
Carbon materialization of ionic liquids: from solvents to materials
Shiguo
Zhang
,
Kaoru
Dokko
and
Masayoshi
Watanabe
*
Department of Chemistry and Biotechnology, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: mwatanab@ynu.ac.jp
First published on 13th November 2014
Abstract
Carbon materials have been extensively used in diverse areas, especially in energy-related applications. Traditionally, these materials have been synthesized by carbonization of low-vapor-pressure natural or synthetic polymers. However, the polymer-related procedures are multistep and time consuming because of the limited solubility and complicated synthesis of polymers. Recently, ionic liquids (ILs), composed of entirely cations and anions, have emerged as a new family of carbon precursors. The carbon-rich nature of ILs, coupled with their attractive properties such as diverse cation–anion combinations, low volatilities, and high thermal stabilities, not only greatly simplifies the entire carbonization process, but also gives rise to carbons with attractive features, which are distinct from those of carbons obtained using conventional polymer precursors, such as very high nitrogen contents and conductivities. In this review, we highlight recent approaches to the preparation of carbon materials using ILs as versatile precursors. We begin with a brief introduction to these novel precursors, discussing the key structures and properties of ILs that enable successful carbonization, and then address synthetic techniques for the fabrication of advanced porous carbons from ILs by either self- or external-template methods, followed by a review of the potential applications of ionic-liquid-derived carbons such as electrocatalysis, Li-ion batteries, supercapacitors, CO2 capture and chemical catalysis. The review concludes with an overview of possible directions for future research in this field.
 Shiguo Zhang | Shiguo Zhang received his Bachelor's degree in Chemistry from Hunan Normal University in 2004. He received his PhD degree in 2010 in Physical Chemistry from the Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences (CAS), and worked as an assistant researcher in LICP from 2010 to 2012. In April 2012, he joined the group of Prof. Masayoshi Watanabe as a postdoctoral at Yokohama National University. His research interests focus on the design and synthesis of nanostructured carbon materials and their applications in energy conversion and storage. |
 Kaoru Dokko | Kaoru Dokko is an Associate Professor of Yokohama National University. He received his PhD degree from Tohoku University in 2001 under the supervision of Prof. Isamu Uchida. He then did his postdoctoral research at Case Western Reserve University with Prof. Daniel A. Scherson and at Tokyo Metropolitan University with Prof. Kiyoshi Kanamura, and joined Yokohama National University in 2008. His current research interests are in the physicochemical properties of concentrated electrolytes, nanostructured materials for energy conversion and storage, and electrochemical reaction processes in electrochemical devices. |
 Masayoshi Watanabe | Masayoshi Watanabe is a Professor of Yokohama National University. He received his PhD degrees from Waseda University in 1983. After working as a visiting scientist with Prof. Royce W. Murray at the University of North Carolina (1988–1990), he joined Yokohama National University in 1992 and was promoted to a full Professor in 1998. He received the Lecture Award for Young Scientists from the Chemical Society of Japan in 1991, the Award for Creative Work from the Electrochemical Society of Japan in 2006, the Award of the Society of Polymer Science, Japan in 2006, and Distinguished Research Award of Yokohama National University in 2012. His research interest has been mainly concerned with “ionics” and “nano-structured materials” for chemical energies such as fuel cells and lithium-ion and lithium–sulfur batteries. He has published more than 300 original research papers and 150 books and reviews in these and related fields; cited more than 14 |
1. Introduction
Ionic liquids (ILs) have been a source of interest to scientists and engineers for many decades.1–4 In particular, the unusual combination of a wide liquid range, substrate-reactant solubility, ionic conductivity, low volatility, and high electrochemical and thermal stabilities has led to an explosion in their use as green, ionic solvents for a host of processes linked to green chemistry and clean technology, and as ideal electrolytes for a number of energy-related applications such as batteries,5 fuel cells,6 supercapacitors,7 and dye-sensitized solar cells.1,2 In contrast, the use of ILs in materials design and synthesis remains less explored, although several novel applications of IL-based fluids and materials as thermal, magnetic, and optical fluids,8–10 lubricants,11 and propellants12 have received more attention in recent years (Fig. 1).3,4 | ||
| Fig. 1 Ionic liquids as novel precursors for carbon materials. | ||
On the other hand, because of their high surface areas, electrical conductivities, low costs, and excellent chemical, mechanical, and thermal stabilities, carbon materials have been extensively used in diverse areas such as environmental treatment, catalysis, sensing, separation, gas capture/storage, and, particularly, as electrode materials or electrocatalysts for energy conversion/storage.13–18 Carbon materials are generally prepared by carbonization of carbon-containing precursors at high temperature. The nature of the organic precursor is crucial to the preparation process (e.g., interactions between the precursor and template19), and the structures, properties, and performances of the final carbons. The chemical compositions of the precursors are often reflected in the carbon structure, and strongly affect characteristics such as the carbon yield, porosity, surface area, graphitization, conductivity, and heteroatom doping. Most organic compounds are completely evaporated or decomposed to gaseous products during high-temperature carbonization, therefore currently available precursors are rather limited, and are mainly low-vapor-pressure natural or synthetic polymers.20 However, polymer-related procedures are multistep and time consuming. Moreover, for carbons derived from “hard carbon” precursors such as sucrose and furfuryl alcohol, the graphitic structure and nitrogen content, which account for high conductivity, are always insufficient for task-specific applications.20 To meet the requirements of most applications, great efforts have therefore been devoted to the development of novel precursors.
Let us return to ILs. The cation–anion combinations of ILs, coupled with their carbon-rich nature, make them potential candidates for carbonaceous precursors with the following merits. (a) The intrinsic ionic nature and strong coulombic interactions between the constituent ions give rise to negligible vapor pressure and high thermal stability; these reduce mass loss before the beginning of the decomposition process and are favorable for carbonization processes without applied pressure. (b) Carbonization of small-molecular precursors is easy to handle compared with polymers that have limited solubility and require complicated syntheses, simplifying the overall carbonization process, and reducing the preparation time and cost. (c) The nitrogen-containing motifs of ILs incorporate carbons with homogeneously distributed nitrogens in situ, without any doping agents. (d) The liquid state under ambient conditions facilitates the production of seamless continuous carbon films. (e) The interactions between the polar IL precursor and the polar surfaces of inorganic materials may assist successful fabrication of advanced carbon materials such as hollow porous carbon or nitrogen-doped carbon-coated materials. (f) The structural diversity of ILs offers additional possibilities for easily controlling the structures and properties of carbon materials at the molecular level.21,22
Based on an analysis of the previous literature, including the most recent articles, the present review is an attempt to highlight recent progress in the study of IL-based carbon precursors. This review is divided into the following sections. First, we will briefly introduce the recent development in the synthesis of carbon materials from ILs, and the IL-based precursors will be roughly classified into five types, i.e., cyano/nitrile-containing ILs, conventional aprotic ILs, poly(ionic liquid)s (poly-ILs), deep-eutectic solvents (DESs), and protic ILs (PILs) and protic salts (PSs); these are then discussed separately. For each class, the structural and physicochemical correlations between the original precursors and resultant carbon, if available, will be concisely reviewed, in terms of the carbonization mechanism and characteristics of the carbon such as carbon yield, conductivity, heteroatom doping, and graphitic structure. Secondly, the use of ILs as surface-coating agents, dopants, and media for the synthesis of carbon materials will be reviewed. Thirdly, synthesis of metal-doped carbons from ILs will be discussed. Fourthly, strategies for the fabrication of porous carbons from ILs such as template-free, nanocasting methods, mainly based on silica and salt templates, will be discussed. Fifthly, the potential applications of IL-derived carbons such as electrocatalysis, supercapacitors, gas adsorption, Li-ion batteries (LIBs), and chemical catalysis are demonstrated. Finally, the challenges and prospects for future research on the development of IL-based carbons are discussed.
2. Ionic liquids as carbon precursors
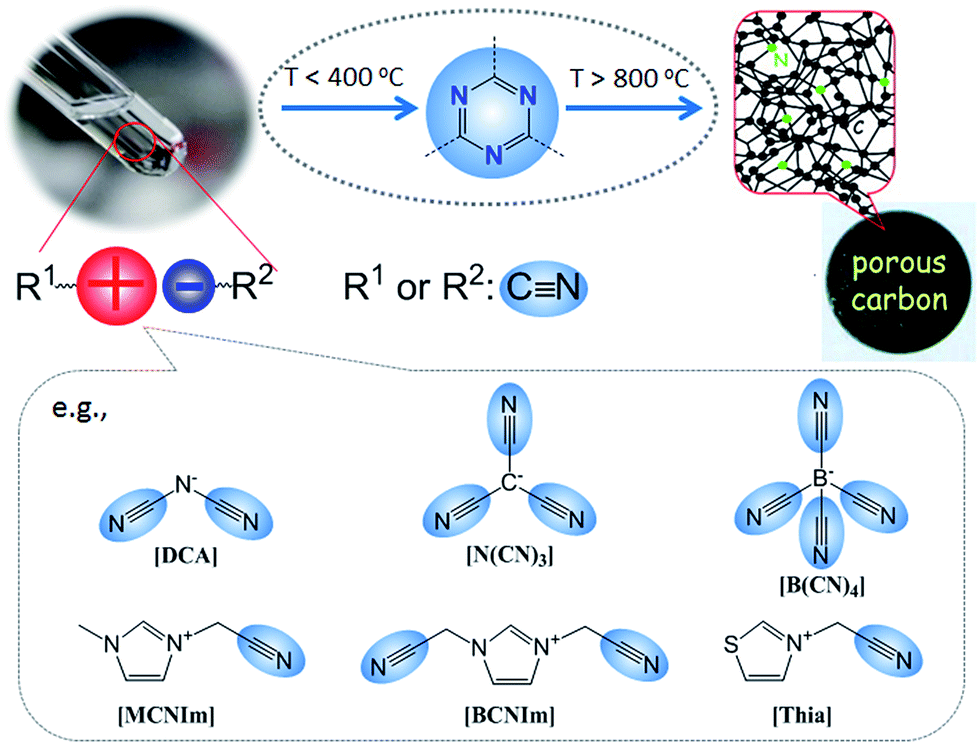
2.1. Cyano/nitrile-containing ILs
Despite the attractive features of IL-based organic carbon precursors, a question still arises: Can their high thermal stability make ILs sufficiently robust to endure harsh carbonization conditions and ensure carbon yields as substantial as those obtained using polymer precursors? The answer is “yes”, but the thermal stability and the extent of decomposition depend strongly on the IL structure. In the very early study performed by MacFarlane and co-workers,23 the thermogravimetric (TG) profiles of dicyanamide ([DCA])- or tricyanomethide ([C(CN)3])-based ILs showed that residual yields remained up to 500 °C. This interesting phenomenon was further investigated in detail by Scott et al.24 They found that the extent of decomposition of the IL depends on the structures of both the cation and anion. ILs with nitrogen-containing cations and [DCA] or [C(CN)3] anions do not decompose completely to volatile products, yielding an intractable char, but phosphonium-based ILs with [DCA] anion and ILs containing the [NTf2] anion decompose completely to volatile products in a single step, leaving no significant residues. Unfortunately, the obtained chars were not investigated in detail, and the pyrolysis temperature was too low to produce fully carbonized samples.The first real connection between ILs and carbon materials was discovered in 2009 by Dai et al.25 Through the synergistic use of the negligible vapor pressures of ILs and incorporation of cross-linkable functional groups into the cationic structures, they developed a high-yield and solvent-free route to nitrogen-rich carbons via direct, atmospheric-pressure carbonization of task-specific imidazolium ILs bearing one or more nitrile side chains ([MCNIm] and [BCNIm], Fig. 2), without any other treatments. Although no carbon yield was achieved from the attempted carbonization of [BMIm][NTf2], in agreement with Scott's conclusion,24 significant yields were observed for all nitrile-functionalized ILs, with the thermal stability depending strongly on the nature of the anion. The resulting textural properties such as pore structure and surface area are highly dependent on the structural motifs of the ions. The carbon yields at 800 °C are very close to the theoretical values, suggesting nearly quantitative transformation of the overall carbon contents of the precursors. Uniform carbon films can be routinely deposited using this novel method, by virtue of the liquid nature.
 | ||
| Fig. 2 Direct synthesis of nitrogen-doped carbon from specific nitrile/cyano-containing IL precursors under ambient pressure. | ||
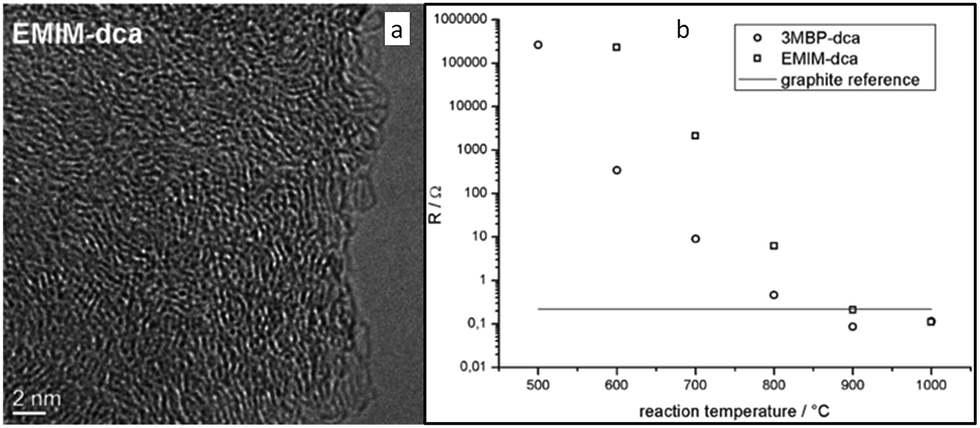 | ||
| Fig. 3 (a) High-resolution transmission electron microscopy (HRTEM) image of the nitrogen-doped carbon material derived from [EMIm][DCA] at 900 °C. (b) Electrical resistances of samples measured using electrical impedance spectroscopy. [Reprinted with permission from ref. 26, copyright 2010 Wiley.] | ||
Nearly parallel with the work done by Dai et al., Antonietti et al. investigated the carbons obtained via direct carbonization of [DCA]-anion-based ILs in detail.26 One distinctive feature of [DCA]-based ILs is their high nitrogen contents; for example, the N/C molar ratio of [EMIm][DCA] (Fig. 2) is 0.63 (39.5 wt% N), which is expected to give highly nitrogen-doped carbons. The resultant carbon is indeed featured by a very high nitrogen content, up to 10.4 wt% at 1000 °C.26 Combining this IL with natural nitrogen-rich nucleobases for carbonization further improves the nitrogen content to 12.0 wt%; this ultrahigh nitrogen content is the highest value for a one-step synthesis of nitrogen-doped carbon reported to date.27 This high nitrogen content is important, considering that nitrogen doping in the carbon lattice can significantly alter or improve the physicochemical properties of carbon materials, such as their conductivity, basicity, oxidative stability, and catalytic activity, for potential applications.28–30 Another important property of carbons derived from [DCA]-based ILs is a partially developed graphite-like structure. As shown in Fig. 3, the materials are featured by graphitic order with bent layer planes and weak edge termination; this and their intrinsic high nitrogen content result in high conductivities, even higher than that of graphite (Fig. 3).26 After these pioneering studies, a series of ILs that are structurally close to well-understood IL precursors, that is, either the cation or anion contains a cyano/nitrile group (Fig. 2), were synthesized and used as carbon precursors.31–34
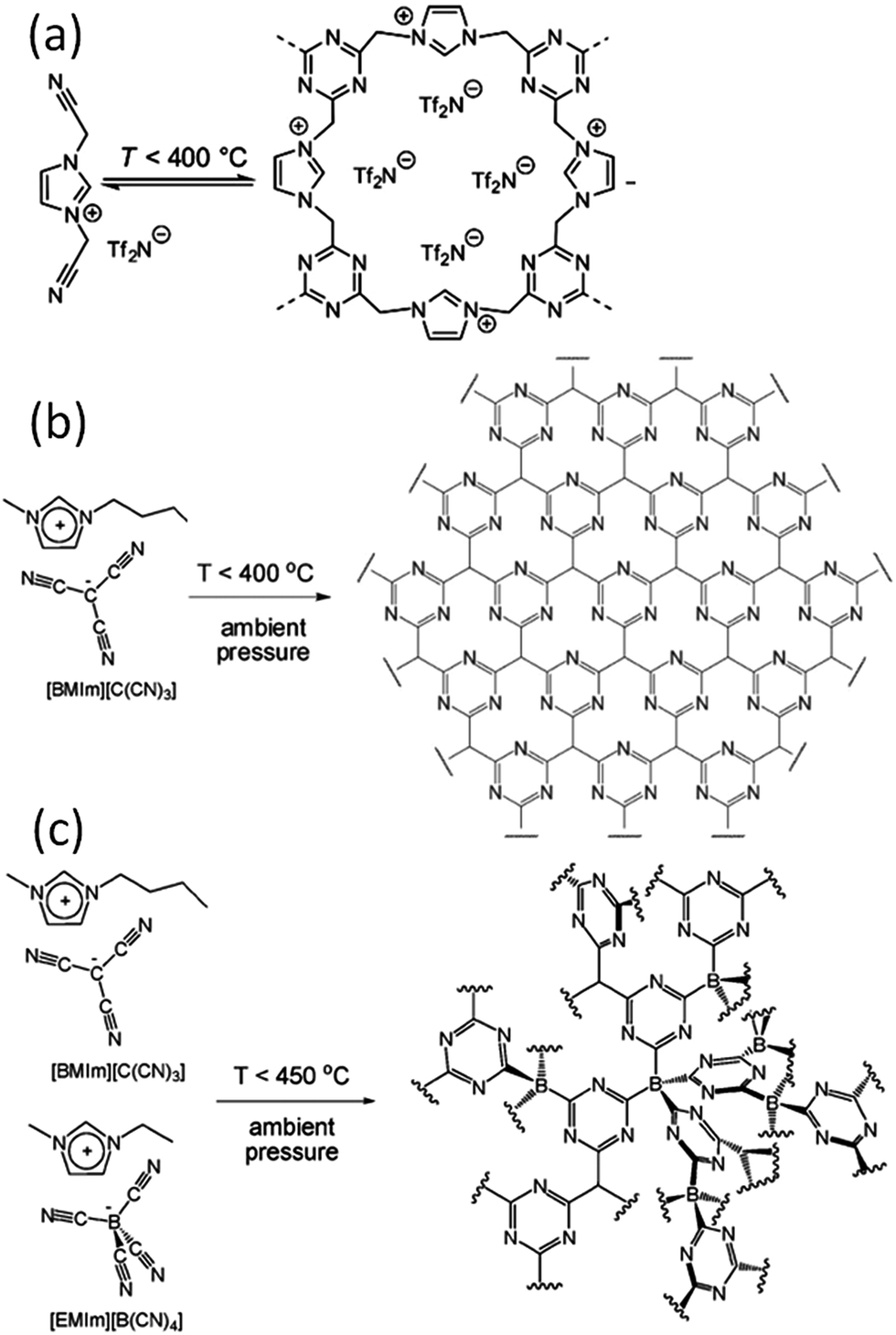 | ||
| Fig. 4 Proposed reaction scheme for trimerization of the nitrile-containing anion, leading to formation of an extended two- or three-dimensional framework. [Panel (a) reprinted with permission from ref. 25, copyright 2009 American Chemical Society. Panel (b) reprinted with permission from ref. 31, copyright 2010 Wiley. Panel (c) reproduced from ref. 41 with permission from the PCCP Owner Societies.] | ||
Based on TG-MS analysis,35 the carbonization of [DCA]-based ILs can be divided into three major steps: at around 300 °C, decomposition is initiated by the cleavage of alkyl chains, elimination of ammonia, and other reactions at the lower-molecular level. This is followed by condensation of the material by trimerization of cyano groups, and aromatic condensation, up to approximately 500 °C. On further increasing the temperature to 1000 °C, the reaction is dominated by condensation of the entire system, with elimination of hydrogen and nitrogen, giving rise to the graphitic domains. The role of cyano functional groups is therefore highly important, permitting the first and essential step of formation of a resin that is preorganized and thermostable. As shown in Fig. 2, this reaction proceeds at lower temperatures (ca. 400 °C) via the formation of dynamic amorphous polytriazine networks,36–38 depressing the evaporative loss of intermediate volatile species and ensuring subsequent carbonization at higher temperatures. Based on the intermediate triazine-based charged structure formed during pyrolysis of cyano-containing ILs, a new class of microporous polymers with ion-exchange capabilities was developed by pyrolysis of specific ILs with bisnitrile-functionalized cations, for the adsorption of perrhenate ions in aqueous solution (Fig. 4a).39 The successful carbonization of ILs based on cross-linking reactions of cyano moieties was further proved by the synthesis of nitrogen-enriched carbons from alkali salts containing [C(CN)3] anions.40 It should be noted that compared with the cations, which are typically based on an imidazolium backbone, the anions are more easily designed to bear multiple cross-linkable components such as two nitrile groups in [DCA], three nitrile groups in [C(CN)3], and four nitrile groups in tetracyanoborate ([B(CN)4]). In particular, [B(CN)4] is expected to be more favorable for the formation of three-dimensional connected intermediate carbonaceous frameworks (Fig. 4).
Multiple doping is a promising method for functionalizing carbon materials. The presence of heteroatoms other than nitrogen, such as boron and sulfur, in the cationic or anionic backbone facilitates the synthesis of codoped carbon from ILs.33,34,41–43 For example, an [EMIm][B(CN)4] IL containing boron, carbon, and nitrogen was investigated as a direct precursor for the synthesis of boron- and nitrogen-rich carbons or boron carbon nitrides at elevated temperatures.33 [EMIm][B(CN)4] decomposes via an unusual process, with thermal stability above 400 °C, which could be related to the low nucleophilicity and low basicity of the anion. At temperatures above 1000 °C, the product starts to become a graphitic boron carbon nitride (BCN) with a weakly ordered structure and a B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N ratio of 1:1. The final material, prepared at 1400 °C, has a composition close to BC3N. In the corresponding reference samples of nitrogen-doped carbon from [DCA]-based ILs, the nitrogen content decreases significantly with increasing temperature, therefore the presence of boron in [EMIm][B(CN)4] stabilizes the nitrogen within the structure and the BCN units rearrange to a more stable aromatic arrangement. The electrical resistivity in the temperature range of 0–950 °C reflects the semiconducting behavior of the BCN, and its electrical resistivity is only 1.75 times less than that of nitrogen-doped carbon.
:N ratio of 1:1. The final material, prepared at 1400 °C, has a composition close to BC3N. In the corresponding reference samples of nitrogen-doped carbon from [DCA]-based ILs, the nitrogen content decreases significantly with increasing temperature, therefore the presence of boron in [EMIm][B(CN)4] stabilizes the nitrogen within the structure and the BCN units rearrange to a more stable aromatic arrangement. The electrical resistivity in the temperature range of 0–950 °C reflects the semiconducting behavior of the BCN, and its electrical resistivity is only 1.75 times less than that of nitrogen-doped carbon.
The presence of sulfur atoms in cations, instead of anions, with nitrile functionalities should alter the carbonization processes in a way that allows the incorporation of not only nitrogen, but also sulfur, into the developing carbonaceous material. For example, direct carbonization of nitrile-functionalized thiazolium-based ILs produces carbons that are intrinsically codoped with nitrogen and sulfur heteroatoms.34 After carbonization at 1000 °C, the nitrogen and sulfur contents are as high as 8.75% and 6.98%, respectively. The powder X-ray diffraction (XRD) patterns of the obtained carbons have prominent diffraction lines at 2θ values between 24° and 26°, representing the typical (002) reflection for graphite-like stacking. Nitrogen and sulfur codoped carbons were also obtained by one-step carbonization of precursor mixtures of common [DCA]-based ILs and an IL containing an isothiocyanate counter-ion ([EMIm][SCN]).42 The sulfur content is easily tunable in the range of 1.5–3 wt% via varying the initial amount of [EMIm][SCN]. It is worth mentioning that direct carbonization of pure [EMIm][SCN] yields negligible amounts of carbon (5 wt% at 800 °C and 0% at 1000 °C).42 However, this is to be expected, as the thiocyanate trimerizes to thiocyanuric acid, which does not form extended polymer networks, but is sublimated instead.
