
Great Prospects for PAF-1 and its derivatives†
Cuiying
Pei
a,
Teng
Ben
*b and
Shilun
Qiu
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Jilin University, Changchun, China. E-mail: sqiu@jlu.edu.cn
bDepartment of Chemistry, Jilin University, Changchun, China. E-mail: tben@jlu.edu.cn
First published on 29th September 2014
Abstract
In materials design and preparative chemistry, it is imperative to understand the thought and logic behind synthesizing a particular kind of material. Computational modelling can help in this regard by not only optimizing the materials but also by simulating their properties. Furthermore, the experimental results fill the gap addressing complicated practical conditions that can't be covered using theoretical calculations. In this work, we focus on PAF-1 and its derivatives in order to analyse the correlations between the nature of the material (e.g. pore size, surface area, pore volume, functional groups, metal sites, interpenetrated frameworks) and their properties such as gas sorption capacity, molecular recognition and separation.
 Cuiying Pei | Cuiying Pei received her B.Sc. degree in chemistry from Harbin Normal University in 2008. In cooperation with the state key laboratory of inorganic synthesis and preparative chemistry, Jilin University, Cuiying Pei worked on a research project and obtained her M.Sc. degree in 2011. After that, she became a Ph.D. student in the group of Prof. Shilun Qiu. Her research interests are the design and synthesis of novel porous organic frameworks for their application in gas storage, separation and nanodevices. The developed methods make it possible to tune the pore size and the electric field density and distribution. |
 Teng Ben | Teng Ben received his Ph.D. in 2002 from Jilin University in polymer science. After graduation, he joined the faculty at Jilin University, working with Prof. Zhongwen Wu as a lecturer. In 2005, he moved to Prof. Eiji Yashima’s group at the Nagoya University in Japan as a postdoctoral researcher. In 2008, he moved back to Jilin University as an Associate Professor and was promoted to a Professor in 2010. His research interests include the fundamental understanding of host–guest interactions in nanoporous materials, gas storage and separation using porous organic frameworks. |
 Shilun Qiu | Shilun Qiu received his Ph.D. in chemistry from Jilin University in 1988. He joined the University De Haute-Alsace, France, and Hokkaido University Sapporo, Japan, for postdoctoral research. In 1994, he was promoted to be a full professor in the Department of Chemistry, Jilin University. He won the Second Grade Award of the State Natural Science Award of China in 2008 and is a fellow of the RSC. His recent research interests focus on studies of molecular engineering, the synthesis and catalysis of micro- and mesoporous materials, the rational synthesis of porous organic framework materials for hydrogen storage, and host–guest chemistry, in particular, nanocomposites in porous materials. |
Introduction
PAF-1, which links tetrahedral rigid building blocks with robust covalent bonds, was synthesized and reported in 2009.1 One year later, Cooper et al. succeeded in producing network 12 with the same structure as PAF-1. In 2011, PAF-1 was resynthesized and renamed as PPN-6 by the Zhou group.3 The most attractive feature of PAF-1 is that it successfully combines an ultrahigh surface area (SBET = 5600 m2 g−1) with high physicochemical stability. Hence, it can be used repeatedly for the storage of hydrogen and methane as a clean energy alternative and also for capturing greenhouse gases like carbon dioxide under harsh conditions. In addition, the high physicochemical stability of PAF-1 makes it tolerant towards the modification conditions, facilitating the formation of functionalized frameworks retaining the structural advantage. This provides scope for modifying PAF-1 thereby extending its range of application.The modifications can be divided into three methods, namely (1) the pre-modification method where the basic monomer is decorated with hybrid atoms4,5 or heterocyclic units,6,7 and linked by organic building blocks of different lengths6–8 or widths;9–11 (2) the post-synthesis modification (PSM) method, which involves the chemical functionalization of PAF-1 with functional groups,3,12 metal atoms or ions,13–15 and (3) the carbonization of PAF-1 (ref. 16 and 17) (Fig. 1, multiple modifications).
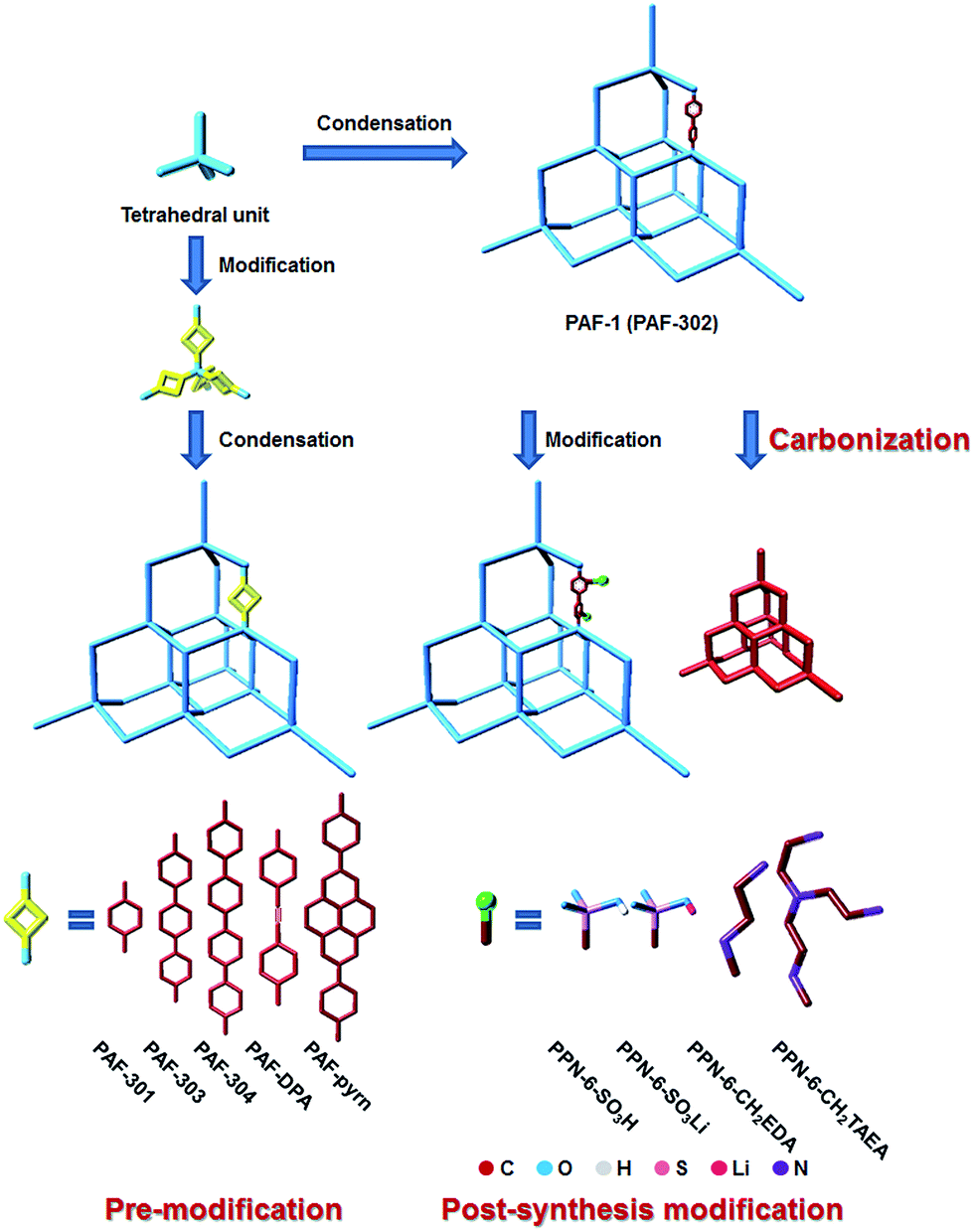 | ||
| Fig. 1 Strategies to construct and modify PAF-1 as well as several typical examples. The cyan sticks represent the constructs of the dia topology frameworks, the yellow square stands for a functionalized link, the dark red stick represents a carbon unit and blue, grey, pink, magenta and purple represent oxygen, hydrogen, sulphur, lithium and nitrogen, respectively. | ||
The study on PAF-1 also successfully demonstrates the general idea of targeted synthesis. Conventionally, the hit and miss approach in synthetic chemistry not only exhausts chemical resources but also cannot predict the characteristics of the obtained products. Theoretical studies can provide reliable calculations that can guide a design strategy and prevent chemists from aimless trials.
