
Heteroannulated acceptors based on benzothiadiazole
Timothy C.
Parker
a,
Dinesh G. (Dan)
Patel
b,
Karttikay
Moudgil
a,
Stephen
Barlow
a,
Chad
Risko†
a,
Jean-Luc
Brédas‡
a,
John R.
Reynolds
ac and
Seth R.
Marder
*a
aGeorgia Institute of Technology, School of Chemistry & Biochemistry, Center for Organic Photonics and Electronics, 901 Atlantic Drive, Atlanta, GA 30332, USA. E-mail: seth.marder@chemistry.gatech.edu
bThe Pennsylvania State University, 76 University Drive, Hazleton, PA 18292, USA. E-mail: dgp15@psu.edu
cGeorgia Institute of Technology, School of Chemistry & Biochemistry, School of Materials Science, Center for Organic Photonics and Electronics, 901 Atlantic Drive, Atlanta, GA 30332, USA. E-mail: reynolds@chemistry.gatech.edu
First published on 6th August 2014
Abstract
Increasing the acceptor strength of the widely used acceptor benzothiadiazole (BT) by extending the heterocyclic core is a promising strategy for developing new and stronger acceptors for materials in organic electronics and photonics. In recent years, such heteroannulated BT acceptors have been incorporated into a wide variety of materials that have been used in organic electronic and photonic devices. This review critically assesses the properties of these materials. Although heteroannulation to form acceptors, such as benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole (BBT), does result in materials with significantly higher electron affinity (EA) relative to BT, in many cases the extended BT systems also exhibit lower ionization energy (IE) than BT. Both the significantly higher EA and lower IE limit the efficacy of these materials in applications such as bulk heterojunction organic photovoltaics (BHJ-OPV) based on C60. Although the relatively high EA may enable some applications such as air stable organic field effect transistors (OFET), more widespread use of heteroannulated BT acceptors will likely require the ability to moderate or retain the high EA while increasing IE.
Introduction
The ability to precisely tune properties such as conductivity, charge-carrier mobility, light absorption, and light emission in π-conjugated materials often impacts the performance of devices such as organic photovoltaics (OPVs), dye-sensitized solar cells (DSSCs), organic field-effect transistors (OFETs), organic light-emitting diodes (OLEDs), and electrochromic devices (ECDs). As a consequence, organic chemists must carefully control, and often achieve a balance between, various redox, optical, and electronic properties in both polymers and small molecules. Fundamental to tuning a material's electronic properties are the energies of both the highest occupied molecular orbital (EHOMO), related to the ionization energy (IE) of the molecule, and the lowest unoccupied molecular orbital (ELUMO), related to the electron affinity (EA) of the molecule or polymer, as well as the difference (Eg) between these energies. Indeed, although other material properties, such as morphology and microstructure play a critical role,1–3 IE and EA generally must be appropriately aligned relative to the work-functions, IEs, or EAs of known electrode materials, interfacial materials, and/or other adjacent organic layers to enable efficient charge-carrier injection or extraction, or photoinduced charge separation in devices.4,5One approach to tuning the properties of π-conjugated small molecules or polymers6 is the covalent coupling of at least one electron donor (D) to at least one electron acceptor (A), either directly or through a π-conjugated bridge (π). Several different structural motifs have been extensively studied during the past two decades, leading to advances in: (i) second-order nonlinear optical (NLO) chromophores (D–π–A);7 (ii) two-photon absorbing (TPA) chromophores (D–A–D, D–π–A, and A–D–A);8,9 (iii) electrochromics (–Dn–A–);10–13 (iv) chromophores for dye-sensitized solar cells (DSSCs, D–π–A and D2A);14–17 (v) polymers (–D–A–)n and small molecules (D–A) for OLEDs;18 (vi) small-molecule donors (D–A)19 and acceptors20 and polymers (–D–A–)n for OPVs;21–26 (vii) polymers (–D–A–)n for OFETs,27,28 and (viii) polymers (–D–A–)n for electrochromism.29,30
Early D–A polymers that demonstrated the utility of the approach in modifying optical absorption energies typically used discrete electron accepting groups such as cyano, nitro, or sulfonyl groups as substituents on an aryl subunit or a vinylene in the polymer backbone.31,32 More recently, heterocycles with high EAs have gained favor as stronger and more synthetically variable acceptors. Although a variety of heterocyclic acceptors have been studied and are covered in extensive reviews,22,23 some of the more often-used acceptors, shown in Fig. 1, include thieno[3,4-c]pyrrole-4,6-dione (TPD, 1),33,34 esters of 3-fluorothieno[3,4-b]thiophene-2-carboxylic acid35 (2), diketopyrrolo[3,4-c]pyrrole36 (DPP, 3), isoindigo (4),37–39 and 2,1,3-benzothiadiazole (BT, 5).40Fig. 1 also shows the heterocyclic numbering scheme for BT, which will be used extensively in this review. In particular, BT and its 5-monofluoro- (MFBT) and 5,6-difluoro- (DFBT) derivatives have been used in a variety of materials including both polymers41–43 and small molecules44,45 in OPVs, in OFETs,46,47 as electron-deficient π-bridges in DSSC chromophores,48,49 TPA chromophores,50–52 electrochromics,53,54 and as emitters in small-molecule and polymer OLEDs that include white light emitters,55 emitters with colors spanning the visible spectrum,56–59 and near-infrared emitters.60 The 5,6-dinitro BT derivative also has exhibited strong electron withdrawing ability.61
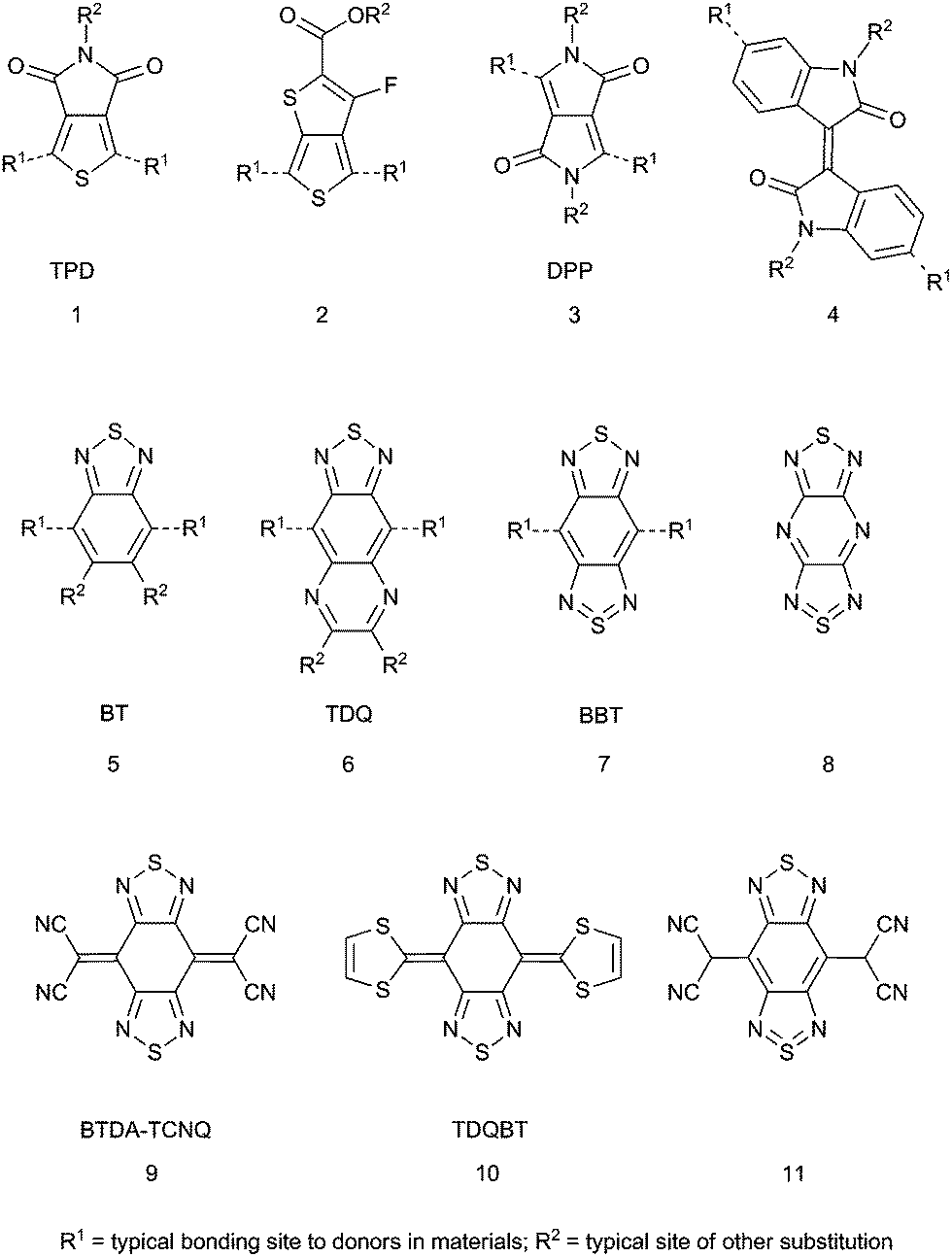 | ||
| Fig. 1 Heterocyclic acceptors used in organic electronics. Dashed lines indicate bonds to donor groups in materials. | ||
Generally, BT is an effective electron acceptor; the presence of the imine functionalities with relatively low energy π*-orbitals gives BT a relatively high EA itself. The molecule can be described as a quasi-quinoidal structure (i.e., with localized, relatively short π-bonds in the benzo ring) rather than a 10π-electron heteroaromatic system; this can increase electronic coupling between substituents in the 4- and 7-positions relative to that found across 1,4-substituted aromatic moieties in both small molecules and polymers. Although BT has proven to be useful, there is still a desire to develop stronger electron acceptors, for example, to decrease the optical gap (Eoptg) in D–A polymers in order to increase light absorption in the near infrared (NIR) and, therefore, to utilize the solar spectrum more efficiently in OPVs,62 to provide electron-accepting materials in OPVs that might replace the currently used fullerene derivatives,63 or to provide air-stable electron-transport materials for n-channel OFETs.64 One way to increase the electron-accepting strength of BT is to heteroannulate at the 5- and 6-positions to give acceptors such as [1,2,5]thiadiazolo[3,4-g]quinoxaline (TDQ, 6) and benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole (benzobisthiadiazole, BBT, 7). BBT is structurally similar to the known strong acceptor 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 65 as well as the core heterocyclic framework of both bis([1,2,5]thiadiazolo)tetracyanoquinodimethane (BTDA-TCNQ, 9) (Fig. 1), which is an electron acceptor that forms conductive charge-transfer crystals with organic donors,66–68 and bis[1,2,5]thiadiazolo-p-quinobis(1,3-dithiole) (TDQBT, 10) (Fig. 1), which forms single-component crystals that have high Hall charge-carrier mobility (3 cm2 V−1 s−1).69–74 In fact, the first known isolated derivative of BBT was 4,8-bis(dicyanomethyl) derivative 11, which was obtained from two-electron reduction of BTDA-TCNQ and protonation of the stable, isolable disodium salt.75 Indeed, in recent years the number of research articles on heteroannulated BT-containing materials has increased significantly, allowing an initial critical assessment of their properties and performance, which is the subject of this current review. Herein, we highlight key aspects of the electronic structure of heteroannulated BT derivatives that give rise to controllable optical and electronic properties and describe how materials containing these acceptors have been employed in organic electronic and photonic applications.
65 as well as the core heterocyclic framework of both bis([1,2,5]thiadiazolo)tetracyanoquinodimethane (BTDA-TCNQ, 9) (Fig. 1), which is an electron acceptor that forms conductive charge-transfer crystals with organic donors,66–68 and bis[1,2,5]thiadiazolo-p-quinobis(1,3-dithiole) (TDQBT, 10) (Fig. 1), which forms single-component crystals that have high Hall charge-carrier mobility (3 cm2 V−1 s−1).69–74 In fact, the first known isolated derivative of BBT was 4,8-bis(dicyanomethyl) derivative 11, which was obtained from two-electron reduction of BTDA-TCNQ and protonation of the stable, isolable disodium salt.75 Indeed, in recent years the number of research articles on heteroannulated BT-containing materials has increased significantly, allowing an initial critical assessment of their properties and performance, which is the subject of this current review. Herein, we highlight key aspects of the electronic structure of heteroannulated BT derivatives that give rise to controllable optical and electronic properties and describe how materials containing these acceptors have been employed in organic electronic and photonic applications.
