 7635–7638 CrossRef CAS PubMed.
- D. Ke, C. Zhan, A. D. Li and J. Yao, Angew. Chem., Int. Ed., 2011, 50, 3715–3719 CrossRef CAS PubMed.
- M. M. Zhang, L. Y. Meng, X. H. Cao, M. J. Jiang and T. Yi, Soft Matter, 2012, 8, 4494–4498 RSC.
- X. Yu, Q. Liu, J. Wu, M. Zhang, X. Cao, S. Zhang, Q. Wang, L. Chen and T. Yi, Chem.–Eur. J., 2010, 16, 9099–9106 CrossRef CAS PubMed.
- B. Adhikari and H. B. Kraatz, Chem. Commun., 2014, 50, 5551–5553 RSC.
- L. Sambri, F. Cucinotta, G. De Paoli, S. Stagni and L. De Cola, New J. Chem., 2010, 34, 2093–2096 RSC.
- S. F. Pan, S. Luo, S. Li, Y. S. Lai, Y. Y. Geng, B. He and Z. W. Gu, Chem. Commun., 2013, 49, 8045–8047 RSC.
- H. Yokoi, T. Kinoshita and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414–8419 CrossRef CAS PubMed.
- H. Okumura and S. G. Itoh, J. Am. Chem. Soc., 2014, 136, 10549–10552 CrossRef CAS PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- C. G. Pappas, Y. M. Abul-Haija, A. Flack, P. W. Frederix and R. V. Ulijn, Chem. Commun., 2014, 50, 10630–10633 RSC.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS PubMed.
- C. Tang, R. V. Ulijn and A. Saiani, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 111 CrossRef PubMed.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- J. Raeburn, B. Alston, J. Kroeger, T. O. McDonald, J. R. Howse, P. J. Cameron and D. J. Adams, Mater. Horiz., 2014, 1, 241–246 RSC.
- D. M. Ryan, T. M. Doran, S. B. Anderson and B. L. Nilsson, Langmuir, 2011, 27, 4029–4039 CrossRef CAS PubMed.
- S. Fleming, P. W. J. M. Frederix, I. R. Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510–9515 CrossRef CAS PubMed.
- J. W. Sadownik, J. Leckie and R. V. Ulijn, Chem. Commun., 2011, 47, 728–730 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mh00223g |
| This journal is © The Royal Society of Chemistry 2015 |