Specifically, PAF-1 was modelled by replacing the C–C covalent bonds of diamond with rigid phenyl rings. Computational studies indicate that the replacement of one phenyl ring yields a P1 structure, which has a lower surface area, while the replacement of three phenyl rings constructs a mesoporous P3 structure.1 In comparison, the replacement of two phenyl rings makes a P2 structure showing the surface area, density, and void framework at just the right level. As expected, the empiric evaluation of the ultrahigh surface area (SBET = 5600 m2 g−1)1 coincides well with the simulated one (SBET = 5640 m2 g−1).1
The mechanism of formation of PAF-1 may explain the ordered crystal oligomer with a dia topology formed at the beginning of the reaction. As condensation proceeds, defects are generated inevitably. Since the Yamamoto18-type Ullmann reaction19 is an irreversible process, the defects are not corrected by reassembly and, ultimately, expand further.
Finally, a hybrid structure consisting of dia topology crystals and a continuous random network (CRN) phase of SiO2 (ref. 20) is obtained. The utilization of a rigid monomer effectively prevents single bond rotation which can block the structural rearrangement which could have caused the collapse of the porous structure.21 Calculations predict a restricted pore size of PAF-1 which is too small for constituent monomer diffusion and results in a non-interpenetrated framework.22 Derivatives with more void space will allow higher levels of interpenetration and systematically minimize the energy.23 Hence, PAF-1 exhibits high thermal stability in the range of porous organic frameworks characterized by their ultrahigh stability such as COF-102 (thermally stable beyond 450 °C)24 and ZIF-300, 301, 302 (stable towards water and 450 °C in dry air).25 Besides, PAF-1 shows a narrow uniform pore size distribution. Its surface area is only exceeded by MOF-210 (SBET = 6240 m2 g−1),26 PPN-4 (SBET = 6461 m2 g−1)27 and NU-110(SBET = 7000 m2 g−1).28 Based on these favourable characteristics, these derivatives which were constructed by tuning the pore size, tailoring the organic building blocks and modifying the frameworks, demonstrate improved properties and an expanded application area. Herein, we summarize both the simulation and experimental studies on PAF-1 (ref. 1) and its derivatives (Fig. 2). We focus on the key structural factors that influence such remarkable properties. This work will provide valuable information for guiding future work on materials design and preparative chemistry.
 | ||
| Fig. 2 The structural summary of PAF-1 and its derivatives. | ||
Design principles
Tailoring the pore size
The effect of pore size on molecule sorption and separation has been widely investigated. Presser et al. reported that pores smaller than 0.8 nm contributed the most to the CO2 uptake at 1 bar, and that 0.5 nm pores exhibited a higher uptake at 0.1 bar.29 For hydrogen storage, a porous material with a pore size of two gas diameters is preferred.27,30 On the other hand, for molecule separation, the material with a pore size between the kinetic diameters of the two different guest molecules can be directly used for separation. Even in cases where the pore size of the material is larger than the kinetic diameters of the molecules, the molecule with the dimension closer to the pore size will be retained while the other molecule can be exhausted. This has been confirmed by Grand Canonical Monte Carlo (GCMC) simulation results on diamondyne and PAF-302.31 Hence, the design and synthesis of porous materials with targeted pore sizes is one of the key strategies in this field. Generally, two effective methods to tune the pore size are: the selection of organic building blocks with different lengths or structures versus the functionalization of the framework.The covalent bonds in the robust hydrocarbon scaffold of PAF-1 contribute not only to the high physicochemical stability, but also to the low bulk density and high pore volume. Based on this topology, a series of porous organic materials has been designed by pore size tuning for different application requirements. PAF-301, PAF-302, PAF-303, PAF-304 (ref. 32 and 33) belong to the cubic space group P1 (Fig. 1). The difference between them is the number of phenyl rings which are connecting two neighbouring tetrahedral building blocks. Without interpenetration, the pore size of PAF-30X widens with the increase in phenyl ring linkages, to the extent that PAF-304 exhibits mesoporosity.32,33 GCMC simulation was chosen to evaluate the hydrogen, methane, and carbon dioxide storage as well as gas separation performance. It shows that PAF-304 possesses the highest gravimetric hydrogen uptake among the others due to the large pore size and pore volume. The value reaches 6.53 wt% at 298 K/100 bar. However, PAF-301 with the smallest pores of 5.2 Å shows not only the highest CO2 uptake (275 mg g−1 at 298 K/1 bar) in the low pressure range, but also exhibits higher selectivity for CO2/H2, CO2/N2, CO2/CH4, and CH4/H2 among PAF-30X.32,33
Another strategy to tune the pore size is to construct PAFs with different widths and numbers of aromatic rings. Kuc et al. studied the mechanical and the hydrogen adsorption properties of these materials using the density functional based tight-binding (DFTB) method and quantized liquid density functional theory (QLDFT).34–37 Exothermic formation energies of the PAFs, calculated with regard to PAFs formed using saturated linkers and CH4 molecules in a dehydrogenation reaction, indicate strong coordinating linkers in the dia topology. The mechanical stability decreases with an increase in length of the organic linkers; however, it strengthens with the expansion of width of the linkers. The aromatic building blocks provide binding sites for hydrogen via London dispersion forces. Long linkers expand the pore volume and decrease the mass density, which offers more space for hydrogen storage and shows a significantly higher hydrogen gravimetric capacity. On the other hand, wider linkers increase the surface area per volume and enhance the interactions between the guest molecules and the host framework.
It should be noted that large open skeletons are always accompanied by interpenetrative structures. The interpenetrated material has minimal entropy due to the filling of void space leading to repulsive forces which enhance the stability of the framework.38 Although interpenetration both reduces the pore size and the pore volume and increases the bulk density, it creates more adsorption sites and increases the volumetric hydrogen uptake due to a higher surface area per unit volume. The surface area of this novel structure can be maximized by decreasing the pore size without blocking the binding sites. For instance, PAF-11 (ref. 8) was expected to have the same structure as PAF-304 and to exhibit a significantly high hydrogen uptake. However, considering that no residual bromine was left after complete formation of the framework, it can be safely concluded that partial interpenetration is responsible for the relatively low surface area and hydrogen uptake capacity. For smaller pore sizes, i.e., pore sizes closer to the kinetic diameter of the guest molecules, the slow diffusion rate of the guest molecules is a potential drawback. Porous materials with hierarchical pore dimensions are suitable candidates to resolve this issue. However, the design and synthesis of hierarchical porous PAFs are a great challenge.