Electronic structure of BBT and derivatives
The stability of 11 and the structural similarity of BBT to the known strong acceptor 8, as well as the intriguing valence-bond representation in which there is a formally tetravalent sulfur atom in an aromatic 14 π-electron ring system, inspired Yamashita and co-workers to develop a synthetically useful route to BBT and TDQ derivatives that provided a variety of compounds 12–20 (Fig. 2) from 1994 to 1997.76–79 More recently, an alternative synthesis of key intermediates 12 and 17 was reported.80 The acceptor strength of BBT and TDQ compared to the parent BT was initially assessed using optical absorption and electrochemistry of 15–17. Both TDQ derivative 16 (λmax = 524 nm) and BBT derivative 17 (λmax = 702 nm) showed marked red shifts compared to BT 15 (λmax = 445 nm). Assuming that these electronic transitions represent excitation from a HOMO delocalized over the thiophene rings and the six-membered ring to a LUMO located on the fused-ring heterocycles, these shifts suggest that TDQ and BBT are strong intramolecular π-acceptors; however, it should be pointed out that substituents can sterically interact with the BBT ring, causing a torsion that may decrease intramolecular charge transfer. This is likely the case for both the methyl group on the pyrrole in 18, which causes a blue shift of 8 nm compared to the weaker thiophene donor in 17, and the proximal methoxy group in 19, which causes a reduced red shift of only 6 nm even though the dimethoxy substituted benzene ring in 19 should be a significantly stronger donor than the phenyl group of 13. The strength of BBT in particular as a intramolecular π-acceptor is manifested by the near-infrared absorption (λmax = 732 nm) of the relatively short chromophore 20. Additionally, electropolymerized 17 and 18—the first reported D–A polymers incorporating BBT—showed Eoptg (taken as the low energy onset of the optical absorption) of 0.5 eV and 0.6 eV, respectively, which was amongst the lowest Eoptg reported for any polymer at the time.78 Reduction potentials (Ered) of TDQ derivative 16 (−0.72 V vs. SCE) and BBT derivative 17 (−0.53 V vs. SCE) are less cathodic than that of BT derivative 15 (−1.22 V vs. SCE), consistent with the trend in acceptor strength deduced from optical data; however, the oxidation potentials (Eox) were also less anodic than that of their BT counterpart (+0.98, +0.95, and +1.23 V vs. SCE, respectively), indicating that although the EA may indeed be increased, the IE may also be lowered in TDQ and BBT compounds relative to their BT analogues. While this might be good for lowering Eoptg, it may have deleterious effects for materials in certain applications such as OPVs as the lower IE can lead to a decrease in the open-circuit voltage (see below).81 Nevertheless, these initial results by Yamashita and coworkers on both small molecules and polymers show the potential of TDQ and BBT as good electron acceptors that might be used for a variety of organic electronic applications where tunable and facile redox properties and/or low-energy optical absorptions are desirable.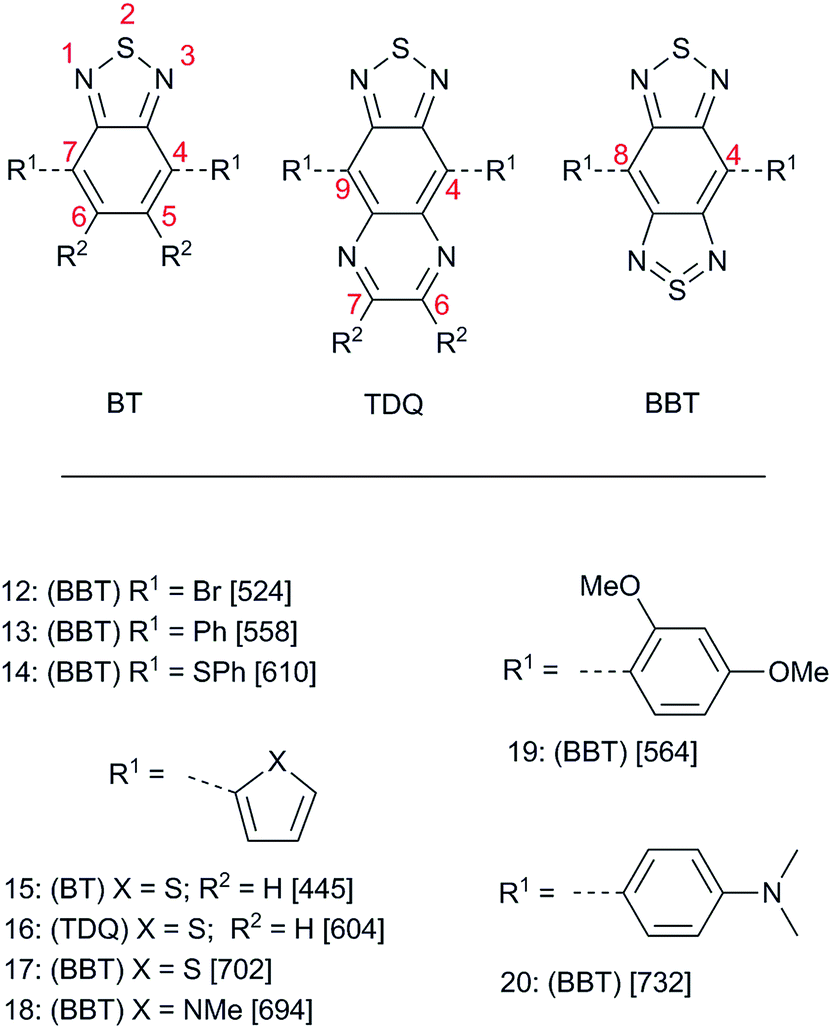 | ||
| Fig. 2 BBT derivatives prepared by Yamashita and coworkers and related compounds. Number in brackets is λmax (nm) in CHCl3. Red numbers indicate heterocyclic numbering scheme for that specific parent. | ||
In addition to the interest in the effects of BBT on material properties, the bonding in BBT is intriguing itself in terms of the “tetravalent sulfur” (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SN–) present in the formal representation shown in Fig. 1, which to maintain a formal charge of zero, must expand its octet to a 10 valence-electron configuration. Other compounds with formally tetravalent sulfur have been reported as a means to probe classical structure and bonding theories including thieno[3,4-c]thiophene derivatives 21 and 2282 (Fig. 3) and thiaphenalene 24 that were reported by Cava and coworkers,83 with 24 also independently reported by Schlessinger and Ponticello.84 Although originally shown as having a tetravalent sulfur, these compounds each have the ylidic resonance structure 27 and the 1,3-dipolar resonance structure 28. Indeed, 21, 22, and 24 each were reportedly unstable and their transient existence as intermediates was only inferred via formation of stable adducts with N-phenylmaleimide (29, Fig. 3), which may occur as a result of a 1,3-dipolar addition of the resonance form 28. The following year, isolable versions were reported by Ponticello and Schlessinger (25)85via sterically deactivating the reactive tetravalent sulfur with adjacent, out-of-plane phenyl rings, and were followed by reports of 2386 and 2687 in back-to-back publications. An X-ray structure of 2388 showed that, compared to thiophene, the length of the S–C bonds shortened by 0.008 Å, nearly within experimental error, that the average CC bond lengths adjacent to S increased by 0.037 Å, and that the fused bond increased by 0.029 Å. These bond length increases are consistent with somewhat less intra thiophene localization in favor of a 10 π-electron annulene-like delocalization. Likewise, relevant bond lengths for [1,2,5]thiadiazole 30, which were determined from gas-phase electron-diffraction82 measurements (and independently from microwave spectroscopy81) and from the X-ray crystal structure of 4,7-diphenyl BT 31,89 can be compared to those determined for 4,8-diphenyl BBT 13 (Fig. 3, Table 1) using X-ray crystallography.79 The diphenyl derivatives were chosen for comparison since the Ph groups are twisted out of plane by 35–43° (BT 31, the range of four independent angles for two independent molecules in the crystal) and 44° (BBT 13), thereby reducing conjugation between the substituents and the heteroaromatic rings that might affect the bonding within the heteroaromatic cores. As seen in Table 1, on going from thiadiazole to BT 31, there is a lengthening of both the imine bonds (“B”) and the C–C bond (“C”; 7a–3a in BT; 8a–3a in BBT, (the lettering scheme for the bond is given in Fig. 3)), indicating a shift in BT away from intra thiadiazole delocalization and toward peripheral annulene-type delocalization; however, note that the benzannulated bonds “D” and “F” are longer than “E” by 0.062 Å and 0.052 Å, respectively (the lettering scheme for the bonds are given in Fig. 3). On going from BT to BBT, the decrease in the S–N “A” bond lengths (0.028 Å) is much more pronounced than that seen going from thiadiazole to BT (0.003 Å), the F bond (equivalent to 8a–3a in BBT (Fig. 3)) lengthens (0.028 Å). Furthermore, in BBT the D bond shortens (0.031 Å), and the E bond lengthens (0.031 Å) such that the bond alternation between D and E is lost and the bonds are equivalent by symmetry at 1.406 Å, similar to the aromatic bonds in benzene. Taken together, such bond distortions on going from BT to BBT are consistent with an increase both in multiple-bond character on the S, which could be expected if there is tetravalent sulfur bonding in BBT, and in annulene-type 14 π-electron delocalization (32) compared to the Kekulé representation (13). To our knowledge, there have been no detailed characterization studies carried out on BBT specifically to test the degree of aromatic delocalization; however, more detailed insight into the bonding in BBT has been explored through computational studies, the results of which are generally useful to rationalize and predict property changes on incorporation of BBT units into small molecules and polymers, as discussed below.
SN–) present in the formal representation shown in Fig. 1, which to maintain a formal charge of zero, must expand its octet to a 10 valence-electron configuration. Other compounds with formally tetravalent sulfur have been reported as a means to probe classical structure and bonding theories including thieno[3,4-c]thiophene derivatives 21 and 2282 (Fig. 3) and thiaphenalene 24 that were reported by Cava and coworkers,83 with 24 also independently reported by Schlessinger and Ponticello.84 Although originally shown as having a tetravalent sulfur, these compounds each have the ylidic resonance structure 27 and the 1,3-dipolar resonance structure 28. Indeed, 21, 22, and 24 each were reportedly unstable and their transient existence as intermediates was only inferred via formation of stable adducts with N-phenylmaleimide (29, Fig. 3), which may occur as a result of a 1,3-dipolar addition of the resonance form 28. The following year, isolable versions were reported by Ponticello and Schlessinger (25)85via sterically deactivating the reactive tetravalent sulfur with adjacent, out-of-plane phenyl rings, and were followed by reports of 2386 and 2687 in back-to-back publications. An X-ray structure of 2388 showed that, compared to thiophene, the length of the S–C bonds shortened by 0.008 Å, nearly within experimental error, that the average CC bond lengths adjacent to S increased by 0.037 Å, and that the fused bond increased by 0.029 Å. These bond length increases are consistent with somewhat less intra thiophene localization in favor of a 10 π-electron annulene-like delocalization. Likewise, relevant bond lengths for [1,2,5]thiadiazole 30, which were determined from gas-phase electron-diffraction82 measurements (and independently from microwave spectroscopy81) and from the X-ray crystal structure of 4,7-diphenyl BT 31,89 can be compared to those determined for 4,8-diphenyl BBT 13 (Fig. 3, Table 1) using X-ray crystallography.79 The diphenyl derivatives were chosen for comparison since the Ph groups are twisted out of plane by 35–43° (BT 31, the range of four independent angles for two independent molecules in the crystal) and 44° (BBT 13), thereby reducing conjugation between the substituents and the heteroaromatic rings that might affect the bonding within the heteroaromatic cores. As seen in Table 1, on going from thiadiazole to BT 31, there is a lengthening of both the imine bonds (“B”) and the C–C bond (“C”; 7a–3a in BT; 8a–3a in BBT, (the lettering scheme for the bond is given in Fig. 3)), indicating a shift in BT away from intra thiadiazole delocalization and toward peripheral annulene-type delocalization; however, note that the benzannulated bonds “D” and “F” are longer than “E” by 0.062 Å and 0.052 Å, respectively (the lettering scheme for the bonds are given in Fig. 3). On going from BT to BBT, the decrease in the S–N “A” bond lengths (0.028 Å) is much more pronounced than that seen going from thiadiazole to BT (0.003 Å), the F bond (equivalent to 8a–3a in BBT (Fig. 3)) lengthens (0.028 Å). Furthermore, in BBT the D bond shortens (0.031 Å), and the E bond lengthens (0.031 Å) such that the bond alternation between D and E is lost and the bonds are equivalent by symmetry at 1.406 Å, similar to the aromatic bonds in benzene. Taken together, such bond distortions on going from BT to BBT are consistent with an increase both in multiple-bond character on the S, which could be expected if there is tetravalent sulfur bonding in BBT, and in annulene-type 14 π-electron delocalization (32) compared to the Kekulé representation (13). To our knowledge, there have been no detailed characterization studies carried out on BBT specifically to test the degree of aromatic delocalization; however, more detailed insight into the bonding in BBT has been explored through computational studies, the results of which are generally useful to rationalize and predict property changes on incorporation of BBT units into small molecules and polymers, as discussed below.
