
Elucidating new structural features of the triazole scaffold for the development of mPGES-1 inhibitors†
Maria Giovanna Chini‡ a, Claudia Ferroni‡ b, Vincenza Cantone a, Paolo Dambruoso b, Greta Varchi b, Antonella Pepe§ c, Katrin Fischer d, Carlo Pergola d, Oliver Werz d, Ines Bruno a, Raffaele Riccio a and Giuseppe Bifulco *a
aDepartment of Pharmacy, Via Giovanni Paolo II 132, 84084 Fisciano (SA), Italy. E-mail: bifulco@unisa.it; Fax: +39 089 969602; Tel: +39 089 969741
bInstitute for the Organic Synthesis and the Photoreactivity, ISOF – CNR Area della Ricerca di Bologna, Via P. Gobetti 101, 40129 Bologna, Italy
cLaboratory of Synthetic Chemistry, Leidos Biomedical Research Inc, Frederick National Laboratory for Cancer Research, 1050 Boyles Street, Frederick, MD 21702, USA
dDepartment of Pharmaceutical/Medicinal Chemistry, Institute of Pharmacy, Friedrich Schiller University, Philosophenweg 14, Jena, D-07743 Jena, Germany
First published on 15th September 2014
Abstract
We report a new potent revisited version of a triazole-based inhibitor obtained by structure-based drug design on the human mPGES-1 crystal structure. Moreover, we disclosed the substitution with a halogen atom at position 5 as a new key factor influencing the biological activity on the mPGES-1 enzyme.
The microsomal prostaglandin E2 synthase-1 (mPGES-1) enzyme has emerged as an attractive target for the discovery and development of new anti-inflammatory and anti-cancer drugs.1 In the arachidonic acid cascade, this enzyme is responsible for the conversion of COX-derived unstable peroxide PGH2 into PGE2,2 and it is over expressed in several inflammatory disorders3 as well as in many human tumors.4 mPGES-1 is a glutathione-dependent trans-membrane enzyme belonging to the MAPEG family that includes leukotriene C4 synthase (LTCS), microsomal glutathione transferase-1 (MGST-1) and 5-lipoxygenase-activating protein (FLAP). Among the three isoforms so far identified for biosynthesis of PGE2, mPGEs-1 seems to be the one most closely involved in pathology development. In fact, this enzyme is functionally coupled with COX-2 and it is directly involved in the biosynthesis of prostaglandins5 induced by inflammatory stimuli, without affecting constitutive prostaglandins implicated in physiological functions. For this reason, the identification of new mPGES-1 inhibitors represents a promising approach for the development of safer drugs devoid of classical NSAID-related side effects. In addition, in humans, mPGES-1 is up-regulated in arthritic synovial tissues,6 in the cartilage and chondrocytes of osteoarthritic patients7 and in inflamed intestinal mucosa of patients with inflammatory bowel diseases.8 Strictly related to inflammation, mPGES-1 has recently emerged to be involved in the pathogenesis of different cancer forms and in the induction of angiogenesis.9 However, despite the fact that several mPGES-1 inhibitors have been identified so far10,11 only a few of them exhibited in vivo anti-cancer and anti-inflammatory activities.12 Interestingly, no selective inhibitors targeting mPGES-1 have been identified and, despite the high number of published patents, none of these drugs have yet made it to the clinic.13 In this framework, the recently published X-ray crystal structure of mPGES-1 by Geschwindner and co-workers14 represents a powerful tool for the rational in silico design of potent and efficient mPGES-1 inhibitors.
As confirmed by Geschwindner’s work,14 the mPGES-1 active site is sub divisible in cofactor (GSH) and substrate (PGH2) binding sites. Moreover, it includes the N-terminal (helices II and IV), the C-terminal (helix I) and an adjacent monomeric cytoplasmic domain. In more detail, the major portion of the active site is occupied by GSH while only the PGH2 ring interacts with it. This pattern of binding is well represented by the co-crystallized structure of mPGES-1 with the GSH analogue, 1-(4-phenylphenyl)-2-(S-glutathionyl)-ethanone and a β-octyl glucoside,14 which discloses key interactions for the rational design of substrate's competitors (Fig. 1).