Although theoretical simulation studies can be used to refine obtained experimental results and thus to direct the synthesis of potential functional materials, these solely depend on the generation of models with proper geometrical and exact physical parameters. It is difficult to obtain the exact values of these mentioned parameters for amorphous materials. The short-range order structure in PPNs and PAFs can be approximately considered as crystalline and can be used to predict their molecular adsorption behaviour. Experimental gas storage results on PAF-1 (ref. 1) coincide well with the simulation data obtained using the crystalline model, in fact, better than with the amorphous model.32,39
Pre-modification
The modification of PAF-1 with different functional groups or building blocks is another strategy to cater to the special application requirements. Both the pre-modification and PSM can implement functionalizations to PAF-1 (ref. 1) with their own pros and cons. Pre-modification means to design functionalized monomers before the efficient and high-yielding cross-coupling reaction. The advantage of the pre-modification method is that it unblocks the pores in order to functionalize the parent material and effectively utilizes terminal groups to introduce specific units. With this strategy, heterocyclic building blocks such as tetrazole groups in PIMs,40 pyridyl and thiophenyl groups in PON-2 and PON-3, respectively,6 B3N3 rings in BLP-12,41 and perylene diimide in Td-PPI,11 can be introduced into the PAF-1 framework. This enhances the interactions between the host adsorbent and the guest molecules by increasing the electron density of the framework. In particular, the interaction between an ion of the heterocyclic rings and the molecular quadrupole of a CO2 molecule is stronger than that between the π conjugated phenyl ring and the molecular quadrupole of a CO2 molecule. This makes the binding sites around the heterocyclic rings more accessible to CO2 molecules. Another pre-modification strategy is the introduction of a hybrid element to an alternative sp3 central carbon in the framework. Examples for the addition could be Si in PAF-3 (ref. 4) and in [D4]PAF3,42 Ge in PAF-4 (ref. 4) (also named as PPN-5 (ref. 27)), P+ in PP-Br,5 and N+ in Ph4N+F−.43 With the advantage of a stable and open framework, the introduction of a hybrid element increases the structural diversity further expanding the area of possible applications.Post-synthesis modification (PSM)
PSM is superior and versatile owing to the possibility of incorporating a wide variety of functional groups and a controllable degree of modification. The introduction of polar functional groups and coordination sites to the framework mainly enhances the binding capacity. One example is the functionalization of PAF-1 (ref. 1) to increase the CO2 capture capacity. Considering the quadrupole moment of the CO2 molecule, the incorporation of polar functional groups into the framework can effectively strengthen the separation efficacy of CO2 over other non-polar or weakly polar molecules such as H2, N2, CH4.The PSM strategy used to modify PAF-1 is accomplished via incorporation of two moieties: (1) chemical functional groups as pendant units such as sulfonic acid,3,44 hydroxyl,45 alkyl and amino12 groups where lone pair donation and H-bonding improve the binding energy between CO2 and the cluster;46 and (2) metals doped into the open framework of PAF-1. The two preparation strategies that have been used for doping are: (1) vaporized fusion or mechanical mixing of the metal atoms with the host material; and (2) ion-exchange or post-coordination of the metal cations via the functional groups in the host skeleton. The former exposes more active sites but lowers the stability of the host framework, while the latter does just the opposite.47–49 However, the chemical introduction of metal ions can prevent atoms from forming a cluster, actuate the design depending on the type and position of the doped metal, and enhance the electrostatic field. Different doping methods are available as per the requirement of specific applications. Several factors should be considered before the introduction of metals into the frameworks in order to enhance gas molecule sorption. For example, apart from the sorption affinity between the gas molecules and the doped host framework, it is crucial to consider the affinity between the metal particles and the host framework.
The other factors that need to be considered in the modification design process are as follows: (1) the weight of the metal ion or atom, (2) the metal doping style and location in the framework, (3) the gas molecule sorption location near the binding site, (4) the binding energy between the metal particles and the gas molecules, (5) the degree of exposure of adsorption sites to the gas molecules, and (6) the binding stability of the metal within the host framework. The latter creates a diminished stability if excess particles form clusters which reduce the available surface.
Considering the above factors, alkali, alkaline-earth and transition metal ions and atoms are preferred for doping in porous organic frameworks to enhance the gas sorption capacity. A multiscale simulation shows that the binding capacity of alkaline-earth metal atoms to the framework is weak while that of transition metal atoms is strong.47 It should be noted that if the binding capacity is in the chemisorption range it will suffer from desorption. Among the alkali metals, Na and K show low binding energies towards organic frameworks and prefer to form clusters. Alkaline-earth metals with larger ionic radii have a binding stability problem. The Li atom has a low mass, binds to the framework in a stable fashion, and easily loses its valence electron to form a Li+ cation which in turn increases the electropositive force and the binding energy between the framework and the gas molecules. Sun et al.'s calculation studies demonstrate that the lithium tetrazolide group is more stable and polarized than a Li atom doped into a porous organic framework.49 In addition, GCMC simulation predicts that the hydrogen gravimetric uptake of PAF-1 containing lithium tetrazolide moieties can exceed the 2010 DOE target and approaches the 2015 US Department of Energy (DOE) target. One major drawback is that it is highly reactive and flammable. Hence, there is a substantial amount of research on the gas sorption behaviour and properties at the simulation stage but few experimental reports of samples doped with lithium atoms or ions in the porous framework exist. This is one of the biggest current challenges for synthetic chemists.
Carbonization
Carbonization of the framework is another effective functionalization method towards enhanced gas sorption capacities. For example, direct annealing of PAF-1 at 450 °C shrinks the pores to match the gas molecule dynamic diameter.16 KOH-activated carbonized PAF-1 exhibits a unique bimodal microporous structure.17 PAF-1 template carbon is another kind of porous material with a high CO2 binding capacity.50 It is prepared by introducing furfuryl alcohols into the PAF-1 framework followed by thermolysis at 900 °C. The pore size of PAF-1/C-900 decreased to 5.4 Å, and the CO2 heat of sorption reached 27 kJ mol−1. Compared with PAF-1-450, the pores of PAF-1/C-900/furfuryl have half the volume but retain an equal surface and ability to adsorb CO2.29Applications
CO2 capture
Carbon capture and sequestration (CCS) of carbon, released from industries and energy-related sources, is a process to deposit carbon for long term isolation from the atmosphere. Greenhouse gas (carbon dioxide) capture from power plants is one of the most important application areas of CCS. Although a power plant with CCS could reduce the released CO2 by 80–90%, the capture and compression of CO2 requires 25–40% additional fuel which, in turn, increases the cost of electricity generation.51 There are mainly three different CO2 capture systems: (1) pre-combustion removal of CO2 from H2 or CH4 for clean energy fuel after the transformation of primary fuel (coal, natural gas, oil or biomass) into syngas (CO2 and H2 or CH4) in the reactor; (2) post-combustion separation of CO2 from flue gases after the combustion of primary fuel in air; and (3) oxy-fuel combustion which isolates CO2 from water, the other combustion product.51 The components of flue gas in the post-combustion process are primarily nitrogen (N2, >70%) and CO2 (10–15%) which accounts for roughly 33–40% of the global CO2 emission. In the post-combustion model, adsorption of CO2 occurs at 1 bar and desorption occurs at 0.1 bar. In comparison with the post-combustion carbon capture, the pre-combustion process is carried out at a higher pressure and also has a higher concentration of CO2 (15–60%). The exhaust from the oxygen combustion has a very high concentration of CO2 requiring sorbents that are stable in high humidity. The ideal CO2 capture system should have sufficient space for CO2 storage and an appropriate binding capacity between the CO2 molecules and the sorbent material. Generally, a large pore volume and a high surface area contribute to the high storage uptake, especially in the high pressure range. Matching the pore size with the kinetic diameter (3.3 Å) of CO2 and a suitable binding energy lead to optimal interactions between the gas molecules and the material. In this regard, PAF-1 and its derivatives show potential for carbon dioxide capture when applied in pressure (PSA), temperature (TSA) or vacuum swing adsorption (VSA) systems. To improve the CO2 sorption capacity; carbonization, amination, metalation and sulfonation are four effective strategies.In relating to PAF-1, the carbonization follows two routes: either direct carbonization, annealing PAF-1 with hybrid compounds as the template, or fabrication of porous carbon with PAF-1 as the template. Generally, in an annealing process, framework shrinkage and decreasing pore sizes contribute to the overlap of the force fields and increase the interactions between the carbon dioxide molecules and the framework.16 In addition, all carbon scaffolds possibly create an electric field around the framework surface which strengthens its interaction with the quadrupole moment of CO2. However, with a hard template, carbonized PAF-1 possesses both small and large pores.17 As a result, it shows a faster diffusion rate of guest molecules as well as a higher CO2 uptake in both low and high pressure ranges than that of PAF-1.