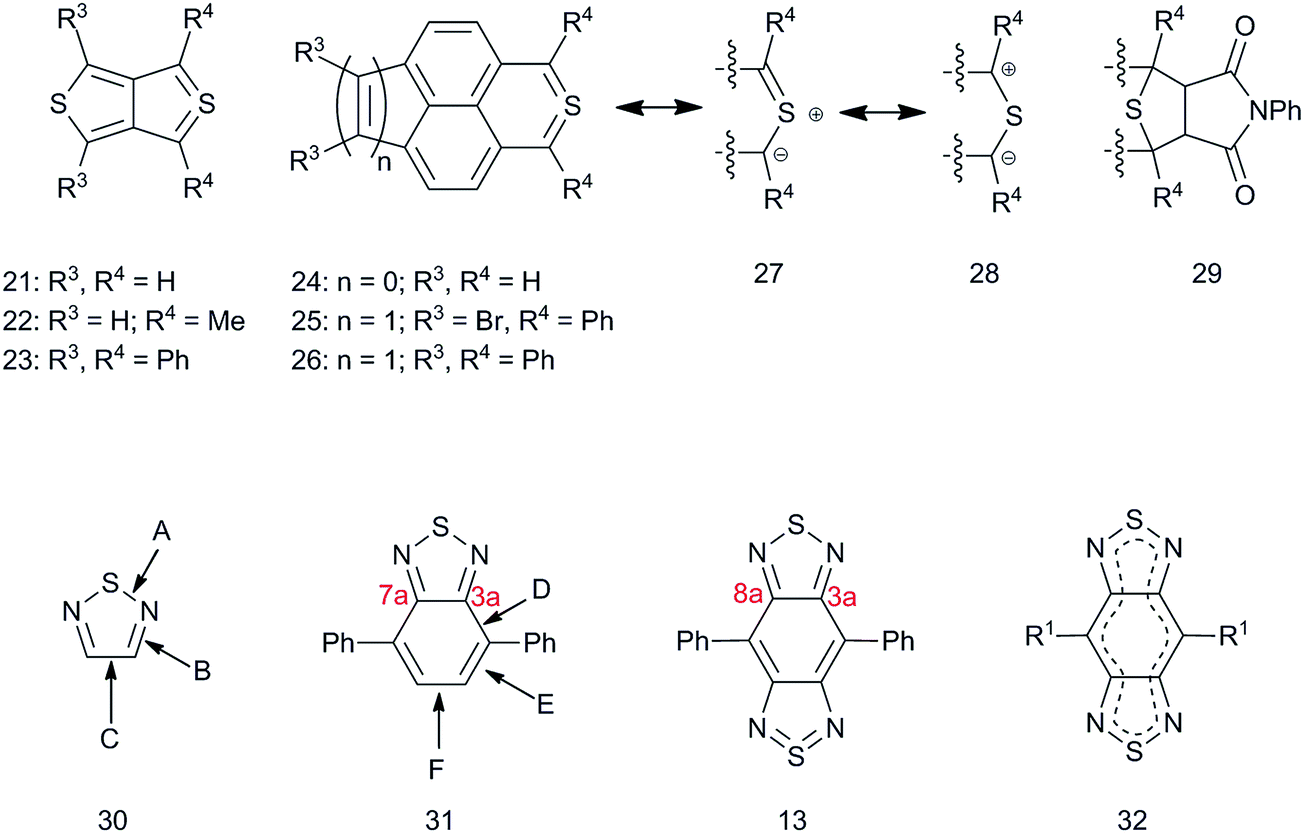 | ||
| Fig. 3 Non-classical bonding in sulfur heterocycles and bonding considerations in thiadiazole, BT, and BBT. | ||
| Compound | A | B | C | D | E | F |
|---|---|---|---|---|---|---|
| a Values from gas-phase electron diffraction. b Two complete molecules in the asymmetric unit, each with approximate non-crystallographic Cs symmetry. c Half a molecule in the asymmetric unit; the molecule has crystallographic inversion symmetry and approximate C2h symmetry. d Averages of multiple chemically equivalent but crystallographically independent bond lengths. e Values in parentheses are the difference between the bond length in the compound and the bond length in the compound immediately above. | ||||||
| 30 (thiadiazole) | 1.632a | 1.329a | 1.413a | — | — | — |
| 31 DiPh(BT)b | 1.629d (−0.003)e | 1.355d (+0.026)e | 1.452 (+0.039)e | 1.437d | 1.375d | 1.427d |
| 13 DiPh(BBT)c | 1.601d (−0.028)e | 1.378d (+0.023)e | 1.455d (+0.003)e | 1.406d (−0.031)e | 1.406d (+0.031)e | 1.455 (+0.028)e |
A long-standing question relevant to the proposed tetravalent sulfur bonding is whether the bonding can be better represented as an ylidic structure with an 8-electron sulfur such as in 33 (Fig. 4) rather than as a 10-electron sulfur, as in 34. Strassner and Fabian90 examined a number of structures for various degrees of tetravalent sulfur bonding including acyclic sulfur diimides, thiadiazole, BT, and BBT using density functional theory (DFT, at the B3LYP/6-31G* level). The main finding was that, generally, for compounds that have tetravalent sulfur in Kekulé representations (such as BBT), there was a higher degree of positive charge on the S atoms and a higher degree of negative charge on the N atoms compared to thiadiazole and BT, which is consistent with an increased contribution from the ylidic valence-bond structures such as 33. This is consistent with d-orbitals on S typically having little direct bonding with p-orbitals on N and is also in agreement with a more recent charge density study on acyclic S–N multiple bond containing compounds.91 Another component of the Strassner and Fabian work was the identification of a relatively small singlet–triplet (S0/T1) energy gap of 20.1 kcal mol−1, which, according to the definition proposed by Wirz,92 put BBTs on the borderline of having some degree of diradicaloid character. Similarly, DFT calculations by Bhanuprakash and coworkers93,94 have also shown that BBT and like-molecules, depending on the nature of the chemical modification and the density functional employed (i.e., the amount of [non-local] Hartree–Fock exchange included in hybrid functionals), can have diradicaloid character. These results for BBT were largely confirmed by Shen and coworkers,95 who additionally showed that the Wiberg bond indices of the bonds A, B, and D (Fig. 3) in BBT indicated a “considerable” degree of conjugation around the BBT periphery; this was associated with an aromatic ring current according to their calculations. The natural charges from Shen et al. are also shown in Fig. 4 (34), and they are consistent with Strassner and Fabian's90 calculated ylidic structure. Indeed, this strongly positive sulfur has recently been identified by Reynolds and coworkers as the source of the tendency of BBT and the BT-heteroannulated derivative benzo(triazole-dithiazole) 35 to lower the (B3PW91/6-31**)-calculated LUMO energy of materials relative to BT.96 It should be noted that Yamashita and coworkers reported formation of the Diels–Alder-like adduct 36 in 89% yield by refluxing BBT derivative 13 with N-phenylmaleimide in xylene;79 however, the existence of 36 should not be used to infer a formal 4 + 2 cycloaddition from a Kekulé-type form such as 32 since symmetry allowed cycloadditions may still follow a stepwise “diradical” cyclization mechanism,97 which may be reasonable to expect from diradicaloid structures.
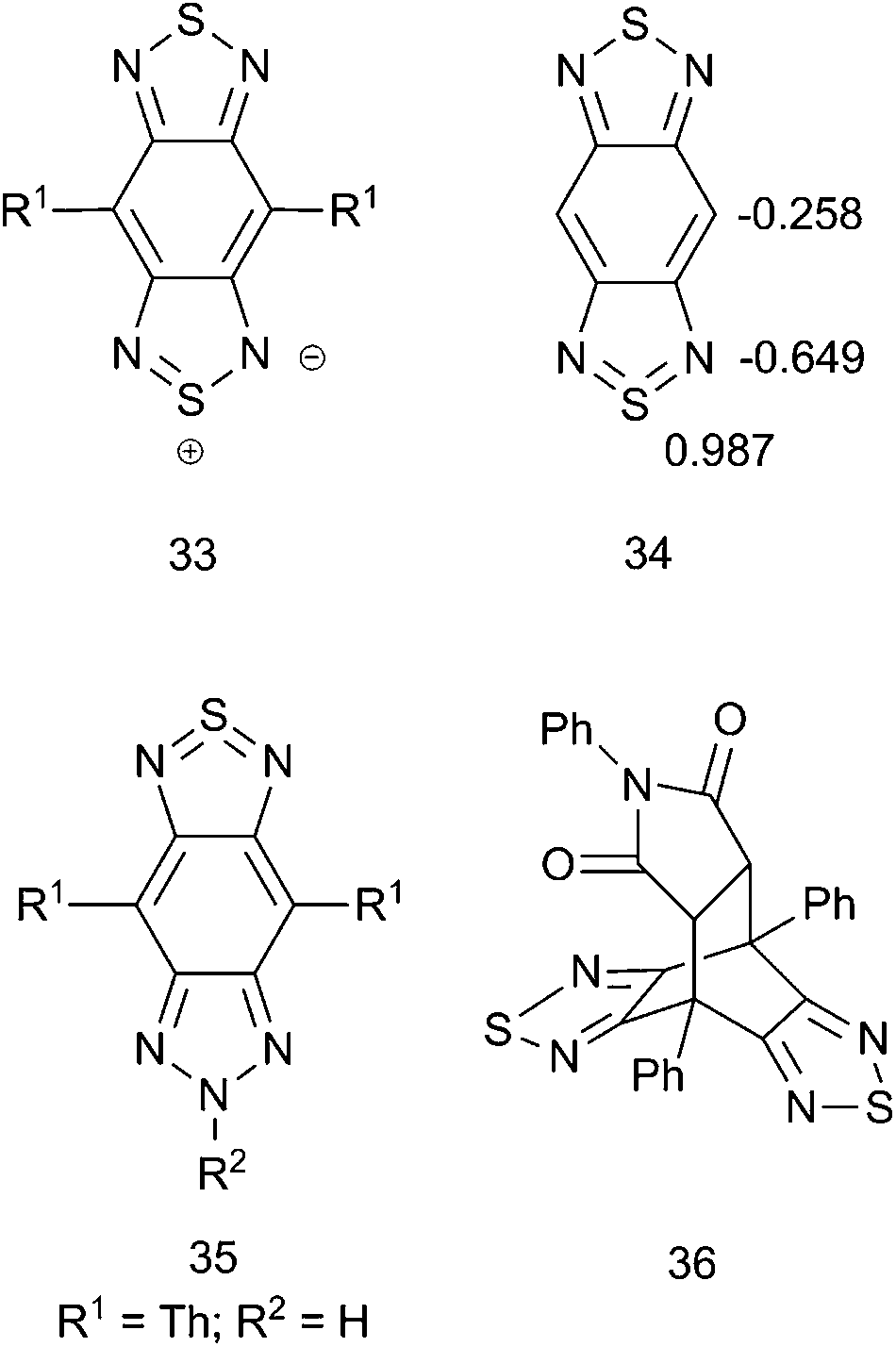 | ||
| Fig. 4 Structural considerations in BBT and BBT cyclization adduct 36. | ||
Although the nature of bonding in BBT is interesting from a theoretical standpoint, a more practical question is to what extent material properties are affected upon extension of BT to afford TDQ, BBT, and other possible BT heteroannulation derivatives. DFT studies on small molecules96,98–100 and oligomers101–108 (to represent polymers) have been carried out to explore the electronic, redox, and optical properties of these moieties in a number of donor–acceptor architectures. These investigations show that a subtle interplay between steric and electronic effects (e.g., relative co-planarity of the subunits) on the electronic coupling between the electron donor and acceptor moieties affects the key energy levels of the materials; however, these studies also indicate that replacement of BT with TDQ and, in particular, with BBT, tends to lead to lower ELUMO and similar or slightly higher EHOMO. These tendencies can be explained using a simple perturbational illustration as shown in Fig. 5, which is similar to the analysis used in a computational approach taken by Pandey et al. on a range of acceptors.109 In Fig. 5, the local B3LYP/6-31G* calculated EHOMO and ELUMO of a “donor” (bithiophene, 37), represented by dashed lines across the graph, along with the EHOMO and ELUMO values of BT, TDQ, and BBT are shown. In D–A–D molecules, mixing of the much lower energy LUMOs of TDQ and BBT with the donor LUMO would be expected to significantly decrease ELUMO of the D–A systems compared to their BT analogue (ΔELUMO); on the other hand, the slightly higher local EHOMO of both TDQ and BBT could be expected to raise EHOMO of the D–A system somewhat relative to BT (ΔEHOMO), and such trends are consistent with the calculated EHOMO and ELUMO values for the D–A–D molecules. This would result in a significant narrowing of the HOMO–LUMO gap (green arrows) across the series BT → TDQ → BBT, and thus, assuming that the lowest lying transition is well-described as a HOMO–LUMO transition, lead to a red shift in the absorption band. Another potential consequence of the relatively low ELUMO for TDQ and BBT is that one might expect the LUMO of a D–A–D system would have higher coefficients in the acceptor portion of the molecule for A = TDQ and BBT than for A = BT. On the other hand, there should be relatively little change in the acceptor contributions to the HOMOs of D–A derivatives, since the change in ELUMO going from BT to BBT is significantly larger than the change in EHOMO. This may mean that, for a given donor, there would be less overlap between the HOMO and LUMO in the TDQ and BBT systems compared to their BT analogues, which can in turn decrease the oscillator strength of the optical transitions, such as was computed and discussed in the work by Köse for a range of acceptors;99 however, as is often the case, these effects will be subject to the modulations of the D–A electronic couplings, and may not be manifested if steric interactions force the D–A moieties significantly out of coplanarity in TDQ- or BBT-containing materials.