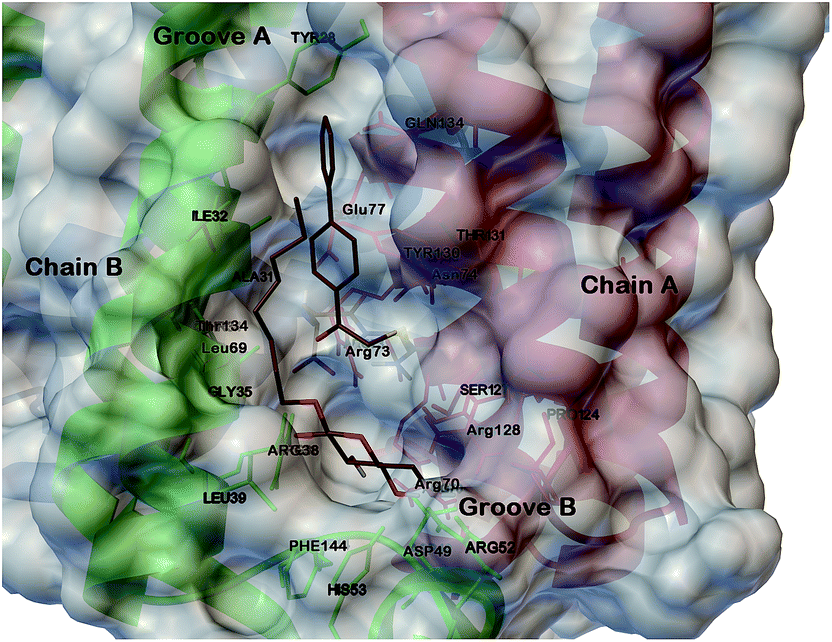 | ||
| Fig. 1 Three dimensional model of mPGES-1 in the complex with GSH analogue and β-octyl glucoside depicted by stick and balls (by atom type: C, brown; O, red; and H, white). The crucial amino acids of the mPGES-1 receptor are depicted by stick and balls, and ribbon colored by chains (A, red; B, green; C, blue). | ||
In the course of previous studies, we identified a novel class of 1,4-disubstitued 1,2,3-triazoles that inhibited mPGES-1 in a cell-free assay with IC50 values in the low μM range.15 In particular, compound 4 (Fig. 2) showed the most promising activity with an IC50 of 0.7 μM in the microsomal fraction of A549 cells that was used as a source for the human mPGES-1 enzyme.15 These active compounds were disclosed by means of structure-based studies using the microsomal glutathione transferase 1 (MGST-1)16 as the model enzyme. Thanks to the recent human mPGES-1 X-ray structure resolution14 and encouraged by the structural differences between the two enzymes, we decided to re-evaluate our lead compounds and to develop a new focused triazole-based collection. For docking studies, we have used Glide software (version 9.6)17 with extra-precision (XP) mode,18 and we have designed the triazole compounds for an optimal placement in the substrate's binding site. On this basis, we analysed our previous results in relation to the model reported above (Fig. 1). As shown by Geschwindner,14 a competitive inhibitor of mPGES-1 should be able to interact with Ile32 and Tyr28 of chain B, and Gln134 and Tyr130 of chain A in groove A, as well as with the cofactor, with Arg126, Ser127 of chain A, and with Asp49, His53, Arg38, Phe144 of chain B in groove B (Fig. 1).
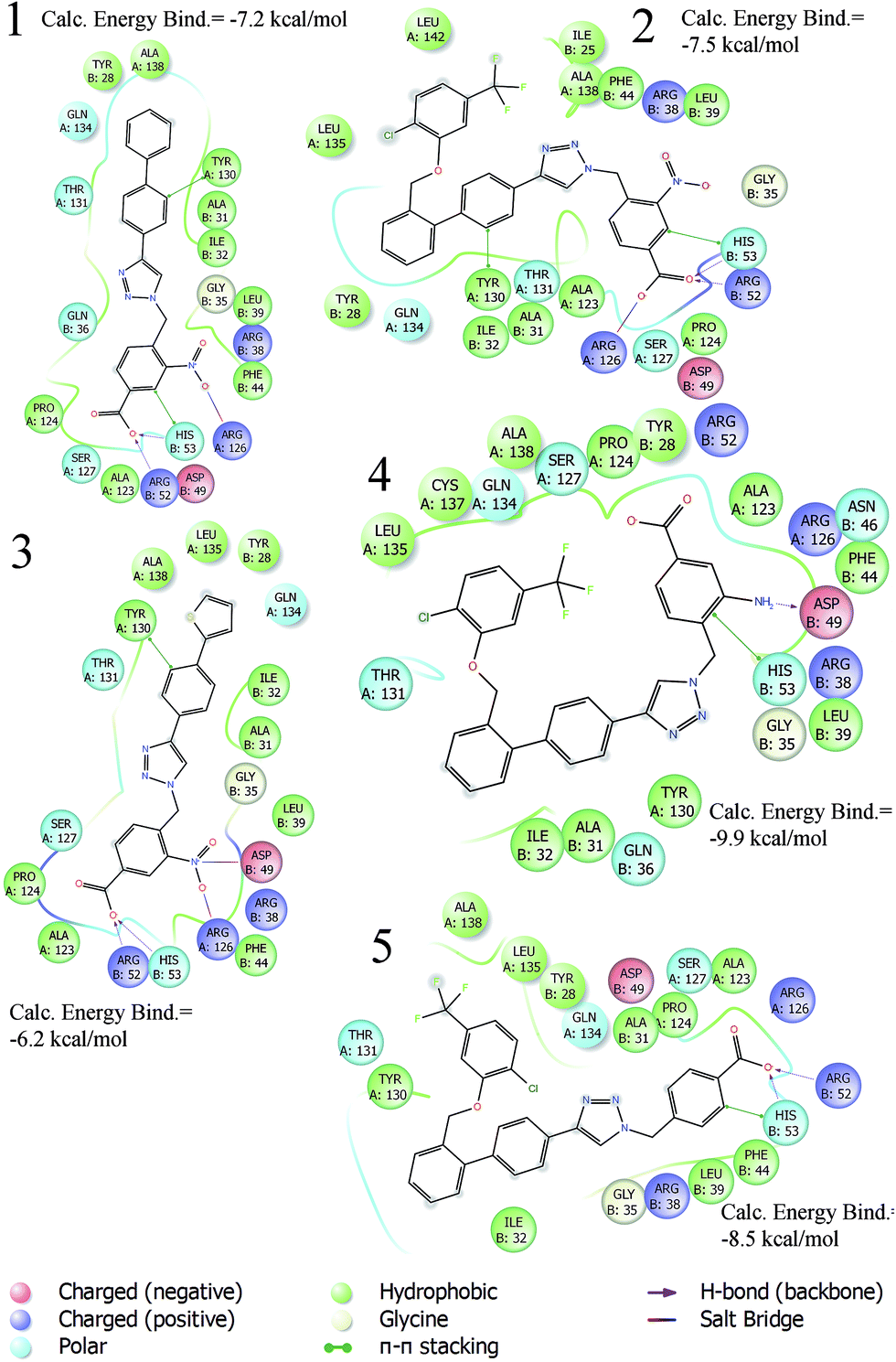 | ||
| Fig. 2 2D diagram interactions of active triazole-based inhibitors 1–5. | ||
The best binding modes of compounds 1–5 are in agreement with the key interactions reported for the putative mPGES-1 inhibitor. Based on these considerations, we have undertaken a new structure drug design with the aim of investigating the influence of the ring-substituent topological position and simplifying the mPGES-1 inhibitor structure. In fact, as can be seen from the 2D diagram interactions (Fig. 2), in our previous triazole