2.2. Conventional ionic liquids
The strategy of direct carbon formation from ILs generally only works for a few, expensive, and, in particular, cyano/nitrile-containing ILs,24,25,31 whereas conventional aprotic ILs without any cross-linkable functional groups under nitrogen and ambient pressure afford no carbon, because of the lack of stable intermediate polymeric structures and the eventual formation of volatile vapors during thermal decomposition of such ILs.24,25 Several strategies have been used to deal with this issue. The first approach is to trap conventional ILs in a nanoconfined framework, which is expected to affect pyrolysis and increase aromatization. Similar to the observed abnormal thermal transitions of nanoconfined ILs compared with those of bulk ILs,44,45 the confinement of ILs makes the impossible possible, i.e., modifies the carbonization of usually non-carbonizable ILs. For example, introducing the same IL, 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([BMIm][NTf2]), which gives no char residue in the bulk phase into an oxide framework via an in situ non-hydrolytic sol–gel process affords improved carbonization yields (Fig. 5).43 In addition, by means of the unique solvation environment offered by ILs through coulombic interactions and nanoscopic phase separation, monolithic silica gels with homogeneously dispersed ILs are liable to produce uniform meso/macropores originating from those of the parent silica–carbon composite, the spaces left by removal of silica, and the interparticle pores between primary carbon particles. The resultant carbon exhibits a surface area of 469 m2 g−1.43 It is believed that aromatization through dehydrogenation reactions is considerably enhanced, and evaporative loss of intermediate species (other than hydrogen gas) is suppressed by the combined effects of nanoconfinement and oxide-catalyzed carbonization. The good energetic interactions between the polar IL precursor and the polar surface of the inorganic template may additionally assist carbonization,26,46,47 as it was reported that commonly non-carbonizable ILs such as [C8MIm][Cl], [RMIm][Br], [EMIm][NTf2], and [C4mpyr][NTf2] yielded carbons through carbonization in the presence of inorganic materials such as Fe3O4, LiFePO4, and Li3V2(PO4)3 nanoparticles, or templates such as monodisperse silica and eutectic salts.47–53 This confinement-induced carbonization opens the way to carbon synthesis from diverse conventional ILs. The intrinsic nitrogen-containing natures and relatively low carbon yields of such ILs are favorable for the formation of a thin, uniform, conductive, nitrogen-doped carbon layer on the particle surface. In particular, ILs with relatively long carbon chains facilitate adhesion to, and encapsulation of, the inorganic surface, leading to the formation of hollow carbon spheres (HCSs).47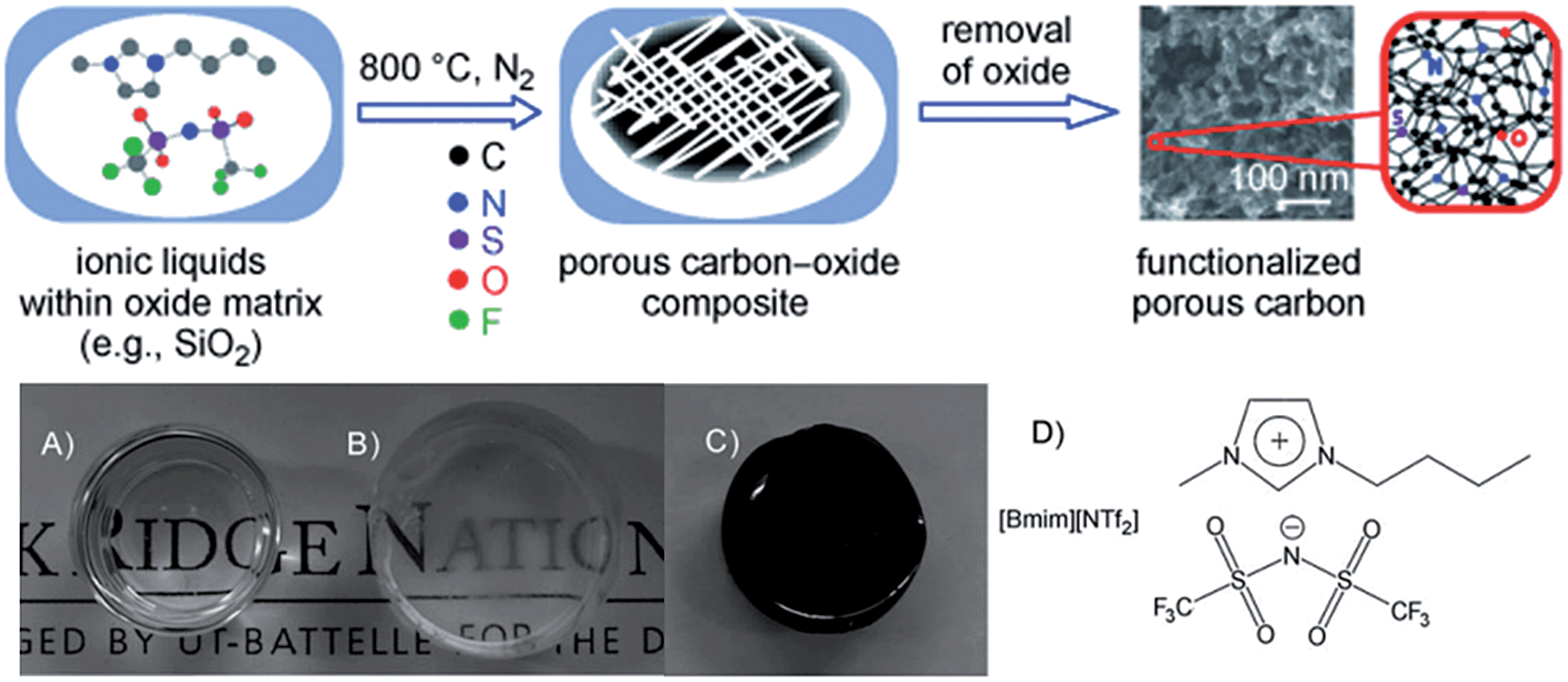 | ||
| Fig. 5 In situ nanoconfinement and carbonization of conventional non-carbonizable ILs. (a and b) Monolithic silica gels before carbonization, (b) after carbonization, and (d) structure of [BMIm][NTf2]. [Reprinted with permission from ref. 43, copyright 2010 Wiley.] | ||
In addition to the nanoconfining method, commonly non-carbonizable ILs can also be transformed into fluorescent carbon dots by fast and simple microwave pyrolysis.54–56 In this case, the IL acts not only as a carbon source, but also as an excellent medium for absorbing microwaves, because of the high polarizability of the ions in ILs.57 The obtained carbon dots are small, with a narrow size distribution, in situ surface functionalization, and a high quantum yield; they have potential applications such as label-free detection of metal ions.56
2.3. Poly(ionic liquid)s
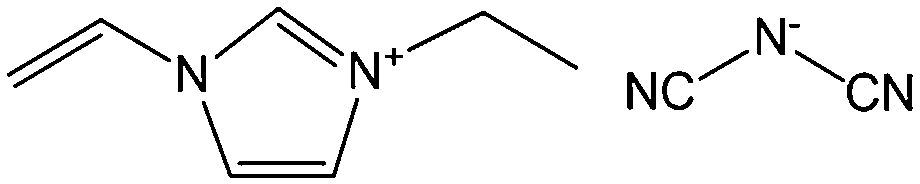
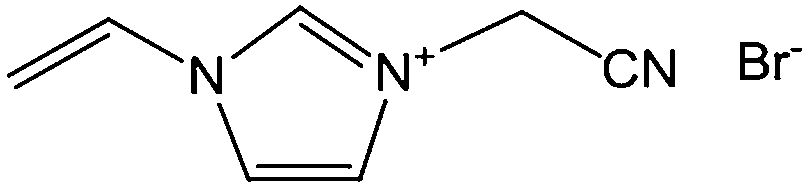
Poly-ILs, which are the polymeric form of ILs, and are commonly prepared by polymerization of a monomeric IL using an initiator,58–61 have also been used as carbon precursors. In spite of the time-consuming polymerization step, poly-ILs show great potential for the preparation of carbon materials. First, unlike the case of IL monomers, for which, because of their fluidic properties, it is difficult to obtain nitrogen-doped carbon materials of designed shapes in the absence of templates,25,41 the polymeric nature of poly-ILs enables them to be easily processed. Well-defined morphologies that are inaccessible via monomers or small-molecule ILs, such as fibers and monoliths, can be obtained using poly-ILs.62,63 Secondly, polymerization makes it possible to improve the carbon yields from common ILs after appending cross-linking moieties. Thirdly, because of the strong interactions between ILs and templates, poly-ILs can homogeneously cover the template surface, by in situ polymerization, giving desirable HCSs or carbon-encapsulated structures.64–68Antonietti and Yuan's group have contributed extensively to the area of poly-IL use as carbon precursors. They first directly carbonized a series of IL monomers containing vinyl or cyano/nitrile groups either in the cation or the anion, and compared their carbon yields.69 The results, summarized in Table 1, show that in the presence of cross-linking moieties, the IL monomers provide good carbonization yields, which are related to the choice of anions and the alkyl chain on the imidazolium ring. It should be noted that other than the expected cyano/nitrile groups, decorating ILs with vinyl groups can also result in substantial carbon yields and make common ILs containing non-carbonizable anions potential carbon precursors.67–69 Compared with the popular [EMIm][DCA] (IL-1),26 IL monomers with an additional vinyl group on the imidazolium ring (IL-3) was found to double the carbonization yield (28.6 wt%). Comparisons of IL-2 and IL-3, and IL-2 and IL-4 suggest that the presence of an additional cross-linkable group significantly improves the carbon yield. However, a comparison of IL-4 and IL-5 shows that a longer methylene spacer between the imidazolium core and the cyano group greatly reduces the carbon yield, from 29.0% to 14.3%, probably because of the decrease in the percentage of non-carbonized components. These results show an interesting effect of the precursor structure on the final carbon yield, namely that certain functional groups that undergo cross-linking reactions under pyrolytic conditions significantly improve the carbon yield. Homopolymers were then prepared by conventional free-radical polymerization of IL-2 and IL-3, and successfully carbonized, with carbon yields very close to those of the IL monomers.69 Thermal polymerization of the IL monomers in the early stages of the reaction may therefore occur prior to pyrolysis. Although there are great and expected similarities between using IL monomers and polymers as precursors for the pyrolytic formation of carbons, starting from a prepolymerized monomer allows typical polymer operations such as molding, extrusion, coating, or casting with preservation of a given shape to be performed (unlike low-molecular-weight liquids). Such a structured polymer film could then be transferred in a ceramic-like processing step into the final carbon structure.
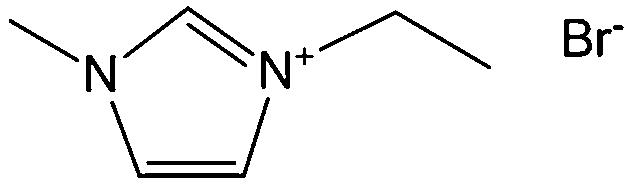
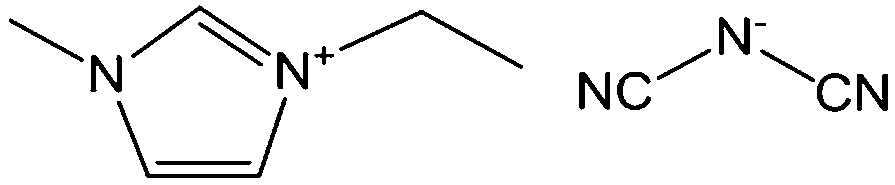
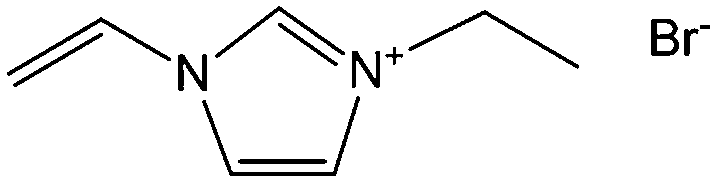

Shaped carbon materials can be easily obtained from poly-ILs. Recently, using the well-established electrospinning technique, nitrogen-doped carbon fibers and membranes were obtained from imidazolium- and pyridinium-based poly-ILs as precursors by Yuan et al. (Fig. 6).62 However, poly-ILs for electrospinning and carbonization should be carefully designed to solve various problems, in particular, the deformation of polymer chains at elevated temperatures and destruction of the anisotropic shape. The introduction of allyl functions was found to enable poly-IL to cross-link and to maintain their fiber morphology at high temperatures before typical condensation reactions of ILs. In addition, to achieve a high degree of cross-linking, a co-cross-linking agent, i.e., trimethylolpropane tris(3-mercaptopropionate), and a radical initiator, namely 4,4′-azobis(4-cyanovaleric acid), were codissolved with the poly-ILs in an organic solvent. The solution was then subjected to electrospinning followed by carbonization. Fig. 6a shows a side view of a monolayer of carbon fibers prepared from an imidazolium-type poly-IL (PAVIm–DCA) on a silicon wafer of diameter 0.2–3 μm. The corresponding carbon membrane, as shown in Fig. 6b, displays a homogeneous cross-section membrane structure, of thickness 3–6 μm. In addition, most of the carbon fibers in the membrane maintain the original morphology of the poly-IL fibers. In contrast, the membrane obtained from a pyridinium-type poly-IL (PAVP–DCA) displays structural characteristics different from those of PAVIm–DCA; it is made up of overlapping individual carbon fibers, in a less compact membrane structure, with all the fibers fusing into neighboring ones at the junctions (Fig. 6c). The above results demonstrate that the superior processability of poly-ILs and the inherent incorporation of IL moieties in the macromolecular structure can be combined to generate advanced materials with desired morphologies and complex architectures. In addition, XRD of the membranes indicated that the carbons are graphitic (inset in Fig. 6c) and elemental analysis revealed that PAVIm–DCA and PAVP–DCA carbon fibers are doped with 6.33% and 8.0% nitrogen, respectively.
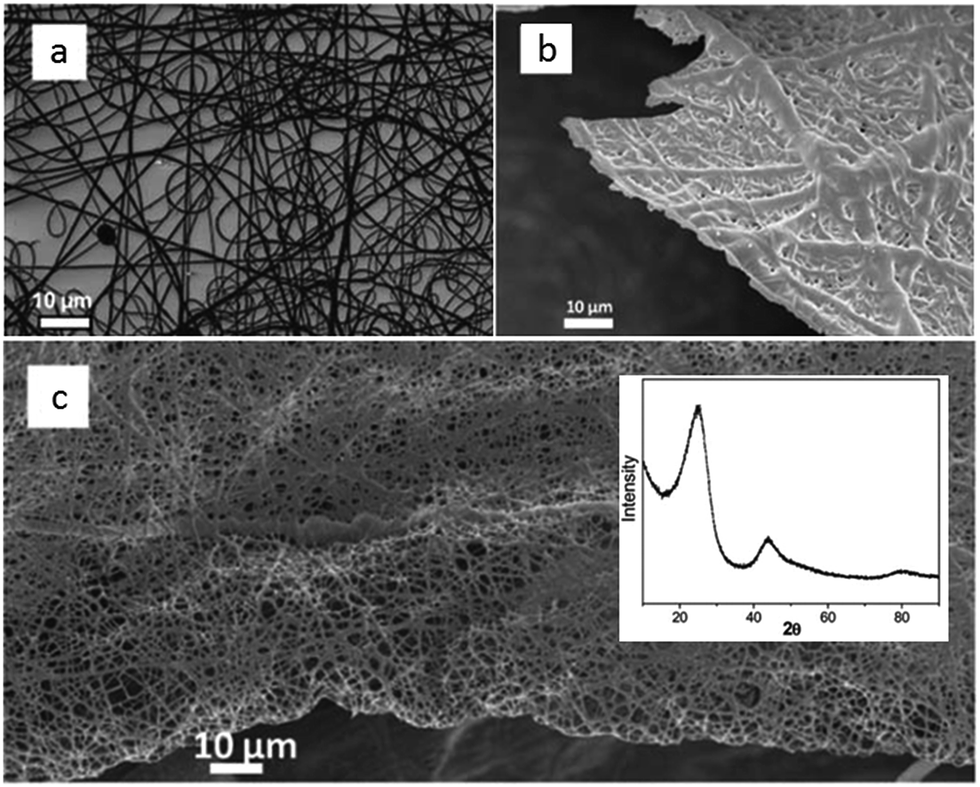 | ||
| Fig. 6 Scanning electron microscopy (SEM) images of nitrogen-doped (a) carbon fibers and (b) membranes prepared from electrospun PAVIm–DCA fibers, and (c) carbon membranes prepared from electrospun PAVP–DCA fibers. Inset shows the XRD pattern. [Reproduced from ref. 62.] | ||
With the help of low fractions of ILs or poly-ILs, sugar-based molecules and polysaccharide biomass can be turned into functional carbonaceous products at low temperature, i.e., 400 °C, under a nitrogen atmosphere.63 As shown in Fig. 7, flexible, shapeable carbonaceous frameworks such as carbonaceous membrane paper, strips, and monoliths were easily obtained by simply coating shaped biotemplates such as cellulose filter membranes, coffee filter paper, or natural cotton with an IL/poly-IL, followed by thermal transformation. These carbonized hybrids exhibit comparably good mechanical properties such as bendability of membranes or shape recovery of foams. Tensile-testing experiments showed that, in contrast to the intact filter membrane, the poly-IL-doped filter membrane gave rise to a carbon membrane with not only a four-fold higher breaking strength, but also a 50% larger rupture strain (Fig. 7b). The nitrogen heteroatoms incorporated into the final products from the IL/poly-IL precursors further improved the oxidation stability in fire-retardant tests (Fig. 7c and d). The “low temperature carbonization/activation” function of ILs/poly-ILs for biomolecules and biomass was further proved by TG analysis of a mixture of IL/β-cyclodextrin (β-CD), which showed a significantly lower onset of mass loss than that of either the IL or β-CD, indicating that a new process resembling chemical activation is involved. Even at a rather low doping ratio (7 wt%), the use of ILs and poly-ILs as activating agents can effectively promote conversion and pore generation in biomaterials, achieving high surface areas of up to 720 m2 g−1. This is attributed to the unique structures of ILs and poly-ILs; both contain a bulky anion, [NTf2], and one or two cyanomethyl groups attached directly to the imidazolium cation (Fig. 7a). Such a chemical combination is known to readily form porous carbons on pyrolysis, without any external template or activating agent.25,39 Carbon monoliths can also be prepared by one-pot solvothermal copolymerization of IL monomers with divinylbenzene and subsequent carbonization in a CO2 atmosphere.65
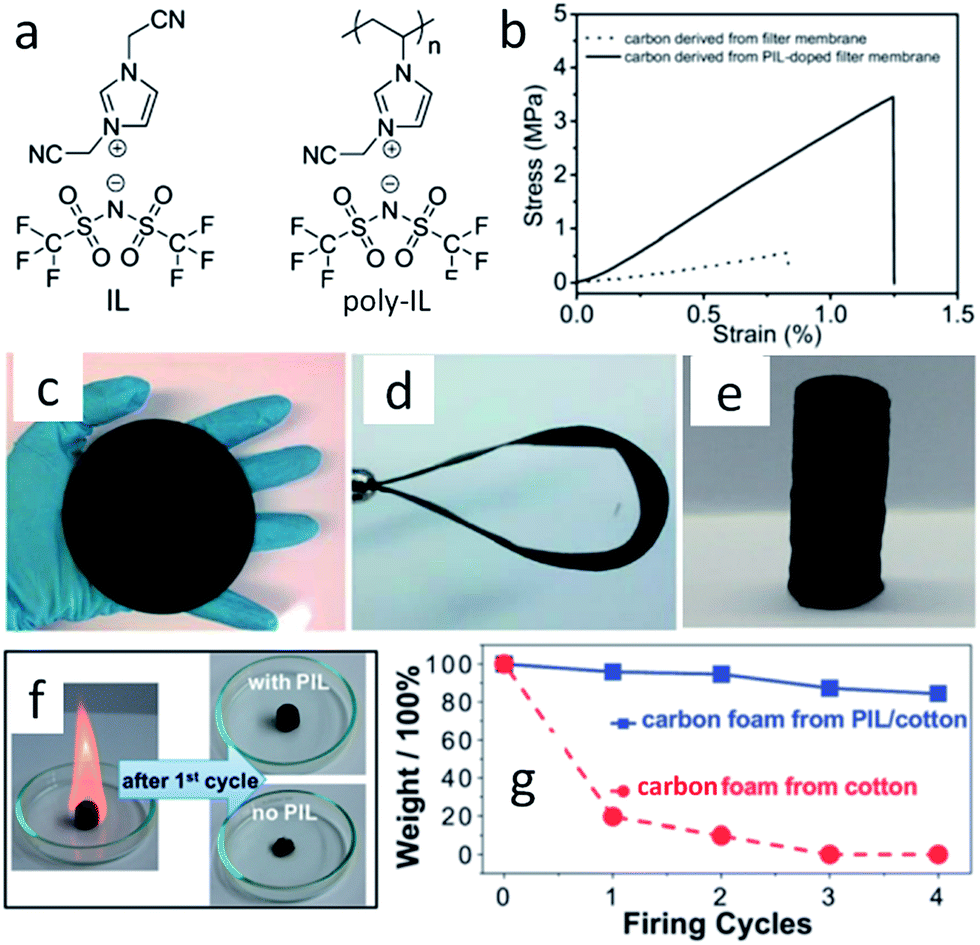 | ||
| Fig. 7 (a) Chemical structures of IL and poly-IL. (b) Tensile tests of carbon membranes derived from the pure cellulose filter membrane (dotted line) and from the poly-IL-doped filter membrane (solid line). (c) Carbonaceous membrane paper, (d) bent strip, and (e) monolith derived from poly-IL wetted polysaccharide biomass. (f) Photographs of first firing cycle test of carbon foams. (g) Plot of carbon foam mass vs. firing cycle. [Adapted from ref. 63.] | ||
The known favorable interactions between the strongly polar IL monomers and polar inorganic silica templates undoubtedly facilitate the synthesis of high-surface-area, nitrogen-doped carbons with various morphologies.46 HCSs can be easily fabricated using poly-ILs as carbon precursors and monodisperse silica particles as templates.67,71 The overall procedure for HCS preparation is presented in Fig. 8a. First, the IL monomer (1-vinyl-3-ethylimidazolium tetrafluoroborate, [VEIm][BF4]) was coated on the surfaces of monodisperse silica particles, accompanied by in situ polymerization. The obtained core/shell composite spheres with a uniform poly-IL layer were then carbonized, with subsequent template removal, generating carbon spheres with hollow structures. Transmission electron microscopy (TEM) images (Fig. 8b and c) show that the HCSs are made up of hollow spheres with shells 7 nm thick, and hollow cores 105 nm in size, consistent with the diameter of the silica template. Based on a very similar strategy, but replacing the monomer IL by one containing both cyano and vinyl groups, Yuan et al. successfully prepared porous nitrogen-doped HCSs with tailored particle sizes, nitrogen contents, and wall thicknesses, which are partially graphitized and exhibit hierarchical pore structures and size-dependent surface areas of up to 570 m2 g−1.64 The strategy for synthesizing nitrogen-doped carbon layers by in situ polymerization of monomer ILs can be expanded to other nanosubstrates such as carbon nanotubes (CNTs).68 Instead of in situ polymerization, the as-synthesized poly-ILs are directly deposited on a template such as colloidal silica using layer-by-layer assembly.66 Subsequent carbonization and template removal produce carbon capsules containing ca. 7 wt% nitrogen, with a surface area of 423 m2 g−1.