Amination is another useful strategy to introduce CO2-philic moieties into porous organic frameworks. The polarizability and high charge density create stronger interactions between the network and the quadrupole moment of the CO2 molecules. For instance, PAF-1-CH2NH2 demonstrates the highest isosteric heat of sorption of 57.6 kJ mol−1 among all PAF-1 derivatives, equalling that of chemical sorption12 (Table S2†). However, the strong interaction demands more energy to regenerate the network. Notably, as the Qst (isosteric heat of adsorption) increases, it becomes less satisfactory. It has been shown that materials with Qst values around 21 kJ mol−1 exhibit the best working capacity in landfill gas separation via a PSA process.52 However, in real world applications, CO2 sorption is usually combined with water vapour even in pre- or post-combustion processes. The stability of the above mentioned derivatives in the presence of water vapour should be considered. It has been reported that all the amine-containing materials are deactivated at different rates compared with urea under humid conditions. Deactivation does not occur under dry CO2 adsorption conditions.53
The metal cation in the framework acts as an open coordination site after full activation, which leads to stronger electrostatic interactions between CO2 and the metal cation.3,13,15,111,112 The light element Li is a commonly used dopant. However, the Li content present should be in an appropriate optimum range. If one Li ion is present per phenyl ring, the Li content is as high as 8 wt%. Extremely high loading levels of Li will make the lithium ions agglomerate, degrade the framework and lead to a diminished CO2 sorption capacity. The introduction of Ni into the framework by coordination interaction with porphyrin has been proven to enhance the CO2 sorption capacity.15
In the case of sulfonate grafted porous polymer networks, two factors enhance the CO2-philic effect of the materials.3 First, the functionalization of the all-carbon-scaffold frameworks creates electric fields on the surface that impart a strong affinity of the networks towards the CO2 molecules by their high quadrupole moment. Second, a small pore size and polar functionalities increase the heat of adsorption. Accurate control of the amount and location of the sulfonate molecules is the same whether performed before or after the polymerization.44,54 Other hybrid elements, such as Si in PAF-3 (ref. 4) (also known as PPN-4,27 network 2 (ref. 2)), Ge in PAF-4,4 P in PP-Br,5 S in PON-3,6 B in BLP,41,55 have been studied for their influence on host–guest interactions. Increases of different degrees are seen in the carbon dioxide enthalpy (Table S2†).
Comparing the four kinds of modification strategies, amination is the most effective method to improve the carbon dioxide sorption enthalpy and PPN-6-CH2DETA56 demonstrates the highest uptake of all the polyamine-tethered PPNs at room temperature and 1 bar. With an increased affinity towards carbon dioxide, the affinity of other gases increases, too. Selectivity is another indicator of an ideal carbon dioxide sorbent. PPN-6-CH2DETA exhibits the highest CO2/N2 selectivity based on the IAST method. It should be noted that a sample with an ultrahigh surface area has an absolute advantage in the high pressure carbon dioxide capture. MOF-200 with the highest uptake is the best example (Fig. 3). Considering the stability and the storage capacity, PAF-1 and its derivatives are the best CO2 adsorbents among the porous organic frameworks.
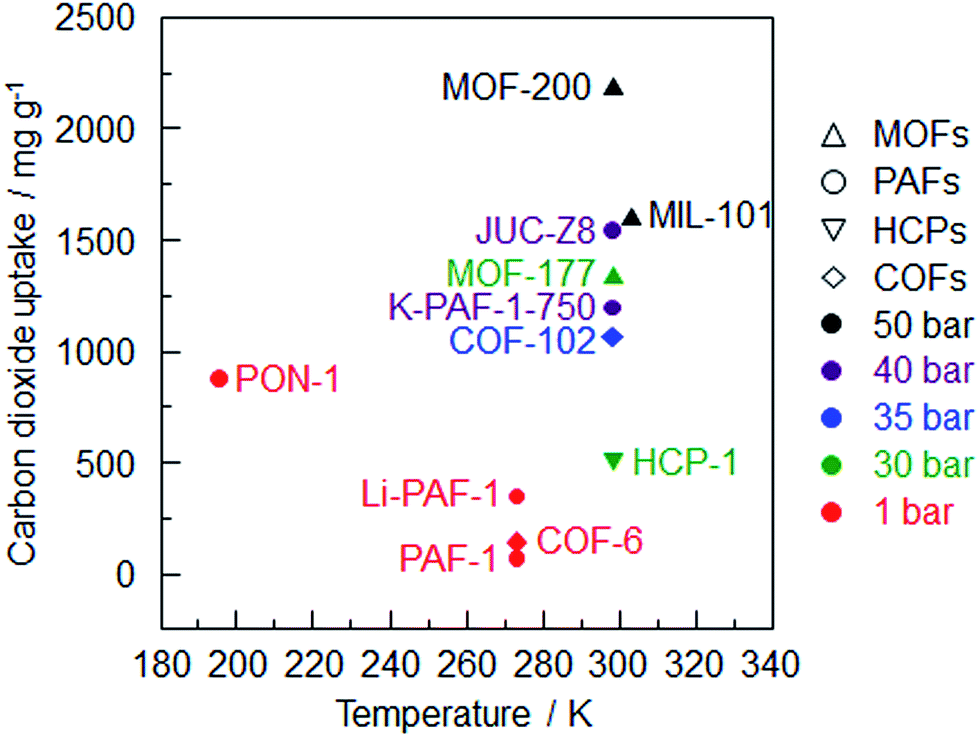 | ||
| Fig. 3 Summary of carbon dioxide uptake of porous organic frameworks vs. sorption temperature (MOF-200,26 MIL-101,82 JUC-Z8,95 MOF-177,83 K-PAF-1-750,17 COF-102,24 PON-1,6 HCP-1,91 Li-PAF-1,22 COF-6,85 PAF-11). | ||
H2 storage
Hydrogen with an energy content of 142 MJ kg−1 is an environmentally friendly energy storage medium in the hydrogen economy. However, the safe transfer and cost-effectiveness of the storage of H2 is still a big challenge in terms of application. The practical 2015 requirement of the U.S. Department of Energy (DOE) sets a storage standard (5.5 wt% in gravimetric capacity, 40 g L−1 in volumetric capacity between 233–333 K).57 With the aid of chemical bonds, hydrides can adsorb as much as the target set by the DOE under ambient conditions.58 However, the strong interactions require high temperatures for hydrogen release, which makes recycling inefficient. For instance, the promising material, MgH2, requires over 350 °C to release hydrogen. In addition, the metal hydride is generally too heavy for gravimetric hydrogen uptake. Hence, lowering the desorption temperature and increasing the amount of gravimetric adsorption is a big challenge in the chemisorption of hydrogen. On the other hand, reversible physisorption allows recycling due to the weak van der Waals forces between the host and guest, requiring very low temperatures to make the kinetics faster and store significant amounts of hydrogen. The challenge for hydrogen storage using physical interactions is how to increase the gas uptake under ambient conditions. Fortunately, although MOFs and PAFs exhibit similar volumetric amounts of hydrogen, a greater gravimetric uptake can be expected due to the light element framework and the low density of PAF-1.PAF-1 adsorbs hydrogen with 186 cm3 g−1 (1.66 wt%) at 77 K/1 bar, and 75.3 mg g−1 (7 wt%) at 77 K/48 bar (Table S2†).1 However, this value decreases dramatically as the temperature rises and the pressure drops. To achieve the DOE target, a high hydrogen uptake at close to ambient temperatures and in a lower pressure range is required. In this regard, improving the hydrogen binding energy of the framework is one of the breakthroughs to resolve this issue. There are three main forces (dispersion interactions, electrostatic interactions, and orbital interactions) between the hydrogen molecules and the adsorbent, which directly determine the strength of the binding energy.59 The ideal enthalpy for the adsorbent should be 15–25 kJ mol−1.60 However, Froudakis et al. revealed that the binding energy between the phenyl rings and a hydrogen molecule is less than 1 kcal mol−1, which corresponds to 4.18 kJ mol−1.61 Moreover, weaker liquid–liquid interactions in empty spaces of the large pores make the enthalpy decrease with an increase in the coverage, which reduces the overall hydrogen uptake.62 Generally, the introduction of a functional group or metal element partly blocks the channel, narrowing the pore size to match the hydrogen kinetic diameter. Significant charge–quadrupole or charge–induced-dipole interactions between the metal ions and the hydrogen molecules enhance the binding energy.45
Substantiated by experimental results, lithiation,13 carbonization16,17 and nickel coordination15 are three effective methods to enhance the hydrogen heat of sorption. It was found that Li is one of the most ideal elements doped into the framework. PAF-1 lithiated with naphthalene shows a hydrogen adsorption enthalpy as high as 9 kJ mol−1. PAF-1 lithiated with naphthalene shows the highest hydrogen adsorption enthalpy of 9 kJ mol−1 among PAF-1 and all its derivatives (Table S2†).