inhibitors, the key features were represented by (i) a benzyl group at the N1 position bearing a para hydrogen bond acceptor able to interact with groove B, responsible for PGH2 recognition and involved in its isomerisation into PGE2; and (ii) bis-aryl substituents at the C4 position, which establish hydrophobic and π–π interactions with groove A. Interestingly, in the crystallized model of the GSH analogue (Fig. 1)14 only the first aromatic ring of the biphenyl portion directly bonded at position 4 is involved in π–π interactions with the key amino acid Tyr130 (A), while the second aromatic ring is involved in hydrophobic interactions with groove A (e.g. Gln134 (A) and Tyr 117 (A)).
Based on this, we present herein the effect of the substituents inversion on the triazole ring. Thus, a phenyl ring was positioned on N1, while a phenoxy-methyl group was attached at the C4 position (scaffold II, Fig. 3). Moreover, as an effort to improve the activity with respect to the previous molecules, we also explored the substitution at position C5 (R7, Fig. 3 and 4). The first step towards the scaffold optimization was the introduction of a p-substituent on ring B (II, Fig. 3). Docking studies performed on compounds 6–8 revealed that none of them was able to interact with groove B (Fig. S1†). Therefore, in order to improve the interaction with its amino acids (e.g. Ser127 and Arg126), we introduced another hydrogen bond acceptor, namely a CF3 group, at R3 of ring B (scaffold II, Fig. 3), in analogy to the ring B of scaffold I (Fig. 3) of the previously reported molecules (Fig. 2).15 Docking studies on derivatives 9–11 showed that, with respect to 10 and 11, scaffold 9 strongly interacts with groove B, being able to simultaneously interact with Arg126 (A), Arg38 (B), Asp49 (B) and Ser127 (A) (Fig. 5 and Fig. S2†), thus 9 was selected for further optimization. By superimposing the binding mode of 9 with respect to the lead compound 4 in the mPGES-1 structure (See Fig. S3†), the para and meta positions of ring A (II, Fig. 3) were identified as the most suitable and attractive for modifications. For this reason, compound 9 was functionalized with four different hydrogen bond acceptors (e.g. CF3, NO2, OH and CN) at meta and para positions, respectively (Fig. 4, compounds 12–19).