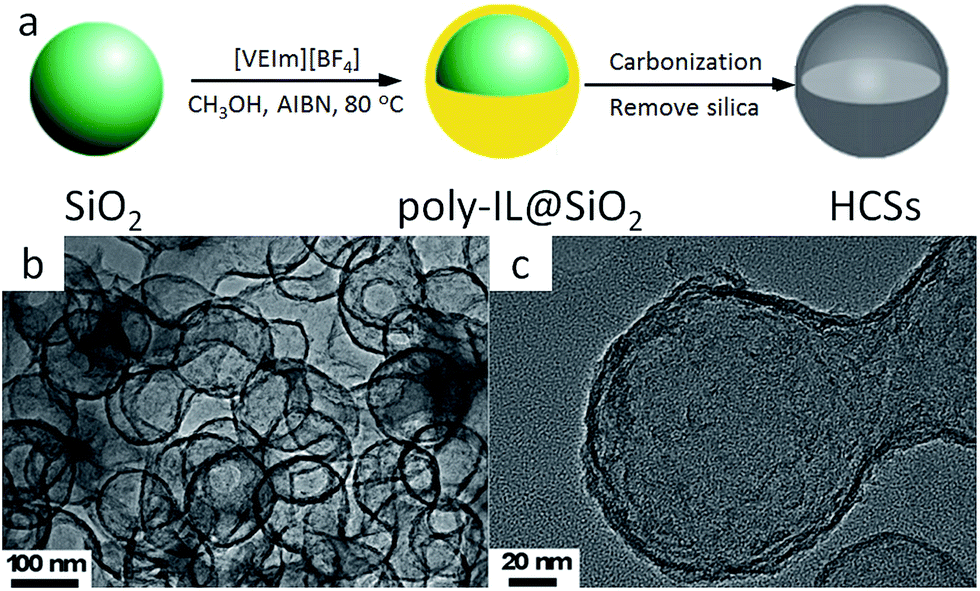 | ||
| Fig. 8 (a) Synthetic procedure and (b and c) TEM images of HCSs. [Reprinted with permission from ref. 67, copyright 2011 Elsevier.] | ||
In addition, in an inverse strategy to encapsulate a silica template by poly-ILs to obtain a nitrogen-doped carbon layer, carbon nanobubbles with controlled dimensions, variable nitrogen doping, superior aqueous dispersibility, high conductivity, and a distinctive atomic graphitic order were obtained using poly-IL nanoparticles with a triple role, namely sacrificial template, carbon precursor, and nitrogen source, with silica as a solidifying agent.72 The structures of the poly-ILs and the overall synthetic routes are shown in Fig. 9. Spherical poly-IL nanoparticles in aqueous solution were obtained via simultaneous polymerization/self-assembly of IL monomers with long alkyl chains.73,74 It was assumed that the as-formed poly-IL nanoparticles were probably assembled in organized mesophases consisting of separate nanodomains of charged IL moieties and long hydrophobic alkyl chains. As seen in Fig. 10a and b, the poly-IL–Br nanoparticles have a number of spontaneously formed bilayers as well as a central “void”, the size of which is independent of the bilayer number.58 Silica nanocasting was performed by dropwise addition of a liquid silica precursor such as tetraethyl orthosilicate, followed by hydrolysis and condensation. The poly-IL/silica composite was then subjected to carbonization and template removal, yielding porous HCSs (Fig. 10c and d). It should be noted that nanoconfinement of the poly-IL in the solidified silica reservoir greatly enhanced the carbonization yields, which were similar to that reported by Dai et al.43 For example, a carbon yield of up to 30% was achieved for poly-IL–DCA nanoparticles, which is more than twice that for non-confined carbonization of poly-IL–DCA (13%). Based on the anion type in the poly-IL nanoparticles ([Br] or [DCA]), and the type of liquid silica precursor, the nitrogen content and the size of the nanobubble are tunable over a wide range. Highly graphitic three-dimensional nanostructures with special local organization were observed in the resultant carbons. As seen in Fig. 10e and f, high-resolution (HR) TEM images of the carbon derived from poly-IL–DCA showed a layer-like pattern stemming from stacking of slightly distorted graphene layers. Independent of the specific position, these graphene layers are essentially oriented perpendicularly to the nanosphere surface, i.e., all the layers point to the nanosphere center. Although each layer appears parallel to its neighbor, the graphene sheet shifts its orientation slightly via step defects, so a short distance of several nanometers produces a detectable tilt. Along the nanosphere circumference, the graphene director basically rotates through 360°.
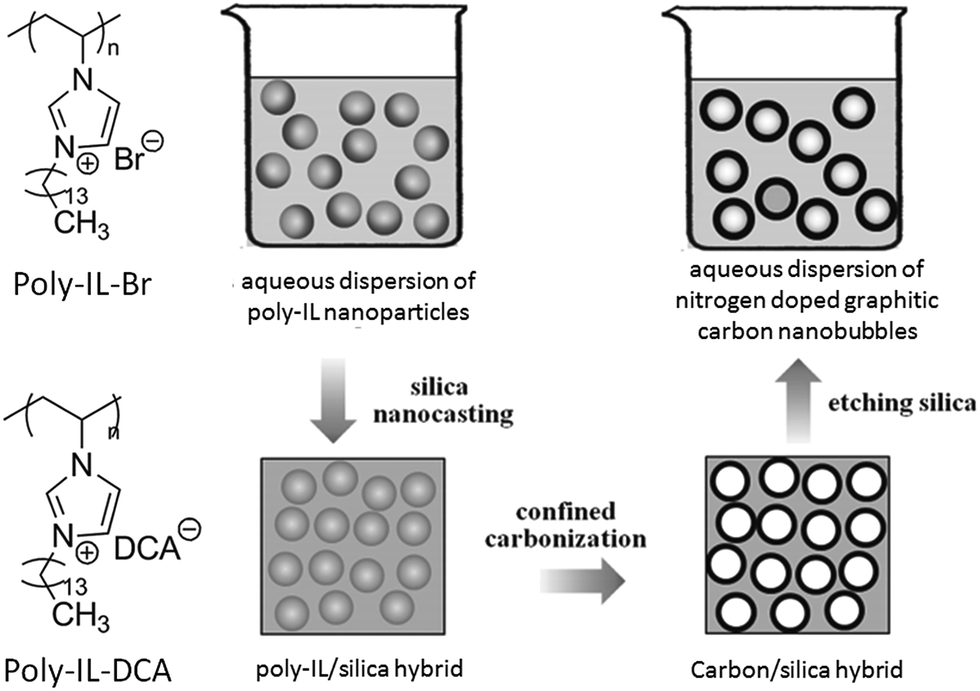 | ||
| Fig. 9 Chemical structures of poly-IL nanoparticles and synthetic route to nitrogen-doped carbon nanobubbles by confined carbonization of poly-IL nanoparticles in a silica matrix. [Reprinted with permission from ref. 72, copyright 2013 Wiley.] | ||
 | ||
| Fig. 10 (a and b) Cryo-TEM images of poly-IL–Br nanoparticles in aqueous solution. [Reprinted with permission from ref. 73, copyright 2011 American Chemical Society.] (c and d) TEM images of nitrogen-doped carbon nanobubbles prepared from poly-IL–Br and poly-IL–DCA, respectively. (e) TEM image of a single nitrogen-doped carbon nanobubble. (f) HRTEM image of part 4 in (e). The white arrows indicate the planar direction of the stacked graphenes. [Reprinted with permission from ref. 72, copyright 2013 Wiley.] | ||
In addition, poly-ILs were shown to improve the hydrothermal carbonization of ordinary sugars in terms of the structures, properties, and functions of the carbon products, which are porous, nitrogen-containing, and composed of primary spherical nanoparticles. In particular, compared with the carbon products derived from pure sugars or with IL additives with very low surface areas (<10 m2 g−1), the use of poly-IL additives, at weight fractions of 0.55–13.3%, significantly increased the surface areas to 161 m2 g−1 for hydrothermal carbonization materials and 572m2 g−1 for post-carbonization materials. The poly-ILs are therefore believed to act not only as nitrogen sources, but also as stabilizers and pore-generating agents.75
2.4. Deep-eutectic solvents
DESs are mixtures for which the freezing point is lower than that of either of its individual components; they can be generally obtained by complexation of quaternary ammonium salts with hydrogen-bond donors such as acids, amines, and alcohols,76,77 or even obtained by direct combination of amides and inorganic salts.78 DESs have recently been described as a new class of ILs, because they share many characteristics with conventional ILs, but their low cost makes them particularly desirable for use in the large-scale synthesis of new functional materials.77,79–81Monte et al. performed pioneering work in the field of synthesis of carbon materials from DESs.82–90 In 2010, they explored the suitability of DESs based on a mixture of ethylene glycol and choline chloride (molar ratio 2:1, DES-1, Fig. 11) to achieve the polycondensation of resorcinol with formaldehyde.82 However, the DES investigated in this work, because of its thermal instability, was demonstrated to be not a real carbon precursor, but a simple solvent and a catalyst that can promote the polycondensation of resorcinol–formaldehyde. A DES based on p-toluenesulfonic acid and choline chloride (DES-2, Fig. 11) was also prepared and used as a solvent, acidic catalyst, and structure-directing agent to assist the condensation of furfuryl alcohol, with subsequent carbonization. This DES is also an excellent solvent for homogeneously dispersing CNTs, finally resulting in hierarchical porous composites with a homogeneous distribution of CNTs throughout the entire monolithic structure.89 Following this work, Monte et al. further synthesized a new type of DES by replacing the previous hydrogen-bond acceptors (ethylene glycol and p-toluenesulfonic acid) with resorcinol, a regular carbonaceous precursor for formation of resorcinol–formaldehyde resins by polycondensation (DES-3 to DES-8, Fig. 11). For example, resorcinol-based DESs with or without urea (DES-3 and DES-4) were directly used to prepare hierarchical porous carbon monoliths via formaldehyde polycondensation and subsequent carbonization.83 The DESs played multiple roles in the synthetic process, i.e., a liquid medium ensuring reagent homogenization, a carbonaceous precursor via formaldehyde polycondensation and subsequent carbonization, and a structure-directing agent via choline chloride segregation in a spinodal-like decomposition process, which is responsible for achievement of the hierarchical structure. One attractive feature is that self-contained resorcinol in the DESs requires no other phenolic additives for polycondensation and carbonization. The resorcinol becomes the material itself, with high carbon conversion, and the choline chloride can be recovered and reused in subsequent reactions, making DES-assisted syntheses very efficient in terms of reagent economy. Carbon conversion of resorcinol-based DESs was as high as 80%. Moreover, carbon monoliths with high surface areas, up to 612 m2 g−1, bimodal pores (micropores and mesopores), and narrow mesopore diameter distributions were obtained. The urea composition in the DESs can further expand the mesopore size to 23 nm. Since the urea component in DES-4 is released during carbonization, Monte et al. synthesized new DESs containing 3-hydroxypyridine as the nitrogen source in the carbon (DES-5, Fig. 11).86 Carbon monoliths with high nitrogen contents, high surface areas, and high carbon conversions (above 70%) were obtained via a formaldehyde polycondensation and carbonization process similar to that used with previous DESs. Scanning electron microscopy (SEM) images showed that the resulting carbon morphology consisted of a bicontinuous porous network built of highly cross-linked clusters that aggregated and assembled into a stiff, interconnected structure (Fig. 12). Furthermore, on addition of phosphoric acid as a catalyst during polycondensation of DES-6, phosphate-functionalized carbon monoliths were obtained after carbonization of the unwashed phenolic resin, which had a phosphorous content of about 8 wt%, determined using energy-dispersive X-ray spectroscopy (EDX), and a micropore surface area of about 600 m2 g−1, calculated using a t-plot, making it highly suitable for electrodes in supercapacitors.84
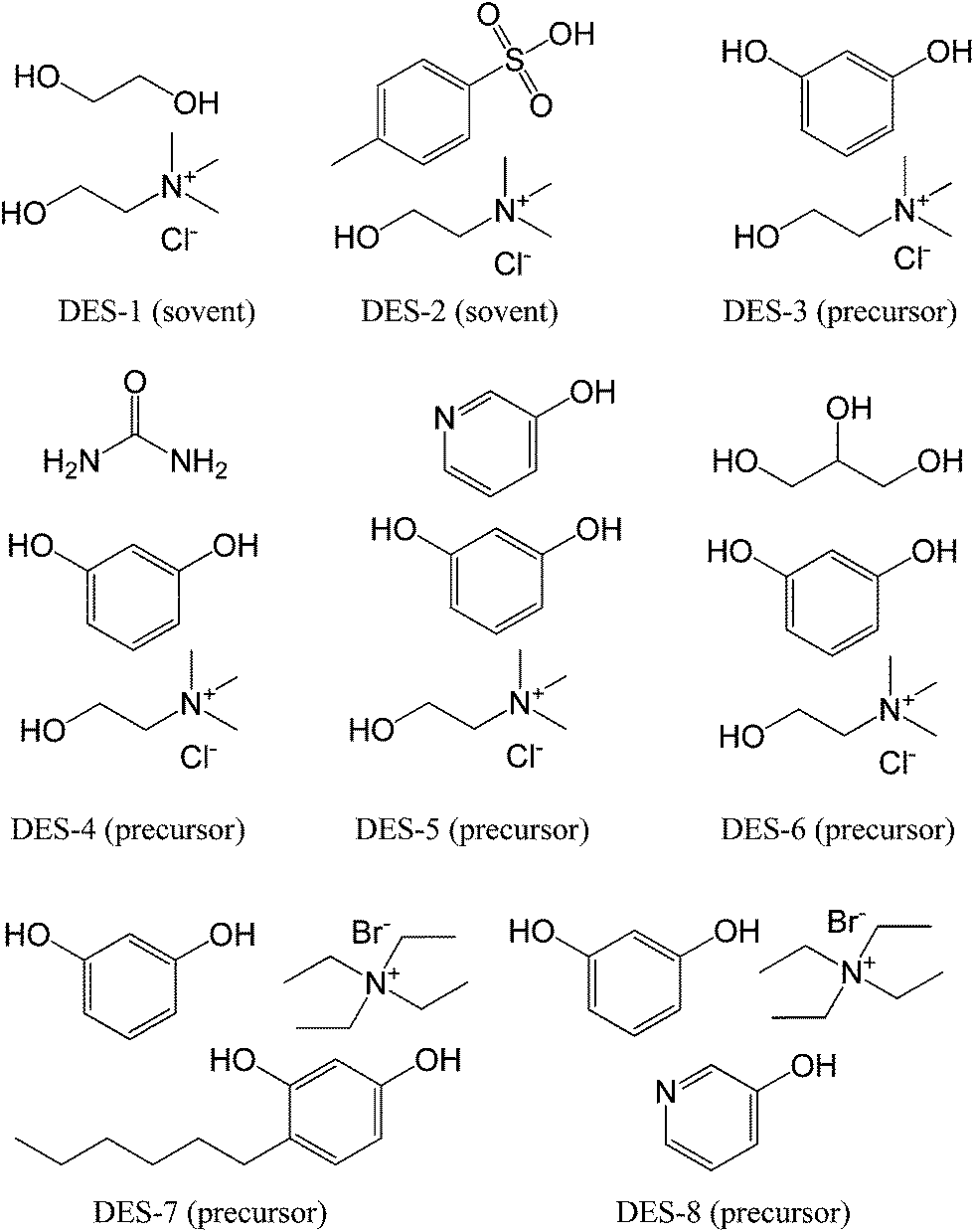 | ||
| Fig. 11 Structures of DESs used for synthesis of carbon materials. Description in parentheses indicates the corresponding role during polycondensation and carbonization. | ||
 | ||
| Fig. 12 SEM micrographs of nitrogen-doped hierarchical carbon obtained from DES-5. Insets show the photograph of the carbon monolith and high-resolution N1s X-ray photoelectron spectra (XPS), respectively. [Adapted from ref. 86.] | ||
Using long-alkyl-chain 4-hexylresorcinol and tetraethylammonium bromide as the co-hydrogen donor and hydrogen acceptor, respectively, the as-formed DES-7 (Fig. 11) was capable of providing microporous carbon monoliths. Notably, all the resultant carbons showed particularly narrow microporosities, with an average micropore diameter of ca. 0.6 nm. This narrow microporosity gave rise to negligible N2 uptake and a low surface area (<7 m2 g−1), but they did exhibit high adsorption of CO2 (2.2 mmol g−1 at 25 °C). The selective adsorption of CO2 was ascribed to the molecular sieve character of these carbons, so gases with small kinetic diameters (e.g., 0.33 nm for CO2) are preferentially adsorbed over those with large ones (e.g., 0.36 nm for N2 or 0.38 nm for CH4).85 Hierarchical porous nitrogen-doped carbons were obtained from DES-8, composed of resorcinol, 3-hydroxypyridine, and tetraethylammonium bromide, in which 3-hydroxypyridine acted as the nitrogen source in the resulting carbons, and tetraethylammonium bromide determined the formation of a molecular sieve structure.87 Varying the carbonization temperature and molar ratio between each component can tailor the hierarchical-type porous structure to give macro- and micro-pores, and to control the nitrogen content up to 8.7 wt%. Finally, a high and selective CO2 adsorption capacity over N2 (2.7 mmol g−1 at 25 °C) was achieved.
Although DESs show great potential as carbon precursors, such as low cost, and ease of production of bulk monolithic carbons with hierarchical porous and interconnected hierarchical structures, the direct carbonization of DESs without polycondensation resulted in no carbon residues. TG analysis (Fig. 13) confirmed that compared with the formed resorcinol–formaldehyde gels, even the DESs containing an aromatic component (resorcinol) are nearly completely decomposed below 300 °C.83 A possible reason is the relatively weak hydrogen-bond strength between the hydrogen donors (amines and alcohols) and acceptors (halogen anions), which makes DESs thermally unstable prior to preliminary polymerization and carbonization at high temperature. The unavoidable use of toxic formaldehyde for polycondensations with very long reaction times (usually 1 week) in the synthesis of porous carbon using the soft template method involving formaldehyde and phenol, resorcinol, or phloroglucinol18 undoubtedly hinder their widespread application.
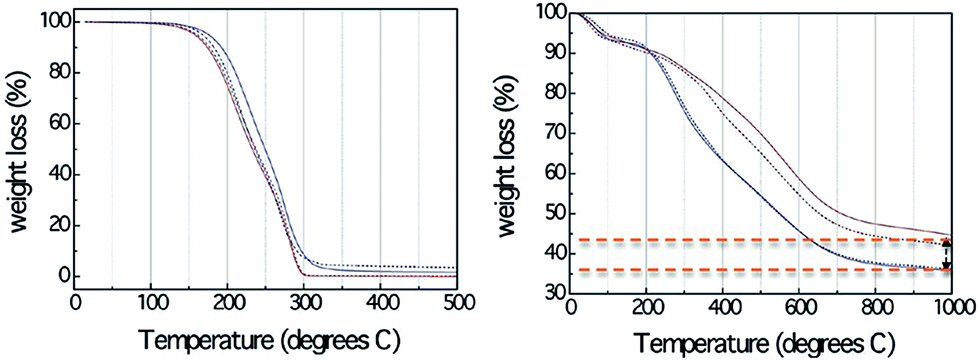 | ||
| Fig. 13 TG profiles of (left) DESs and (right) corresponding resorcinol–formaldehyde gels derived from DES-3 and DES-4. [Reprinted with permission from ref. 83, copyright 2010 American Chemical Society.] | ||
2.5. Protic ionic liquids and salts
As mentioned above, although cyano/nitrile-containing ILs show great potential for carbon materials, very few cyano/nitrile-containing aprotic ILs yield carbons.24,25 Moreover, the multistep and time-consuming synthesis, rigorous purification, metal impurities (e.g., Li, Na, or Ag, possibly from metathesis), and high cost complicate their manufacture and inevitably hinder large-scale production.Based on the previous finding that 1,8-diazabicyclo[5.4.0]undec-7-ene-based PILs can bear relatively high-temperature treatment (500 °C), with residual yields of up to 25%,92 Watanabe et al. recently found that nitrogen-containing PILs and PSs have great potential as single, low-cost, and versatile precursors for the facile one-step synthesis of carbon materials, without any complicated synthesis, catalyst, prepolymerization or other complex techniques.91,93 Unlike the specific aprotic ILs limited to those with cyano/nitrile moieties, PILs/PSs that tolerate harsh carbonization conditions can be easily prepared and are cheap. As shown in Fig. 14, the whole process is simple and rapid, involving only two steps, i.e., neutralization and carbonization. Instead of the expected complete weight loss of the parent bases below 400 °C, the carbon yields from the PILs/PSs depended strongly on the base structures. The unique compositions of all the PILs/PSs discussed here enable in situ fabrication of intrinsic nitrogen-doped carbons, with nitrogen contents varying significantly, depending on the precursors. In particular, the carbon derived from [3-CNPy][HSO4] has a very high nitrogen content of 11.1 wt% after carbonization at 1000 °C. Considering that [3-CNPy][HSO4] has a N/C molar ratio of 0.33, only half that of [EMIm][DCA] (0.63),26 the high nitrogen content observed in the resulting carbon is surprising, and indicates that at least 30% of the nitrogen atoms originally present in the [3-CNPy][HSO4] precursor were finally incorporated within the carbon architecture.
 | ||
| Fig. 14 Schematic representation of synthesis of carbon materials by direct carbonization of PILs/PSs and structures of the typical PILs/PSs. [Reprinted with permission from ref. 91, copyright 2014 American Chemical Society.] | ||
Most of the highly nitrogen-doped carbons, i.e., [3-CNPy][HSO4]-, [Phen][2HSO4]-, and [CNC2phIm][HSO4]-based carbons, despite their very high nitrogen contents, are more graphitic than carbons with lower N contents, shown by the more positive shift of the diffraction position, higher intensity, and narrower line width of the (002) peak (Fig. 15a). In particular, the [CNC2phIm][HSO4]-derived carbon shows a high-intensity (002) diffraction peak centered at 26.2°. The calculated interlayer distance (d002) of 0.339 nm is comparable to that of graphite (0.335 nm).94 HRTEM showed local organization of graphitic carbons, in which bent graphite-like sheets are uniformly present throughout the entire carbon structure. Interestingly, the graphene layers in the [CNC2phIm][HSO4]-based carbon are mostly arranged in one direction, because of its high degree of graphitization, whereas in the case of [3-CNPy][HSO4], the graphene layers are much more randomly oriented (arrows in Fig. 15c and d). In contrast, the [oBPy][2HSO4]-based carbon, with a high nitrogen content, is more amorphous, as revealed by the weak, broad (002) signal. The differences were checked using XPS. The results showed that the carbons from [3-CNPy][HSO4], [CNC2phIm][HSO4], and [Phen][2HSO4] have high percentages of pyridinic nitrogen but low percentages of quaternary nitrogen. The former is known to be planar, whereas the latter has an out-of-plane bonding configuration.95 The above three carbons with high percentages of pyridinic nitrogen thus have short interlayer distances and are more graphitic.
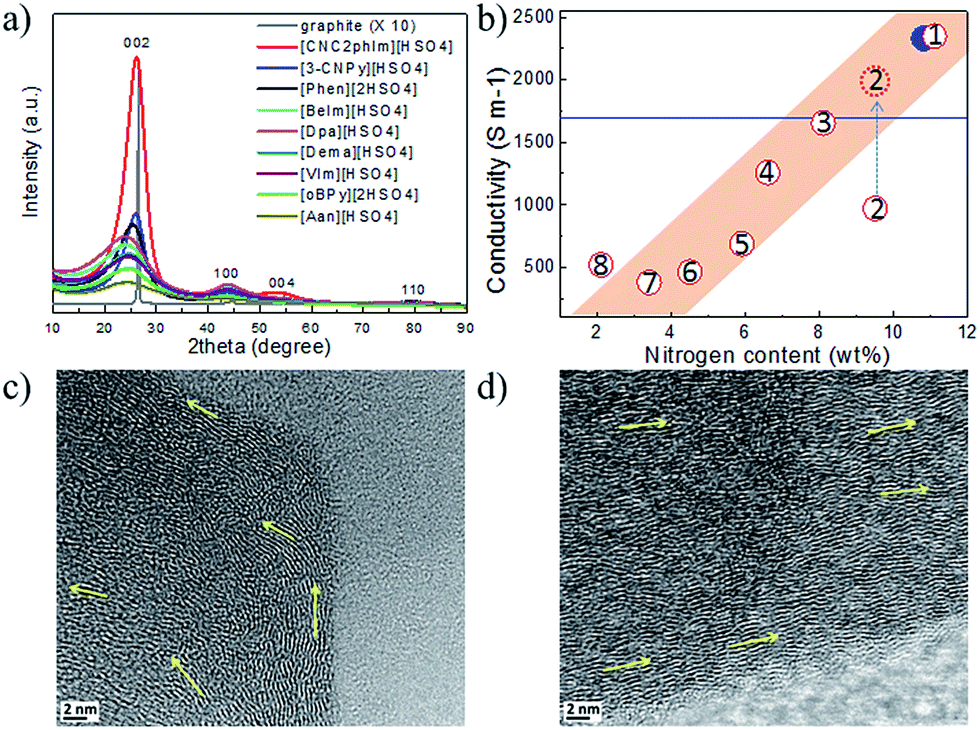 | ||
| Fig. 15 (a) XRD patterns and (b) relative conductivities of carbons derived from PILs/PSs. (1) [3-CNPy][HSO4], (2) [oBPy][2HSO4], (3) [CNC2phIm][HSO4], (4) [Phen][2HSO4], (5) [BeIm][HSO4], (6) [Dpa][HSO4], (7) [VIm][HSO4], and (8) [Dema][HSO4]. Red dashed circle indicates the estimated conductivity of carbon 2 from the general relationship between nitrogen content and conductivity. [EMIm][DCA]-based carbon and graphite are shown by the blue solid circle and line, respectively. HRTEM images of carbons derived from (c) [3-CNPy][HSO4] and (d) [CNC2phIm][HSO4]. [Reprinted with permission from ref. 93, copyright 2014 American Chemical Society.] | ||
Interestingly, a nearly linear relationship between the nitrogen contents and electrical conductivities of the PIL/PS-based carbons was observed. As shown in Fig. 15b, the conductivity is roughly proportional to the nitrogen content (orange region in Fig. 2d), because of the doping effect of electron-rich nitrogen, which induces a donor state above the Fermi energy level and increases its electron density.30,96,97 For [3-CNPy][HSO4]-based carbon, which has the highest nitrogen content and relatively high degree of graphitization, the conductivity is even higher than that of graphite, showing similar results to those for [EMIm][DCA]-based carbon.26 In contrast, [oBPy][2HSO4]-based carbon, despite its high nitrogen content (9.5 wt%), exhibits much lower conductivity than expected (Fig. 15b). The graphitic structures and nitrogen contents of PIL/PS-derived carbons can both affect the thermal stability against oxidation, although the observed trend in the thermal stabilities of PIL/PS-derived carbons is slightly different from that of the conductivities. In the case of the thermal stabilities, graphitization of the carbon material significantly improves its thermal stability in air. As seen in Fig. 16, the [CNC2phIm][HSO4]-based carbon, which has the highest degree of graphitization and a relatively high nitrogen content (8.1 wt%), has a much higher decomposition temperature (ca. 773 °C) than the others. The effect of graphitization is further shown by the lower decomposition temperature (635 °C) for [oBPy][HSO4]-based carbon, although its nitrogen content (9.5 wt%) is higher than that of [CNC2phIm][HSO4]-based carbon. [Dema][HSO4]- and [Aan][HSO4]-based carbons, which both have low nitrogen contents (2.1 and 2.7 nitrogen wt%) and graphitization, showed lower-temperature weight-loss profiles, at 577 and 534 °C, as expected.91
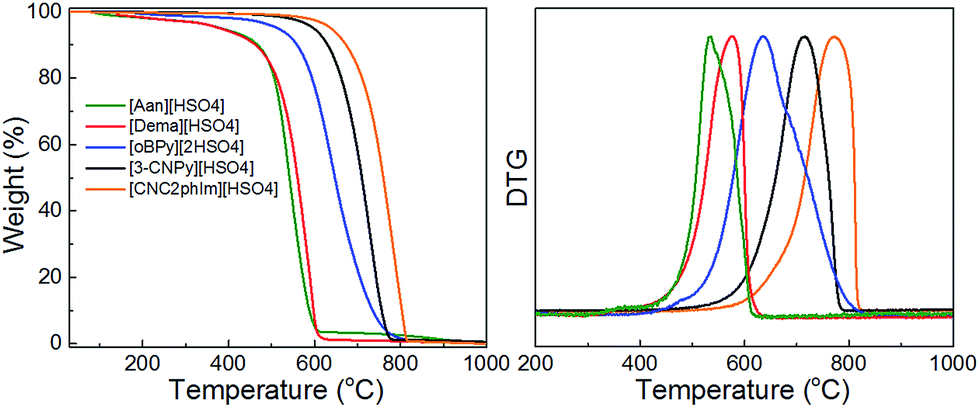 | ||
| Fig. 16 TG curves of PIL/PS-derived carbon materials in air (heating rate: 10 °C min−1). Decomposition temperature was determined from the differential TG peak. [Reprinted with permission from ref. 91, copyright 2014 American Chemical Society.] | ||
In addition, depending on the precursor structure, the obtained carbon materials vary from non-porous to microporous. Some of them exhibit high surface areas of up to 1380 m2 g−1. Highly porous carbon can only be obtained via nanocasting or activation, therefore this facile one-step method is of great significance. The carbonization mechanism was investigated using [CNC2phIm][HSO4] as a typical precursor containing various distinct but common moieties present in most PILs/PSs. It was found that during carbonization, the thermally unstable moieties such as alkyl chains, cyano functional groups, anions, and heterocyclic imidazolium rings decomposed at low temperature, and the fragments formed in situ then underwent a number of transformations such as cross-linking polymerization and recombination, partially condensed and/or fused with the thermally stable domains to form nitrogen-containing polycyclic aromatic compounds, and were finally transformed into nitrogen-doped carbon. Notably, the high proton-transfer energy between the protic acid and base, which is much higher than those of DESs, is essential to improve the thermal stabilities of the PILs/PSs and ensure high carbon yields under harsh carbonization conditions.92
This strategy was used to synthesize and directly carbonize a series of PILs/PSs with various nitrogen-containing bases and acids.91 The nitrogen-containing bases investigated were aliphatic amines, aromatic amines, and heterocycles, with or without unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds or cyano groups, and the acids were common inorganic and organic acids. Because of the wide availability of the PIL/PS precursors and their diverse structures, the general correlations between the carbonaceous precursors and resultant carbon materials, in terms of carbon yield, graphitic structure, nitrogen content, and porosity, have been investigated in detail; the results are summarized below.