Until now, MOF-210 (ref. 26) exhibited the highest hydrogen uptake under high pressure (48 bar), while Li-CMP87 shows the highest value under low pressure (1 bar) among the porous organic frameworks (Fig. 4). Unfortunately, none of them reach the DOE target. One can expand hydrogen sorption by adopting a new strategy involving a combination of high surface area and pore volume PAF-1 with high stoichiometric hydrogen content (19.6 wt%) and moderate dehydrogenation temperature using ammonia borane (AB).63 As expected, hydrogen release is substantially improved by AB-PAF-1 which operates at a temperature of 358 K compared with the 77 K operating temperature of other porous organic frameworks. Moreover, nano-dispersed AB in PAF-1 provides a method to increase the hydrogen gravimetric capacity which is an important aspect to consider in chemical hydrogen storage processes.
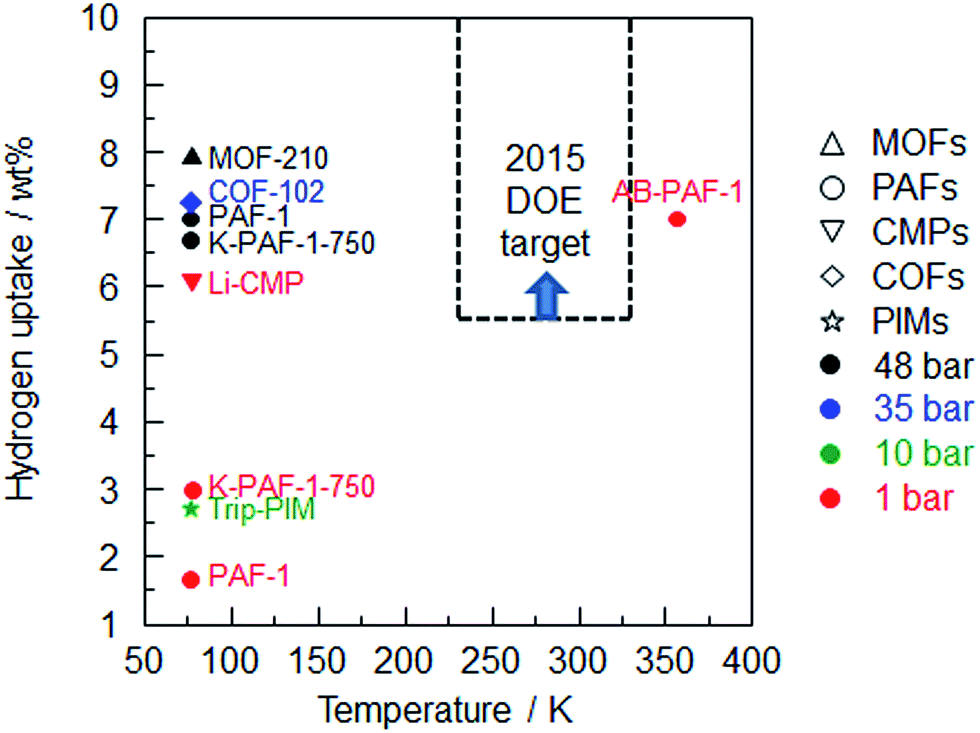 | ||
| Fig. 4 Summary of hydrogen uptake of the porous organic frameworks vs. sorption temperature (MOF-210,26 COF-102,24 PAF-1,1 K-PAF-1-750,17 Li-CMP,87 Trip-PIM,94 AB-PAF-163). | ||
CH4 storage
Methane benefits from its high gravimetric heat of combustion (55.7 MJ kg−1 compared with 46.4 MJ kg−1 of gasoline), the smallest amount of CO2 release per unit of heat produced among fossil fuels and large natural reserves. This makes methane an attractive fuel option in cars and other automobiles as an alternative to petroleum-based fuels. The DOE initiated a methane storage program with 0.5 gmethane/gsorbent for the gravimetric capacity requirement and 263 vmethane (STP)/vsorbent for the volumetric capacity requirement. Considering 25% packing loss, the volumetric capacity should be up to 330 vmethane (STP)/vsorbent.64 In addition, the ideal sorbent should be stable to impurities in natural gas sources with a lifetime of at least 100 adsorption–desorption cycles.The adsorption behaviour of methane towards the adsorbents is the same as that of hydrogen. The physisorption mainly depends on the surface area, pore volume and affinity towards methane and it is preferred to the chemisorption due to its reversible nature. However, it does not mean that a larger surface area would ensure better adsorption. It has been calculated for MOFs that the gravimetric uptake increases with an increase in surface area up to 2500–3000 m2 g−1; above this point, the only effect is a decrease in volumetric uptake.65 Until the measurement pressure exceeds 100 bar, the accessible volume plays a major role in methane storage. In this case, the effect of adsorbent is minimal, almost equivalent to the directly compressed methane at the same pressure.65 Van der Waals forces are the driving force in physisorption. Tuning the pore size in order to match one or two kinetic diameters (3.8 Å) of methane is the optimal strategy. The incorporation of large amounts of aromatic building blocks or electron-donating species improves the methane uptake because of enhanced electrostatic interactions. For example, the statistical simulation results from 137![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 953 hypothetical MOFs show that methyl, ethyl and propyl functional groups could dramatically improve the methane uptake above 205 vmethane (STP)/vsorbent.65 The methane uptake can also be improved by lithiation of PAF-1 and its derivatives. London dispersion and induced dipole interactions between a Li+ cation and a methane molecule can strengthen the binding capacity.66 For example, the methane uptake of 5% Li-PAF-1 is 20.8 mg g−1, which is about two times higher than that of PAF-1 under the same conditions (273 K/1 bar).14 Fortunately, the heat of methane sorption is reasonable, but the enhancement of the packing density and the amount of desorption pose two main challenges that need to be addressed.
953 hypothetical MOFs show that methyl, ethyl and propyl functional groups could dramatically improve the methane uptake above 205 vmethane (STP)/vsorbent.65 The methane uptake can also be improved by lithiation of PAF-1 and its derivatives. London dispersion and induced dipole interactions between a Li+ cation and a methane molecule can strengthen the binding capacity.66 For example, the methane uptake of 5% Li-PAF-1 is 20.8 mg g−1, which is about two times higher than that of PAF-1 under the same conditions (273 K/1 bar).14 Fortunately, the heat of methane sorption is reasonable, but the enhancement of the packing density and the amount of desorption pose two main challenges that need to be addressed.
PAF-1 can adsorb 185 mg g−1 CH4 at 298 K/35 bar, but this value is still far away from the DOE target (Fig. 5). Even PPN-4,27 which shows the highest methane uptake of 273.6 mg g−1 (295 K/35 bar) among the PAF-1 derivatives, can't reach the DOE target (Table S2†). The volumetric uptake calculated for PPN-4 with a network density of 0.2 g cm−3 is only 77 v/v. Fortunately, PPN-4 can be compressed to half of its volume without a significant loss of porosity in order to improve its density and volumetric uptake.67 Thus, it can be concluded that excessively large pores are not mandatory for a high methane volumetric uptake capacity. Additionally, the interaction between methane and the pore surface area decreases with an increase in surface coverage, possibly because the excessive space cannot be effectively utilized for methane loading.