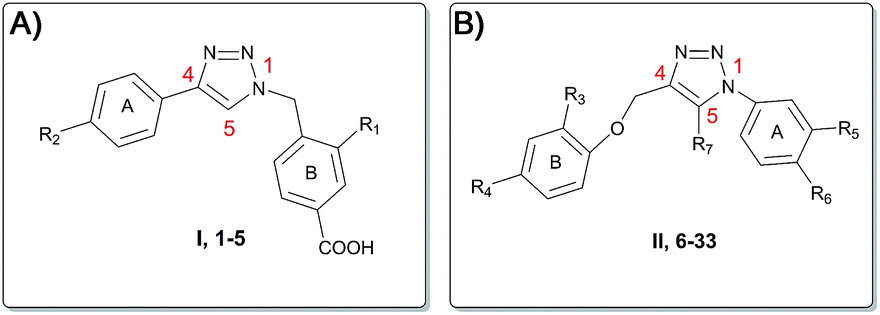 | ||
| Fig. 3 Scaffold hopping of the triazole pharmacophore. (A) Scaffold I, compounds 1–5. (B) Scaffold II, compounds 6–33. | ||
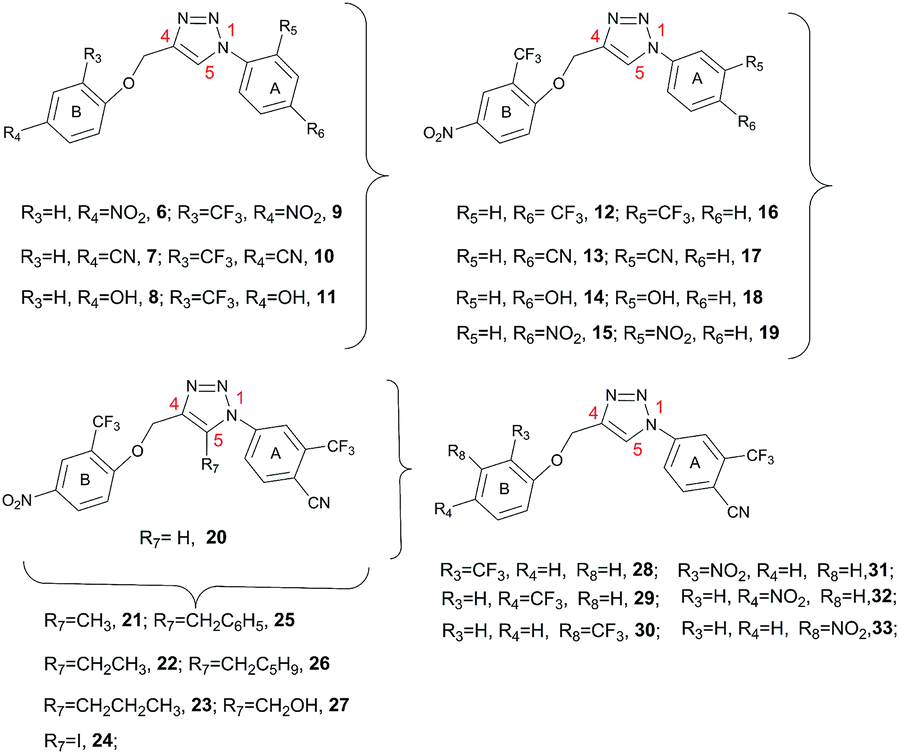 | ||
| Fig. 4 Triazoles 6–33 structures. | ||
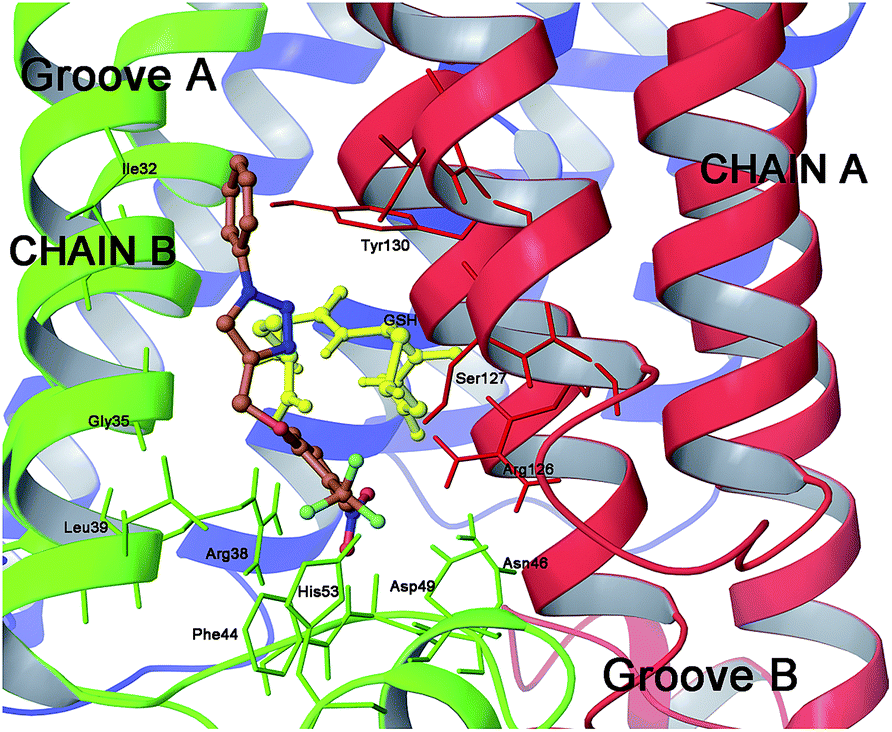 | ||
| Fig. 5 Three dimensional model of scaffold 9 (orange sticks) in mPGES-1 binding site. | ||
From the analysis of the docking results (Fig. 6), the best substitutions were represented by the CF3 group at meta and para positions (12, predicted energy of binding = −7.4 kcal mol−1 and 16, predicted energy of binding = −7.2 kcal mol−1), and by the CN group at the para position (17, predicted energy of binding = −6.4 kcal mol−1), displaying an energy gain of ca. 1.5 kcal mol−1 in comparison to those of the other complexes.