C bonds or cyano groups, and the acids were common inorganic and organic acids. Because of the wide availability of the PIL/PS precursors and their diverse structures, the general correlations between the carbonaceous precursors and resultant carbon materials, in terms of carbon yield, graphitic structure, nitrogen content, and porosity, have been investigated in detail; the results are summarized below.
(a) High carbon yields can be obtained from PILs/PSs with high proton-transfer energies, which prevent the reverse reaction to form neutral acids and bases. In addition, thermally stable domains such as fused aromatic moieties, benzene groups, or unsaturated bonds help to improve carbon yields.
(b) Carbons with high nitrogen contents can be obtained from PIL/PS precursors with relatively high C/N ratios (usually C/N is 3–5), but not from those with extremely high nitrogen contents (e.g., C/N < 1). The incorporation of structural nitrogen such as pyridine, imidazole, CN, and thermally stable CN moieties may additionally increase the number of the expected functional nitrogen species, but aliphatic amines containing unstable C–N bonds do not contribute significantly to the nitrogen content.
(c) Carbons with high surface areas can be obtained from PIL/PS precursors containing unstable moieties such as aliphatic chains or heterocyclic rings; most of the nitrogen-containing bases and anions are sacrificed as porogens or templates to generate large amounts of pores.
(d) Carbons with high degrees of graphitization can be obtained from PIL/PS precursors simultaneously containing a heterocycle, a CN group, and an aromatic structure; at least a heterocycle and a CN group are required. However, establishing a firm correlation between the chemical structures of PILs/PSs and the carbon characteristics remains a challenge for the future.
These conclusions show that these low-cost and versatile precursors pave the way for the design and synthesis of task-specific carbon materials through rational selection of bases, acids, and functional groups. In fact, most of the above points hold true for aprotic ILs and neutral precursors.
3. Ionic liquids as surface-coating agents, dopants, and media for carbon materials
3.1. Ionic liquids as surface-coating agents
The promising liquid features of IL precursors coupled with the high nitrogen contents and graphitic structures of the resultant carbons make [DCA]-based ILs ideal precursors. However, the relatively low carbon yield (<13% at 1000 °C) and high cost strongly hinder large-scale production. In particular, compared with carbon materials derived from conventional precursors, those formed from ILs are expensive. The carbon yield can be improved by using the aforementioned nanoconfinement method. Regarding the high cost, an alternative method is to decorate the surfaces of bulk materials with a thin layer of IL-derived nitrogen-doped carbons; this reduces the absolute amount of IL required, but maintains the attractive features of IL-derived carbons, such as high conductivities and nitrogen contents.Hu et al. recently used ILs as precursors for the formation of carbon coatings on Li4Ti5O12 electrodes for LIBs.53,98 The composite material was obtained by simply mixing [EMIm][DCA] with pristine Li4Ti5O12, accompanied by carbonization. The HRTEM image in Fig. 17a shows that a very thin uniform amorphous layer is formed on the surface of the anode material. The EDX mapping images (Fig. 17b) show that the distribution of carbon and nitrogen elements is consistent with that of Ti, indicating a uniform nitrogen-doped carbon coating on/in the particles. The resultant composite Li4Ti5O12 electrode coated with a thin, conductive, nitrogen-doped carbon layer has a superior rate capability and cycling performance versus Li compared with the pristine sample.98 [EMIm][DCA] was also used to coat a Pt/XC-72 catalyst with a nitrogen-doped carbon layer; the coating structure not only improved the charge-transfer rate at the metal-support interface, but also promoted dispersion and reduced agglomeration of Pt nanoparticles. The resultant catalysts exhibited considerable electrochemical surface areas and significant electrocatalytic activity in methanol oxidation compared with the uncoated catalysts.99 In addition, nitrogen-doped-carbon-coated CNTs were obtained through pyrolyzing IL precursors on the surfaces of commercial CNTs, using either a typical carbonizable IL ([EMIm][DCA])100 or in situ polymerization of IL monomers.68 In the latter case, the TEM image revealed that after polymerization and carbonization, a thin amorphous carbon layer (CNx) of thickness ca. 1.5 nm covered the CNT side walls (Fig. 18). The resultant surface-nitrogen-enriched CNTs (SNE-CNTs) were demonstrated to be potential supports for noble-metal nanoparticles for electrocatalysis. This strategy for obtaining SNE-CNTs introduces a high content and uniform distribution of nitrogen atoms in the shell, enabling the CNT surface to anchor and grow noble-metal nanoparticles, while maintaining the electronic conductivity and structure of the core CNT.
 | ||
| Fig. 17 (a) HRTEM and (b) SEM and EDX mapping images of Ti, carbon, and nitrogen elements in coated Li4Ti5O12. [Reprinted with permission from ref. 98, copyright 2011 Wiley.] | ||
 | ||
| Fig. 18 Schematic diagram of preparation of SNE-CNTs and PtRu nanoparticle/SNE-CNT nanohybrids, and TEM images of and SNE-CNT nanocomposites. [Adapted from ref. 68.] | ||
3.2. Ionic liquids as heteroatom dopants
Another method for reducing the IL amount but retaining the performance of nitrogen-doped carbon is to use ILs as heteroatom dopants in the synthesis of carbon materials from other inorganic (e.g., graphene or graphene oxide (GO)) or organic carbonaceous precursors (e.g., a polymer).101–105 For example, ILs can function as both nitrogen dopants and shape-directing agents for the synthesis of microporous carbon nanosheets from GO and resorcinol–formaldehyde polymers.101 As shown in Fig. 19a, the carbon sheets obtained in the presence of IL consist of two parts, with the inner graphene layer coated with carbon on both sides. No free carbon particles or naked graphene sheets are observed in SEM images, suggesting perfect assembly of resorcinol, formaldehyde, and the ILs on the surfaces of the GO sheets; this was confirmed by the uniform distribution of nitrogen on the carbon surface indicated by EDX mapping images. In contrast, in the absence of an IL, the carbon sheets stack on top of each other, resulting in serious aggregation (Fig. 19b).101 Nitrogen-doped graphene with a N/C molar ratio and electrical conductivity of 22.1% and 7.15 × 104 S m−1, respectively, can be synthesized by IL-assisted electrolysis with subsequent thermal annealing of the IL-functionalized graphene sheets. The surface properties such as the nitrogen configuration can be controlled by changing the anion ([Br], [AC], and [PF6]).104 In addition, nitrogen and boron codoped porous carbons with fully connected three-dimensional hierarchical porosities and local graphitic textures were derived from poly(benzoxazine-co-resol) polymers, using the IL [C16MIm][BF4] as an additive. Use of the IL not only leads to heteroatom incorporation into the framework, but also modulates the carbon microstructure.105 | ||
| Fig. 19 Field-emission SEM images of NMCS samples synthesized in the (a) presence and (b) absence of 1-butyl-3-methylimidazolium imidazolide ([BMIm][Im]). [Reprinted with permission from ref. 101, copyright 2014 Wiley.] | ||
3.3. Ionic liquids as media, solvents, and soft templates
As well as acting as dopants, ILs can also be used as media, solvents, or even soft templates in the formation of carbon materials.106–112 The ILs may be partially incorporated into the final carbon materials during carbonization. For example, cothermal condensation of [BMIm][BF4] and dicyandiamide was used to synthesize a new type of boron- and fluorine-enriched polymeric carbon nitride. Notably, the resulting materials have the technically advantageous “morel-like” mesoporous structure, with a narrow pore size distribution (Fig. 20); this originates from the IL as a soft template. These materials exhibit good photoactivities under visible-light illumination and good catalytic performances in the selective oxidation of cycloalkanes.106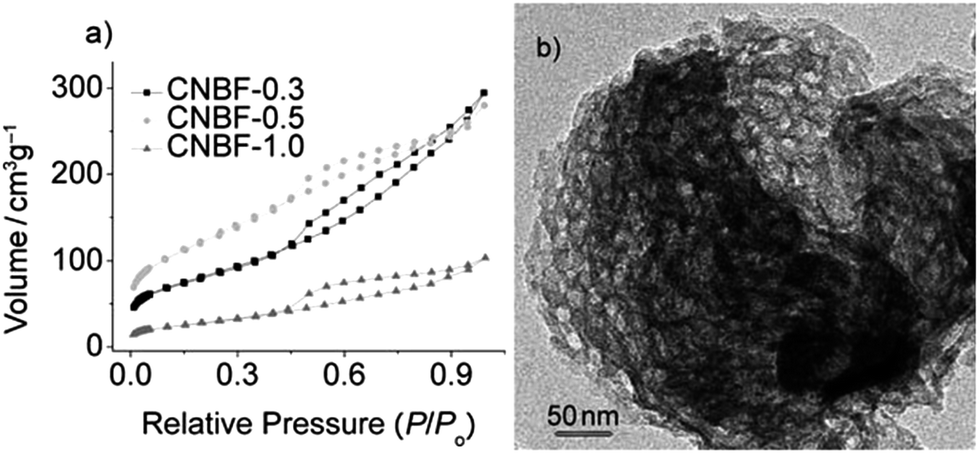 | ||
| Fig. 20 Nitrogen adsorption–desorption isotherms and TEM image of boron- and fluorine-enriched polymeric carbon nitride. [Reprinted with permission from ref. 106, copyright 2010 Wiley.] | ||
ILs can be used as alternatives to traditional reaction media (e.g., H2O) in the ionothermal synthesis of carbon materials from biomass at ambient pressure.110,113 For example, mesoporous carbons have been synthesized by ionothermal carbonization of glucose or fructose in PILs at low temperatures under ambient pressure.110 Three factors are considered to be key to the success of the ionothermal approach. First, the low vapor pressure of the PIL maintains a stable reaction environment, without solvent vaporization, and avoids the use of autogenic reaction systems. Secondly, the latent acidity of the PIL catalyzes the dehydration of fructose and glucose. Finally, the PIL significantly enhances the solubility of sugars with abundant hydroxyl groups via enhanced hydrogen-bonding interactions.110 Lewis acidic metal-containing ILs such as [BMIm][FeCl4] can also play a “three-in-one” role (soft template, solvent, and catalyst) in the synthesis of hierarchical porous carbonaceous materials.113 Porous carbon aerogels were obtained from resorcinol–formaldehyde resin without a supercritical drying process, using ILs as soft templates.108 Both the structure (different side chains or anions) and amount of the IL were found to strongly affect the porosity of the carbon aerogel. Interestingly, mixing with 10% [BMIm][BF4] not only increases the surface area from 371 to 559 m2 g−1, but also introduces additional mesopores (of pore size around 7.0 nm) compared with the original carbon aerogel without an IL template (Fig. 21).108 The porous structures and surface areas of the IL-templated carbon aerogels can be further developed by KOH activation of the as-synthesized dry resorcinol–formaldehyde gel.107 Use of a combination of phenol and formaldehyde as precursors and [BMIm][PF6] as a pore generator leads to the formation of macroporous carbon.109
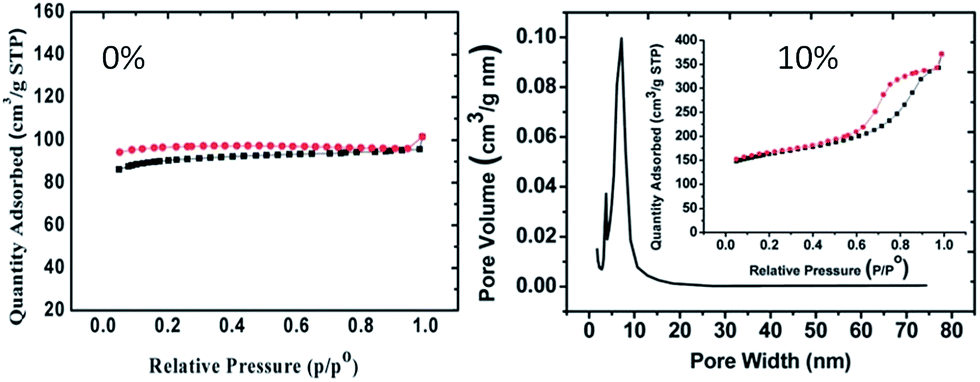 | ||
| Fig. 21 N2 adsorption–desorption isotherms of carbon aerogels with variable amounts of IL ([BMIm][BF4]). [Adapted from ref. 108.] | ||
4. Synthesis of metal-doped carbons from ionic liquids
ILs in the presence of metal salts or metal-containing ILs can be directly carbonized to carbon materials supporting metal nanoparticles; this approach has the following advantages. First, the presence of metal salts during carbonization greatly improves the graphitic structure of the obtained carbon materials,69,114 as observed in the case of catalytic growth of CNTs from metal particles.115 Secondly, mesopores can be developed by migration and aggregation of metal nanoparticles during carbonization.116 Thirdly, the resultant metal-particle-doped carbon materials have potential applications in areas such as electrocatalysis and chemical catalysis. The carbon materials containing metal particles synthesized from ILs can be classified into two types, according to the supported metal nanoparticles: one is carbon materials doped with transition metals such as Fe,69,113,117 Co,118 and Ni,118 and the other is carbon materials doped with precious metals such as Au, Pd, and Pt.119–121 It should be noted that carbon materials containing magnetic metal particles such as Fe can be easily transferred and separated by applying a magnetic field.117We first consider the case of transition-metal nanoparticles supported on carbon materials. Direct carbonization of IL monomers containing cross-linking groups, or their corresponding poly-ILs, in the presence of FeCl2 was reported to yield highly conductive, mesoporous, graphitic carbon nanostructures.69 As shown in Fig. 22, the threshold temperature for producing a large surface area is 900 °C. Above 900 °C, the nitrogen sorption isotherms exhibit a type H2 hysteresis loop, with a pronounced desorption step. This indicates delayed capillary evaporation and the presence of constrictions in the mesoporous structure (cage-like or bottle-like pores), and a broad pore size distribution. Moreover, HRTEM shows that all these textures are made up of stacked graphene sheets with a spacing of 0.338 nm (Fig. 22B), very close to the interplanar spacing in graphite,94 confirming that the lamellar-like graphitic nanostructures survived the HCl etching process. The pyrolysis of block copolymers comprising a hydrophilic poly-IL segment in the presence of metal salts such as FeCl2·4H2O and CrCl3·6H2O enables the preparation of mesoporous carbon nanomaterials with comparatively well-developed graphitic structures.122 Metal-containing ILs such as [BMIm][FeCl4] can be directly carbonized to mesoporous carbon/iron carbide hybrids by confining the IL precursor within a monolithic mesoporous silica matrix. The high fractions of Fe3C and γ-Fe2O3 render these materials magnetically responsive for facile separation, and they have, from the silica exotemplate, high surface areas of up to 800 m2 g−1.117 Other transition metals such as Co and Ni can also be deliberately added during carbonization of IL precursors.118 The content and type of transition metal were found to affect the sp2 carbon network and nitrogen configuration slightly, thus influencing the final oxygen reduction reaction (ORR) activity.
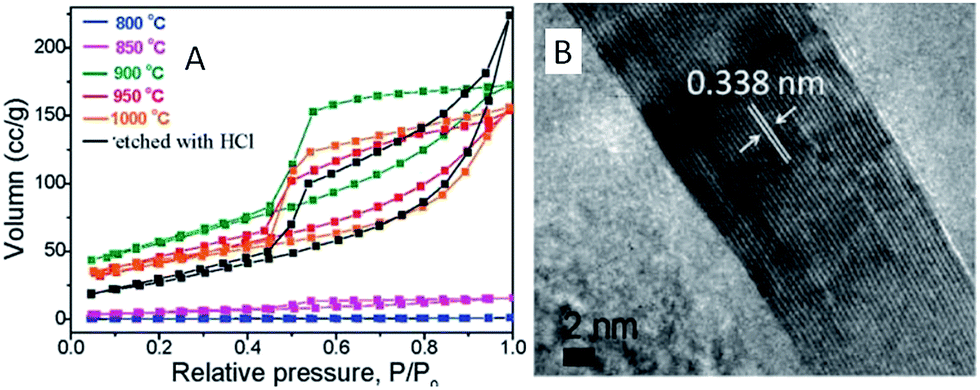 | ||
| Fig. 22 (A) Nitrogen sorption isotherms of carbons prepared by carbonization of IL monomers at different temperatures in the presence of FeCl2·4H2O and (B) HRTEM images of carbon pyrolyzed at 1000 °C after HCl etching. [Reprinted with permission from ref. 69, copyright 2010 American Chemical Society.] | ||
Binary and ternary metal nitride@nitrogen-doped carbon composites were obtained by template-free, one-pot carbonization of mixtures containing ILs and the corresponding metal precursors, TiCl4 or VOCl3; the ILs act as both carbon and nitrogen sources.123 The composites were produced simply by dissolving the respective metal precursors in the IL, i.e., [BMP][DCA], and subsequent calcination of the mixture. The introduction of metal ions into the precursor solution results in the formation of metal nitride nanoparticles with a narrow diameter distribution of around 7 nm, which are homogeneously distributed in a nitrogen-doped carbon matrix. Not only the amount of metal nitride in the composite but also the metal ratio in the ternary compound can be easily controlled by varying the metal precursor concentration in the starting solution. Nitrogen sorption measurements show that the composites have accessible micropores with surface areas of 350 and 550 m2 g−1 for TiN@N-dC and VN@N-dC, respectively. The introduction of simple salts, i.e., cesium and zinc acetate, as porogens, into the IL mixture increases the surface area to 2400 m2 g−1.124
In the case of precious-metal nanoparticles supported by carbon materials, we only discuss the one-pot synthesis from composite ILs;119–121 the postdeposition of metal nanoparticles on as-synthesized IL-based carbons will not be considered.68,100 Typical IL-based metal-containing precursors are shown in Fig. 23. Clearly, most of the structures feature metal-containing anions. For example, anion exchange between a polymerizable IL, i.e., poly(1-butyl-3-vinylimidazolium bromide-co-acrylonitrile), and a Pt precursor (H2PtCl6), and direct carbonization of the obtained sample (Fig. 23a) produced nitrogen-doped carbon–Pt nanohybrids. It can be clearly seen from the TEM image that the produced carbon material had a high loading of well-dispersed Pt nanoparticles, with a relatively narrow size distribution and an average size of ca. 3.5 nm. This hybrid showed high catalytic activity and stability in the electrocatalytic oxidation of methanol.119 Pt–Au nanoparticle–carbon was obtained via carbonization of triethoxysilane-derived ILs containing [PtCl6]x[AuCl4]y anions (Fig. 23b). The triethoxysilane group was transferred in situ into a silica template, and the exchanged anions provided Pt and Au sources. The resultant Pt–C hybrids show high electroactivity in methanol oxidization in an acidic medium, and Pt4Au1–C shows high activity in 4-nitrophenol reduction.120 Analogously, Pd-nanoparticle-supported porous nitrogen-doped carbon was prepared by direct carbonization of an IL-like salt (Fig. 23c); the product gave a catalytic performance even higher than that of a typical Pd2+ homogeneous catalyst.121 Although all the IL-based precursors give a homogeneous distribution of metal nanoparticles in the carbon materials, the one-pot method is not cost effective, because it requires large amounts of precious-metal salts for anion exchange.