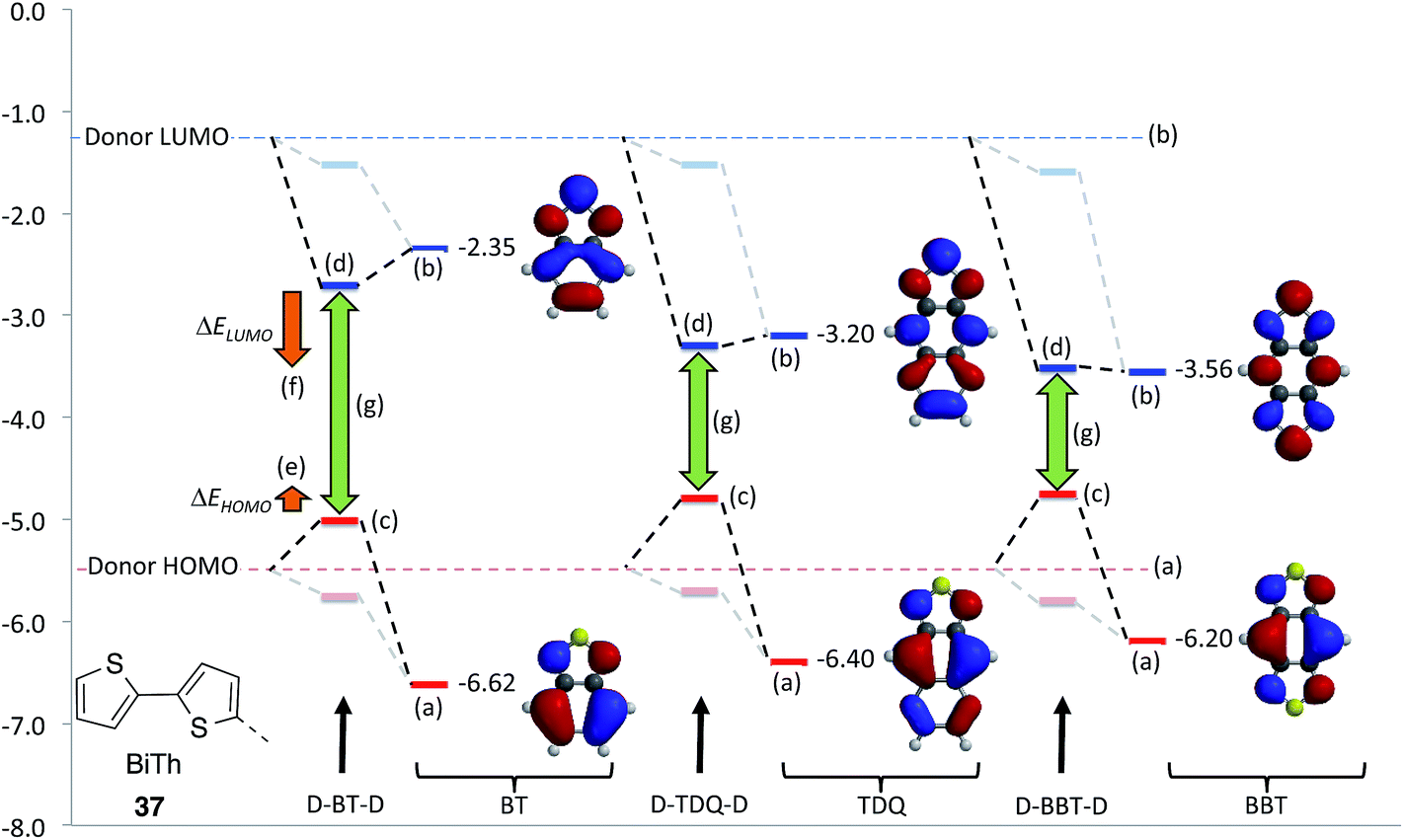 | ||
| Fig. 5 Perturbational analysis of bonding D–A–D systems with BT, TQD, and BBT acceptors and BiTh donors. (a) EHOMO levels of acceptors; (b) ELUMO levels for the acceptors; (c) EHOMO levels of the D–A–D compound; (d) ELUMO levels for the D–A–D compounds; (e) EHOMO trend across the series; (f) ELUMO trend across the series; (g) HOMO–LUMO gaps. Calculations performed at the B3LYP/6-31G* level. | ||
To examine the extent to which the above-mentioned trends are supported by experiment, examples of small molecules with a D–A–D general structure are compared in Fig. 6. Care should be taken in comparing data between the six groups of compounds in Fig. 6 since different measurements conditions and assumptions were used by the various authors (see figure notes). The porphyrin derivatives 38–40 reported by Therien and coworkers,100 the thiophene donor molecules 41 and 42 reported by Yamashita and coworkers110,111 and 43 and 44 reported by Wang and coworkers112 all largely show the general trends discussed above in that EA is substantially increased while IE either decreases or is approximately the same. Other D–A–D derivatives of TDQ such as 45 and 46 reported by Reynolds and coworkers113–116 also follow these trends; however, other compounds such 47 and 48 that are substituted with bulkier 3,4-ethylenedioxythiophene (EDOT),117 display higher IE than the BT counterpart (ΔIE = +0.13 eV from BT → TDQ). This is presumably due to a greater distortion from planarity in the case of the TDQ–EDOT system than its BT–EDOT analogue, which was seen in the EDOT derivative 49, where a torsion between the plane of the BBT and the EDOT thiophenes (53° in the X-ray crystal structure)113 results in a blue shift (λmax 650 nm) compared to the weaker donor thiophene 17 (Fig. 1, λmax = 702 nm in CH2Cl2).77
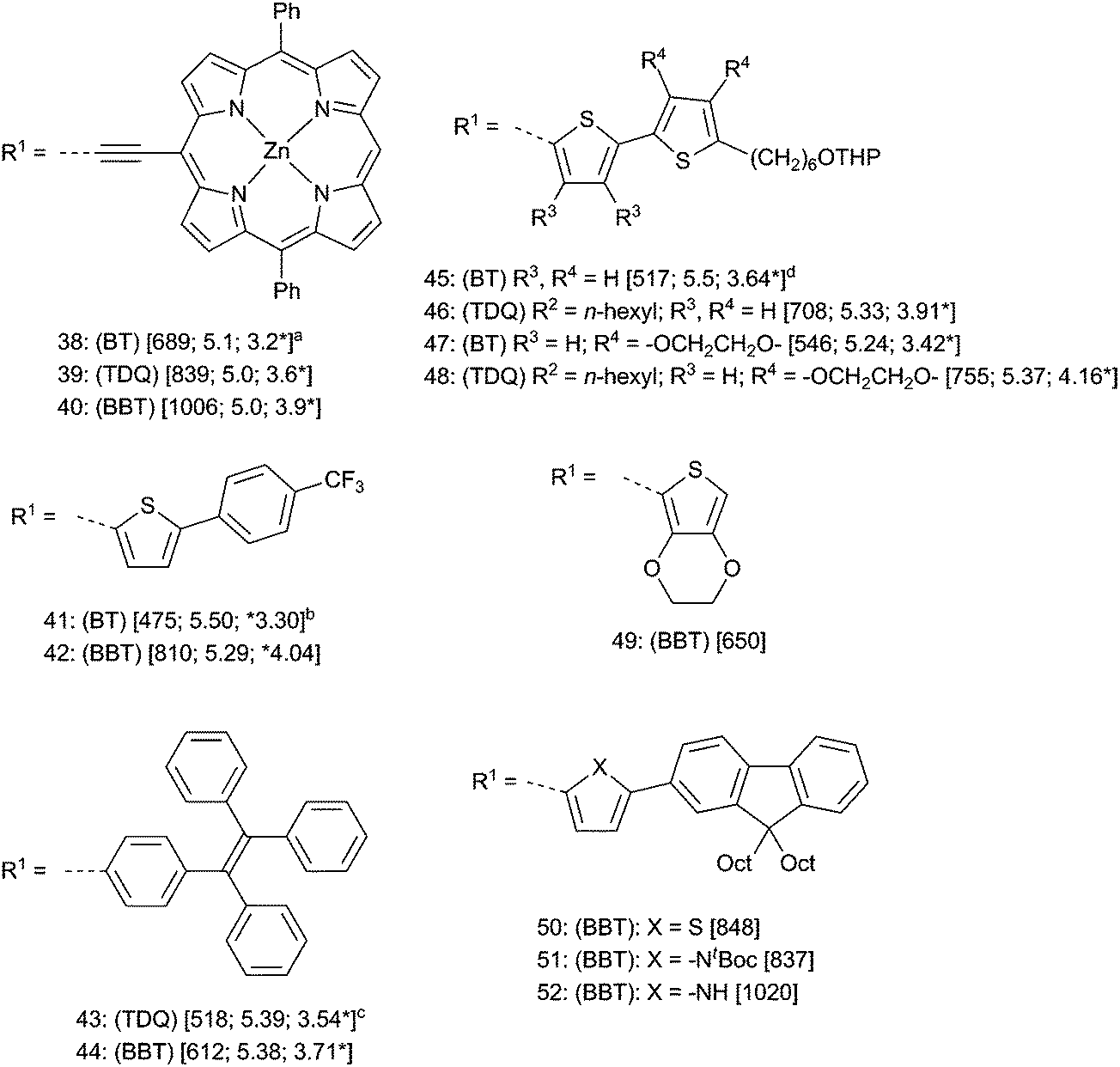 | ||
| Fig. 6 Small molecule D–A–D compounds for A = BT, TDQ, or BBT. For all compounds: (BT, TDQ, or BBT) refer to the parent compounds and R1; R2 refer to parent substituents in Fig. 2. Data are [λmax (nm); IE (eV); EA* (eV)]. IE and EA given are: a reported E1/2ox or E1/2red + 4.8 eV. beEox onset or eEred onset +4.8 eV. ceE1/2ox or eE1/2red + 4.34 eV. deE1/2ox or eE1/2red + 5.1 eV. | ||
The importance of coplanarity is also demonstrated in the series of D–A–D compounds 50–52,111,112,118–121 where there is a small blue shift on going from thiophene donor 50 to the more electron-donating pyrrole donor in 51, but the expected large red shift when the t-Boc group is removed (52), allowing for planarization. Thus, it is clear from the available small-molecule data and computations that, in the absence of large donor–acceptor torsions in TDQ and BBT derivatives, substituting BT with TDQ and BBT results in a red-shift of λmax arising mostly from a relatively large decrease in ELUMO (increase in EA) and, in many cases, a relatively small increase in EHOMO (decrease in IE); however, the effect on IE is subtle and may differ depending on the donors and the specifics of bonding.
Although many polymers containing BT, TDQ, and BBT have been prepared, relatively few studies have directly compared polymers containing each of the three acceptors. Again, comparisons among studies are difficult due to differences in conditions and methods for measuring properties such as λmax, Eg from the optical absorption onset (Eoptg), IE, and EA. Because of these difficulties, we restrict discussion of acceptor trends in polymers to a few studies that have incorporated each of BT, TDQ, and BBT units into comparable polymers, and other TDQ and BBT containing polymers will be discussed below in the sections on device properties. Early on, Yamashita and coworkers electropolymerized monomers 15, 16, and 17 (Fig. 2) using an ITO electrode to give corresponding polymers 53–55 (Fig. 7), respectively.77,78 Polymers 53–55 were intractable solids with Eoptg of 1.2 eV, 0.7 eV, and 0.5 eV, respectively, in the solid state, and with EA increased markedly by TDQ and BBT, both of which are roughly in line with what is seen in BT, TDQ, and BBT small molecules and this trend is consistent with calculations on oligomeric systems. Although the 0.5 eV Eoptg was among the narrowest optical gaps reported at the time, the extremely poor solubility of 53–55 limited more complete characterization. More recently, Marder, Reynolds, and coworkers reported soluble polymers 56–58 (Fig. 7) with the strong DTP donor, which were prepared via Stille cross coupling reactions.107,108 The solubility of these polymers allowed more thorough characterization, and the data in Fig. 7 show both increasing λmax and decreasing Eoptg across the series BT → TDQ → BBT while the electrochemically estimated IE decreases between BT polymer 56 and BBT polymer 58 (4.9 eV to 4.7 eV). The EA increase was more pronounced across the series BT → TDQ → BBT (3.2 eV → 3.5 eV → 4.0 eV). Again, the properties of polymers in Fig. 7 were generally consistent with those discussed above for small molecules and for computations. The implications of these property trends for materials and device performance in OPVs, NIR OLEDs, and OFETs are discussed below.