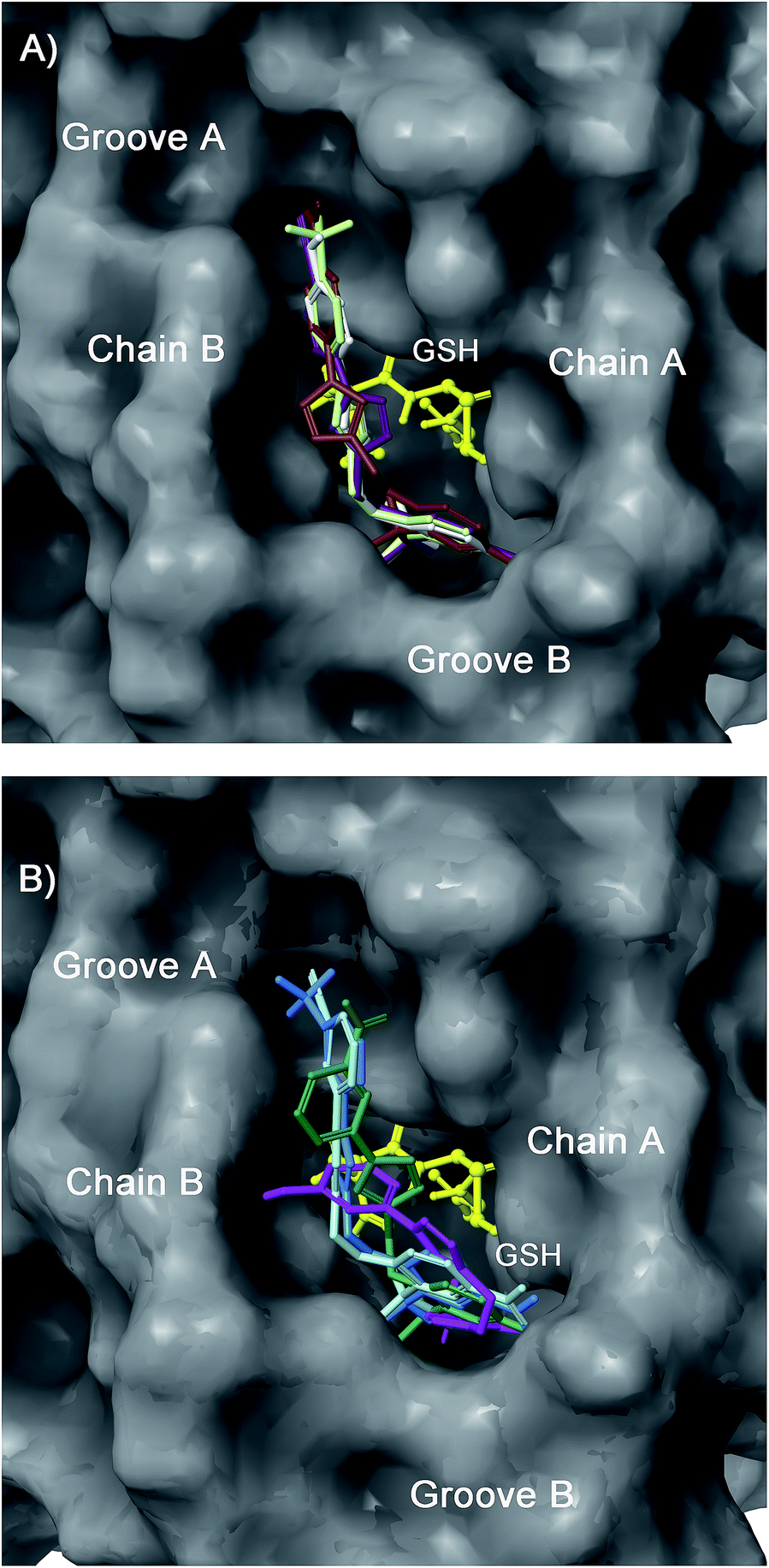 | ||
| Fig. 6 Superimposition of 12 (light green), 13 (purple), 14 (white), and 15 (brown) (panel A), and 16 (light blue), 17 (cyan), 18 (magenta), and 19 (dark green) (panel B) in the mPGES-1 binding site. | ||
In order to further extend the compounds’ virtual library, we inserted a chimeric substituent at N1 of the triazole scaffold with CF3 at meta and a CN group at para position of ring A (compound 20, Fig. 4), which provided a calculated binding energy of −7.6 kcal mol−1.
Moreover, the analysis of the binding mode of 20 (Fig. 7) suggested that a substituent at position C5 of the triazole ring should protrude towards a left hydrophobic side (Ile32, Ala31 chain B) and a right side where the polar amino acids Thr131 and Ser127 of chain A are located. As a consequence, we explored the effect on the binding energy of properly selected substitutions at this position (compounds 21–27, Fig. 4). As a result of the docking studies, derivative 24, which bears an iodine atom at C5, displayed the most promising binding mode (Fig. 8).