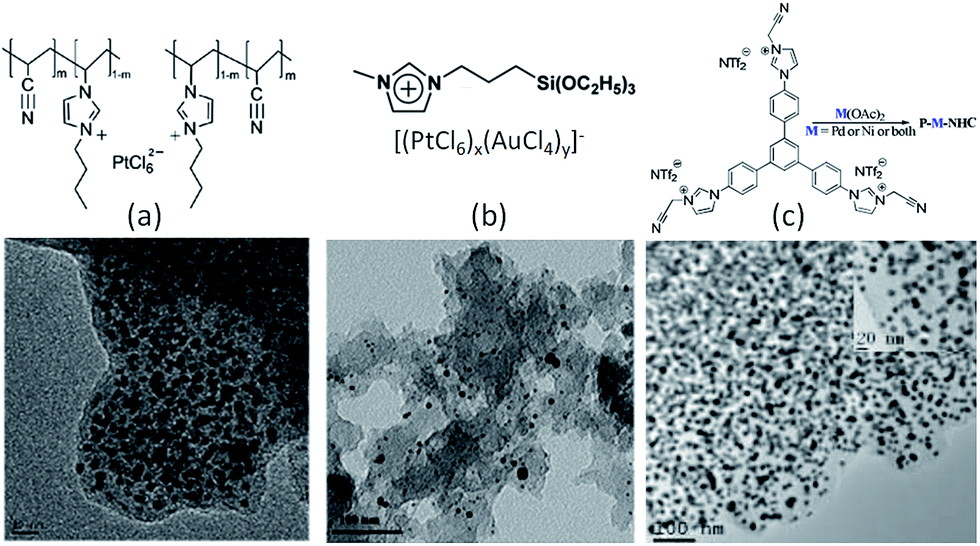 | ||
| Fig. 23 Typical IL-based precursors for one-pot synthesis of precious-metal nanoparticles supported by carbon materials. [Panel (a) adapted from ref. 119. Panel (b) adapted from ref. 120. Panel (c) reprinted with permission from ref. 121, copyright 2013 American Chemical Society.] | ||
5. Introducing pores into IL-derived carbons
Although IL-derived carbons have attractive features such as high nitrogen contents and graphitic structures, most of the directly carbonized samples are unsuitable for immediate applications, mainly because of non-porous structures or low surface areas. It is therefore desirable to maximize the potential applications of IL-derived carbons by introducing adequate porosity into the IL-derived carbon structure. As expected, high porosity facilitates mass transport and diffusion, providing abundant active sites for efficient processes such as chemical catalysis and electrocatalysis.5.1. Template-free method
As shown in Fig. 24, several methods have been used to produce porous carbons from ILs. Among them, direct carbonization of the precursor, without additional templates and complex procedures, i.e., the template-free method, is the simplest, but it is challenging, mainly because of the following two issues. First, most organic compounds, because of their low thermal stabilities, are completely evaporated/decomposed to volatile products during carbonization; secondly, even for those precursors that can survive severe carbonization conditions, pores are generally unavailable because of the lack of suitable and stable self-sacrificial porogens.25,93 Identification of appropriate carbon precursors, which could simultaneously address these two issues, is the key to guaranteeing successful fabrication of porous carbons via a template-free approach. The pioneering work by Dai et al. showed that a series of cyano-containing imidazolium ILs, if combined with bulky anions such as [NTf2] or bis(perfluoroethylsulfonyl)imide ([BETI]), could give rise to porous carbon via direct carbonization.25 For instance, simply replacing [NTf2] by chloride for a fixed cation [BCNIm] results in a decrease in the surface area from 640 to 15.5 m2 g−1, nearly complete loss of porosity. The cationic structure was found to affect the textural properties of the resulting carbon slightly. As shown in Fig. 25a, although carbon derived from [BCNIm][NTf2] with bisnitrile-functionalized cations was strictly microporous (type I isotherm), [MCNIm][NTf2], containing a single-nitrile-bearing cation, produced carbon with a typical type IV isotherm, with an associated H2-type hysteresis loop, indicating the presence of significant mesoporosity.125 The generation of mesoporosity with a surface area of up to 780 m2 g−1, without a templating phase, is remarkable; the non-cross-linked parts such as bulky anions are believed to act as self-templates during carbonization.25 Poly-ILs (Fig. 7a) derived from such IL monomers containing both nitrile-bearing cations and bulky anions also give rise to micro/mesoporous nitrogen-doped carbons, even at unexpectedly low temperatures (ca. 500 °C); the polymer backbone acts as the carbon precursor as well as the nitrogen source, and the counter-ion acts as a molecular template.126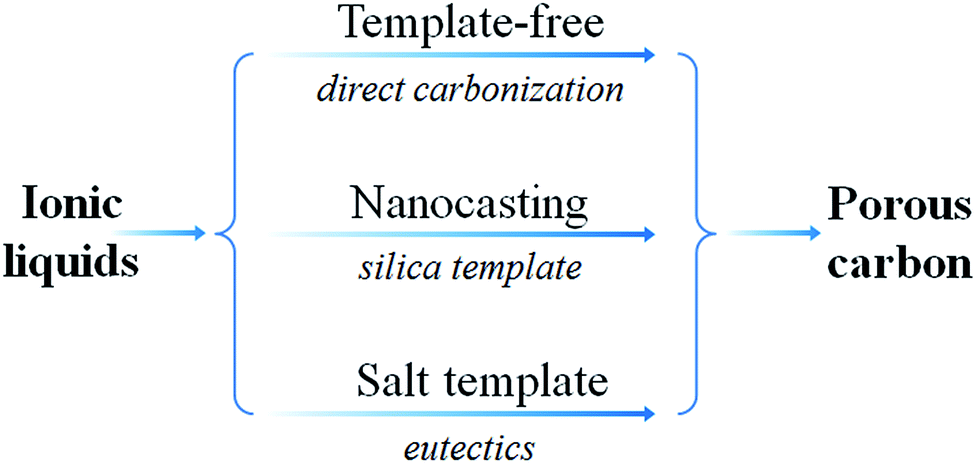 | ||
| Fig. 24 General routes for preparation of porous carbon from IL precursors. | ||
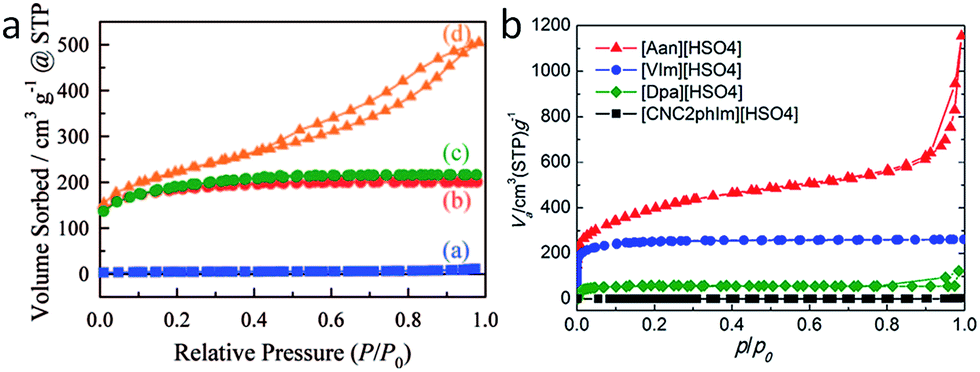 | ||
| Fig. 25 Nitrogen sorption isotherms of carbons from (left) cyano/nitrile-containing aprotic ILs ((a) [BCNIm][Cl]; (b) [BCNIm][NTf2]; (c) [BCNIm][BETI]; and (d) [MCNIm][NTf2]) and (right) from PILs and salts. [Panel a reprinted with permission from ref. 25, copyright 2010 American Chemical Society. Panel (b) reprinted with permission from ref. 93, copyright 2014 American Chemical Society.] | ||
The mesoporous structures of specific nitrile-containing ILs with bulky anions can be further developed by careful design of the cationic component.127,128 For example, modification of the cationic structure using a bisimidazolium motif (Fig. 26c) enabled synthesis of hierarchical nanoporous carbons via direct carbonization. Not only the length, but also the type, of the linker located between the two symmetric imidazolium cores was found to have a profound influence on the final porous structure. As shown in Fig. 26c and Table 2, when the number of methylene spacers is above four, a longer linker greatly decreases the surface area, from 870 m2 g−1 for [BBI][NTf2] to 588 m2 g−1 for [NBI][NTf2], mainly because of depression of micropore formation. Replacing the methylene spacer by a p-xylyl group yields a porous carbon with very high microporosity. Interestingly, for the IL [EBI][NTf2], with a very short methylene spacer, the resultant carbon exhibits a high surface area of 854 m2 g−1 and a very low microporosity (Smic = 269 m2 g−1), suggesting a mainly mesoporous structure. Changing the anion of [EBI][NTf2] from [NTf2] to a more bulky one, i.e., [BETI], can further improve the surface areas up to 1300 m2 g−1; however, this is mainly the result of micropore development.127
 | ||
| Fig. 26 Structures of ILs or IL mixtures that can generate mesoporous carbons. | ||
| IL | S BET [m2 g−1] | V tot [cm3 g−1] | S mic [m2 g−1] | V mic [cm3 g−1] |
|---|---|---|---|---|
| a Data from ref. 127. For structures of ILs, please refer to Fig. 26c. | ||||
| [EBI][NTf2] | 854 | 1.00 | 269 | 0.12 |
| [EBI][BETI] | 1277 | 1.28 | 757 | 0.34 |
| [BBI][NTf2] | 870 | 0.52 | 716 | 0.33 |
| [HBI][NTf2] | 643 | 0.33 | 601 | 0.28 |
| [NBI][NTf2] | 588 | 0.34 | 520 | 0.24 |
| [XBI][NTf2] | 706 | 0.33 | 686 | 0.32 |
In addition, certain PILs/PSs can directly yield porous carbons with porous structures, depending on the precursor structures (Fig. 25b).91,93 In particular, [Aan][HSO4]-based carbon has a surface area of 1380 m2 g−1. As shown in Fig. 25b, its nitrogen sorption shows a typical type I isotherm with two sharp uptakes at low (P/P0 < 0.15) and high (p/p0 < 0.9) relative pressures, respectively, indicating that it is a microporous material with some small macropores. A mixture of [BMIm][C(CN)3]/[BMIm][B(CN)4] (1:2) was also reported to give rise to porous carbon with slit-like pores and a specific surface area exceeding 500 m2 g−1via direct carbonization (Fig. 26b).41 Moreover, copyrolysis of IL monomers and IL homopolymers or block copolymers with metal salts enables the preparation of mesoporous carbon nanomaterials.69,122 In this case, the mesopores are thought to develop by migration and aggregation of metal nanoparticles during carbonization.116 Considering that porous carbons generally can only be obtained via a nanocasting method or activation, the generation of porosity, especially mesoporosity, from certain ILs, based on a template-free method, is significant and should be of wide interest.
5.2. Nanocasting method, mainly based on silica templates
Although certain ILs give porous carbons via template-free carbonization, as noted above, most IL-derived carbons have either no pores or very low surface areas in the absence of templates.26,35,118 In particular, cyano-containing ILs, because of the intrinsically high nitrogen contents, high conductivities, and graphitic structures of the resultant carbons, have been intensively investigated as carbonaceous precursors. However, the materials produced by direct carbonization have small or negligible surface areas, probably because of strong cross-linking polymerization and condensation. For example, the carbons obtained at 800 °C from [EMIm][DCA], [EMIm][C(CN)3], and [EMIm][B(CN)4] have surface areas as low as 3.41,118 3.8,31 and 65 m2 g−1,41 respectively. In most cases, therefore, sophisticated nanocasting techniques based on hard templates are needed to prepare porous carbons from ILs; the morphology of the obtained material can be controlled by manipulating the template structure, as in the case of traditional precursors. Unlike traditional precursors such as sucrose,129 furfuryl alcohol,130,131 and polyacrylonitrile,132 ILs can be directly carbonized at ambient pressure without any acidic or metal catalysts for prepolymerization prior to carbonization. This favors their easy incorporation into the voids of (inorganic or polymeric) templates to produce advanced nanoporous carbon materials. Other advantages of a liquid state and high thermostability, as well as the good energetic interactions between most ILs and inorganic surfaces,35,46,133 may also facilitate homogeneous coverage of ILs on the template surface, without the necessity of using additional solvents or pressure. It has been reported that favorable interactions of liquid salts with polar inorganic structures enables the synthesis of titania nanocrystals134 or hollow microspheres,135 mesoporous silica,136 photoluminescent EuF3 nanospheres,137 and other functional nanostructures.46The facile synthesis of nitrogen-doped mesoporous carbons was achieved via direct infiltration of ordered mesoporous silica (SBA-15) with a pure IL, i.e., [3MBP][DCA], followed by carbonization and template removal. The TEM in Fig. 27 shows good replication of the two-dimensional pore structure of the SBA-15 template in the nitrogen-doped carbon. Nitrogen sorption measurements revealed that this replica is indeed mesoporous, with a maximum pore size of 3.8 nm (compared with 4.2 nm for the SBA-15 template) and a specific surface area of 906 m2 g−1 (compared with 844 m2 g−1 for the SBA-15 template).26 SBA-15 was then used with other ILs for the synthesis of mesoporous carbon materials for various potential applications.138–142
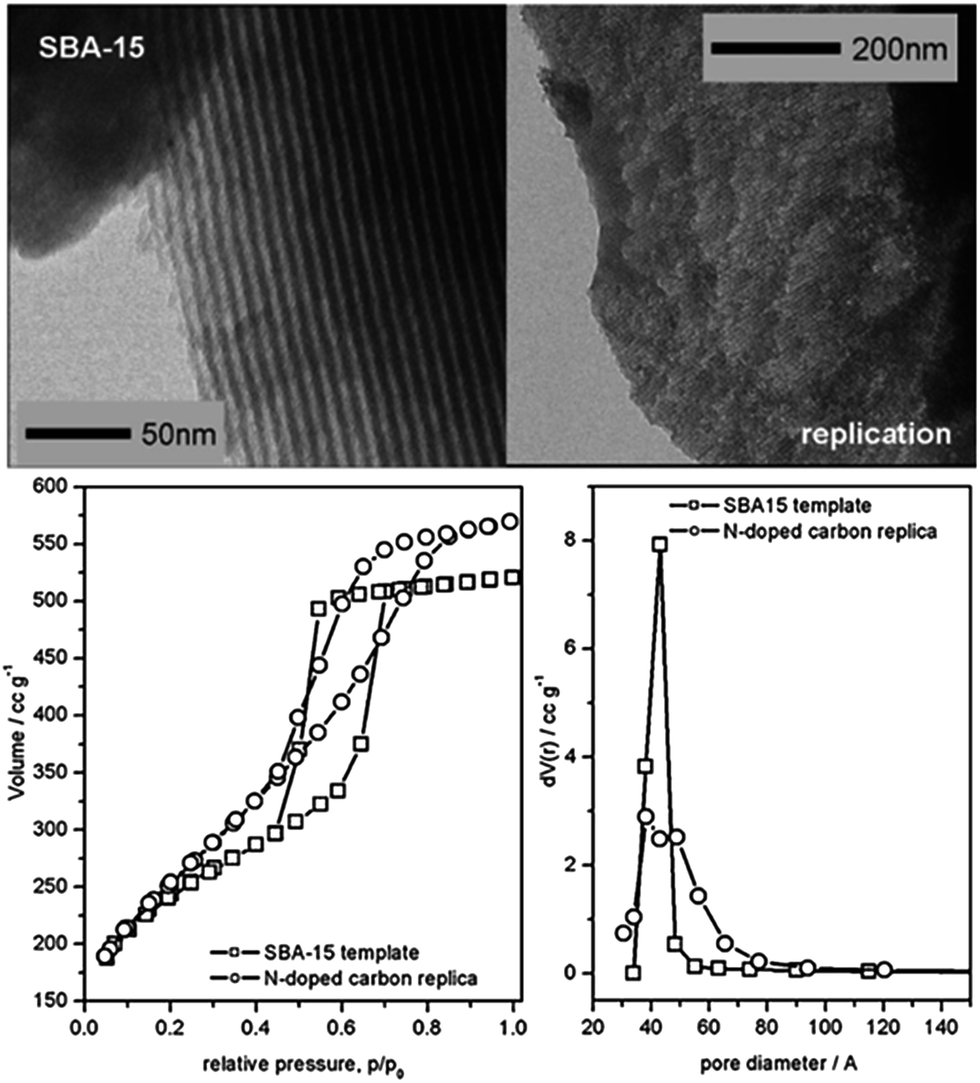 | ||
| Fig. 27 TEM images and nitrogen sorption isotherms of SBA-15 template and nitrogen-doped carbon replica from [3MBP][DCA]. [Reprinted with permission from ref. 26, copyright 2010 Wiley.] | ||
Although an SBA-15 template, which has periodically ordered hexagonal mesopores, can produce ordered porous carbons from ILs, the preparation of such ordered silica templates involves complex steps and is therefore expensive and time consuming.143,144 Alternatively, disordered, but low-cost and commercially available, colloidal silica materials such as LUDOX®HS-40 (12 nm silica nanoparticles) are ideal templates for use in nanocasting approaches to the generation of nanostructured, mesoporous carbons from IL precursors.26,27,34,35,145–148 For example, cocarbonization of [EMIm][DCA] with nucleobases in the presence of LUDOX®HS-40 yielded highly nitrogen-doped carbons with high surface areas and large pore volumes of up to 1553 m2 g−1 and 2.58 cm3 g−1, respectively.27 In contrast, the carbon obtained from a single IL precursor such as [BMP][DCA] has a much lower surface area, 320 m2 g−1,145 suggesting that the porosity of the templated carbon derived from ILs depends on the precursor structure.
A combination of a sol–gel process and an IL appended with a prolonged aliphatic chain ([NHP][DCA]) was used to synthesize mesoporous composite materials containing nitrogen-doped carbon and silica, by controlled condensation and subsequent thermal treatment.149 [NHP][DCA] played a bifunctional role during the synthesis: as an active cationic surfactant capable of inducing mesostructures, and as a precursor for advanced chemical modifications of the targeted materials. The concentration of [NHP][DCA] strongly affected the porosity and micrographs of the resultant carbons. With increasing concentration from 1.2 to 1.6 mmol, the surface area decreased from 994 to 717 m2 g−1, with bimodality appearing at high concentrations. The composite materials consisted of submicron-sized particles, and the mesopores tended to be more ordered when higher concentrations of [NHP][DCA] were used. However, after removal of the silica from the high-concentration composite, the obtained pure carbonaceous component did not exhibit any specific order and the surface area decreased to 467 m2 g−1. The collapse of the structure could be caused by mutual stabilization of the silica and nitrogen-doped carbon components.149 ILs with long aliphatic chains, such as 1-hexadecyl-3-methylimidazolium bromide ([C16MIm][Br]), can be used as both the carbon source and structure-directing agent, in combination with an in situ sol–gel process and simple one-step assembly.150 Partially graphitic, mesoporous carbon with a Brunauer–Emmett–Teller (BET) surface area of 372 m2 g−1 and mesopore volume of 0.61 cm3 g−1 was obtained. The nitrogen in the IL precursor was incorporated into the carbon framework, resulting in a content of 3.2 wt%.150 Impregnation of monodisperse silica spheres with [CnMIm][Br]-based ILs without cross-linkable groups gave nitrogen-doped, uniform HCSs, as a result of interactions between the ILs and the silica sphere surfaces.47 The morphology of the HCSs, including the size and shell thickness, can be adjusted by controlling the IL/silica sphere molar ratio. Considering that these ILs are generally viewed as non-carbonizable precursors, because of the lack of stable polymeric intermediates or formation of volatile decomposition intermediates, the interactions between the IL precursors and silica template are expected to be the key to inducing a carbon yield from the IL precursor and forming carbon spheres.43 It should be noted that this method is simpler than that using polymerizable IL monomers mentioned above.64,66,67 In addition, through simple coating of ILs on the surfaces of silica nanospheres and an annealing process, nitrogen-doped carbon nanobubbles52 or HCSs47 can be easily fabricated. The obtained nanobubbles have multiscale nanopores and abundant few-layer graphitic zones in the thin shells, which exhibit excellent ion-storage capabilities and enhanced cyclability in high-rate kinetic-dominated processes for LIBs.52
The nanocasting method can be extended to other hard templates such as porous alumina membranes,35 silica monoliths,35 and magadiite.114 In particular, in the presence of magadiite as a layered template, high-surface-area graphene-type nanosheets were obtained in high yields from imidazolium-based eutectic ILs containing certain transition-metal salts as carbon precursors.114 The proposed mechanism involves the intercalation of the ILs within the magadiite layers; intercalation proceeds until exfoliation of the silicate into few-layer organic–inorganic nanocomposites, carbonization, and magadiite dissolution. This facile method may enable the synthesis of dispersible carbon nanosheets from two-dimensional templates for energy-storage and -conversion devices.
5.3. Salt templates
The widely used silica-based templating method generally requires hazardous chemicals for template removal, so it is time and energy consuming. To overcome these drawbacks, Antonietti et al. recently developed a new technique called salt templating for preparing porous carbons using various low-melting point eutectic salt mixtures (Fig. 28).42,124,151–154 The overall procedure for salt templating is very simple (Fig. 28a).151 An IL (precursor) is mixed with a non-carbonizable inorganic eutectic salt (template), forming a glassy beige solid, which is condensed and scaffolded in the presence of the molten salt at elevated temperatures. After carbonization in a nitrogen atmosphere and simple aqueous removal of the salt porogen, highly porous nitrogen- or nitrogen/boron-doped carbons are obtained. The porogen can be recovered for further use, which results in a closed-loop process including salt recycling. The following two factors are believed to facilitate the homogeneity of the precursor mixtures and the generation of highly porous heteroatom-doped carbons. First, strong coulombic interactions exist between the ILs and salt molecules, which are proven by the exothermicity of the process of mixing of the precursor and template prior to carbonization. Secondly, the melting points of the eutectics are below the onset temperature of cross-linking of the IL units.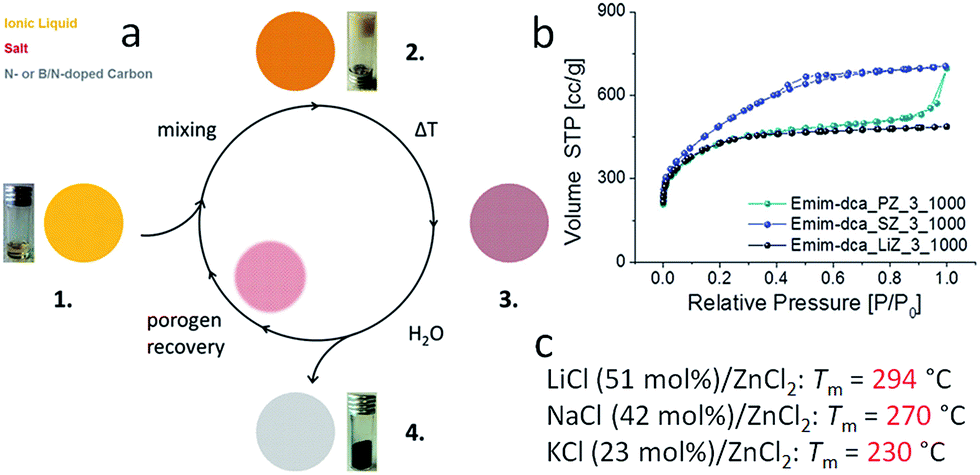 | ||
| Fig. 28 (a) Diagram of salt-templating approach, (b) nitrogen sorption isotherms of [EMIm][DCA]-derived carbons templated with LiZ (LiCl–ZnCl2), SZ (NaCl–ZnCl2), and PZ (KCl–ZnCl2), and (c) compositions and melting points of eutectic-based salt templates. [Reprinted with permission from ref. 151, copyright 2013 Wiley.] | ||
The pore morphology and porosity of the resultant carbon were shown to depend solely on the nature of the templating salt, and not on the IL. The nitrogen sorption isotherms of [EMIm][DCA]-based carbons are shown in Fig. 28b as an example. In the case of eutectic LiZ (LiCl–ZnCl2), the isotherm is type I, which implies that the carbon has a solely microporous structure, i.e., the salt presumably acts as a molecular template, either forming ion pairs or small salt clusters of minimal free energy (Fig. 29a). For carbons templated with SZ (NaCl–ZnCl2), the sorption isotherms of the materials show a further uptake in the medium relative pressure region, as well as a small hysteresis (Fig. 28b), reflecting a substantial contribution from additional supermicropores and small mesopores. This could be attributed to the lower melting point of SZ and larger salt clusters, resulting from the onset of phase demixing in the later stages of condensation, giving structures in the 4 nm range, which now act as a template (Fig. 29b). Finally, for materials templated with PZ (KCl–ZnCl2) which has the lowest melting point, additional uptake in the high relative pressure region is observed (Fig. 28b). This is typical of macroporous materials. Particle formation is ascribed to more pronounced demixing in earlier phases of the cross-linking reaction, resulting in a continuous salt phase (Fig. 29c), therefore interstitial voids between the micro- and meso-porous particles become visible. Based on a combination of the effects of salt concentration on pore formation, highly micro-, meso-, and macro-porous carbon materials can be easily obtained, depending on the salt nature and amount. The resultant carbon can be tuned to be highly porous, with surface areas up to 2000 m2 g−1, even higher than those of zeolites or activated carbons, and approaching the value for single-layer graphene.151 This newly developed salt-templating method has been used to prepare highly porous carbon/metal nitride nanocomposites,124 and nitrogen and sulfur codoped hierarchical porous carbons from non-carbonizable ILs.50 For example, by calcining a mixture of an IL ([Emim][DCA]) as both the nitrogen and carbon sources, a metal precursor (VOCl3 or NH4VO3), and simple salts (cesium and zinc acetate) as porogens, vanadium nitride nanoparticles embedded in a porous nitrogen-doped carbon matrix were prepared; the surface area, pore volume, pore size, and nanoparticle size can be tuned based on the amount and nature of the porogen salt.124
 | ||
| Fig. 29 Schematic representation of pore formation for carbons templated with LiZ (a), SZ (b), and PZ (c) at equal mass ratios. The upper image depicts the carbon (grey)/salt (red) composite, and the lower images show carbon after washing. [Reprinted with permission from ref. 151, copyright 2013 Wiley.] | ||
However, compared with the carbon obtained via direct carbonization of [EMIm][DCA], the salt-templating approach leads to a slight depression of the chemical functionality and structure. For example, the highest nitrogen content from [Emim][DCA]-based carbons is 6.1 wt% at 1000 °C,151 which is lower than the value of 10.4 wt% obtained in the absence of a template at the same temperature.26 In addition, instead of the well-developed and sharp stacking 002 peak observed for the directly carbonized carbon, the salt-templated carbon exhibits a broad, lower-shifted peak (Fig. 30), indicating that the graphitic structure becomes amorphous.