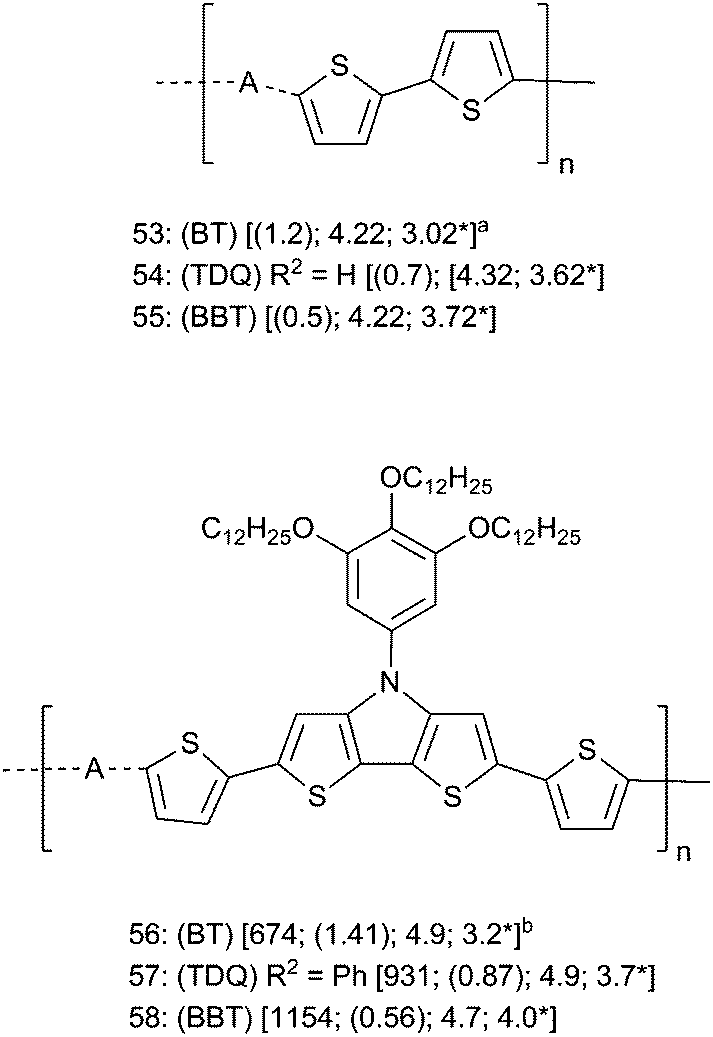 | ||
| Fig. 7 Polymers comparing A = BT, TDQ, and BBT. For all compounds: (BT, TDQ, or BBT) refer to the parent compounds and R2 refers to parent substituents in Fig. 2. Data are [λmax (nm); (Eoptg (eV)); IE (eV); EA* (eV)]. a EA = reported eEpc + 4.4, IE = EA + Eoptg. b IE = eEonsetox + 4.4; EA = eEonsetred + 4.4. | ||
OPVs
Blends of conjugated donor–acceptor hole-transporting polymers with electron-transporting fullerenes are widely studied as active layers in organic bulk-heterojunction (BHJ) photovoltaic cells. Numerous optical, electronic, and morphological criteria are required by a candidate polymer–fullerene system and have been discussed elsewhere.26,122–124 For the present discussion it is important to note that increasing the power conversion efficiency (PCE) depends on maximizing both the short-circuit current density, JSC, and the open-circuit voltage, VOC. Three factors that influence these quantities will be considered here. Firstly, large JSC requires efficient dissociation of the polymer excited states to form well-separated holes and electrons in polymer and fullerene, respectively. It should be noted that such a hole–electron pair is often referred to as a “charge-separated” state, whereas an electrostatically bound geminate ion pair is referred to as a “charge-transfer” state.125 To be thermodynamically feasible (ΔGCS < 0), charge separation from the polymer excited state (P*) requires that the EA of the fullerene, EA(F), exceeds the excited-state IE of the polymer, IE(P*) = IE(P) − Eoptg, where Eoptg is the energy of the relaxed excited state relative to the ground state (generally estimated from the low-energy onset of the absorption spectrum in a film). Secondly, minimizing Eoptg can potentially increase JSC by allowing more photons (over a wider range of the solar spectrum) to be absorbed assuming absorbance is not substantially lost from other parts of the absorption spectrum. Thirdly, VOC is limited by IE(P)–EA(F). Thus, there is a tradeoff between these quantities; increasing IE(P) to increase VOC will, for a given Eoptg, lead to a reduced driving force for charge separation. Indeed, to optimize the tradeoff between VOC and JSC in a single-layer BHJ cell, it has been proposed that Eoptg for the polymer should be ca. 1.3–1.5 eV (ca. 830–950 nm).126The low-lying LUMOs of the TDQ and BBT acceptors lead to lower energy optical absorption in donor–acceptor polymers incorporating these moieties than in analogous BT systems (see Table 2). Examples examined in OPVs as blends with fullerenes include (Fig. 8): oligothiophene donor polymers 59, 60,127,128 and 61;129 the rigid thiophene–phenylene–thiophene (TPT) donor polymers 62 and 63;130 the dithiophene–pyrrole donor polymer 64;131 the thiophene–fluorene–thiophene donor polymers 65132 and 66;133 and the DTP donor polymers 56–58108 (Fig. 7). As well as optical and OPV data, the table also lists IE and EA values, along with driving forces for formation of a well-separated polymer/fullerene ion pair from the polymer excited state (−ΔGCS). These values are estimates and comparison between studies is complicated by the use of different assumptions and approximations; however, we have attempted to standardize the methods used for estimation for all compounds; moreover, the trends, if not the absolute numbers, within a given study can provide useful insight.
| X | A | λ max/nm | E optg /eV | IE/eV | EA/eV | −ΔGCSb/eV | J SC/mA cm−2 | V OC/V | PCE/% |
|---|---|---|---|---|---|---|---|---|---|
| a From onset of absorption. b Driving force for charge separation from X to [60]PCBM estimated by ΔG = IE(X) − Eoptg(X) − EAPCBM, using an EA[60]PCBM value of 3.8 eV from IPES.144 A similar value of 3.7 eV is also obtained for both [60]- and [70]PCBM using electrochemical data145 and assuming EA = eE1/2(PCBM) + 4.8 eV (potential vs. FeCp2+/0). c From UPS. d Estimated from electrochemical onset oxidation potentials using the assumption that IE = eEox + 4.8 eV where the potential is quoted vs. FeCp2+/0 (equivalent to offsets of 4.4 eV for SCE or AgCl/Ag reference or 4.7 eV for Ag+/Ag reference). e Estimated from electrochemical onset reduction potential using EA = eEred + 4.8 eV (potential vs. FeCp2+/0) or an equivalent relation. f Driving force given applies to PCBM; a somewhat larger driving force is expected for the fullerene employed in the OPV work, the EA of which was estimated to be 4.2 eV. g Estimated from electrochemical data, but assumptions used not specified. h Reduction potentials vs. AgCl/Ag used to estimate EAs using an offset of 4.4 eV, rather than the 4.72 eV in the original publication, for consistency with other data here. No oxidation potentials given; lower limit for IEs estimated from values of EA and Eoptg. i Values refer to devices with [60]PCBM. j Values refer to devices with [70]PCBM. k higher VOC, JSC, and PCE (up to 2.02%) were obtained with a related acceptor that presumably exhibits a similar EA, but contributes increased light absorption. | |||||||||
| 59 | BT | 597 | 1.7 | 4.7c | — | 0.8 | 2.87i | 0.60 | 0.55 |
| 60 | BBT | 902 | 0.7 | 4.3c | — | 0.1 | 0.00719i | 0.04 | — |
| 62 | BT | 514 | 1.7 | 5.3d | 3.5e | 0.2 | 10.1j | 0.8 | 4.3 |
| 63 | TDQ | ∼875 | 1.0 | 5.2d | 3.7e | −0.4 | 3.6j | 0.54 | 0.84 |
| 56 | BT | 674 | 1.4 | 4.9d | 3.2e | 0.3 | 3.9i | 0.510 | 1.3 |
| 57 | TDQ | 931 | 0.9 | 4.9d | 3.7e | −0.2 | 1.7i | 0.268 | 0.22 |
| 58 | BBT | 1154 | 0.6 | 4.7d | 4.0e | −0.3 | 0.20i | 0.109 | 0.001 |
| 65 | TDQ | 815 | 1.2 | 5.1d | 3.9e | −0.1f | 3.4 | 0.58 | 0.70 |
| 66 | TDQ | 788 | 1.3 | 5.2g | 3.9g | −0.1g | 7.35j | 0.82 | 2.36 |
| 61 | TDQ | 703 | 1.4 | 5.0d | 3.8e | 0.2 | 1.58i | 0.58 | 0.48 |
| 64 | TDQ | 756 | 1.1 | 4.8d | 3.7e | 0.1 | 3.41j | 0.39 | 0.43 |
| 67 | TDQ | 820 | 1.5 | 5.5d | 3.6e | −0.2 | 5.75i | 0.77 | 2.44 |
| 68 | TDQ | 833 | 1.4 | 5.4d | 3.6e | −0.1 | 3.50i | 0.72 | 1.32 |
| 69 | TDQ | 872 | 1.4 | 5.3d | 3.5e | −0.1 | 4.25i | 0.65 | 1.42 |
| 70 | TDQ | 776 | 1.7 | >5.2h | 3.5h | <0.3h | ∼3.04j | ∼0.72 | — |
| 71 | BBT | 810 | 1.5 | >5.2h | 3.6h | <0.2h | 4.95j | 0.66 | 1.02 |
| 72 | BBT | 636 | 1.6 | 5.3d | 3.5e | 0.2 | 3.50i | 0.72 | 1.05k |
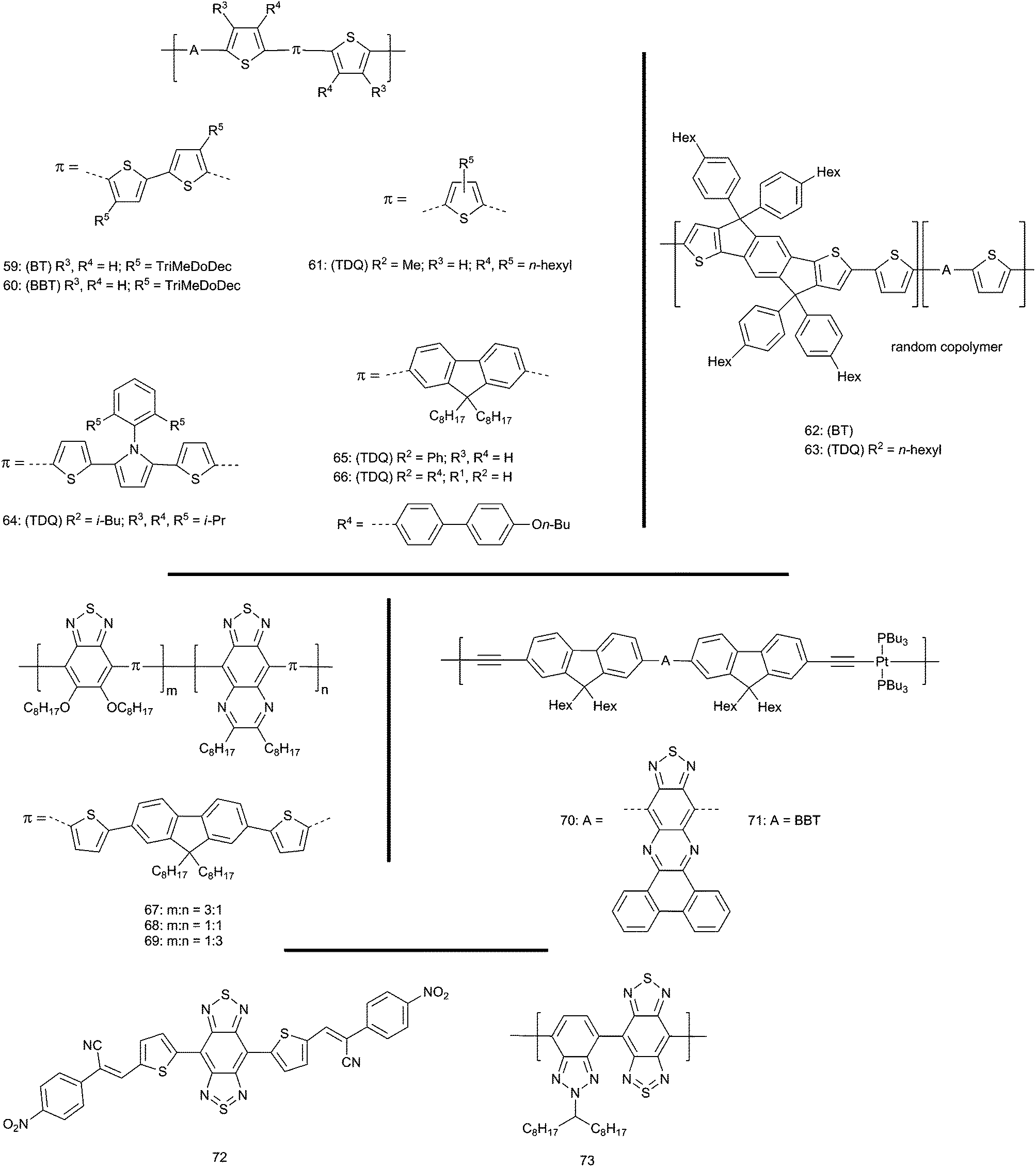 | ||
| Fig. 8 Polymers containing A = BT, TDQ, and BBT used to fabricate BHJ OPVs. For all compounds: (BT, TDQ, or BBT) refer to the parent compounds and R2 refers to parent substituents in Fig. 2. | ||
Although the low-energy absorptions possible in donor–acceptor polymers incorporating BBT and TDQ acceptors are attractive from the point of view of extending solar harvesting into the near-IR, in many cases, especially those where these acceptors are coupled with electron-donating quaterthiophene (60), dithiophene-pyrrole (64), or dithiophene DTP donors (57, 58), these absorptions extend to longer wavelengths than the optimal 830–950 nm referenced above. In several studies, this results in BBT and TDQ derivatives performing much more poorly than the corresponding BT derivative (60vs.59; 63vs.62, 57 and 58vs.56). Multiple factors may contribute to this; for example, a significantly lower absorptivity is found for 60vs.59, and open-circuit voltages are often smaller for the BBT and TDQ derivatives (this effect being much more significant than the effect attributable to the slightly lower estimated IEs relative to BT analogues). However, an important role is presumably played by the lower, in some cases negative, driving forces estimated for charge-separation from the excited states of the BBT and TDQ materials. Indeed, for some compounds (e.g., 58) the electrochemically estimated EAs exceed that of [60]PCBM. Reasonably efficient devices with large open-circuit voltages are obtained for the TDQ derivative, 66; however, in this particular case the assumptions used to estimate the IE (and, therefore, −ΔGCS) and EA were not described, meaning these values are difficult to reliably compare to those for other materials.
The remainder of the table summarizes the properties of materials with less straightforward alternating donor–acceptor structures: copolymers containing both TDQ- and BT-containing monomers (67–69),134 polymers containing platinum in the main chain (70, 71),135 and a small-molecules with electron-withdrawing substituents on the periphery (72).136 Other more recent polymers incorporating dithienosilole137 and benzodithiophene138 donors showed similar results. Although some of these materials lead to moderate efficiencies, charge separation is again estimated to be of marginal thermodynamic feasibility.
Overall, the relatively low fundamental HOMO–LUMO gaps of the TDQ and, especially, BBT building blocks create a challenge in achieving both efficient charge separation and a moderate open-circuit voltage in simple single-layer BHJ devices. The best prospects are for materials with less electron-donating co-monomers or substituents in the case of small molecules. However, it is worth noting that, given the very low-energy absorptions seen for some of these materials, TDQ and BBT materials may be useful in tandem cells,139–141 in which a low-voltage cell that harvests longer wavelength light is sandwiched with, and connected in series to, a higher voltage cell that harvests shorter wavelengths. TDQ and BBT materials with near-IR absorptions could potentially be used in the low-voltage portion either as hole-transport materials, perhaps in conjunction with higher EA electron-transport materials than [60]- or [70]PCBM to ensure efficient charge separation, or, given that electron-transport has been observed in some BBT derivatives (see below), as a light-harvesting electron-transporting component in conjunction with a more electron-donating hole-transporting polymer. Indeed, Wong and coworkers recently reported a dual acceptor polymer 73 containing both BBT and benzotriazole that was used as a PCBM replacement in conjunction with P3HT as a donor, albeit with low reported efficiencies so far (PCE = 0.4%).142 Moreover, some of these materials, in conjunction with appropriate partner materials, may be useful in near-IR photodetectors,143 where a measureable efficient photocurrent generation is required, but a photovoltage is not.