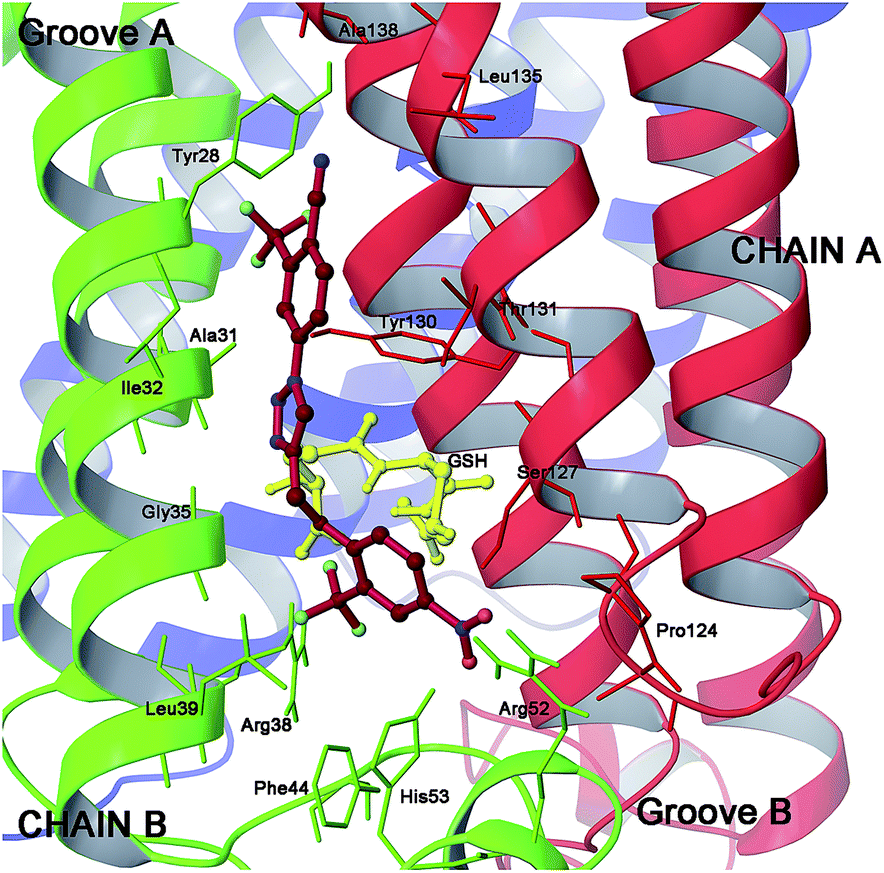 | ||
| Fig. 7 Three dimensional model of scaffold 20 (red sticks) in mPGES-1 binding site. | ||
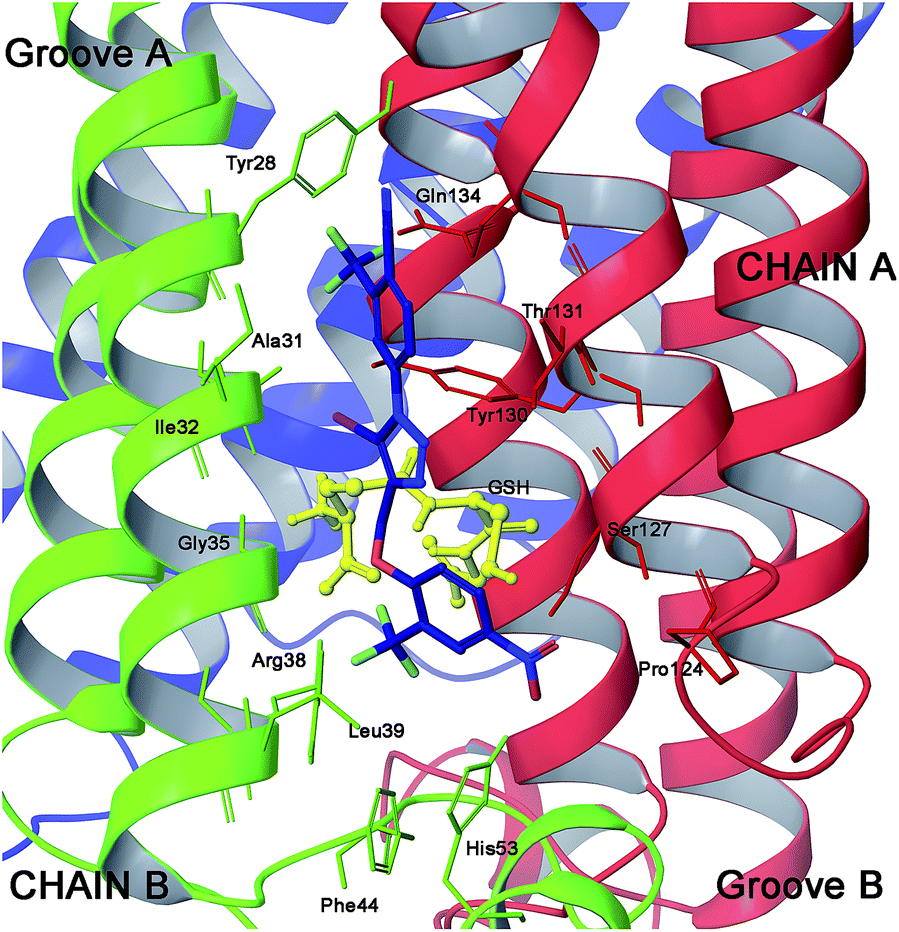 | ||
| Fig. 8 Three dimensional model of 24 (blue sticks) in the mPGES-1 binding site. | ||
In more detail, the portions at C1 and C4 of the triazole ring maintained similar binding modes of 20 (Fig. 9), while the iodine atom at C5 is involved in a halogen bond with CO of Ala31 (B). These optimal interactions were associated with a calculated binding energy of −8.8 kcal mol−1 for 24, which was in good agreement with the calculated affinities of our lead compound15 (Fig. 2). Moreover, the iodine atom at position C5 presents several advantages with respect to the other halogens especially in terms of halogen bonding strength, which decreases from iodine to chlorine.19 In summary, our docking studies suggested that this revisited version of triazole based inhibitors should be able to interact with mPGES-1 in the same manner of the previously reported 2, 4 and 5 inhibitors, and that the halogen atom should play a fundamental role in the inhibition activity.