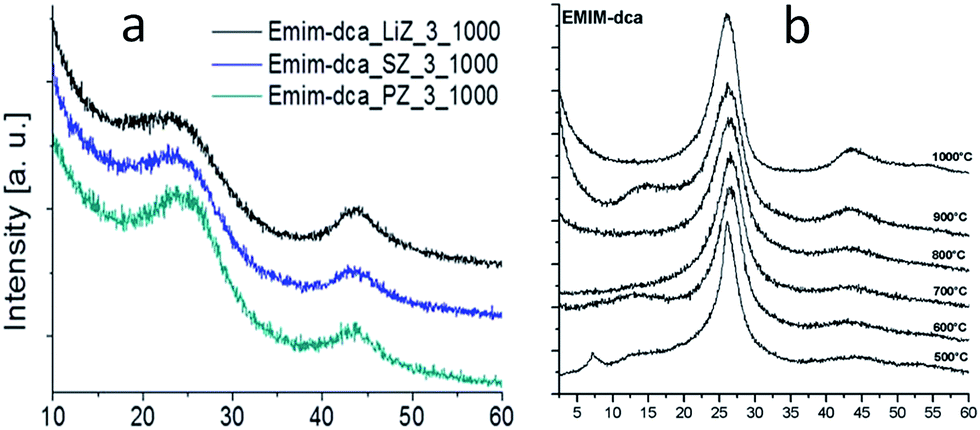 | ||
| Fig. 30 XRD patterns of carbon derived from [EMIm][DCA] (a) with or (b) without salt template. [Panel a reprinted with permission from ref. 151, copyright 2013 Wiley. Panel b reprinted with permission from ref. 26, copyright 2010 Wiley.] | ||
5.4. Other methods
IL-derived carbons prepared using hard templates such as mesoporous silica still suffer from low surface areas. Metal–organic frameworks (MOFs), because of their exceptionally large surface areas and controlled pore textures, have potential for use as templates for the synthesis of nanoporous carbon materials.155 Recently, Xu et al. demonstrated a simple method for fabricating high-surface-area, uniformly nitrogen- and boron–nitrogen-decorated nanoporous carbons by impregnation of cyano-containing ILs within a MOF to produce a precursor/template composite, followed by carbonization (Fig. 31).156 The obtained heteroatom-doped microporous carbons have narrow pore size distributions and high surface areas of up to 2397 m2 g−1. | ||
| Fig. 31 Schematic illustration of the procedure for nanoporous carbon synthesis from MOF and ILs. [Reproduced from ref. 156.] | ||
Incorporation of phosphorus-containing additives such as tetraalkylphosphonium bromide into the [BMP][DCA] IL precursor not only yielded nitrogen- and phosphorus-codoped carbons, but also promoted the formation of porous carbons with surface areas up to 283 m2 g−1.157 The properties of the alkyl chains and the additive concentration both determine the achieved surface area, indicating a templating effect of the phosphonium salts. In addition, Watanabe et al. found that blending common PILs/PSs such as [Dema][HSO4] with a non-carbonaceous [H2PO4]-containing PIL with only 12 wt% doping content greatly increased the surface area, from 690 to 1355 m2 g−1;91 this result is very similar to the activation effect of phosphoric acid.158–160
6. Applications of ionic-liquid-derived carbons
The use of IL precursors for carbon formation has developed rapidly in the past five years, and the properties of the obtained carbon materials make them highly promising for potential applications in diverse areas such as energy storage and conversion,27,145 gas storage,32 adsorption,126 sensing,56 and catalysis.146 However, it should be noted that the target applications depend strongly on the carbon structure, and therefore on the original precursor. For example, DES-derived carbons with highly microporous structures are mainly used as solid absorbents for CO2 capture or as electrode materials for supercapacitors, whereas [EMIm][DCA]-derived porous carbons with very high nitrogen contents are expected to be efficient electrocatalysts. More specifically, the applications can be divided into five main classes, i.e., electrocatalysis, LIBs, supercapacitors, CO2 capture, and chemical catalysis; these are discussed separately.6.1. Electrocatalysis
Electrocatalysis is a highly attractive application for electrochemical energy conversion and storage. The ORR is currently the technological bottleneck for the industrial development of fuel cells.161 Pt-based materials have long been investigated, as they are the most efficient catalysts for the ORR.162 However, the scarcity, high cost, sluggish ORR process, intolerance to fuel cross-over, and poor long-term stabilities of Pt-based catalysts are major obstacles. Nitrogen-doped carbon materials were recently shown to be efficient ORR electrocatalysts;28,163–165 they exhibit significant ORR performances, and have long-term stability and excellent resistance to cross-over effects.27,166 The highly nitrogen-doped carbons intrinsically obtained from IL precursors are therefore expected to find potential applications as metal-free electrocatalysts for the ORR.167,168 For example, via carbonization of nucleobases in [EMIm][DCA] in the presence of LUDOX®HS-40, mesoporous carbon materials with nitrogen contents of up to 12.0 wt% and high surface areas of up to 1553 m2 g−1 were obtained; they exhibited high ORR activities and durability.27 The abundant mesopores in the carbons facilitate efficient diffusion/mass transfer and provide a large amount of accessible active sites for electrocatalysis. Notably, the cathodic currents in alkaline media are comparable to those of commercial Pt/C catalysts. A mechanistic analysis based on the Koutecky–Levich equation revealed a four-electron process (n = 4.1), indicating selective reduction of oxygen to water. IL-derived nanostructured carbon materials such as nitrogen-doped hollow macroporous carbon spheres,71 carbon nanobubbles,72 and nitrogen- and sulfur-dual-doped porous carbons50 also have high ORR activities. In particular, one of the nitrogen-doped porous carbons (A-NPC), obtained by direct carbonization of a PS, exhibited excellent electrocatalytic activity in the ORR, as shown by cyclic voltammetry (CV) and rotating ring–disk electrode (RRDE) measurements (Fig. 32). A-NPC has an onset potential and half-wave potential very close to those of commercial Pt/C. The calculated peroxide yield was <15%, and the average electron-transfer number (n) was >3.7.93 Clearly, the ORR performance of this catalyst is much better than most of those reported for carbon-based catalysts that require special templates and complicated synthetic steps,165,169 probably because of its high surface area (1380 m2 g−1), high content of pyridinic nitrogen, and possible synergistic activation by sulfur and nitrogen dual doping.169 In addition, instead of water as the main product, as observed for most IL-derived carbon electrocatalysts obtained at 1000 °C, a pure two-electron reduction process from oxygen to H2O2 was achieved using nitrogen-doped carbon synthesized by silica-templated carbonization of a [DCA]-based IL at a relatively low temperature (800 °C).145 The obtained porous carbon has a very high nitrogen content, 14.2 wt%, as determined by XPS, and has higher catalytic activity in the ORR than commercial carbon (Vulcan XC 72R), even in an acidic medium. The electron-transfer number was calculated to be very close to 2, indicating a pure two-electron mechanism. This high selectivity was verified by comparison of the CVs obtained in a deoxygenated electrolyte before and after the addition of H2O2. In both cases, no reduction peak or faradaic current was observed, but subsequent oxygen purging led to strong reduction currents. More recently, nitrogen-doped carbon aerogels, made from [EMIm][DCA] using the salt-templating strategy, with surface areas up to 1800 m2 g−1, were found to catalyze four-electron oxygen reduction to water. An outstanding ORR performance with a well-defined diffusion-limited region was achieved in both acidic and alkaline media, comparable to those of Pt-based commercial catalysts.154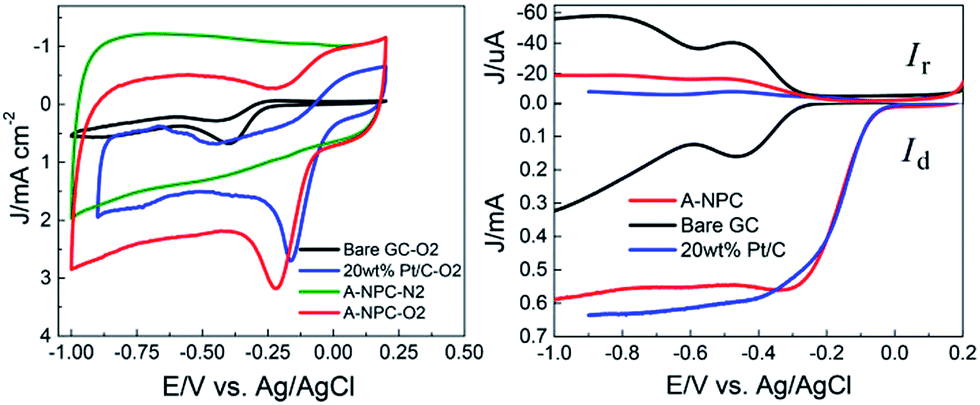 | ||
| Fig. 32 CV and RRDE results for bare glassy carbon, A-NPC, and commercial 20 wt% Pt/C in O2-saturated 0.1 M KOH solution. [Reprinted with permission from ref. 93, copyright 2014 American Chemical Society.] | ||
In order to understand if and how the porosities and pore size distributions of carbon materials affect the ORR activity, a series of nitrogen-doped carbons were obtained by direct carbonization of specific ILs containing a bis[N-(2-cyanoethyl)]imidazole core and bulky anions.128 As shown in Fig. 33a, the nitrogen contents and N/C ratios in all the catalysts are affected by the carbonization temperature rather than the type of starting material. In addition, the resultant carbons exhibit various porous structures (i.e., micro- and/or meso-pores), depending on the IL structure (Fig. 33b), with surface areas varying in the range of 588–1277 m2 g−1. These totally metal-free carbons were tested in the ORR. Interestingly, a comparison of three catalysts carbonized at the same temperature showed that the half-wave potentials (E1/2) increase monotonically with the surface areas (Fig. 33d). These results indicate that the porous structure of the catalyst is a key factor in ORR activity. The pore limiting currents calculated from Koutecky–Levich plots are 5.41, 6.90, and 3.92 mA for EBi-T/950, EBi-B/950, and NBi-T/950, respectively. These results show that similar to nitrogen doping, the pore size distribution is of significance in the ORR activity of a carbon-based catalyst. Hierarchical micro- and meso-porous structures may increase the activity.
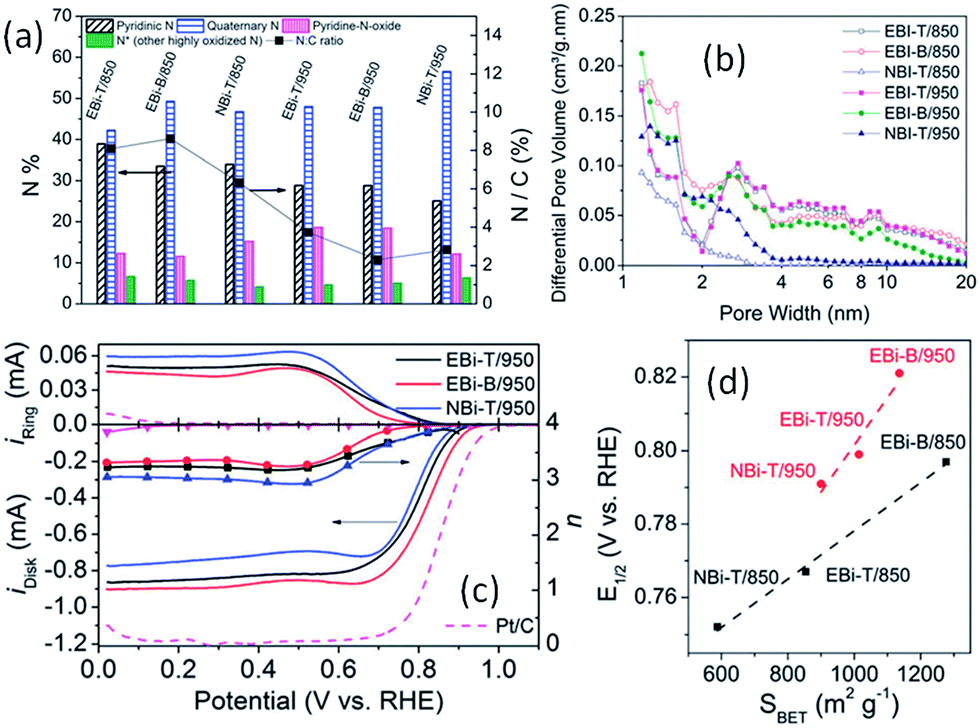 | ||
| Fig. 33 (a) Nitrogen composition and (b) pore size distribution of different carbons derived from ILs containing a bis[N-(2-cyanoethyl)]imidazole core. (c) Disk (lower part) and ring current (upper part), and number of transferred electrons (n, symbol line) of IL-derived carbon catalysts. (d) Correlation between the half-wave potentials (E1/2) and surface areas (SBET) of different carbons. [Adapted from ref. 128.] | ||
In addition to having intrinsic electrocatalytic activities, IL-derived carbon materials supporting metal nanoparticles are also particularly promising for electrocatalysis such as ORR or methanol oxidization. These nanoparticle-containing carbons are synthesized either by in situ one-pot carbonization or a post-deposition method.67,68,119,120,147 Most of the catalysts show activities comparable to those of benchmark commercial Pt/C catalysts, and, sometimes, significantly increased Pt electrochemical active surface areas are observed.
6.2. Li-ion batteries
LIBs are extensively used in portable electronic devices, because of their high energy densities and long cycling life-times. However, graphite, the most commonly used anode material in commercial LIBs, has a very limited Li-intercalation capacity (LiC6, 372 mA h g−1), which hinders its applications in transportation and large-scale energy storage. As alternatives to graphite, nanostructured porous carbons have been studied as candidates for anode materials in LIBs, because of their good chemical and thermal stabilities and high Li+-storage capacities. In addition, the presence of heteroatoms such as nitrogen on the carbon surface can enhance the reactivity and electrical conductivity.170,171 IL-based nitrogen-doped carbons can be directly used as anode materials for LIBs.40,52 For example, IL-derived carbon nanobubbles exhibit an initial discharging capacity of more than 3000 mA h g−1 at a current density of 1 A g−1, as well as excellent rate performances of up to 20 A g−1.52 Almost 100% capacity retentions were achieved for galvanostatic discharging at 1, 10, and 20 A g−1, with a coulombic efficiency of nearly 100% from the fifth cycle. This superhigh capacity compared with the theoretical capacity of graphite can be explained by the presence of other Li-storage mechanisms such as storage in cavities and nanopores, in addition to the classical graphite intercalation compound mechanism.170,171 However, as observed for most porous carbons, only half of the initial discharging capacity is reversibly achieved in the charging process, suggesting a very low initial coulombic efficiency (<50%). This can be attributed to their high specific surface areas and irreversible Li insertion into special sites, such as in the vicinity of residual hydrogen atoms in carbonaceous materials, which can result in decomposition of the electrolyte and the formation of solid–electrolyte interphase (SEI) films at the electrode–electrolyte interface.170,171,175In contrast to nitrogen-doped porous carbons, spinel Li4Ti5O12, a zero-strain insertion material, gives an excellent cycling performance and a relatively high lithiation voltage plateau, at 1.54 V versus Li/Li+, which avoids SEI formation, making it a promising anode material for large-scale, long-life, energy-storage batteries. However, its low electronic conductivity and moderate Li+ diffusion coefficient severely limit the high rate performance.176 Nitrogen-doped-carbon-coated Li4Ti5O12 was obtained by carbonization of [EMIm][DCA] as a single IL precursor at 600 °C in an Ar atmosphere.98 The nitrogen-doped carbon coating derived from an IL is effective in improving the rate capability and charge/discharge properties. Compared with either pristine Li4Ti5O12, or a solely carbon-coated sample produced using sugar as a precursor, the nitrogen-doped-carbon-coated sample has a much better rate capability at high current rates of 5 C and 10 C (Fig. 34a and b). Capacities of 145 and 129 mA h g−1, respectively, were achieved; the capacities of pristine Li4Ti5O12 at the same rates were 60 and 15 mA h g−1, respectively. In addition, the coated samples exhibit excellent cycling performances compared with pristine Li4Ti5O12. Even after 2200 cycles at 2 C, nearly 83% of the initial capacity was retained (Fig. 34d). The carbon contents of the coated samples were found to affect the specific capacity at high current rates (Fig. 34c). A moderate carbon content of 7.0 wt% (sample 2) provides a thin but uniform nitrogen-doped carbon layer, which enhances the electrochemical performance. This is ascribed to significant electron transfer from the Li4Ti5O12 surface to the nitrogen-doped-carbon coating layers and the improved interfacial stability and electrical conductivity.53 This IL-based carbon coating method can be extended to other cathode or anode materials such as NiO nanocrystals,173 rod-like LiFePO4,49 Fe2O364 and Fe3O448 nanoparticles, TiO2 nanofibers,172 and Li3V2(PO4)3,51 even using non-carbonizable ILs.173Table 3 summarizes the corresponding battery performance. Clearly, most of them were carbon-coated metal oxides and were explored as anodes for LIBs. The resulting battery performances are somewhat related to the intrinsic capacity of the metal oxide. Although cyano/nitrile-containing aprotic ILs generally result in carbons with low surface area and low reversible capacity for LIBs, they are likely to give rise to high initial coulombic efficiency and good rate capability at high current rates.40,98 Instead of aprotic ILs, some of the above-mentioned specific PILs may be also suitable organic precursors to coat metal oxide with a thin layer of N-doped carbon in the future, because the resultant carbons possess simultaneously high N content, high graphitic degree, and high conductivity. More importantly, they are easily available and low cost.
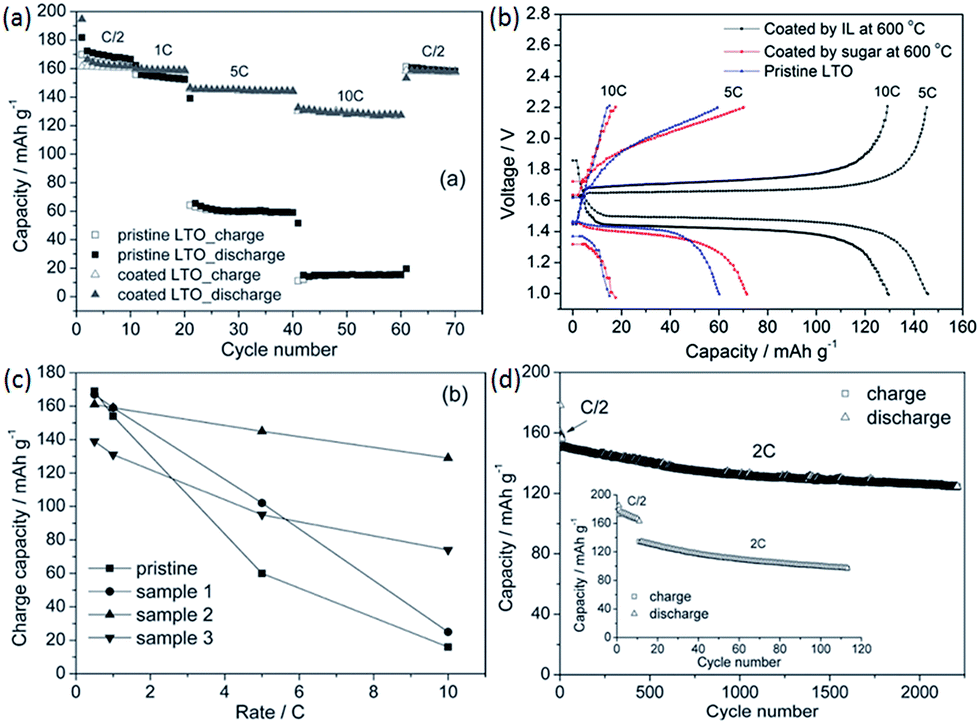 | ||
| Fig. 34 (a) Discharge/charge capacities at different current rates, (b) discharge/charge curves at 5 C and 10 C rates, and (c) specific capacities at different current rates. Samples 1, 2, and 3 are Li4Ti5O12 coated with 3.6, 7.0, and 9.6 wt% carbon, respectively. (d) Cycling performances of pristine Li4Ti5O12 and nitrogen-doped-carbon-coated Li4Ti5O12. [Reprinted with permission from ref. 98, copyright 2011 Wiley.] | ||
| ILs | Electrode materials | Anode/cathode | Reversible capacity | Cycling stability | Ref. |
|---|---|---|---|---|---|
| [EMIm][DCA] | N-doped carbon coated Li4Ti5O12 | Anode | 150 mA h g−1 at 2 C | 124 mA h g−1 at 2 C after 2000 cycles | 98 |
| 145 mA h g−1 at 5 C | |||||
| 129 mA h g−1 at 10 C | |||||
| [EMIm][DCA] | N-doped mesoporous carbon decorated TiO2 nanofibers | Anode | 200 mA h g−1 at 1 C | 264 mA h g−1 at 0.1 C after 100 cycles | 172 |
| 150 mA h g−1 at 5 C | |||||
| [EMIm][BF4] | N-doped carbon coated NiO nanocrystals | Anode | 690 mA h g−1 at 1 C | 610 mA h g−1 at 0.3 C after 50 cycles | 173 |
| 600 mA h g−1 at 5 C | |||||
| 470 mA h g−1 at 10 C | |||||
| LiC(CN)3 | Li ion-doped carbon | Anode | 500 mA h g−1 at 50 mA h g−1 | 500 mA h g−1 at 50 mA h g−1 after 100 cycles | 40 |
| LiC(CN)3 | Lt-600 coated mesoporous carbon | Anode | 620 mA h g−1 at 50 mA h g−1 | 540 mA h g−1 at 50 mA h g−1 after 100 cycles | 40 |
| [C8MIm][Cl] | N-doped carbon-coated Fe3O4 | Anode | 980 mA h g−1 at 100 mA h g−1 | 880 mA h g−1 at 100 mA h g−1 after 100 cycles | 48 |
| [EMIm][NTf2] | N-doped carbon-coated LiFePO4 | Cathode | 116 mA h g−1 at 2 C | 122 mA h g−1 at 0.1 C after 30 cycles | 49 |
| 98 mA h g−1 at 5 C | |||||
| 129 mA h g−1 at 10 C | |||||
| [CMVIm][Br] | N-doped hollow carbon loaded with Fe2O3 | Anode | 1120 mA h g−1 at 100 mA h g−1 | 700 mA h g−1 at 100 mA h g−1 after 65 cycles | 64 |
| [AMIm][Br] | N, O-codoped carbon nanobubbles | Anode | 1394 mA h g−1 at 1 A h g−1 | 1308 mA h g−1 at 1 A h g−1 after 130 cycles | 52 |
| 903 mA h g−1 at 2 A h g−1 | |||||
| 622 mA h g−1 at 5 A h g−1 | |||||
| [C4mpyr][NTf2] | Carbon-coated Li3V2(PO4)3 | cathode | 120 mA h g−1 at 1 C | 108 mA h g−1 at 10 C after 100 cycles | 51 |
| 116 mA h g−1 at 10 C | |||||
| 110 mA h g−1 at 20 C |
6.3. Supercapacitors
Supercapacitors are attracting increasing interest because of their high power density, which are highly desirable for applications in electric vehicles. Hierarchical porous monoliths were obtained by carbonization of a composite of multiwalled CNTs (MWCNTs) and poly(furfuryl alcohol) gels formed in DES-based ILs.89 The DES-assisted in situ condensation of furfuryl alcohol not only promoted the formation of monoliths, but also provided a carbon shell that coats every MWCNT and forms strong junctions between them (see Fig. 35), resulting in a high conductivity of up to 4.8 S cm−1 and a high elasticity (Young's modulus, 11 MPa). The resultant carbon monolith retains 75% of the initial capacitance at a high current density of 765 mA cm−2 (ca. 29 A g−1), much better than most porous carbon monoliths. The rapid electrical response of the monolithic composite was also reflected in the preservation of the rectangular CV shape at scan rates of up to 300 mV s−1 (Fig. 35e). It should be noted that direct processing of the monolithic carbon materials enables their direct assembly (without any further processing, such as using a binder to form pellets and/or adding carbon black to improve the electrode electrical conductivity) into supercapacitor cells (Fig. 35d). Phosphate-functionalized carbon monoliths derived from DES-based ILs were reported to have high surface areas, micropores capable of accommodating protons and ions from aqueous electrolytes, and macropores that allow mass transport and accessibility to the micropores.84 The phosphate functionalization of carbon materials broadens the operational voltage window of the electrode to 1.4 V,177 and high energy densities of up to 13 W h kg−1 were achieved.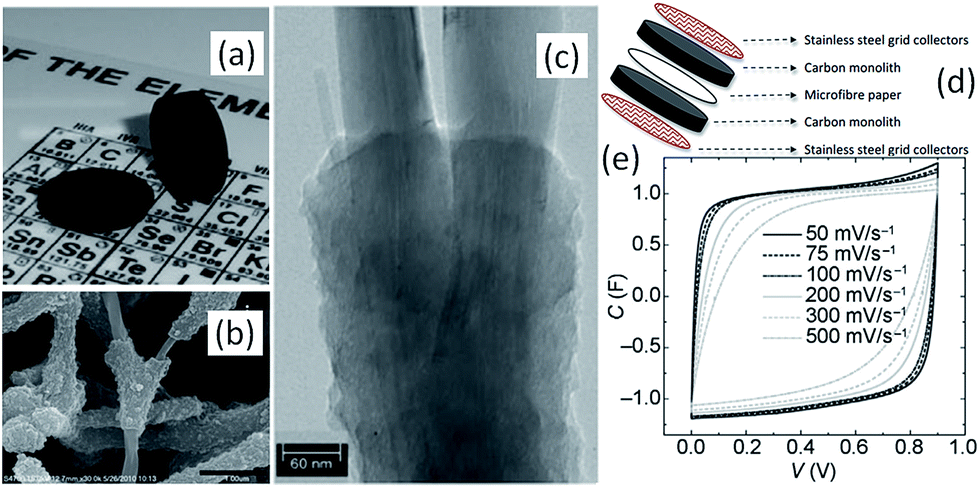 | ||
| Fig. 35 (a) Photograph, (b) SEM, and (c) TEM of MWCNT-based carbon monolith, (d) schematic diagram of the assembly of the supercapacitor cell from carbon monolith electrodes, and (e) CV recorded in H2SO4 (2 M) at voltage scan rates between 50 and 500 mV s−1. [Reprinted with permission from ref. 89, copyright 2011 Wiley.] | ||
In comparison with nitrogen-free carbons obtained from DESs, the nitrogen-doped porous carbons from classic IL precursors involving nitrogen-containing cores such as imidazolium or cyano/nitrile groups are considered to be promising electrode materials for supercapacitors,127,142 because of their wettability by electrolytes, high conductivity,178 and additional pseudo-capacitance, especially in acidic electrolytes,179,180e.g., nitrogen-doped hierarchical nanoporous carbons obtained by direct, template-free carbonization of ILs with a bisimidazolium motif and bulky anion displayed capacitance values above 100 F g−1 at slow scanning rates.127 Carbonization of a fluidic IL, [EMIm][DCA], in an SBA-15 template at 900 °C gave a nitrogen-doped mesoporous carbon material with a high nitrogen content of 8.27 wt% and high surface area of 931 m2 g−1; it has a high specific capacitance of 210 F g−1 and excellent cycling stability, even after 1000 cycles at a current density of 1 A g−1.142 Other nanostructured carbon materials obtained through IL-assisted synthesis have been widely explored as electrode materials for supercapacitors; in these, the ILs have one or multiple roles, e.g., as nitrogen dopants and shape-directing agents in microporous carbon nanosheets,101 as nitrogen and boron sources in hierarchically porous carbons,105 as templates,107 and as nitrogen-rich carbon precursors in graphene/N-doped porous carbon composites.103 Two-dimensional nitrogen-doped carbon sheets obtained using IL-functionalized GO as a shape-directing agent and a resorcinol–formaldehyde polymer as a carbon precursor have a microporous structure with a pore size distribution centered at 0.53 nm and a hydrophilic surface (contact angle ∼69°). The assembled supercapacitor has a specific capacitance of 213 F g−1 at 0.5 A g−1, excellent capacitance retention, and a long cycling life.101 To give easy reference to the reader, we listed the supercapacitor performance based on different types of ILs/DES-derived carbons. As shown in Table 4, it appears that direct comparison between these carbon materials is impossible because the specific capacitances were obtained at different current densities or from different methods (cyclic voltammogram or galvanostatic charge/discharge). However, the nitrogen doped structure formed during in situ carbonization of ILs is expected to be important to the excellent performances, which not only improves the conductivity, but also sometimes contributes pseudocapacitance to the double-layer capacitance. In addition to this point, desirable porous structure, which is essential to supercapacitor electrodes, is required to be carefully designed by introducing a suitable template.