NIR OLEDs and related materials
Near-infrared emitting organic light-emitting diodes (NIR-OLEDs) have applications in telecommunications, defense, and biomedicine. The long wavelength absorption of the TDQ and BBT derivatives make them potentially good candidates for NIR-OLEDs. Fortunately, there are many small molecule and polymer hosts that have been developed for OLEDs that may be used to match the energy levels in the TDQ and BBT derivatives, which is advantageous compared to state-of-the-art BHJ solar cell applications where, as discussed above, the various energy levels of the TDQ and BBT donor polymers often must be aligned to those of C60 or C70 derivatives. However, one drawback is that NIR luminescence of organic molecules is limited by the energy gap law of radiationless transitions,146 meaning that quantum yields of luminescence for NIR emitters will likely be low. Nevertheless, some TDQ and BBT derivatives and polymers have been used in NIR-OLEDs: selected materials are shown in Fig. 9, and materials and device properties are summarized in Table 3. Included in these tabulations are TDQ copolymers 74 and 75 studied by Cao147 and Sun and coworkers,148 a series of triarylamine donor D–A–D compounds reported by Wang and coworkers including examples 76–78,118,120 a series of tetraphenylethene D–A–D-type compounds also reported by Wang and coworkers112 including examples 43 and 44, and the EDOT D–A–D compounds 80 and 49 (Fig. 6) reported by Xue, Reynolds, and coworkers.114,115 Most of these materials electroluminescence in the NIR, but solution photoluminescent quantum yields (ϕf) are mostly under 15%. Consequently, the external quantum efficiency (EQE) of optimized devices are mostly under 1%, although Xue, Reynolds, and coworkers were able to increase the EQEs of 80 and 49 to 3.1% and 1.5%, respectively, by using sensitized fluorescence with organometallic dopants.115 Although these materials do not approach the EQEs of the most efficient small-molecule and polymer OLEDs due to fundamental limitations in the luminescent yields at these long wavelengths, the variety of host materials available to match the high EA and relatively low IE of TDQ and especially BBT materials may allow sufficient optimization for certain applications. In addition to NIR OLEDs, another potential application was reported by Wang and coworkers; the NIR photoluminescence of D–A–D chromophore 79 was found to be sensitive to cyanide concentrations, which was proposed to attack one of the partially positively charged sulfur atoms of BBT as shown in 34 (Fig. 4), suggesting its use as a potential sensing agent.119 In related work, Wang and coworkers demonstrated NIR chemiluminescence from D–A–D chromophores structurally similar to 76–79.121 Finally, BBT was used as a core to give NIR-absorbing and emitting hexagonal columnar liquid crystals (82) that, among other properties, showed the expected red-shift in λmax from the BT compound 81.149 This demonstrated the ability of BBT to provide materials that are NIR fluorescent with reasonable quantum yields, which may be useful in applications outside of organic electronics such as NIR biosensing; however, the EQEs presented in Table 3 are still about 10 times less than those reported for Pt–porphyrin OLEDs.150,151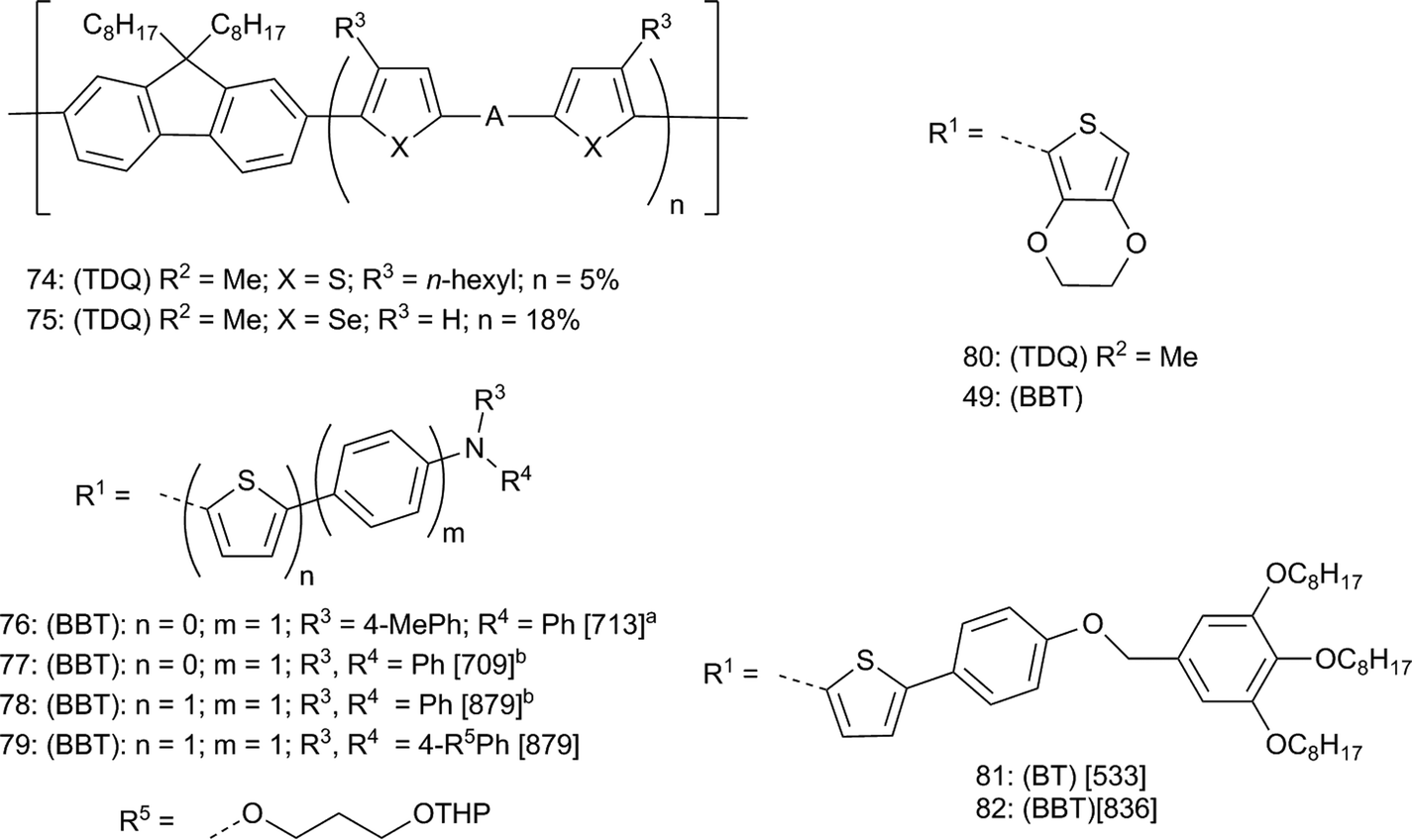 | ||
| Fig. 9 A = BT, TDQ, or BBT polymers 74,75 and D–A–D compounds 76–82 studied for NIR electroluminescence and fluorescence. Data are [λmax (nm)]. a In toluene. b In CH2Cl2. For all compounds: (BT, TDQ, or BBT) refer to the parent compounds and R1; R2 refer to parent substituents in Fig. 2. | ||
| X | A | IE/eV | EA/eV | ϕ f/% | λ El/nm | EQE/% |
|---|---|---|---|---|---|---|
| a Estimated from electrochemical oxidation potential using IE = eEox + 4.8 eV where the potential is quoted vs. FeCp2+/0 (equivalent to offsets of 4.3–4.4 eV for SCE or AgCl/Ag references). b Estimated from EA = eEred + 4.8 eV (potential vs. FeCp2+/0) or equivalent expression. c eE 1/2ox or eE1/2red + 4.34 eV. d Estimated from electrochemical data and optical absorption data, but assumptions used are not specified. | ||||||
| 74 | TDQ | 5.7a | 4.2b | — | 849 | 0.013 |
| 75 | TDQ | 5.7a | 4.3b | — | 859 | — |
| 76 | BBT | 5.2a | 3.8d | 5.8 | 1080 | 0.73 |
| 77 | BBT | 5.2a | 3.9d | 7.4 | 1050 | 0.05 |
| 78 | BBT | 5.0a | 4.0c | 4.9 | — | |
| 43 | TDQ | 5.4a | 3.5c | 10.1 | 706 | 0.89 |
| 44 | BBT | 5.4a | 3.7c | 13.0 | 802 | 0.43 |
| 80 | TDQ | 5.6d | 3.7d | 21 | 692 | 1.6 |
| 49 | BBT | 5.6d | 4.1d | 7.6 | 815 | 0.51 |
OFETs
The sizable increase of EA typically seen when replacing BT with TDQ and BBT raises the possibility of TDQ and BBT materials exhibiting oxygen-stable electron-transporting properties.152 Several BBT containing materials, mostly D–A polymers (Fig. 10), have been studied in OFETs and properties of selected materials are summarized in Table 4. Of the small molecules studied by Yamashita and coworkers, the fluorinated derivative 42 (Fig. 6) shows an air-stable electron mobility110,153 (μe, Table 4) of 0.40 cm2 V−1 s−1, optimized to 0.77 cm2 V−1 s−1 by modifying deposition conditions, which was 10-fold higher than that of the corresponding BT derivative 41.154 Additionally, Th–BBT–Th 17 (Fig. 2) showed ambipolar transport behavior under similar conditions. The D–A polymers 56–58107,108 (Fig. 7) and the TDQ polymer 83 (Fig. 10) showed mostly hole transport, but BBT polymer 58 showed ambipolar charge transport. More recently, ambipolar behavior was also observed in a series of BBT-containing polymers 84, 86, and 87 reported by the groups of Heeger and Wudl,155 and in the structurally similar 85 reported by the groups of Lee and Prasad.156 However, in contrast the fluorene-bridged polymer 87, no μe was reported for structurally similar polymer 88,156 which may be yet another example of how changes in structure and/or processing conditions can lead to differing properties. Another example of this is found in work by the Heeger and Wudl groups with the quaterthiophene-bridged polymer 89, which shows quite high μh relative to other BBT-containing polymers yet very low μe;157 in contrast, substitution of two of the thiophene units in 89 with the thienylthiophene unit in polymer 90 results in optimized ambipolar behavior.158 Once more, an example of the ability of BBT to both increase EA and lower IE, and to provide ambipolar charge transport, is demonstrated with the mixed acceptor diketopyrrolopyrrole (DPP)–BT/BBT polymers 91 and 92, where substitution of BT in 91 with BBT to give 92 results in an increase in μh and gives rise to measurable μe.159 Generally, this tendency of BBT to raise EA significantly while either maintaining or lowering IE is probably closely related to the ability of BBT polymers to exhibit relatively high μe while also exhibiting relatively high μh, although inter- and intrachain electronic coupling must also play a role, perhaps with some contribution from the larger π-surface of BBT compared to BT leading to better A–A interchain overlap in BBT polymers.