 | ||
| Fig. 9 Superimposition of 24 (blue sticks) with LVJ (34, light violet) (panel B) in the mPGES-1 binding site. | ||
To prove our hypothesis, starting from compound 20, we have synthesized compounds 16, 20 and 24.
Moreover, starting from scaffold 20, to prove the influence of topology of selected hydrogen bond acceptors on ring B (NO2 and CF3 group), we have designed another small pool of compounds (28–33), and synthesized among them the most significant compounds (29 and 30) (See ESI†).
The biological evaluation of the representative compounds (16, 20, 24, 29, and 30) on the mPGES-1 activity in a cell-free assay12b showed efficient inhibitory activity for 24 (IC50 = 0.7 ± 0.2 μM) (Fig. S9†) and significant but incomplete suppression of mPGES-1 activity by the other compounds (20% inhibition at 10 μM, IC50 > 30 μM, see ESI†).
To further corroborate the influence of an iodine atom at C5 on a triazole scaffold on the biological activity, we have compared the putative binding mode of 24 with respect to the known inhibitor LVJ (34, Fig. S8†) recently co-crystallized with the mPGES-1 enzyme.20
Fig. 9 clearly shows the good superimposition of the aromatic ring B of 24 (Fig. 4) with respect to the bis-chlorophenyl ring of 34 in groove B of the mPGES-1 surface, even if a more potent inhibitor (2.4 nM) makes an optimal π–π stacking with Phe44.
On the other hand, peculiar interactions of the benzimidazole portion of 34 with Ile132, Val128, Ala123, and Arg52 are partially balanced by the halogen bonding of 24 with Ala31, and by the contacts with Tyr28, Gln134 accounting for the minor inhibitory potency of 24 with respect to 34. In conclusion, even if 24 shows a relatively simple skeleton in comparison to 34, the docking results suggest that the different patterns of interactions established with mPGES-1 are sufficient to support its biological activity in occupying and inhibiting the enzyme binding site.
Conclusions
In conclusion, thanks to the recent disclosure of the mPGES-1 X-ray crystal structure, we were able to perform a structure-based design of a novel class of potential mPGES-1 triazole inhibitors. In particular, compound 24 was identified as the most promising of the series, enabling the interaction with the membrane protein and occupying the PGH2 binding site, and inhibiting mPGES-1 activity in a cell-free assay with IC50 = 0.7 μM. The smaller dimension and different interactions of the rings A and B with respect to the lead compound (4) are balanced by the presence of an iodine atom at position C5 and of precise positions of the substituents on these two rings. In fact, the halogen bonding of the iodine atom with the receptor backbone resulted as a new key interaction suitable for the design of new mPGES-1 inhibitors, proved by the complete inactivity of compounds lacking this atom.Acknowledgements
Financial support by the University of Salerno, and by Associazione Italiana Ricerca sul Cancro (AIRC) Grant IG 2012 – IG_12777 – Bifulco Giuseppe is gratefully acknowledged. We thank Dr Massimo Capobianco (ISOF-CNR) for MS measurements.Notes and references
- H.-H. Chang and E. Meuillet, Future Med. Chem., 2011, 3, 1909 CrossRef CAS PubMed.
- C. D. Funk, Science, 2001, 294, 1871 CrossRef CAS PubMed.
- D. Claveau, M. Sirinyan, J. Guay, R. Gordon, C. C. Chan, Y. Bureau, D. Riendeau and J. A. Mancini, J. Immunol., 2003, 170, 4738 CrossRef CAS.
- (a) M. Nakanishi, V. Gokhale, E. J. Meuillet and D. W. Rosenberg, Biochimie, 2010, 92, 660 CrossRef CAS PubMed; (b) O. Rådmark and B. Samuelsson, J. Intern. Med., 2010, 268, 5 Search PubMed; (c) K. Yoshimatsu, D. Golijanin, P. B. Paty, R. A. Soslow, P. J. Jakobsson, R. A. De Lellis, K. Subbaramaiah and A. J. Dannenberg, Clin. Cancer Res., 2001, 7, 3971 CAS; (d) K. Yoshimatsu, N. K. Altorki, D. Golijanin, F. Zhang, P. J. Jakobsson, A. J. Dannenberg and K. Subbaramaiah, Clin. Cancer Res., 2001, 7, 2669 CAS; (e) S. Mehrotra, A. Morimiya, B. Agarwal, R. Konger and S. Badve, J. Pathol., 2006, 208, 356 CrossRef CAS PubMed.