| ILs/DES | Electrode materials | Surface area [m2 g−1] | Electrolyte | Specific capacitance | Ref. |
|---|---|---|---|---|---|
| DES-2 | Hierarchical porous MWCNTs | 415 | 2 M H2SO4 | 90 F g−1 at 765 mA cm−2 | 89 |
| [EMIm][DCA] | Graphene/N-doped porous carbon composite | 404 | 1 M H2SO4 | 206 F g−1 at 0.1 A g−1 | 103 |
| DES-6 | P-functionalized carbon monolith | 687 | 2 M H2SO4 | 197 F g−1 at 1.27 mA cm−2 | 84 |
| [BMIm][DCA] | N-doped mesoporous carbon | 931 | 6 M KOH | 210 F g−1 at 1 A g−1 | 142 |
| [EBI][BETI] | N-doped hierarchical nanoporous carbon | 1277 | 6 M KOH | Ca. 93 F g−1 at 100 mV s−1 | 127 |
| Ca. 143 F g−1 at 10 mV s−1 | |||||
| [C16MIm][BF4] | Hierarchical porous carbons | 376 | 6 M KOH | 247 F g−1 at 0.5 A g−1 | 105 |
| [BMIM][Br] | Microporous carbon nanosheet | 791 | 6 M KOH | 341 F g−1 at 5 mV s−1 | 101 |
| 213 F g−1 at 0.5 A g−1 |
6.4. CO2 capture
The selective capture and storage of CO2 has attracted considerable attention in recent years, because CO2 is a major greenhouse gas and a renewable carbon source. Nitrogen-doped porous carbons with less basic sites have emerged as promising sorbent solids for physisorption and regeneration of CO2, because of their exceptionally high surface areas, low cost, chemical inertness, and wide availability.181–188 Strong binding interactions such as dipole–quadrupole interactions,187 acid–base interactions,185 and hydrogen bonding189 between CO2 molecules and the nitrogen-modified heterogeneous pore walls within the carbon framework significantly improve the CO2 affinity of nitrogen-doped adsorbents. Nitrogen-doped porous carbons derived from ILs are therefore promising materials for CO2 capture. For example, a high CO2 adsorption capacity of 4.39 mmol g−1 at 0 °C and 1 atm was recently achieved using a microporous, nitrogen-doped porous carbon obtained by direct carbonization of an imidazolium-based IL containing nitrile group, at 500 °C.32 Notably, highly nitrogen-doped porous carbon materials obtained from low-cost, easily available, PILs/PSs are potential candidates for CO2 capture.91 In particular, [Dema][HSO4]-based carbon, despite its low surface area (690 m2 g−1) and nitrogen content (2.1%), showed a higher CO2 uptake (2.63 mmol g−1 at 25 °C and 1 atm) than other carbons. Taking into account that both nitrogen content and microporous surface area are key issues for efficient CO2 capture,187–190 the remarkably high CO2 uptake capacity is ascribed to its intensive and narrow pore size distribution of 1.12 nm, which is reported to be effective for efficient CO2 adsorption.191–193 In contrast, [Aan][HSO4]-based carbon, despite having the highest surface area (1380 m2 g−1), showed the lowest CO2 uptake, with a value of 1.63 mmol g−1; this is probably because of its large micropores and wide pore size distribution, which extends to mesopores and macropores.Nitrogen- and nitrogen/boron-doped nanoporous carbons with high nitrogen contents and large surface areas, obtained by cocarbonization of cyano-containing ILs and MOFs, exhibited excellent CO2 adsorption capacities.156 As shown in Fig. 36a, sample NC800, obtained from [EMIm][DCA] at 800 °C, which has the highest basic nitrogen content (14.8 wt%), but a lower surface area (2397 m2 g−1) than undecorated carbon C800 (2731 m2 g−1), showed much higher CO2 uptakes at 1 atm (5.7 mmol g−1 at 273 K and 3.8 mmol g−1 at 298 K) compared with C800 (4.1 mmol g−1 at 273 K and 2.6 mmol g−1 at 298 K). The high microporosity and high charge density at the nitrogen sites, which might facilitate interactions with polarizable acidic CO2 molecules through local dipole–quadrupole interactions,187,194 were predominantly responsible for the marked improvement in CO2 uptake capacity. DES-based carbons are also potential candidates for efficient CO2 capture, but in this case, nitrogen-containing components such as tetraethylammonium bromide or 3-hydroxypyridine are often needed for nitrogen doping into the final carbon to improve the CO2 capacity.85–87 Nitrogen-doped carbon monoliths with high microporous surface areas, obtained from a DES containing resorcinol, 3-hydroxypyridine, and tetraethylammonium bromide, are particularly suitable for CO2 capture, and have outstanding adsorption capacities of up to 3.3 mmol g−1 at 25 °C, along with excellent recyclabilities and stabilities.86 It is known that the adsorption performance of CO2 capture for N-doped porous carbon materials is generally evaluated by the adsorption capacity at 0 °C and 25 °C, adsorption heat, selectivity, etc.Table 5 summarizes these values for ILs/DES-based carbons. Both ILs and DES-derived carbons exhibited moderate heats of adsorption for CO2, thus avoiding the energy penalties that are typically required for regeneration of functionalized adsorbents. In addition, IL-derived carbons obviously exhibit a much higher selectivity than DES-derived carbons. Very interestingly, carbon monoliths with a tailor-made narrow microporosity obtained from specific DES (DES-7 and DES-8), despite their very low surface areas (ca. 4 m2 g−1, determined by N2 sorption at 77 K), exhibit good capacities, even comparable to the IL-derived carbons with very high surface areas, as listed in Table 5. This unexpected result could be ascribed to the small micropores (ca. 0.6 nm) of the carbon material. Having a molecular sieve network. Such a molecular sieve network with narrow pore dimension restricted the diffusion of N2 at 77 K, while selectively adsorbed CO2 with a smaller molecular size.85
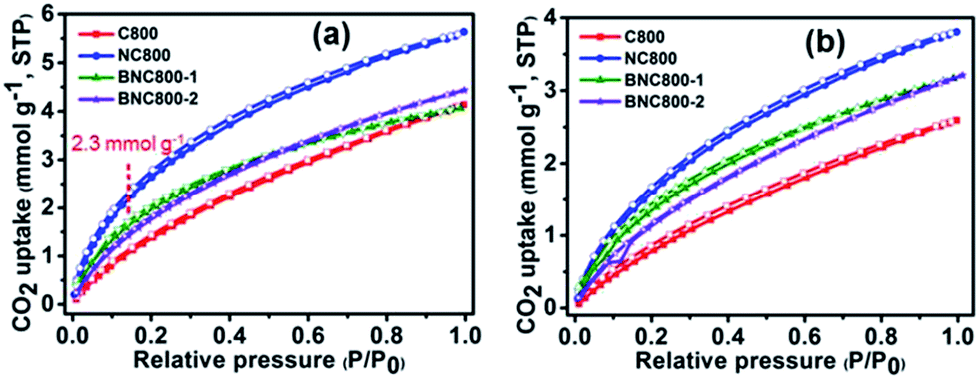 | ||
| Fig. 36 CO2 uptakes at (a) 273 K and (b) 298 K by N- and BN-decorated nanoporous carbons derived from IL and MOF. [Reproduced from ref. 156.] | ||
| ILs/DES | Surface area [m2 g−1] | Heat [KJ mol−1] | Capacitya [mmol g−1] | Selectivity CO2/N2 | Ref. | |
|---|---|---|---|---|---|---|
| 0 °C | 25 °C | |||||
| a Capacity for CO2 adsorption was measured at 1 atm. | ||||||
| [BCNIm][NTf2] | 942 | 32 | 4.39 (19.3 wt%) | 13.5 wt% | N/A | 32 |
| EBI-B | 1013 | 31.5 | 4.07 | 2.74 | 30 | 174 |
| [Dema][HSO4] | 690 | N/A | N/A | 2.63 | N/A | 91 |
| [EMIm][DCA] + MOF | 2397 | 26 | 5.7 | 3.8 | 57 | 156 |
| DES-5 + formaldehyde | 666 | 30–31 | N/A | 3.3 | 4.3–5.7 | 86 |
| DES-7 + formaldehyde | 4 | 29.1 | 3.0 | 2.2 | 8.4 | 85 |
| DES-8 + formaldehyde | 4 | N/A | 3.7 | 2.7 | N/A | 87 |
6.5. Chemical catalysis
It is known that precious metals such as Pt coordinated within a highly nitrogen-rich environment are stable enough to survive several recycling steps.195 Their combination of high nitrogen content and excellent porosity makes IL-derived porous carbons promising candidates for catalyst supports for stabilizing highly dispersed precious-metal nanoparticles (e.g., Pd, Au, and Ru) for various applications such as biomass refining,146 Ullmann coupling reactions,141,148 hydrogenation,111,196 aerobic oxidation,139 carbonylation,121 and phenylacetylene reduction.75,150 The heterojunction between nitrogen-doped carbon and metal nanoparticles is believed to lead to very stable and uniform dispersion of metal nanoparticles and additional electronic activation of the metal nanoparticles, resulting in high catalytic performances. For example, a task-specific IL, [3BMP][DCA], as a precursor and colloidal silica as a hard template, accompanied by a post-deposition method, were used to form heterogeneous mesoporous nitrogen-doped carbon with a high nitrogen content (12 wt%) as a support for Pd nanoparticles.146 The HRTEM images show that Pd nanoparticles of average size 4.1 nm were well dispersed on the surface of the nitrogen-doped carbon (Fig. 37). Two Pd crystal planes, i.e., (111) and (200), are clearly observed, with a plane angle of 55°. The resulting novel Pd catalyst shows very high catalytic activity in hydrodeoxygenation of vanillin (a typical model compound for lignin) at low hydrogen pressure under mild conditions, using water as a clean solvent. Excellent catalytic results (100% conversion of vanillin and 100% selectivity for 2-methoxy-4-methylphenol) were achieved without loss of catalytic activity after six cycles.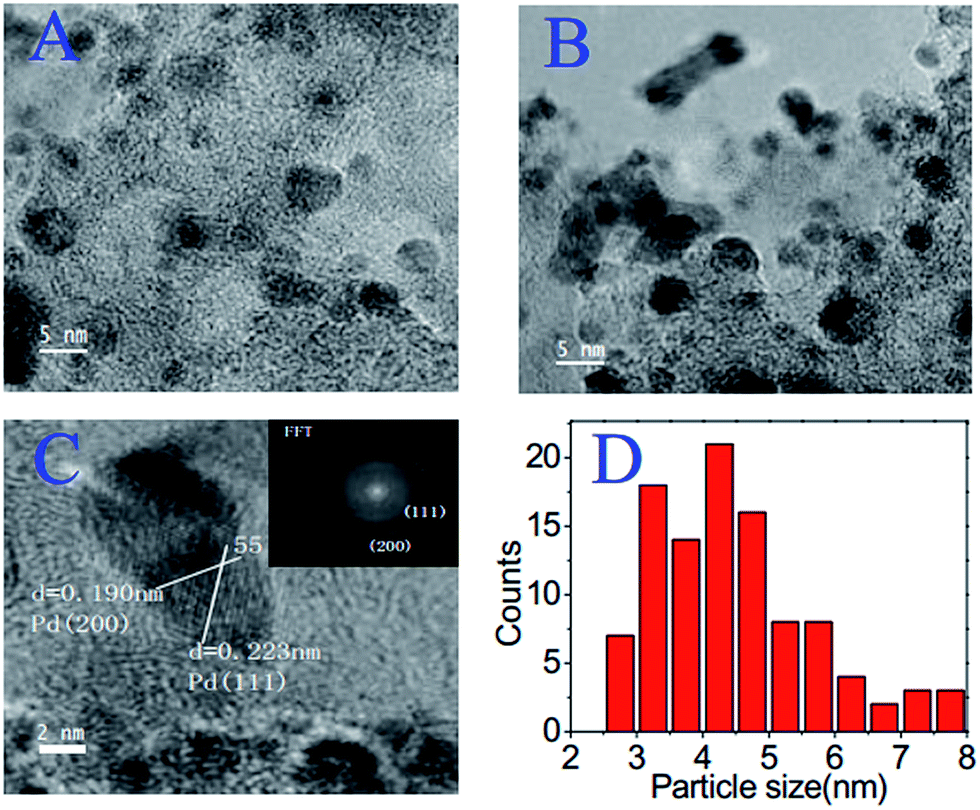 | ||
| Fig. 37 HRTEM mages and particle size distribution of Pd nanoparticles supported on IL-derived mesoporous nitrogen-doped carbon. [Reprinted with permission from ref. 146, copyright 2012 American Chemical Society.] | ||
Using ILs as precursors also provided us with a novel in situ method other than the wet chemical reduction method to prepare carbon supported precious-metal nanoparticles.139,146 For example, precious-metal nanoparticles supported on porous carbon can be obtained by “one-pot” carbonization of metal-containing ILs,75,111,119,121 wherein ILs and metal salt behave as the organic carbon precursor and metal source, respectively. In contrast to the traditional postdeposition and reduction method, the in situ one-step method may give rise to a more homogenous description of the metal nanoparticles. Most work stressed the important effect of high N content on catalysis for IL-derived carbons, however, fully understanding the real role of the doped N remains challenging. In addition, despite the significant advance in the catalytic application of IL-derived carbons, most of the reported catalysts were limited to precious-metal nanoparticles, only very few studies related to nonprecious metals.148 It should be noted that in addition to behaving as a catalyst support to immobilize metal nanoparticles, N-doped carbon itself can also be directly used as a metal-free catalyst. In fact, this topic has attracted considerable attention in recent years.197–199 Doping with heteroatoms makes it possible to control the physicochemical properties and therefore the catalytic performances of nanocarbons by introducing electron acceptors or donors.197 For example, N-doped carbon materials were reported to be effective reusable catalysts for reactions such as Knoevenagel condensation198,199 and oxidative dehydrogenation.200 In this respect, nitrogen-doped carbons obtained from IL precursors provide abundant active sites for catalysis. Shi et al. prepared carbon aerogels, using an IL as a soft template, and used them directly as catalysts in the selective oxidation of tetralin under mild conditions.108 A typical carbon aerogel as a catalyst gave 30% conversion of tetralin to 1,2,3,4-tetrahydronaphthalen-1-ol and 3,4-dihydronaphthalen-1-one, with 78% selectivity. However, until now, little work has been done on the use of IL-based carbons as metal-free catalysts. Clearly, the investigation of IL-derived carbons in chemical catalysis is in its infancy. Further development of new catalytic applications and exploration of the correlation between carbon structure and catalytic activity are needed. For example, IL-derived N-doped porous carbon obtained at moderate temperatures with both high surface area and high nitrogen content may be a potential solid base for base-catalyzed reactions.198,199 In fact, the abundant basic active sites possessed by such carbon materials have already been confirmed by their excellent ability for CO2 adsorption.32,174
7. Conclusion and outlook
Successful carbonization of novel IL precursors represents a breakthrough in the fields of both ILs and carbon materials. This opens up a novel, direct, promising research direction for materialization of ILs, yielding carbons with distinct features for potential applications. We have reviewed the latest progress in the synthesis of carbon materials from ILs, which are alternative organic precursors that lead to a variety of carbonaceous materials. Carbon materials can be facilely obtained by direct carbonization of specific ILs at ambient pressure, without any complicated synthesis, catalyst, prepolymerization, vacuum system, or other complex techniques. The scope of IL-based carbonaceous precursors has been greatly expanded from the first cyano/nitrile-containing ILs to poly-ILs, DESs, and, more recently, PILs and PSs. As summarized in Fig. 38, each of these possesses clear merits and disadvantages as carbon precursors. For general aprotic IL precursors, the key structural prerequisite is the presence of certain functional groups such as cyano/nitrile or vinyl groups that can undergo cross-linking reactions under pyrolysis conditions. Carbon materials derived from most [DCA]-based ILs have useful properties such as high nitrogen contents, developed graphitic structures, and high conductivities. However, the lower carbon yields, non-porous structures, and high costs of such ILs limit their use. The carbon yield can be improved by nanoconfinement and nanopores can be introduced using sophisticated nanocasting techniques. The high cost can be addressed by using them as dopants or surface-covering agents to reduce the amount of IL used. The poly-IL-based route is attractive, because carbons with morphologies such as tubes, fibers, membranes, monoliths, and spheres are easily obtained via different polymer-processing techniques. These shapeable carbon materials are expected to have applications in supported catalysis, as adsorbents, in gas separation, and in water treatment. However, complicated syntheses and additional polymerization of the IL monomers are required. DESs are cheap and can be easily synthesized, but they generally require special polymeric monomers such as resorcinol, and long reaction times and toxic formaldehyde are necessary for condensation prior to carbonization. In contrast, PILs and PSs can be easily synthesized and are very low cost, as cheap as DESs, and are widely available. Importantly, depending on the precursor structure, the resultant carbons can be readily tuned to have a high specific surface area or high nitrogen content, high degree of graphitization, and high conductivity. However, nearly all porous carbons formed from PILs and PSs are microporous. The development of self-templated mesoporous carbons, as found in cyano/nitrile-containing ILs, is therefore highly desirable. It should be noted that non-carbonizable ILs can also be successfully transformed into carbon materials by nanoconfinement, energetic IL-template interactions, or simple microwave pyrolysis. | ||
| Fig. 38 Summary of recent developments of IL-based carbon precursors. | ||
This review provides useful information on how to select and design suitable IL precursors for the preparation of task-specific carbon materials for “on-demand” applications. The large number of possible combinations of cations and anions provides a great opportunity for creating novel carbon materials with tailor-made structural morphologies, porosities, and surface areas from ILs. Moreover, when these novel precursors are used with established templating methods, advanced porous carbons with desirable porosities and the excellent intrinsic properties observed in IL-derived carbons can be fabricated, for high-performance applications in fields such as catalysis, gas capture and storage, electrochemistry, and energy.
Although this IL-based approach has proved to be extremely successful in the production of many distinctive carbon materials, the use of ILs for the synthesis of carbon materials is an emerging field and there are still many aspects to be studied and considered in the future.
(a) In-depth investigation of the correlation between precursor structure and final carbon material is required to modulate the properties of carbon nanomaterials by changing the chemical compositions of the precursors.
(b) Very few ILs containing both nitrile groups and bulky anions such as [NTf2] and [BETI] give mesoporous carbons via direct template-free carbonization, therefore further mechanistic analysis of mesopore formation is needed to enable rational design of specific IL precursors at the molecular level to obtain mesoporous structures.
(c) New IL precursors that may produce carbon with a simultaneously high surface area, high nitrogen content, and mesoporous structure via direct carbonization need to be developed. PILs and PSs are promising candidates, because of their low cost, easy synthesis, diverse structures, and wide availability.
(d) Functionalization of ILs to enable preparation of well-defined mesoporous carbon nanomaterials via combination with soft templates needs to be investigated.
(e) Advanced porous carbon materials need to be developed through rational combination of ILs and suitable templates. For example, it is possible to produce hierarchical porous carbon by combining ILs that can intrinsically yield micropores with macroscopic or mesoscopic silica templates.
(f) Expansion of the applications of IL-derived carbons is desirable.
All these further studies will extend the scope of this strategy and make it a really useful method that materials scientists will use on a regular basis to prepare carbon nanomaterials.