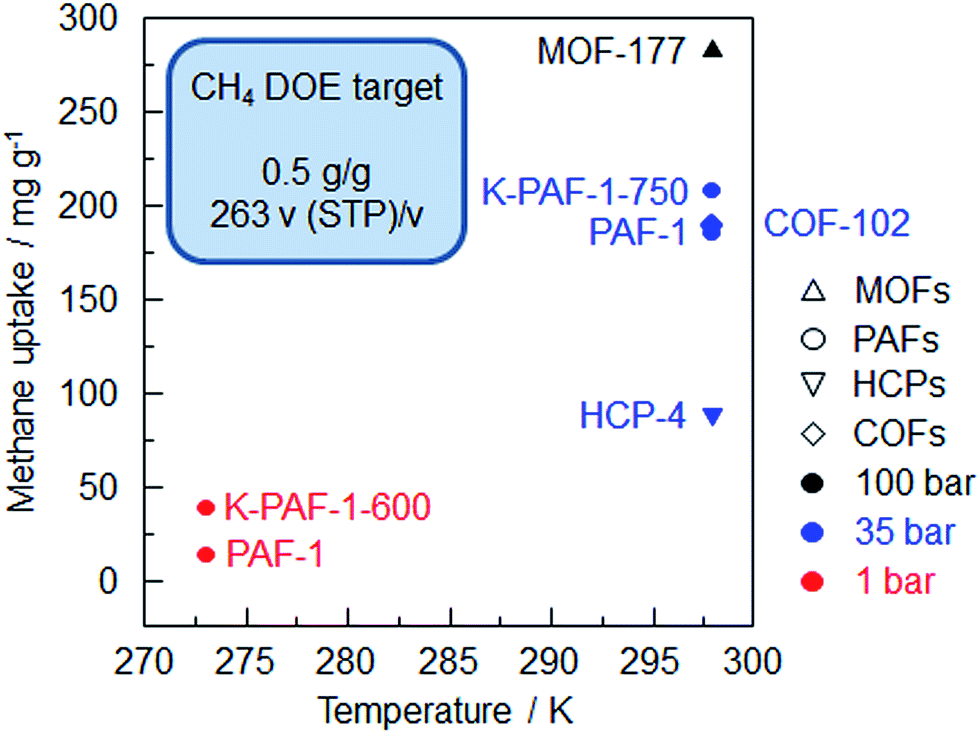 | ||
| Fig. 5 Summary of methane uptake of the porous organic frameworks vs. sorption temperature (K-PAF-1-750,17 PAF-1,1 HCP-4,91 K-PAF-1-600,17 COF-10224). | ||
Small hydrocarbon molecule sorption
C1 to C3 light hydrocarbons (CH4, C2H2, C2H4, C2H6, C3H6 and C3H8) are very important raw materials for industrial products and fine chemicals. Cryogenic distillation is the traditional large scale technology implemented for the separation of these chemicals. From an energy saving point of view, adsorption separation with solid sorbents stands as a potential alternative technology owing to the high operating temperature. Porous organic frameworks characterized with tunable pore sizes for molecule-selective sieving and functionalized frameworks for the specific recognition of molecules, attract attention. Based on the combined simulated breakthrough research, involving IAST experiments, and single component gas sorption measurements on several MOFs,68–73 three conclusions can be draw. First, van der Waals forces predominate in interactions between MOFs and hydrocarbon molecules, so the longer the hydrocarbon chain the greater the interaction capacity.71 As expected, methane shows weaker interactions than C2 and C3 hydrocarbons and, so, can be easily separated. Second, the high density of open metal sites in MOFs plays an important role in the separation of light hydrocarbons.68 It suggests that, based on the high surface area and pore volume, immobilized PAF-1 or its derivatives with different functional sites such as open metal sites, –NH2, –OH, and Lewis pyridine sites can enhance the selectivity towards alkyl, olefin, and alkyne chains. Third, a chemically robust framework is essential. MCOF-1 is one of the few examples of PAF-1 derivatives which is applied in light hydrocarbon separation.74 Using gas sorption measurements at 273 K and 298 K at 1 bar, the zero-point Qst of MCOF-1 was calculated to be 15 kJ mol−1 for CH4, and 41 kJ mol−1 for C2H6, which is about three times higher than that of CH4. Using the IAST method, it was found that the selectivities of MCOF are 1800 for C3H8/CH4, 88 for C2H6/CH4, and 26 for C2H4/CH4. The latter two values surpass the corresponding values of the previously reported porous adsorbents.Other small molecule recognition
The small molecule recognition of the PAF-1 derivatives results from their different binding capacities towards different molecules. Generally, the interactions between the host frameworks and the molecules are non-covalent in nature such as hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, π–π interactions, and electrostatic and/or electromagnetic effects.44,75,76 To detect the binding intensity, several methods such as single component sorption measurements,1,8 spectroscopy,77 chromatography,77 and breakthrough10,78 have been applied. PAF-1 can adsorb large amounts of benzene and toluene vapour at 298 K/1 bar; the values are as high as 1306 mg g−1and 1357 mg g−1, respectively.1 Substantiated using theoretical analysis, all of the PAF-1 derivatives8,38,79,80 contain aromatic building blocks which are advantageous for the selective recognition of small aromatic hydrocarbons from aliphatic hydrocarbons due to the π–π interactions between the aromatic rings. Tuning the hydrophilic–hydrophobic ratio of the channel with sulfonate groups makes the material recognize small molecules with different polarities.44 On the other hand, PAF-1 derivatives could be used to detect toxic small molecules. Examples are: ammonia, octane, cyanogen chloride and sulphur dioxide;10 nuclear waste iodine under dry and humid vapour conditions or in solvent systems;77 and organic small molecules such as methanol and chloroform in n-hexane as the solvent.77Three conclusions are summarized here. First, a large surface area of the material leads to a high small molecule uptake. Second, a nonselective pore size decrease enhances the affinity between all guest molecules and the network. Third, an increased content of aromatic building blocks and specific functional groups modified on PAF-1 improve the binding capacity towards particular molecules, which strengthens selective recognition.
Conclusion and outlook
The synthesis of PAF-1 was inspired by its classic porous contemporaries such as zeolites,81 metal–organic frameworks (MOFs),26,28,82,83 covalent organic frameworks (COFs),24,84,85 conjugated microporous polymers (CMPs),86–88 hyper-crosslinked polymers (HCPs),89–91 triazine-based organic frameworks (CTFs),92 porous aromatic frameworks (PAFs)1,4,8,110 and polymers of intrinsic microporosity (PIMs).93,94 PAF-1 and its derivatives exhibit impressive properties and, therefore, attract great attention. A diversity of building blocks and a variety of synthetic methods gave rise to a series of materials with different structures and functions. It can be confirmed that the nature of the materials was determined via the synergy between various factors and parameters, which was aptly described by Snurr and co-workers as “only one structure characteristic has to be wrong for a material to perform poorly, but many characteristics must be optimal for a material to perform well”.50For future experiments, a co-condensation reaction using tetrahedral building blocks and other types of units, is an attractive method for synthesizing advanced functional materials. For example, the condensation with tetrahedral TBPM and triangular TBPA in different ratios can give a series of porous organic frameworks named C-POFs.95 All of these retain the advantage of PAF-1, but show stronger binding capacities as in JUC-Z2. Compared with PAF-1 and JUC-Z2, the C-POFs show excellent low-pressure gas uptakes and high-pressure gas storage capacities.
In addition, the hybrid building blocks can be pre- or post-functionalized to construct novel structures thus expanding their potential application in catalysis.96–99 Catechol-functionalized porous organic polymers, which are synthesized using the cobalt-catalysed acetylene trimerization (CCAT) strategy, attract much attention in this area. Modified PAF-1 exhibits broad prospects in catalysis for three reasons: (1) PAF-1 and its derivatives, constructed with covalent bonds and highly cross-linked frameworks, lead to high physicochemical stability even under harsh conditions (acid, base or organic solvent system); (2) PAF-1 and its derivatives possess designable structures, porosities and functionalities, which can satisfy the various activity and selectivity requirements of a specific catalytic reaction; and (3) the high surface area and physical separation of coordinating groups can stabilize and isolate catalytically active metal sites.
The large free volume and robust light architecture of porous organic frameworks not only act as storage capsules for molecules,100 but also provide an ideal venue for molecular rotor operation.101 The p-phenylene groups in deuterated PAF-3 spin at ultrafast speed, even at temperatures as low as 200 K.42 In particular, the rotor dynamics can be regulated by guest molecules which easily diffuse through the open framework. The intensive dynamics make these kinds of materials suitable for engineering oscillating dipoles. Moreover, their sensitive responses to chemical stimuli make these porous polymers suitable candidates for responsive materials with switchable ferroelectricity and for applications in devices such as sensors, and actuators which require capture and release of chemicals on command.
Acknowledgements
This work was supported by the National Basic Research Program of China (2011CB808703, 2012CB821700), the National Natural Science Foundation of China (Grant no. 91022030, 21261130584, 21390394), the “111” project (B07016), the Award Project of KAUST (CRG-1-2012-LAI-009) and the Ministry of Education, Science and Technology Development Center Project (20120061130012).Notes and references
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457 CrossRef CAS PubMed.
- J. R. Holst, E. Stöckel, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8531 CrossRef CAS.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H. Zhou, J. Am. Chem. Soc., 2011, 133, 18126 CrossRef CAS PubMed.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991 CAS.
- Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981 CrossRef CAS.
- H. J. Jeon, J. H. Choi, Y. Lee, K. M. Choi, J. H. Park and J. K. Kang, Adv. Energy Mater., 2012, 2, 225 CrossRef CAS.
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239 CAS.
- Y. Yuan, F. Sun, H. Ren, X. Jing, W. Wang, H. Ma, H. Zhao and G. Zhu, J. Mater. Chem., 2011, 21, 13498 RSC.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2012, 24, 1511 CrossRef CAS.
- G. W. Peterson, O. K. Farha, B. Schindler, P. Jones, J. Mahle and J. T. Hupp, J. Porous Mater., 2012, 19, 261 CrossRef CAS.
- K. V. Rao, R. Haldar, C. Kulkarni, T. K. Maji and S. J. George, Chem. Mater., 2012, 24, 969 CrossRef CAS.
- S. J. Garibay, M. H. Weston, J. E. Mondloch, Y. J. Colón, O. K. Farha, J. T. Hupp and S. T. Nguyen, CrystEngComm, 2013, 15, 1515 RSC.
- H. Ma, H. Ren, X. Zou, F. Sun, Z. Yan, K. Cai, D. Wang and G. Zhu, J. Mater. Chem. A, 2013, 1, 752 CAS.
- K. Konstas, J. W. Taylor, A. W. Thornton, C. M. Doherty, W. X. Lim, T. J. Bastow, D. F. Kennedy, C. D. Wood, B. J. Cox, J. M. Hill, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2012, 51, 6639 CrossRef CAS PubMed.
- Z. Wang, S. Yuan, A. Mason, B. Reprogle, D. Liu and L. Yu, Macromolecules, 2012, 45, 7413 CrossRef CAS.
- T. Ben, Y. Li, L. Zhu, D. Zhang, D. Cao, Z. Xiang, X. Yao and S. Qiu, Energy Environ. Sci., 2012, 5, 8370 CAS.
- Y. Li, T. Ben, B. Zhang, Y. Fu and S. Qiu, Sci. Rep., 2013, 3, 2420 Search PubMed.
- T. Yamamoto, Bull. Chem. Soc. Jpn., 1999, 72, 621 CrossRef CAS.
- G. Zhou, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2007, 129, 12211 CrossRef CAS PubMed.
- A. B. Cairns and A. L. Goodwin, Chem. Soc. Rev., 2013, 42, 4881 RSC.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163 CrossRef CAS.
- R. L. Martin, M. N. Shahrak, J. A. Swisher, C. M. Simon, J. P. Sculley, H. Zhou, B. Smit and M. Haranczyk, J. Phys. Chem. C, 2013, 117, 20037 CAS.
- H. Jiang, T. A. Makal and H. Zhou, Coord. Chem. Rev., 2013, 257, 2232 CrossRef CAS.
- H. M. El-Kaderi, J. R. Hunt, J. L. Medoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS PubMed.
- N. T. T. Nguyen, H. Furukawa, F. Gándara, H. T. Nguyen, K. E. Cordova and O. M. Yaghi, Angew. Chem., Int. Ed., 2014, 53, 10645 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed.
- D. Yuan, W. Lu, D. Zhao and H. Zhou, Adv. Mater., 2011, 23, 3723 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016 CrossRef CAS PubMed.
- V. Presser, J. McDonough, S. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059 CAS.
- Y. Yan, I. Telepeni, S. Yang, X. Lin, W. Kockelmann, A. Dailly, A. J. Blake, W. Lewis, G. S. Walker, D. R. Allan, S. A. Barnett, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 4092 CrossRef CAS PubMed.
- L. Huang and D. Cao, J. Mater. Chem. A, 2013, 1, 9433 CAS.
- J. Lan, D. Cao, W. Wang, T. Ben and G. Zhu, J. Phys. Chem. Lett., 2010, 1, 978 CrossRef CAS.
- Z. Yang, X. Peng and D. Cao, J. Phys. Chem. C, 2013, 117, 8353 CAS.
- B. Lukose, M. Wahiduzzaman, A. Kuc and T. Heine, J. Phys. Chem. C, 2012, 116, 22878 CAS.
- A. F. Oliveira, G. Seifert, T. Heine and H. A. Duarte, J. Braz. Chem. Soc., 2009, 20, 1193 CrossRef CAS.
- G. Seifert, D. Porezag and T. Frauenheim, Int. J. Quantum Chem., 1996, 58, 185 CrossRef CAS.
- S. Patchkovskii and T. Heine, Phys. Rev. E, 2009, 80, 031603 CrossRef.
- H. Ren, T. Ben, E. Wang, X. Jing, M. Xue, B. Liu, Y. Cui, S. Qiu and G. Zhu, Chem. Commun., 2010, 46, 291 RSC.
- R. Babarao, S. Dai and D. E. Jiang, Langmuir, 2011, 27, 3451 CrossRef CAS PubMed.
- N. Y. Du, H. B. Park, G. P. Robertson, M. M. Dal-Cin, T. Visser, L. Scoles and M. D. Guiver, Nat. Mater., 2011, 10, 372 CrossRef CAS PubMed.
- K. T. Jackson, M. G. Rabbani, T. E. Reich and H. M. El-Kaderi, Polym. Chem., 2011, 2, 2775 RSC.
- A. Comotti, S. Bracco, T. Ben, S. Qiu and P. Sozzani, Angew. Chem., Int. Ed., 2014, 126, 1061 CrossRef.
- A. Trewin, G. R. Darling and A. I. Cooper, New J. Chem., 2008, 32, 17 RSC.
- Y. Yang, Q. Zhang, Z. Zhang and S. Zhang, J. Mater. Chem. A, 2013, 1, 10368 CAS.
- M. H. Weston, O. K. Farha, B. G. Hauser, J. T. Hupp and S. T. Nguyen, Chem. Mater., 2012, 24, 1292 CrossRef CAS.
- Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 2691 CAS.
- J. Lan, D. Cao, W. Wang and B. Smit, ACS Nano, 2010, 4, 4225 CrossRef CAS PubMed.
- L. Wang, Y. Sun and H. Sun, Faraday Discuss., 2011, 151, 143 RSC.
- Y. Sun, T. Ben, L. Wang, S. Qiu and H. Sun, J. Phys. Chem. Lett., 2010, 1, 2753 CrossRef CAS.