- M. Nakanishi and D. W. Rosenberg, Semin. Immunopathol., 2013, 35, 123 CrossRef CAS PubMed.
- M. Korotkova, M. Westman, K. R. Gheorghe, E. af Klint, C. Trollmo, A. K. Ulfgren, L. Klareskog and P. J. Jakobsson, Arthritis Rheum., 2005, 52, 3439 CrossRef CAS PubMed.
- (a) F. Kojima, H. Naraba, S. Miyamoto, M. Beppu, H. Aoki and S. Kawai, Arthritis Res. Ther., 2004, 6, R355 CrossRef CAS PubMed; (b) K. Masuko-Hongo, F. Berenbaum, L. Humbert, C. Salvat, M. B. Goldring and S. Thirion, Arthritis Rheum., 2004, 50, 2829 CrossRef CAS PubMed; (c) X. Li, H. Afif, S. Cheng, J. Martel-Pelletier, J. P. Pelletier, P. Ranger and H. Fahmi, J. Rheumatol., 2005, 32, 887 CAS.
- K. Subbaramaiah, K. Yoshimatsu, E. Scherl, K. M. Das, K. D. Glazier, D. Golijanin, R. A. Soslow, T. Tanabe, H. Naraba and A. J. Dannenberg, J. Biol. Chem., 2004, 279, 12647 CrossRef CAS PubMed.
- M. Nakanishi, D. C. Montrose, P. Clark, P. R. Nambiar, G. S. Belinsky, K. P. Claffey, D. Xu and D. W. Rosenberg, Cancer Res., 2008, 68, 3251 CrossRef CAS PubMed.
- F. Rörsch, I. Wobst, H. Zettl, M. Schubert-Zsilavecz, S. Grösch, G. Geisslinger, G. Schneider and E. Proschak, J. Med. Chem., 2010, 53, 911 CrossRef PubMed.
- (a) T. Shiro, H. Takahashi, K. Kakiguchi, Y. Inoue, K. Masuda, H. Nagata and M. Tobe, Bioorg. Med. Chem. Lett., 2012, 22, 285 CrossRef CAS PubMed; (b) A. Wiegard, W. Hanekamp, K. Griessbach, J. Fabian and M. Lehr, Eur. J. Med. Chem., 2012, 48, 153 CrossRef CAS PubMed.
- (a) J. Bauer, A. Koeberle, F. Dehm, F. Pollastro, G. Appendino, H. Northoff, A. Rossi, L. Sautebin and O. Werz, Biochem. Pharmacol., 2011, 81, 259 CrossRef CAS PubMed; (b) A. Koeberle, A. Rossi, H. Zettl, C. Pergola, F. Dehm, J. Bauer, C. Greiner, S. Reckel, C. Hoernig, H. Northoff, F. Bernhard, V. Dötsch, L. Sautebin, M. Schubert-Zsilavecz and O. Werz, J. Pharmacol. Exp. Ther., 2010, 332, 840 CrossRef CAS PubMed.
- J. K. Norberg, E. Sells, H.-H. Chang, S. R. Alla, S. Zhang and E. J. Meuillet, Pharm. Pat. Anal., 2013, 265, 12 Search PubMed.
- T. Sjögren, J. Nord, M. Ek, P. Johansson, G. Liu and S. Geschwindner, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3806 CrossRef PubMed.
- (a) M. G. Chini, R. De Simone, I. Bruno, R. Riccio, F. Dehm, C. Weinigel, D. Barz, O. Werz and G. Bifulco, Eur. J. Med. Chem., 2012, 54, 311 CrossRef CAS PubMed; (b) R. De Simone, M. G. Chini, I. Bruno, R. Riccio, D. Mueller, O. Werz and G. Bifulco, J. Med. Chem., 2011, 54, 1565 CrossRef CAS PubMed.
- P. J. Holm, P. Bhakat, C. Jegerschold, N. Gyobu, K. Mitsuoka, Y. Fujiyoshi, R. Morgenstern and H. Hebert, J. Mol. Biol., 2006, 360, 934 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363 CrossRef CAS PubMed.
- D. Li, N. Howe, A. Dukkipati, S. T. A. Shah, B. D. Bax, C. Edge, A. Bridges, P. Hardwicke, O. M. P. Singh, G. Giblin, A. Pautsch, R. Pfau, G. Schnapp, M. Wang, V. Olieric and M. Caffrey, Cryst. Growth Des., 2014, 14, 2034 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Computational details and synthetic and biological procedures. See DOI: 10.1039/c4md00319e |
| ‡ These authors contributed equally to this work. |
| § Current address: Purdue University Center for Cancer Research, 1203 West State Street, West Lafayette, Indiana, 47907. |
| This journal is © The Royal Society of Chemistry 2015 |