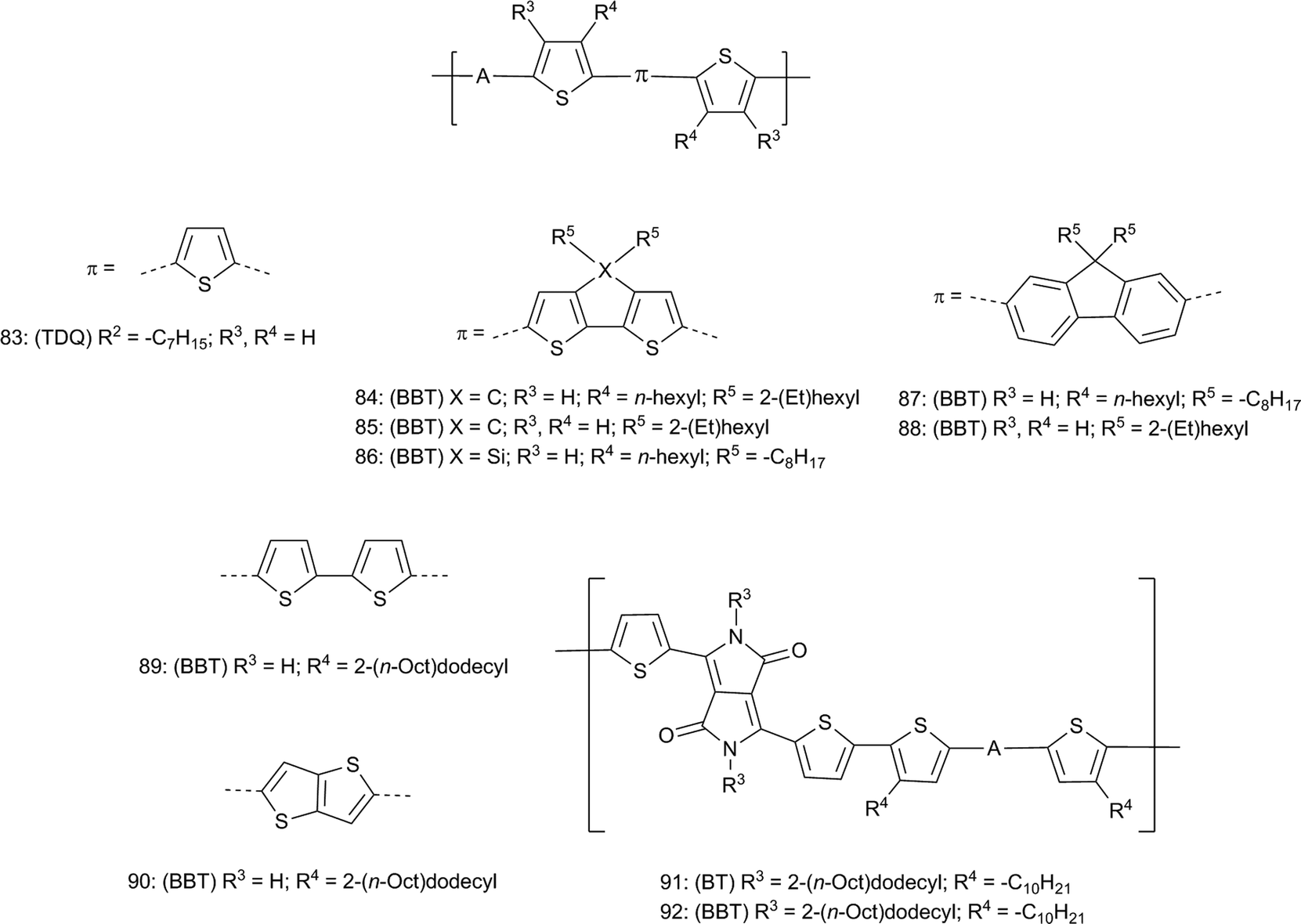 | ||
| Fig. 10 Polymers with A = BT, TDQ, or BBT studied in OFETs. For all compounds: (BT, TDQ, or BBT) refer to the parent compounds and R2 refers to parent substituents in Fig. 2. | ||
| X | A | IE/eV | EA/eV | μ h/cm2 V−1 s−1 | μ e/cm2 V−1 s−1 |
|---|---|---|---|---|---|
| a Estimated from electrochemical onset oxidation potentials using the assumption that IE = eEox + 4.8 eV where the potential is quoted vs. FeCp2+/0 (equivalent to offsets of 4.4 eV for SCE or AgCl/Ag reference or 4.7 where the reference is Ag+/Ag). b Estimated from electrochemical onset reduction potential using EA = eEred + 4.8 eV (potential vs. FeCp2+/0) or an equivalent relation. c Method for determining IE and EA not specified. d Estimated from onset of eEox or eEred, but assumptions used not specified. e From UPS. f Estimated from EA − Eoptg (determined from the absorption onset). | |||||
| 41 | BT | 5.50a | 3.30b | — | 0.04 |
| 42 | BBT | 5.29a | 4.04b | — | 0.40 |
| 17 | BBT | 5.32a | 3.96b | 3.4 × 10−7 | 1.6 × 10−4 |
| 56 | BT | 4.9a | 3.2b | 1.2 × 10−4 | — |
| 57 | TDQ | 4.9a | 3.7b | 2.2 × 10−3 | — |
| 58 | BBT | 4.7a | 4.0b | 1.6 × 10−3 | 7.9 × 10−4 |
| 83 | TDQ | 4.84a | 3.63b | 3.8 × 10−3 | — |
| 84 | BBT | 4.8c | 4.0c | 1.1 × 10−1 | 7.4 × 10−2 |
| 86 | BBT | 4.8c | 4.1c | 1.9 × 10−3 | 1.1 × 10−2 |
| 87 | BBT | 5.1c | 3.9c | 5.6 × 10−3 | 7.0 × 10−4 |
| 85 | BBT | 5.33d | 4.32d | 7.1 × 10−4 | 3.3 × 10−3 |
| 88 | BBT | 5.12d | 3.92d | 3.1 × 10−4 | — |
| 89 | BBT | 4.6e | 3.8b | 2.5 | Low |
| 90 | BBT | 4.36f | 3.8b | 1.0 | 0.7 |
| 91 | BT | 4.75f | 3.4b | 0.17 | — |
| 92 | BBT | 4.55f | 3.9b | 0.89 | 0.99 |
Conclusion
The many small-molecule and polymer examples given above show that TDQ and BBT can be considered strong acceptors compared to BT, lowering Eg and increasing EA. However, they can also generally be considered as weaker acceptors in that they often lead to lower IE than in BT analogues, although this effect is subtle and subject to bonding specifics in a system. This has materials property implications, particularly for use in donors in OPVs relying on PCBM for charge separation and electron transport, where high EA of the donor polymer (assuming it closely approximates the excited state oxidation potential) may lead to much lower JSC compared to BT, while at the same time lower IE may lead to lower VOC compared to BT polymers near the “ideal” EA and IE values.26,124 However, for NIR OLEDs, the strong tendency for heteroannulation to decrease Eoptg from BT → TDQ → BBT is less of a limitation since many OLED host materials have been developed with varying IE and EA, although the energy gap law tends to limit overall performance for NIR emitters. For OFETs, the tendency for BBT in particular to lower the IE and raise the EA imparts BBT-based materials with a strong potential for ambipolar charge transport, and the strong lowering of EA by BBT makes it an intriguing option in the development of air-stable electron-transport materials. Thus, much caution should be exercised when developing OPV materials using heteroannulated BT acceptors; but certain applications in NIR OLEDs, ambipolar OFETs, and air-stable electron transport materials may prove feasible with careful materials and device design.References
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed.
- D. A. daSilvaFilho, E. G. Kim and J. L. Brédas, Adv. Mater., 2005, 17, 1072 CrossRef PubMed.
- V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 CrossRef CAS.
- D. G. Patel, Y. Y. Ohnishi, Y. X. Yang, S. H. Eom, R. T. Farley, K. R. Graham, J. G. Xue, S. Hirata, K. S. Schanze and J. R. Reynolds, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 557 CrossRef CAS PubMed.
- Y. Yang, Q. Pei and A. J. Heeger, J. Appl. Phys., 1996, 79, 934 CrossRef CAS PubMed.
- J. Roncali, P. Leriche and A. Cravino, Adv. Mater., 2007, 19, 2045 CrossRef CAS PubMed.
- L. Dalton, P. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25 CrossRef CAS PubMed.
- M. Rumi, S. Barlow, J. Wang, J. Perry and S. Marder, Adv. Polym. Sci., 2008, 213, 1 CAS.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS PubMed.
- P. M. Beaujuge, H. N. Tsao, M. R. Hansen, C. M. Amb, C. Risko, J. Subbiah, K. R. Choudhury, A. Mavrinskiy, W. Pisula, J.-L. Brédas, F. So, K. Müllen and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 8944 CrossRef CAS PubMed.
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2010, 23, 397 CrossRef.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396 CrossRef CAS PubMed.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed.
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC.
- C. Teng, X. Yang, S. Li, M. Cheng, A. Hagfeldt, L. Wu and L. Sun, Chem.–Eur. J., 2010, 16, 13127 CrossRef CAS PubMed.
- A. Mishra, M. K. R. Fischer and P. Bauerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed.
- C. A. Richard, Z. Pan, H. Y. Hsu, S. Cekli, K. S. Schanze and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2014, 6, 5221 CAS.
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- J. E. Anthony, Chem. Mater., 2011, 23, 583 CrossRef CAS.
- L. Bian, E. Zhu, J. Tang, W. Tang and F. Zhang, Prog. Polym. Sci., 2012, 37, 1292 CrossRef CAS PubMed.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- C. L. Chocos and S. A. Choulis, Prog. Polym. Sci., 2011, 36, 1326 CrossRef PubMed.
- Y. Li, Acc. Chem. Res., 2011, 45, 723 CrossRef PubMed.
- H. J. Son, F. He, B. Carsten and L. Yu, J. Mater. Chem., 2011, 21, 18934 RSC.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296 CrossRef CAS PubMed.
- P. M. Beaujuge and J. M. J. Frechet, J. Am. Chem. Soc., 2011, 133, 20009 CrossRef CAS PubMed.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed.
- A. L. Dyer, E. J. Thompson and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 3, 1787 CAS.
- H. A. M. van Mullekom, J. Vekemans, E. E. Havinga and E. W. Meijer, Mater. Sci. Eng., R, 2001, 32, 1 CrossRef.
- J. Roncali, Macromol. Rapid Commun., 2007, 28, 1761 CrossRef CAS PubMed.
- T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345 CrossRef CAS PubMed.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. Frechet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656 CrossRef CAS PubMed.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859 CrossRef CAS PubMed.
- L. A. Estrada, R. Stalder, K. A. Abboud, C. Risko, J.-L. Brédas and J. R. Reynolds, Macromolecules, 2013, 46, 8832 CrossRef CAS.
- E. D. Glowacki, G. Voss and N. S. Sariciftci, Adv. Mater., 2013, 25, 6783 CrossRef CAS PubMed.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660 CrossRef CAS PubMed.
- B. A. D. Neto, A. A. M. Lapis, E. N. da Silva Júnior and J. Dupont, Eur. J. Org. Chem., 2013, 2013, 228 CrossRef CAS PubMed.
- Y. Zhang, J. Y. Zou, C. C. Cheuh, H. L. Yip and A. K. Y. Jen, Macromolecules, 2012, 45, 5427 CrossRef CAS.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- L. Y. Lin, Y. H. Chen, Z. Y. Huang, H. W. Lin, S. H. Chou, F. Lin, C. W. Chen, Y. H. Liu and K. T. Wong, J. Am. Chem. Soc., 2011, 133, 15822 CrossRef CAS PubMed.
- T. S. van der Poll, J. A. Love, T. Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646 CrossRef CAS PubMed.
- W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437 CrossRef CAS PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinsky, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605 CrossRef CAS PubMed.
- M. Velusamy, K. R. J. Thomas, J. T. Lin, Y.-C. Hsu and K.-C. Ho, Org. Lett., 2005, 7, 1899 CrossRef CAS PubMed.
- W. Zhu, Y. Wu, S. Wang, W. Li, X. Li, J. Chen, Z.-s. Wang and H. Tian, Adv. Funct. Mater., 2011, 21, 756 CrossRef CAS PubMed.
- S.-I. Kato, T. Matsumoto, M. Shigeiwa, H. Gorohmaru, A. Maeda, T. Ishi-i and S. Mataka, Chem.–Eur. J., 2006, 12, 2303 CrossRef CAS PubMed.
- J.-Z. Cheng, C.-C. Lin, P.-T. Chou, A. Chaskar and K. T. Wong, Tetrahedron, 2011, 67, 734 CrossRef CAS PubMed.
- K. Susumu, J. A. N. Fisher, J. Zheng, D. N. Beratan, A. G. Yodh and M. J. Therien, J. Phys. Chem. A, 2011, 115, 5525 CrossRef CAS PubMed.
- P. M. Beaujuge, S. Ellinger and J. R. Reynolds, Adv. Mater., 2008, 20, 2772 CrossRef CAS PubMed.
- P. M. Beaujuge, S. Ellinger and J. R. Reynolds, Nat. Mater., 2008, 7, 795 CrossRef CAS PubMed.
- J. Luo, X. Li, Q. Hou, J. B. Peng, W. Yang and Y. Cao, Adv. Mater., 2007, 19, 1113 CrossRef CAS PubMed.
- J. Liu, Q. Zhou, Y. Cheng, Y. Geng, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Mater., 2005, 17, 2974 CrossRef CAS PubMed.
- L. Chen, B. Zhang, Y. Cheng, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Funct. Mater., 2010, 20, 3143 CrossRef CAS PubMed.
- Q. Fang, B. Xu, B. Jiang, H. Fu, X. Chen and A. Cao, Chem. Commun., 2005, 1468 RSC.
- Z. H. Li, M. S. Wong, H. Fukutani and Y. Tao, Chem. Mater., 2005, 17, 5032 CrossRef CAS.
- S. Ellinger, K. R. Graham, P. Shi, R. T. Farley, T. T. Steckler, R. N. Brookins, P. Taranekar, J. Mei, L. A. Padilha, T. R. Ensley, H. Hu, S. Webster, D. J. Hagan, E. W. Van Stryland, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805 CrossRef CAS.
- Y. He, X. Wang, J. Zhang and Y. Li, Macromol. Rapid Commun., 2009, 30, 45 CrossRef CAS PubMed.
- D. Muhlbacher, M. C. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. J. Brabec, Adv. Mater., 2006, 18, 2884 CrossRef PubMed.
- C. L. Chocos, N. Tagmatarchis and V. G. Gregoriou, RSC Adv., 2013, 3, 7160 RSC.
- S. Kola, J. Sinha and H. E. Katz, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1090 CrossRef CAS PubMed.
- Y. Yamashita, K. Saito, T. Suzuki, C. Kabuto, T. Mukai and T. Miyashi, Angew. Chem., Int. Ed. Engl., 1988, 27, 434 CrossRef PubMed.
- Y. Yamashita, T. Suzuki, T. Mukai and G. Saito, Chem. Commun., 1985, 1044 RSC.
- C. Kabuto, T. Suzuki, Y. Yamashita and T. Mukai, Chem. Lett., 1986, 1433 CrossRef CAS.
- T. Suzuki, H. Fujii, Y. Yamashita, C. Kabuto, S. Tanaka, M. Harasawa, T. Mukai and T. Miyashi, J. Am. Chem. Soc., 1992, 114, 3034 CrossRef CAS.
- H. Inokuchi and K. Imaeda, Acta Phys. Pol., A, 1995, 88, 1161 CAS.
- Y. Yamashita, S. Tanaka, K. Imaeda and H. Inokuchi, Chem. Lett., 1991, 1213 CrossRef CAS.
- Y. Yamashita, S. Tanaka, K. Imaeda, H. Inokuchi and M. Sano, J. Org. Chem., 1992, 57, 5517 CrossRef CAS.
- Y. Yamashita, K. Ono, S. Tanaka, K. Imaeda, H. Inokuchi and M. Sano, Chem. Commun., 1993, 1803 RSC.
- M. Takada, H. Graaf, Y. Yamashita and H. Tada, Jpn. J. Appl. Phys., Part 2, 2002, 41, L4 CAS.
- J. Huang and M. Kertesz, J. Phys. Chem. B, 2005, 109, 12891 CrossRef CAS PubMed.
- Y. Yamashita, T. Suzuki and T. Mukai, Chem. Commun., 1987, 1184 RSC.
- K. Ono, S. Tanaka and Y. Yamashita, Angew. Chem., 1994, 106, 2030 CrossRef CAS PubMed.
- M. Karikomi, C. Kitamura, S. Tanaka and Y. Yamashita, J. Am. Chem. Soc., 1995, 117, 6791 CrossRef CAS.
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570 CrossRef CAS.
- Y. Yamashita, K. Ono, M. Tomura and S. Tanaka, Tetrahedron, 1997, 53, 10169 CrossRef CAS.
- T. L. Tam, H. Li, F. Wei, K. J. Tan, C. Kloc, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2010, 12, 3340 CrossRef CAS PubMed.
- M. C. Scharber, D. Muhlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS PubMed.
- M. P. Cava and N. M. Pollack, J. Am. Chem. Soc., 1967, 89, 3639 CrossRef CAS.
- M. P. Cava, N. M. Pollack and D. A. Repella, J. Am. Chem. Soc., 1967, 89, 3640 CrossRef CAS.
- R. H. Schlessinger and I. S. Ponticello, J. Am. Chem. Soc., 1967, 89, 3641 CrossRef CAS.
- R. H. Schlessinger and I. S. Ponticello, J. Am. Chem. Soc., 1968, 90, 4190 CrossRef.
- M. P. Cava and G. E. M. Husbands, J. Am. Chem. Soc., 1969, 91, 3952 CrossRef CAS.
- R. H. Schlessinger and J. M. Hoffman, J. Am. Chem. Soc., 1969, 91, 3953 CrossRef.
- M. D. Glick and R. E. Cook, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 1336 CrossRef CAS.
- S. Kato, T. Matsumoto, T. Ishi-i, T. Thieman, M. Shigeiwa, H. Gorohmaru, S. Maeda and Y. Yamashita, Chem. Commun., 2004, 2342 RSC.
- T. Strassner and J. Fabian, J. Phys. Org. Chem., 1997, 10, 33 CrossRef CAS.
- D. Leusser, J. Henn, N. Kocher, B. Engels and D. Stalke, J. Am. Chem. Soc., 2004, 126, 1781 CrossRef CAS PubMed.
- J. Wirz, Pure Appl. Chem., 1984, 56, 1289 CrossRef CAS.
- A. Thomas, K. Bhanuprakash and K. M. M. K. Prasad, J. Phys. Org. Chem., 2011, 24, 821 CAS.
- A. Thomas, C. G. Krishna, K. Bhanuprakash and P. K. M. M. Krishna, ChemPhysChem, 2011, 12, 3458 CrossRef CAS PubMed.
- W. Shen, M. Li, H. Huang, Y. Li and S. Wang, Mol. Simul., 2006, 32, 457 CrossRef CAS.
- D. G. Patel, F. Feng, Y.-y. Ohnishi, K. A. Abboud, S. Hirata, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 2599 CrossRef CAS PubMed.
- R. A. Firestone, Int. J. Chem. Kinet., 2013, 45, 415 CrossRef CAS PubMed.
- A. Thomas and K. Bhanuprakash, ChemPhysChem, 2012, 13, 597 CrossRef CAS PubMed.
- M. E. Köse, J. Phys. Chem. A, 2012, 116, 12503 CrossRef PubMed.
- K. Susumu, T. V. Duncan and M. J. Therien, J. Am. Chem. Soc., 2005, 127, 5186 CrossRef CAS PubMed.
- U. Salzner, O. Karalti and S. Durdagi, J. Mol. Model., 2006, 12, 687 CrossRef CAS PubMed.
- L. Pandey, C. Risko, J. E. Norton and J. L. Brédas, Macromolecules, 2012, 45, 6405 CrossRef CAS.
- Y. Fu, W. Shen and M. Li, Polymer, 2008, 49, 2614 CrossRef CAS PubMed.
- Y. Fu, W. Shen and M. Li, Polym. Int., 2011, 60, 211 CrossRef CAS PubMed.
- M. Garcia, L. Fomina and S. Fomine, Synth. Met., 2010, 160, 2515 CrossRef CAS PubMed.
- C.-L. Pai, C.-L. Liu, W.-C. Chen and S. A. Jenekhe, Polymer, 2006, 47, 699 CrossRef CAS PubMed.
- T. T. Steckler, X. Zhang, J. Hwang, R. Honeyager, S. Ohira, X.-H. Zhang, A. Grant, S. Ellinger, S. A. Odom, D. Sweat, D. B. Tanner, A. G. Rinzler, S. Barlow, J.-L. Brédas, B. Kippelen, S. R. Marder and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 2824 CrossRef CAS PubMed.
- X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage Jr, S. P. Tiwari, S. Coppee, S. Ellinger, S. Barlow, J.-L. Brédas, B. Kippelen, J. R. Reynolds and S. R. Marder, J. Mater. Chem., 2010, 20, 123 RSC.
- L. Pandey, C. Risko, J. E. Norton and J.-L. Brédas, Macromolecules, 2012, 45, 6405 CrossRef CAS.
- T. Kono, D. Kumaki, J.-i. Nishida, S. Tokito and Y. Yamashita, Chem. Commun., 2010, 46, 3265 RSC.
- H. Li, T. L. Tam, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 46 CrossRef CAS PubMed.
- X. Du, J. Qi, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2012, 24, 2178 CrossRef CAS.
- T. T. Steckler, K. A. Abboud, M. Craps, A. G. Rinzler and J. R. Reynolds, Chem. Commun., 2007, 4904 RSC.
- Y. Yang, R. T. Farley, T. T. Steckler, S.-H. Eom, J. R. Reynolds, K. S. Schanze and J. Xue, Appl. Phys. Lett., 2008, 93, 163305 CrossRef PubMed.
- Y. Yang, R. T. Farley, T. T. Steckler, S.-H. Eom, J. R. Reynolds, K. S. Schanze and J. Xue, J. Appl. Phys., 2009, 106, 044509 CrossRef PubMed.
- S. Ellinger, K. R. Graham, P. Shi, R. T. Farley, T. T. Steckler, R. N. Brookins, P. Taranekar, J. Mei, L. A. Padilha, T. R. Ensley, H. Hu, S. Webster, D. J. Hagan, S. E. W. Van, K. S. Schanze and J. R. Reynolds, Chem. Mater., 2011, 23, 3805 CrossRef CAS.
- J. M. Raimundo, P. Blanchard, H. Brisset, S. Akoudad and J. Roncali, Chem. Commun., 2000, 939 RSC.
- G. Qian, B. Dai, M. Luo, D. Yu, J. Zhan, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2008, 20, 6208 CrossRef CAS.
- G. Qian, X. Li and Z. Y. Wang, J. Mater. Chem., 2009, 19, 522 RSC.
- G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, Z. Y. Wang and D. Ma, Adv. Mater., 2009, 21, 111 CrossRef CAS PubMed.
- G. Qian, J. P. Gao and Z. Y. Wang, Chem. Commun., 2012, 48, 6426 RSC.
- M. C. Scharber and N. S. Sariciftci, Prog. Polym. Sci., 2013, 38, 1929 CrossRef CAS PubMed.
- C. J. Brabec, S. Gowrisanker, J. J. Halls, D. Laird, S. Jia and S. P. Williams, Adv. Mater., 2010, 22, 3839 CrossRef CAS PubMed.
- P. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456 CrossRef CAS.
- J. L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691 CrossRef PubMed.
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. de Boer, Polym. Rev., 2008, 48, 531 CrossRef CAS.
- E. Bundgaard and F. C. Krebs, Macromolecules, 2006, 39, 2823 CrossRef CAS.
- E. Bundgaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 1019 CrossRef CAS PubMed.
- Y. Lee, T. P. Russell and W. H. Jo, Org. Electron., 2010, 11, 846 CrossRef CAS PubMed.
- C.-Y. Yu, C.-P. Chen, S.-H. Chan, G.-W. Hwang and C. Ting, Chem. Mater., 2009, 21, 3262 CrossRef CAS.
- V. Tamilavan, M. Song, S.-H. Jin and M. H. Hyun, Synth. Met., 2011, 161, 1199 CrossRef CAS PubMed.
- X. Wang, E. Perzon, W. Mammo, F. Oswald, S. Admassie, N.-K. Persson, F. Langa, M. R. Andersson and O. Inganaes, Thin Solid Films, 2006, 511–512, 576 CrossRef CAS PubMed.
- T. L. D. Tam, T. Salim, H. Li, F. Zhou, S. G. Mhaisalkar, H. Su, Y. M. Lam and A. C. Grimsdale, J. Mater. Chem., 2012, 22, 18528 RSC.
- J. Song, C. Zhang, C. Li, W. Li, R. Qin, B. Li, Z. Liu and Z. Bo, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2571 CrossRef CAS PubMed.
- C. Qin, Y. Fu, C.-H. Chui, C.-W. Kan, Z. Xie, L. Wang and W.-Y. Wong, Macromol. Rapid Commun., 2011, 32, 1472 CrossRef CAS PubMed.
- J. A. Mikroyannidis, D. V. Tsagkournos, S. S. Sharma, Y. K. Vijay and G. D. Sharma, J. Mater. Chem., 2011, 21, 4679 RSC.
- Q. Hou, W. Hong, Y. Zhang, J. Liu, Y. Chen and G. Shi, J. Mater. Sci.: Mater. Electron., 2012, 24, 536 CrossRef PubMed.
- M. L. Keshtov, L. Toppare, D. V. Marochkin, V. S. Kochurov, D. Y. Parashchuk, V. A. Trukhanov and A. R. Khokhlov, Polym. Sci., Ser. B, 2013, 55, 360 CrossRef CAS.
- T. Ameri, G. Dennler, C. Lungenschmied and C. J. Brabec, Energy Environ. Sci., 2009, 2, 347 CAS.
- T. Ameri, N. Li and C. J. Brabec, Energy Environ. Sci., 2013, 6, 2390 CAS.
- S. Sista, Z. Hong, L.-M. Chen and Y. Yang, Energy Environ. Sci., 2011, 4, 1606 CAS.
- J. L. Banal, J. Subbiah, H. Graham, J. K. Lee, K. P. Ghiggino and W. W. H. Wong, Polym. Chem., 2013, 4, 1077 RSC.
- H. Xu, J. Li, B. H. Leung, C. C. Poon, B. S. Ong, Y. Zhang and N. Zhao, Nanoscale, 2013, 5, 11850 RSC.
- Z. L. Guan, J. B. Kim, H. Wang, C. Jaye, D. A. Fischer, Y.-L. Loo and A. Kahn, Org. Electron., 2010, 11, 1779 CrossRef CAS PubMed.
- F. B. Kooistra, V. D. Mihailetchi, L. M. Popescu, D. Kronholm, P. W. M. Blom and J. C. Hummelen, Chem. Mater., 2006, 18, 3068 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Mill Valley, CA, 1991 Search PubMed.
- M. Sun, X. Jiang, L. Wang, C. He, B. Du, R. Yang and Y. Cao, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3007 CrossRef CAS PubMed.
- M. Sun, X. Jiang, W. Liu, T. Zhu, F. Huang and Y. Cao, Synth. Met., 2012, 162, 1406 CrossRef CAS PubMed.
- X. Li, A. Liu, S. Xun, W. Qiao, X. Wan and Z. Y. Wang, Org. Lett., 2008, 10, 3785 CrossRef CAS PubMed.
- J. R. Sommer, R. T. Farley, K. R. Graham, Y. Yang, J. R. Reynolds, J. Xue and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 274 CAS.
- K. R. Graham, Y. Yang, J. R. Sommer, A. H. Shelton, K. S. Schanze, J. Xue and J. R. Reynolds, Chem. Mater., 2011, 23, 5305 CrossRef CAS.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586 CrossRef CAS PubMed.
- Y. Fujisaki, Y. Nakajima, D. Kumaki, T. Yamamoto, S. Tokito, T. Kono, J.-i. Nishida and Y. Yamashita, Appl. Phys. Lett., 2010, 97, 133303 CrossRef PubMed.
- T. Kono, D. Kumaki, J.-i. Nishida, T. Sakanoue, M. Kakita, H. Tada, S. Tokito and Y. Yamashita, Chem. Mater., 2007, 19, 1218 CrossRef CAS.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780 CAS.
- H. S. Oh, T.-D. Kim, Y.-H. Koh, K.-S. Lee, S. Cho, A. Cartwright and P. N. Prasad, Chem. Commun., 2011, 47, 8931 RSC.
- J. Fan, J. D. Yuen, W. Cui, J. Seifter, A. R. Mohebbi, M. Wang, H. Zhou, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 6164 CrossRef CAS PubMed.
- J. Fan, J. D. Yuen, M. Wang, J. Seifter, J.-H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186 CrossRef CAS PubMed.
- J. D. Yuen, J. Fan, J. Seifter, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799 CrossRef CAS PubMed.
Footnotes |
| † New permanent address: Department of Chemistry, University of Kentucky, Lexington, KY, 40506-0055, USA. |
| ‡ New permanent address: Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal, 23955-6900, Saudi Arabia. |
| This journal is © The Royal Society of Chemistry 2015 |