- Y. Zhang, B. Li, K. Williams, W. Gao and S. Ma, Chem. Commun., 2013, 49, 10269 RSC.
- B. Metz, O. Davidson, H. C. de Coninck, M. Loos and L. A. Meyer, [IPCC, 2005] IPCC special report on Carbon Dioxide Capture and Storage, Prepared by working group III of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, USA, p. 442 Search PubMed.
- C. E. Wilmer, O. K. Farha, Y. Bae, J. T. Hupp and R. Q. Snurr, Energy Environ. Sci., 2012, 5, 9849 CAS.
- A. Sayari, A. Heydari-Gorji and Y. Yang, J. Am. Chem. Soc., 2012, 134, 13834 CrossRef CAS PubMed.
- Z. Zhang, L. Wu and T. Xu, J. Mater. Chem., 2012, 22, 13996 RSC.
- T. E. Reich and H. M. El-Kaderi, J. Nanopart. Res., 2013, 15, 1368 CrossRef.
- W. Lu, J. P. Sculley, D. Yuan, R. Krishna, Z. Wei and H. Zhou, Angew. Chem., Int. Ed., 2012, 51, 7480 CrossRef CAS PubMed.
- DOE. U.S. Department of Energy, Target for onboard hydrogen storage systems for light-duty vehicles, 2012, http://www1.eere.energy.gov/hydrogenandfuelcells/storage/pdfs/targets_onboard_hydro_storage.pdf Search PubMed.
- Z. Li, G. Zhu, G. Lu, S. Qiu and X. Yao, J. Am. Chem. Soc., 2010, 132, 1490 CrossRef CAS PubMed.
- R. C. Lochan and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 1357 RSC.
- A. W. C. van den Berg and C. O. Areán, Chem. Commun., 2008, 668 RSC.
- E. Klontzas, E. Tylianakis and G. E. Froudakis, J. Phys. Chem. C, 2008, 112, 9095 CAS.
- S. B. Kalidindi and R. A. Fischer, Phys. Status Solidi B, 2013, 250, 1119 CrossRef CAS.
- Y. Peng, T. Ben, Y. Jia, D. Yang, H. Zhao, S. Qiu and X. Yao, J. Phys. Chem. C, 2012, 116, 25694 CAS.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887 CrossRef CAS PubMed.
- C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2012, 4, 83 CrossRef CAS PubMed.
- J. Lan, D. Cao and W. Wang, Langmuir, 2009, 26, 220 CrossRef PubMed.
- T. A. Makal, J. Li, W. Lu and H. Zhou, Chem. Soc. Rev., 2012, 41, 7761 RSC.
- Y. He, R. Krishna and B. Chen, Energy Environ. Sci., 2012, 5, 9107 CAS.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606 CrossRef CAS PubMed.
- Y. He, Z. Zhang, S. Xiang, H. Wu, F. R. Fronczek, W. Zhou, R. Krishna, M. O'Keeffe and B. Chen, Chem.–Eur. J., 2012, 18, 1901 CrossRef CAS PubMed.
- Y. He, Z. Zhang, S. Xiang, F. R. Fronczek, R. Krishna and B. Chen, Chem. Commun., 2012, 48, 6493 RSC.
- J. Duan, M. Higuchi, S. Horike, M. L. Foo, K. P. Rao, Y. Inubushi, T. Fukushima and S. Kitagawa, Adv. Funct. Mater., 2013, 23, 3525 CrossRef CAS.
- H. Xu, B. Chen and G. Qian, J. Mater. Chem. A, 2013, 1, 9916 CAS.
- H. Ma, H. Ren, S. Meng, Z. Yan, H. Zhao, F. Sun and G. Zhu, Chem. Commun., 2013, 49, 9773 RSC.
- A. Dhotel, Z. Chen, L. Delbreilh, B. Youssef, J. Saiter and L. Tan, Int. J. Mol. Sci., 2013, 14, 2303 CrossRef CAS PubMed.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS PubMed.
- C. Pei, T. Ben, S. Xu and S. Qiu, J. Mater. Chem. A, 2014, 2, 7179 CAS.
- A. Torres-Knoop, R. Krishna and D. Dubbeldam, Angew. Chem., Int. Ed., 2014, 53, 7774 CrossRef CAS PubMed.
- V. R. Pedireddi, S. Chatterjee, A. Ranganathan and C. N. R. Rao, J. Am. Chem. Soc., 1997, 119, 10867 CrossRef CAS.
- A. Ranganathan, V. R. Pedireddi, S. Chatterjee and C. N. R. Rao, J. Mater. Chem., 1999, 9, 2407 RSC.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. D. Weireld, J. Chang, D. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245 CrossRef CAS PubMed.
- D. Saha, Z. Bao, F. Jia and S. Deng, Environ. Sci. Technol., 2010, 44, 1820 CrossRef CAS PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS PubMed.
- J. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826 CrossRef CAS PubMed.
- A. Li, R. Lu, Y. Wang, X. Wang, K. Han and W. Deng, Angew. Chem., Int. Ed., 2010, 49, 3330 CrossRef CAS PubMed.
- C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034 CrossRef CAS.
- M. P. Tsyurupa and V. A. Davankov, React. Funct. Polym., 2002, 53, 193 CrossRef CAS.
- C. F. Martin, E. Stöckel, R. Clowes, D. J. Adams, A. I. Cooper, J. J. Pis, F. Rubiera and C. Pevida, J. Mater. Chem., 2011, 21, 5475 RSC.
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675 RSC.
- B. S. Ghanem, K. J. Msayib, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67 RSC.
- C. Pei, T. Ben, Y. Li and S. Qiu, Chem. Commun., 2014, 50, 6134 RSC.
- S. J. Kraft, R. H. Sánchez and A. S. Hock, ACS Catal., 2013, 3, 826 CrossRef CAS.
- K. K. Tanabe, N. A. Siladke, E. M. Broderick, T. Kobayashi, J. F. Goldston, M. H. Weston, O. K. Farha, J. T. Hupp, M. Pruski, E. A. Mader, M. J. A. Johnson and S. T. Nguyen, Chem. Sci., 2013, 4, 2483 RSC.
- Z. Xie, C. Wang, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056 CrossRef CAS PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819 CrossRef CAS.
- A. Comotti, S. Bracco, M. Mauri, S. Mottadelli, T. Ben, S. Qiu and P. Sozzani, Angew. Chem., Int. Ed., 2012, 51, 10136 CrossRef CAS PubMed.
- A. Comotti, S. Bracco, P. Valsesia, M. Beretta and P. Sozzani, Angew. Chem., Int. Ed., 2010, 49, 1760 CrossRef CAS PubMed.
- Y. Yang, Q. Zhang, J. Zheng and S. Zhang, Polymer, 2013, 54, 3254 CrossRef CAS.
- H. A. Patel, S. H. Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz and A. Coskun, Nat. Commun., 2013, 4, 1357 CrossRef PubMed.
- P. Pandey, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Mater. Chem., 2011, 21, 1700 RSC.
- H. Zhao, Z. Jin, H. Su, J. Zhang, X. Yao, H. Zhao and G. Zhu, Chem. Commun., 2013, 49, 2780 RSC.
- D. Zhang, Z. Chang, Y. Lv, T. Hu and X. Bu, RSC Adv., 2012, 2, 408 RSC.
- H. Qian, J. Zheng and S. Zhang, Polymer, 2013, 54, 557 CrossRef CAS.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H. Zhou, Chem. Mater., 2010, 22, 5964 CrossRef CAS.
- E. Stöckel, X. Wu, A. Trewin, C. D. Wood, R. Clowes and A. I. Cooper, Chem. Commun., 2009, 212 RSC.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17 RSC.
- D. A. Yang, H. Y. Cho, J. Kim, S. T. Yang and W. S. Ahn, Energy Environ. Sci., 2012, 5, 6465 CAS.
- Z. Zhang, Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mh00163j |
| This journal is © The Royal Society of Chemistry 2015 |