Acknowledgements
This work was supported by the Japan Science and Technology Agency (JST) – Advanced Low Carbon Technology Research and Development Program (ALCA) of Japan.References
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed
.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS
- M. Smiglak, A. Metlen and R. D. Rogers, Acc. Chem. Res., 2007, 40, 1182–1192 CrossRef CAS PubMed
- T. Torimoto, T. Tsuda, K. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS PubMed
- H. Xing, C. Liao, Q. Yang, G. M. Veith, B. Guo, X. G. Sun, Q. Ren, Y. S. Hu and S. Dai, Angew. Chem., Int. Ed., 2014, 53, 2099–2103 CrossRef CAS PubMed
- S.-Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda and M. Watanabe, J. Am. Chem. Soc., 2010, 132, 9764–9773 CrossRef CAS PubMed
- E. Frackowiak, G. Lota and J. Pernak, Appl. Phys. Lett., 2005, 86, 164104 CrossRef PubMed
- B. Mallick, B. Balke, C. Felser and A. V. Mudring, Angew. Chem., Int. Ed., 2008, 47, 7635–7638 CrossRef CAS PubMed
- T. Peppel, M. Köckerling, M. Geppert-Rybczyńska, R. V. Ralys, J. K. Lehmann, S. P. Verevkin and A. Heintz, Angew. Chem., Int. Ed., 2010, 49, 7116–7119 CrossRef CAS PubMed
- A. J. Boydston, C. S. Pecinovsky, S. T. Chao and C. W. Bielawski, J. Am. Chem. Soc., 2007, 129, 14550–14551 CrossRef CAS PubMed
- F. Zhou, Y. Liang and W. Liu, Chem. Soc. Rev., 2009, 38, 2590–2599 RSC
- S. Schneider, T. Hawkins, M. Rosander, G. Vaghjiani, S. Chambreau and G. Drake, Energy Fuels, 2008, 22, 2871–2872 CrossRef CAS
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS
- C. D. Liang, Z. J. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed
- L. L. Zhang and X. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC
- M.-M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC
- T.-Y. Ma, L. Liu and Z.-Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC
- A. Stein, S. G. Rudisill and N. D. Petkovich, Chem. Mater., 2014, 26, 259–276 CrossRef CAS
- Y. P. Zhai, Y. Wan, Y. Cheng, Y. F. Shi, F. Q. Zhang, B. Tu and D. Y. Zhao, J. Porous Mater., 2008, 15, 601–611 CrossRef CAS
- J. P. Paraknowitsch and A. Thomas, Macromol. Chem. Phys., 2012, 213, 1132–1145 CrossRef CAS
- T. P. Fellinger, A. Thomas, J. Yuan and M. Antonietti, Adv. Mater., 2013, 25, 5838–5855 CrossRef CAS PubMed
- S. A. Forsyth, S. R. Batten, Q. Dai and D. R. MacFarlane, Aust. J. Chem., 2004, 57, 121–124 CrossRef CAS
- T. J. Wooster, K. M. Johanson, K. J. Fraser, D. R. MacFarlane and J. L. Scott, Green Chem., 2006, 8, 691–696 RSC
- J. S. Lee, X. Q. Wang, H. M. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596–4597 CrossRef CAS PubMed
- J. P. Paraknowitsch, J. Zhang, D. S. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS PubMed
- W. Yang, T. P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed
- Y. Y. Shao, J. H. Sui, G. P. Yin and Y. Z. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS PubMed
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., 2005, 117, 7215–7221 CrossRef
- J. S. Lee, X. Q. Wang, H. M. Luo and S. Dai, Adv. Mater., 2010, 22, 1004–1007 CrossRef CAS PubMed
- X. Zhu, P. C. Hillesheim, S. M. Mahurin, C. M. Wang, C. C. Tian, S. Brown, H. M. Luo, G. M. Veith, K. S. Han, E. W. Hagaman, H. L. Liu and S. Dai, ChemSusChem, 2012, 5, 1912–1917 CrossRef CAS PubMed
- T.-P. Fellinger, D. S. Su, M. Engenhorst, D. Gautam, R. Schlögl and M. Antonietti, J. Mater. Chem., 2012, 22, 23996–24005 RSC
- J. P. Paraknowitsch, B. Wienert, Y. Zhang and A. Thomas, Chem.–Eur. J., 2012, 18, 15416–15423 CrossRef CAS PubMed
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746–6758 RSC
- P. Kuhn, A. Forget, D. S. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef CAS PubMed
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed
- E. Irran, B. Jürgens and W. Schnick, Chem.–Eur. J., 2001, 7, 5372–5381 CrossRef CAS
- J. S. Lee, H. Luo, G. A. Baker and S. Dai, Chem. Mater., 2009, 21, 4756–4758 CrossRef CAS
- B. Guo, X. G. Sun, G. M. Veith, Z. Bi, S. M. Mahurin, C. Liao, C. Bridges, M. P. Paranthaman and S. Dai, Adv. Energy Mater., 2013, 3, 708–712 CrossRef CAS
- P. F. Fulvio, J. S. Lee, R. T. Mayes, X. Wang, S. M. Mahurin and S. Dai, Phys. Chem. Chem. Phys., 2011, 13, 13486–13491 RSC
- N. Fechler, T.-P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2013, 1, 14097–14102 CAS
- X. Q. Wang and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 6664–6668 CrossRef CAS PubMed
- S. M. Chen, G. Z. Wu, M. L. Sha and S. R. Huang, J. Am. Chem. Soc., 2007, 129, 2416–2417 CrossRef CAS PubMed
- J. Im, S. D. Cho, M. H. Kim, Y. M. Jung, H. S. Kim and H. S. Park, Chem. Commun., 2012, 48, 2015–2017 RSC
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS PubMed
- A. Chen, Y. Yu, H. Lv, Y. Wang, S. Shen, Y. Hu, B. Li, Y. Zhang and J. Zhang, J. Mater. Chem. A, 2013, 1, 1045–1047 CAS
- Y. Ma, C. Zhang, G. Ji and J. Y. Lee, J. Mater. Chem., 2012, 22, 7845–7850 RSC
- S. Yoon, C. Liao, X.-G. Sun, C. A. Bridges, R. R. Unocic, J. Nanda, S. Dai and M. P. Paranthaman, J. Mater. Chem., 2012, 22, 4611–4614 RSC
- Z. Cui, S. Wang, Y. Zhang and M. Cao, J. Power Sources, 2014, 259, 138–144 CrossRef CAS PubMed
- X. Zhang, N. Böckenfeld, F. Berkemeier and A. Balducci, ChemSusChem, 2014, 7, 1710–1718 CrossRef CAS PubMed
- H. Song, N. Li, H. Cui and C. Wang, Nano Energy, 2014, 4, 81–87 CrossRef CAS PubMed
- Z. Ding, L. Zhao, L. Suo, Y. Jiao, S. Meng, Y.-S. Hu, Z. Wang and L. Chen, Phys. Chem. Chem. Phys., 2011, 13, 15127–15133 RSC
- A. Safavi, F. Sedaghati, H. Shahbaazi and E. Farjami, RSC Adv., 2012, 2, 7367–7370 RSC
- D. Xiao, D. Yuan, H. He and M. Gao, J. Lumin., 2013, 140, 120–125 CrossRef CAS PubMed
- A. Zhao, C. Zhao, M. Li, J. Ren and X. Qu, Anal. Chim. Acta, 2014, 809, 128–133 CrossRef CAS PubMed
- N. E. Leadbeater and H. M. Torenius, J. Org. Chem., 2002, 67, 3145–3148 CrossRef CAS PubMed
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS PubMed
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS PubMed
- J. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS PubMed
- O. Green, S. Grubjesic, S. Lee and M. A. Firestone, J. Macromol. Sci., Polym. Rev., 2009, 49, 339–360 CAS
- J. Yuan, A. G. Márquez, J. Reinacher, C. Giordano, J. Janek and M. Antonietti, Polym. Chem., 2011, 2, 1654–1657 RSC
- Y. Men, M. Siebenbürger, X. Qiu, M. Antonietti and J. Yuan, J. Mater. Chem. A, 2013, 1, 11887–11893 CAS
- J. Balach, H. Wu, F. Polzer, H. Kirmse, Q. Zhao, Z. Wei and J. Yuan, RSC Adv., 2013, 3, 7979–7986 RSC
- D. Kuzmicz, P. Coupillaud, Y. Men, J. Vignolle, G. Vendraminetto, M. Ambrogi, D. Taton and J. Yuan, Polymer, 2014, 55, 3423–3430 CrossRef CAS PubMed
- Q. Zhao, T. P. Fellinger, M. Antonietti and J. Yuan, Macromol. Rapid Commun., 2012, 33, 1149–1153 CrossRef CAS PubMed
- X. Bo, J. Bai, J. Ju and L. Guo, J. Power Sources, 2011, 196, 8360–8365 CrossRef CAS PubMed
- B. Wu, Y. Kuang, Y. Zhang, X. Zhang and J. Chen, J. Mater. Chem., 2012, 22, 13085–13090 RSC
- J. Yuan, C. Giordano and M. Antonietti, Chem. Mater., 2010, 22, 5003–5012 CrossRef CAS
- J.-H. Jeon, K. Tanaka and Y. Chujo, J. Mater. Chem. A, 2014, 2, 624–630 CAS
- F. Zheng, G. Mu, Z. Zhang, Y. Shen, M. Zhao and G. Pang, Mater. Lett., 2012, 68, 453–456 CrossRef CAS PubMed
- S. Soll, T. P. Fellinger, X. Wang, Q. Zhao, M. Antonietti and J. Yuan, Small, 2013, 9, 4135–4141 CrossRef CAS PubMed
- J. Yuan, S. Soll, M. Drechsler, A. H. Müller and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 17556–17559 CrossRef CAS PubMed
- J. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS
- P. Zhang, J. Yuan, T. P. Fellinger, M. Antonietti, H. Li and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 6028–6032 CrossRef CAS PubMed
- D. Carriazo, M. C. Serrano, M. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Soc. Rev., 2012, 41, 4996–5014 RSC
- Q. Zhang, K. D. O. Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC
- A. P. Abbott, J. C. Barron, K. S. Ryder and D. Wilson, Chem.–Eur. J., 2007, 13, 6495–6501 CrossRef CAS PubMed
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC
- E. R. Parnham, E. A. Drylie, P. S. Wheatley, A. M. Slawin and R. E. Morris, Angew. Chem., 2006, 118, 5084–5088 CrossRef
- D. V. Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef CAS PubMed
- M. a. C. Gutiérrez, F. Rubio and F. del Monte, Chem. Mater., 2010, 22, 2711–2719 CrossRef
- D. Carriazo, M. a. C. Gutiérrez, M. L. Ferrer and F. del Monte, Chem. Mater., 2010, 22, 6146–6152 CrossRef CAS
- D. Carriazo, M. C. Gutiérrez, F. Picó, J. M. Rojo, J. L. Fierro, M. L. Ferrer and F. del Monte, ChemSusChem, 2012, 5, 1405–1409 CrossRef CAS PubMed
- J. Patiño, M. C. Gutiérrez, D. Carriazo, C. O. Ania, J. B. Parra, M. L. Ferrer and F. del Monte, Energy Environ. Sci., 2012, 5, 8699–8707 Search PubMed
- M. C. Gutiérrez, D. Carriazo, C. O. Ania, J. B. Parra, M. L. Ferrer and F. del Monte, Energy Environ. Sci., 2011, 4, 3535–3544 Search PubMed
- J. Patiño, M. Gutiérrez, D. Carriazo, C. Ania, J. Fierro, M. Ferrer and F. del Monte, J. Mater. Chem. A, 2014, 2, 8719–8729 Search PubMed
- D. Carriazo, M. C. Gutiérrez, R. Jiménez, M. L. Ferrer and F. del Monte, Part. Part. Syst. Charact., 2013, 30, 316–320 CrossRef CAS
- M. C. Gutiérrez, D. Carriazo, A. Tamayo, R. Jiménez, F. Picó, J. M. Rojo, M. L. Ferrer and F. del Monte, Chem.–Eur. J., 2011, 17, 10533–10537 CrossRef PubMed
- N. López-Salas, M. C. Gutiérrez, C. O. Ania, J. L. G. Fierro, M. L. Ferrer and F. del Monte, J. Mater. Chem. A, 2014, 2, 17387–17399 Search PubMed
- S. Zhang, K. Dokko and M. Watanabe, Chem. Mater., 2014, 26, 2915–2926 CrossRef CAS
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. Susan and M. Watanabe, Phys. Chem. Chem. Phys., 2012, 14, 5178–5186 RSC
- S. Zhang, M. S. Miran, A. Ikoma, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2014, 136, 1690–1693 CrossRef CAS PubMed
- Y. Gogotsi, A. Nikitin, H. H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591–594 CrossRef CAS PubMed
- W. Ding, Z. Wei, S. Chen, X. Qi, T. Yang, J. Hu, D. Wang, L.-J. Wan, S. F. Alvi and L. Li, Angew. Chem., Int. Ed., 2013, 52, 11755–11759 CrossRef CAS PubMed
- R. Czerw, M. Terrones, J. C. Charlier, X. Blase, B. Foley, R. Kamalakaran, N. Grobert, H. Terrones, D. Tekleab, P. M. Ajayan, W. Blau, M. Ruhle and D. L. Carroll, Nano Lett., 2001, 1, 457–460 CrossRef CAS
- J. D. Wiggins-Camacho and K. J. Stevenson, J. Phys. Chem. C, 2009, 113, 19082–19090 CAS
- L. Zhao, Y. S. Hu, H. Li, Z. X. Wang and L. Q. Chen, Adv. Mater., 2011, 23, 1385–1388 CrossRef CAS PubMed
- J. Cao, Y. Chu and X. Tan, Mater. Chem. Phys., 2014, 144, 17–24 CrossRef CAS PubMed
- J. PeteráParaknowitsch, Phys. Chem. Chem. Phys., 2012, 14, 6444–6447 RSC
- Z. Y. Jin, A. H. Lu, Y. Y. Xu, J. T. Zhang and W. C. Li, Adv. Mater., 2014, 26, 3700–3705 CrossRef CAS PubMed
- Y. Yan, Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Chem. Commun., 2012, 48, 10663–10665 RSC
- M. Zhou, X. Li, J. Cui, T. Liu, T. Cai, H. Zhang and S. Guan, Int. J. Electrochem. Sci., 2012, 7, 9984–9996 CAS
- Q. DucáTruong, J. Mater. Chem. C, 2013, 1, 1713–1716 RSC
- D.-C. Guo, J. Mi, G.-P. Hao, W. Dong, G. Xiong, W.-C. Li and A.-H. Lu, Energy Environ. Sci., 2013, 6, 652–659 CAS
- Y. Wang, J. Zhang, X. Wang, M. Antonietti and H. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS PubMed
- G. Wang, Z. Ling, C. Li, Q. Dong, B. Qian and J. Qiu, Electrochem. Commun., 2013, 31, 31–34 CrossRef CAS PubMed
- H. Yang, X. Cui, Y. Deng and F. Shi, J. Mater. Chem., 2012, 22, 21852–21856 RSC
- H. Byun, G. Nam, Y. Rhym and S. Shim, Carbon Lett., 2012, 13, 94–98 CrossRef
- J. S. Lee, R. T. Mayes, H. Luo and S. Dai, Carbon, 2010, 48, 3364–3368 CrossRef CAS PubMed
- X. Cui, F. Shi and Y. Deng, ChemCatChem, 2012, 4, 333–336 CrossRef CAS
- Y. Yao, T. Zhou, T. Yang, R. Xiang and Y. Wu, Carbon, 2013, 58, 249–251 CrossRef CAS PubMed
- Z.-L. Xie, R. J. White, J. Weber, A. Taubert and M. M. Titirici, J. Mater. Chem., 2011, 21, 7434–7442 RSC
- P. F. Fulvio, P. C. Hillesheim, J. C. Bauer, S. M. Mahurin and S. Dai, J. Mater. Chem. A, 2013, 1, 59–62 CAS
- J. H. Hafner, M. J. Bronikowski, B. R. Azamian, P. Nikolaev, A. G. Rinzler, D. T. Colbert, K. A. Smith and R. E. Smalley, Chem. Phys. Lett., 1998, 296, 195–202 CrossRef CAS
- T. Kyotani, Carbon, 2000, 38, 269–286 CrossRef CAS
- R. Göbel, Z.-L. Xie, M. Neumann, C. Günter, R. Löbbicke, S. Kubo, M.-M. Titirici, C. Giordano and A. Taubert, CrystEngComm, 2012, 14, 4946–4951 RSC
- K. H. Lim and H. Kim, Appl. Catal., B, 2014, 158, 355–360 CrossRef PubMed
- C. Pan, L. Qiu, Y. Peng and F. Yan, J. Mater. Chem., 2012, 22, 13578–13584 RSC
- Y. Peng, X. Wu, L. Qiu, C. Liu, S. Wang and F. Yan, J. Mater. Chem. A, 2013, 1, 9257–9263 CAS
- Z. Li, J. Liu, Z. Huang, Y. Yang, C. Xia and F. Li, ACS Catal., 2013, 3, 839–845 CrossRef CAS
- J. Yuan, H. Schlaad, C. Giordano and M. Antonietti, Eur. Polym. J., 2011, 47, 772–781 CrossRef CAS PubMed
- N. Fechler, T.-P. Fellinger and M. Antonietti, Chem. Mater., 2012, 24, 713–719 CrossRef CAS
- N. Fechler, G. A. Tiruye, R. Marcilla and M. Antonietti, RSC Adv., 2014, 4, 26981–26989 RSC
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS
- Q. Zhao, T.-P. Fellinger, M. Antonietti and J. Yuan, J. Mater. Chem. A, 2013, 1, 5113–5120 CAS
- P. F. Fulvio, P. C. Hillesheim, Y. Oyola, S. M. Mahurin, G. M. Veith and S. Dai, Chem. Commun., 2013, 49, 7289–7291 RSC
- Z. Zhang, G. M. Veith, G. M. Brown, P. F. Fulvio, P. C. Hillesheim, S. Dai and S. H. Overbury, Chem. Commun., 2014, 50, 1469–1471 RSC
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS
- A. J. Zarbin, R. Bertholdo and M. A. Oliveira, Carbon, 2002, 40, 2413–2422 CrossRef CAS
- S. Tabata, Y. Isshiki and M. Watanabe, J. Electrochem. Soc., 2008, 155, K42–K49 CrossRef CAS PubMed
- A. Lu, A. Kiefer, W. Schmidt and F. Schüth, Chem. Mater., 2004, 16, 100–103 CrossRef CAS
- Z. Ma, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef CAS PubMed
- Y. Zhou and M. Antonietti, J. Am. Chem. Soc., 2003, 125, 14960–14961 CrossRef CAS PubMed
- T. Nakashima and N. Kimizuka, J. Am. Chem. Soc., 2003, 125, 6386–6387 CrossRef CAS PubMed
- Y. Zhou, J. H. Schattka and M. Antonietti, Nano Lett., 2004, 4, 477–481 CrossRef CAS
- D. Zhang, T. Yan, H. Li and L. Shi, Microporous Mesoporous Mater., 2011, 141, 110–118 CrossRef CAS PubMed
- B. Karimi, H. Behzadnia, M. Rafiee and H. Vali, Chem. Commun., 2012, 48, 2776–2778 RSC
- B. Karimi, H. Behzadnia, M. Bostina and H. Vali, Chem.–Eur. J., 2012, 18, 8634–8640 CrossRef PubMed
- S. H. Kazemi, B. Karimi, A. Fashi, H. Behzadnia and H. Vali, J. Solid State Electrochem., 2014, 1–6 Search PubMed
- B. Karimi, H. Behzadnia and H. Vali, ChemCatChem, 2014, 6, 745–748 CrossRef CAS
- B. Qiu, C. Pan, W. Qian, Y. Peng, L. Qiu and F. Yan, J. Mater. Chem. A, 2013, 1, 6373–6378 CAS
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, science, 1998, 279, 548–552 CrossRef CAS
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS
- T. P. Fellinger, F. Hasche, P. Strasser and M. Antonietti, J. Am. Chem. Soc., 2012, 134, 4072–4075 CrossRef CAS PubMed
- X. Xu, Y. Li, Y. T. Gong, P. F. Zhang, H. R. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed
- F. Hasché, T. P. Fellinger, M. Oezaslan, J. P. Paraknowitsch, M. Antonietti and P. Strasser, ChemCatChem, 2012, 4, 479–483 CrossRef
- P. Zhang, J. Yuan, H. Li, X. Liu, X. Xu, M. Antonietti and Y. Wang, RSC Adv., 2013, 3, 1890–1895 RSC
- J. P. Paraknowitsch, Y. Zhang and A. Thomas, J. Mater. Chem., 2011, 21, 15537–15543 RSC
- C. Liu, P. Tang, A. Chen, Y. Hu, Y. Yu, H. Lv and D. Ma, Mater. Lett., 2013, 108, 285–288 CrossRef CAS PubMed
- N. Fechler, T. P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75–79 CrossRef CAS PubMed
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237–8265 RSC
- X. Liu and M. Antonietti, Carbon, 2014, 69, 460–466 CrossRef CAS PubMed
- K. Elumeeva, N. Fechler, T. Fellinger and M. Antonietti, Mater. Horiz., 2014, 1, 588–594 RSC
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed
- A. Aijaz, T. Akita, H. Yang and Q. Xu, Chem. Commun., 2014, 50, 6498–6501 RSC
- J. P. Paraknowitsch, Y. Zhang, B. Wienert and A. Thomas, Chem. Commun., 2013, 49, 1208–1210 RSC
- A. M. Puziy, O. I. Poddubnaya, A. Martinez-Alonso, F. Suarez-Garcia and J. M. D. Tascon, Carbon, 2003, 41, 1181–1191 CrossRef CAS
- A. M. Puziy, O. I. Poddubnaya, A. Martinez-Alonso, F. Suarez-Garcia and J. M. D. Tascon, Carbon, 2002, 40, 1493–1505 CrossRef CAS
- A. M. Puziy, O. I. Poddubnaya, A. Martinez-Alonso, F. Suarez-Garcia and J. M. D. Tascon, Carbon, 2002, 40, 1507–1519 CrossRef CAS
- Y. J. Wang, D. P. Wilkinson and J. J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed
- Z. Chen, M. Waje, W. Li and Y. Yan, Angew. Chem., Int. Ed., 2007, 46, 4060–4063 CrossRef CAS PubMed
- H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed
- Y. H. Bing, H. S. Liu, L. Zhang, D. Ghosh and J. J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC
- N.-D. O. M. G. Arrays, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef PubMed
- K. Q. Jian, H. S. Shim, A. Schwartzman, G. P. Crawford and R. H. Hurt, Adv. Mater., 2003, 15, 164–167 CrossRef CAS
- C. Han, J. Wang, Y. Gong, X. Xu, H. Li and Y. Wang, J. Mater. Chem. A, 2014, 2, 605–609 CAS
- N. Ranjbar Sahraie, J. P. Paraknowitsch, C. Goebel, A. Thomas and P. Strasser, J. Am. Chem. Soc., 2014, 136, 14486–14497 CrossRef CAS PubMed
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664–2680 CrossRef CAS
- S. Yang, X. Feng, L. Zhi, Q. Cao, J. Maier and K. Müllen, Adv. Mater., 2010, 22, 838–842 CrossRef CAS PubMed
- M.-H. Ryu, K.-N. Jung, K.-H. Shin, K.-S. Han and S. Yoon, J. Phys. Chem. C, 2013, 117, 8092–8098 CAS
- Y. Ni, Y. Yin, P. Wu, H. Zhang and C. Cai, ACS Appl. Mater. Interfaces, 2014, 6, 7346–7355 CAS
- S. M. Mahurin, P. F. Fulvio, P. C. Hillesheim, K. M. Nelson, G. M. Veith and S. Dai, ChemSusChem, 2014 DOI:10.1002/cssc.201402338
- L. Qie, W. M. Chen, Z. H. Wang, Q. G. Shao, X. Li, L. X. Yuan, X. L. Hu, W. X. Zhang and Y. H. Huang, Adv. Mater., 2012, 24, 2047–2050 CrossRef PubMed
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed
- D. Hulicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suárez-García, J. M. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026–5027 CrossRef CAS PubMed
- L.-F. Chen, X.-D. Zhang, H.-W. Liang, M. Kong, Q.-F. Guan, P. Chen, Z.-Y. Wu and S.-H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed
- L. Zhao, L. Z. Fan, M. Q. Zhou, H. Guan, S. Qiao, M. Antonietti and M. M. Titirici, Adv. Mater., 2010, 22, 5202–5206 CrossRef CAS PubMed
- R. Mysyk, E. Raymundo-Piñero, M. Anouti, D. Lemordant and F. Béguin, Electrochem. Commun., 2010, 12, 414–417 CrossRef CAS PubMed
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef CAS PubMed
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed
- A.-H. Lu and G.-P. Hao, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2013, 109, 484–503 RSC
- G. P. Hao, W. C. Li, D. Qian, G. H. Wang, W. P. Zhang, T. Zhang, A. Q. Wang, F. Schuth, H. J. Bongard and A. H. Lu, J. Am. Chem. Soc., 2011, 133, 11378–11388 CrossRef CAS PubMed
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS PubMed
- X. Zhu, C. Tian, S. Chai, K. Nelson, K. S. Han, E. W. Hagaman, G. M. Veith, S. M. Mahurin, H. Liu and S. Dai, Adv. Mater., 2013, 25, 4152–4158 CrossRef CAS PubMed
- M. Sevilla, P. Valle-Vigon and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS
- V. Chandra, S. U. Yu, S. H. Kim, Y. S. Yoon, D. Y. Kim, A. H. Kwon, M. Meyyappan and K. S. Kim, Chem. Commun., 2012, 48, 735–737 RSC
- W. Xing, C. Liu, Z. Y. Zhou, L. Zhang, J. Zhou, S. P. Zhuo, Z. F. Yan, H. Gao, G. Q. Wang and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 7323–7327 CAS
- M. Nandi, K. Okada, A. Dutta, A. Bhaumik, J. Maruyama, D. Derks and H. Uyama, Chem. Commun., 2012, 48, 10283–10285 RSC
- A. Vishnyakov, P. I. Ravikovitch and A. V. Neimark, Langmuir, 1999, 15, 8736–8742 CrossRef CAS
- J. M. Martinmartinez, R. Torregrosamacia and M. C. Mittelmeijerhazeleger, Fuel, 1995, 74, 111–114 CrossRef CAS
- D. CazorlaAmoros, J. AlcanizMonge and A. LinaresSolano, Langmuir, 1996, 12, 2820–2824 CrossRef CAS
- H. Choi, Y. C. Park, Y.-H. Kim and Y. S. Lee, J. Am. Chem. Soc., 2011, 133, 2084–2087 CrossRef CAS PubMed
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef CAS PubMed
- X. Xu, H. Li and Y. Wang, ChemCatChem, 2014 DOI:10.1002/cctc.201402561R1
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed
- S. Dommele and K. P. áde Jong, Chem. Commun., 2006, 4859–4861 RSC
- N. Kan-nari, S. Okamura, S. i. Fujita, J. i. Ozaki and M. Arai, Adv. Synth. Catal., 2010, 352, 1476–1484 CrossRef CAS
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlögl and D. S. Su, Science, 2008, 322, 73–77 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |