
Computationally motivated synthesis and enzyme kinetic evaluation of N-(β-D-glucopyranosyl)-1,2,4-triazolecarboxamides as glycogen phosphorylase inhibitors†
Jaida Begum a, Gergely Varga b, Tibor Docsa c, Pál Gergely c, Joseph M. Hayes *a, László Juhász *b and László Somsák *b
aCentre for Materials Science, Division of Chemistry, University of Central Lancashire, Preston PR1 2HE, UK. E-mail: jhayes@uclan.ac.uk; Fax: +44 (0)172894981; Tel: +44 (0)172894334
bDepartment of Organic Chemistry, University of Debrecen, POB 20, H-4010 Debrecen, Hungary. E-mail: juhasz.laszlo@science.unideb.hu; somsak.laszlo@science.unideb.hu; Fax: +36 52512744; Tel: +36 52512900 ext. 22474 Tel: +36 52512900 ext. 22348
cDepartment of Medical Chemistry, Medical and Health Science Centre, University of Debrecen, Egyetem tér 1, H-4032 Debrecen, Hungary
First published on 15th September 2014
Abstract
Following our recent study of N-(β-D-glucopyranosyl)oxadiazolecarboxamides (Polyák et al., Biorg. Med. Chem. 2013, 21, 5738) revealed as moderate inhibitors of glycogen phosphorylase (GP), in silico docking calculations using Glide have been performed on N-(β-D-glucopyranosyl)-1,2,4-triazolecarboxamides with different aryl substituents predicting more favorable binding at GP. The ligands were subsequently synthesized in moderate yields using N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-tetrazole-5-carboxamide as starting material. Kinetics experiments against rabbit muscle glycogen phosphorylase b (RMGPb) revealed the ligands to be low μM GP inhibitors; the phenyl analogue (Ki = 1 μM) is one of the most potent N-(β-D-glucopyranosyl)-heteroaryl-carboxamide-type inhibitors of the GP catalytic site discovered to date. Based on QM and QM/MM calculations, the potency of the ligands is predicted to arise from favorable intra- and intermolecular hydrogen bonds formed by the most stable solution phase tautomeric (t2) state of the 1,2,4-triazole in a conformationally dynamic system. ADMET property predictions revealed the compounds to have promising pharmacokinetic properties without any toxicity. This study highlights the benefits of a computationally led approach to GP inhibitor design.
Introduction
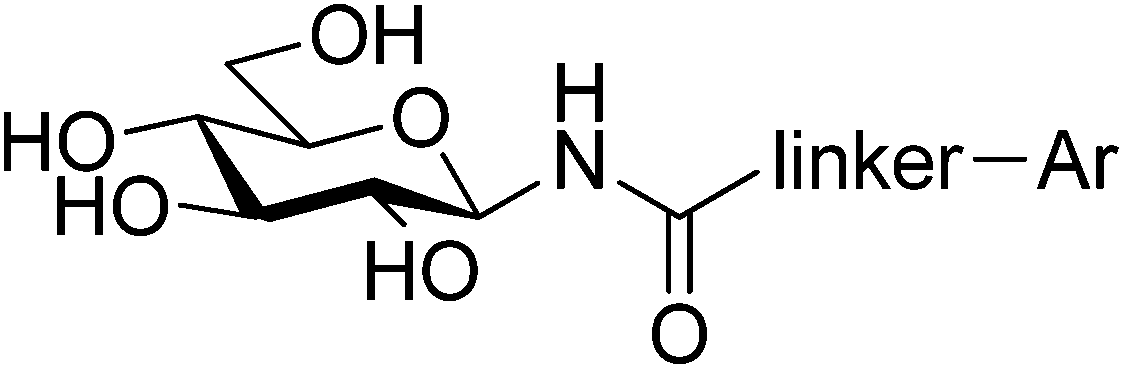
Type 2 diabetes mellitus (T2DM) is characterized by hyperglycemia and peripheral insulin resistance, and represents more than 90% of all diabetic cases. The significant increase in the global incidence of diabetes is a major cause for concern, having doubled over the previous 3 decades to 347 million people in 2008.1 Without adequate control of blood glucose levels, T2DM has several long term complications such as neuropathy and nephropathy, as well as an increased risk of blindness and cardiovascular disease.2 Glycogen phosphorylase (GP; EC 2.4.1.1) is a validated target for T2DM having a direct influence on blood glucose levels through the glycogenolysis pathway.3 It is an allosteric enzyme with a number of different binding sites,4,5 the catalytic, allosteric, new allosteric, inhibitor, glycogen storage, benzimiadazole6 and the very recently identified quercetin binding site.7 Significant efforts have been afforded to the design of GP inhibitors in recent years, with catalytic site inhibitors the most explored.4,8,9 The physiological inhibitor of GP is α-D-glucose (Ki = 1.7 mM), but β-substitutions at the anomeric carbon of D-glucose have led to the most effective GP catalytic site inhibitors and have demonstrated blood sugar lowering effects in vivo.10,11An evaluation of the current status of β-D-glucopyranosyl analogues as GP inhibitors can be found in recent reviews.4,8 The four most potent discovered to date are shown in Fig. 1, all of which possess a 2-naphthyl moiety exploiting favorable interaction of aryl groups in the catalytic β-cavity, a pocket lined by both polar and non-polar groups. In general, the 2-naphthyl moiety has proved the most effective in terms of potency, but our recent studies on N-(β-D-glucopyranosyl)-oxadiazole-carboxamides with linkers 2–4 (Table 1) have revealed that this is not always the case.16 However, these ligands only demonstrated moderate potency at best, 2b and 3c the most potent with Ki's ∼ 30 μM. The 1,2,4-triazole moiety in the form of 3-(β-D-glucopyranosyl)-5-substituted-1,2,4-triazoles has very recently revealed some of the most potent inhibitors of the GP catalytic site,13,14 with the 2-naphthyl derivative (1c in Fig. 1) the most potent. In the current work, we have investigated N-(β-D-glucopyranosyl)-3-substituted-1,2,4-triazole-5-carboxamides, replacing the oxadiazole moiety of linkers 2–4 with a 1,2,4-triazole (5, Table 1) in an attempt to improve ligand potency. Prior to undertaking synthesis, in silico calculations in the form of Glide docking, quantum mechanics (QM) and quantum mechanics/molecular mechanics (QM/MM) calculations were performed to probe the binding potential of these ligands at GP.
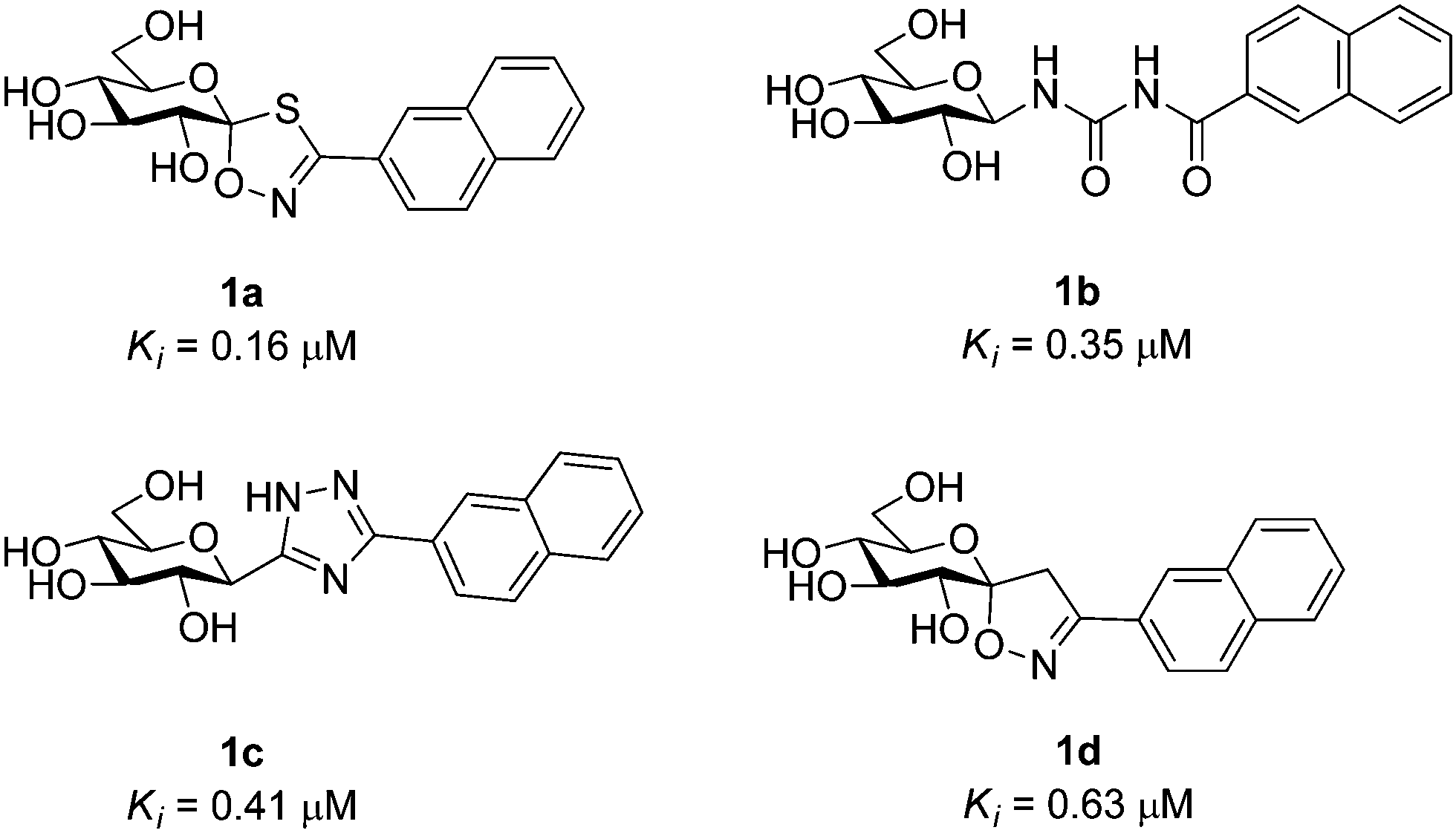 | ||
| Fig. 1 The four most potent glucose analogue inhibitors of GP discovered to date (1a,121b,51c13,14 and 1d15) together with their Ki's for RMGPb inhibition. All ligands possess a 2-naphthyl substituent exploiting favorable interactions in the GP catalytic site β-cavity. | ||
|
| Scaffold | Ar | ||
|---|---|---|---|---|
|
|
|
| ||
| Linker | a | b | c | |
| a No inhibition. b Calculated from the IC50 values by the Cheng–Prusoff equation: Ki = IC50/(1 + [S]/Km).17 | ||||
| Oxadiazoles | ||||
 | 2 | 545b | 30 | 172b |
| −92.31(−3.69) | −116.15(−4.01) | −112.17(−3.87) | ||
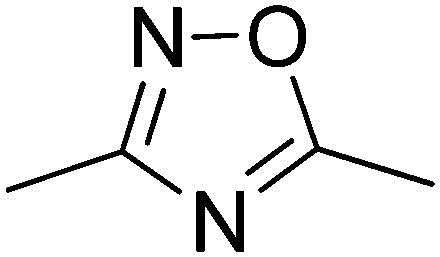 | 3 | 136b | N.I.a | 33 |
| −100.68(−4.03) | −115.78(−3.99) | −117.65(−4.06) | ||
 | 4 | 104 | N.I.a | 145b |
| −100.15(−4.01) | −111.92(−3.86) | −113.96(−3.93) | ||
| 1,2,4-Triazoles | ||||
 | 5 | 1 | 9.2 | — |
| −102.34(−4.09) | −116.52(−4.02) | −120.43(−4.15) | ||

Results & discussion
In silico binding studies
All ligands in Table 1 were prepared for calculations using Maestro and the LigPrep 2.5 program18 generating minimized favorable tautomeric and ionization states of all ligands at pH = 7 ± 2. All ligands were assigned as neutral, but three tautomeric forms (t1, t2 and t3) of the 1,2,4-triazole (linker 5) were produced, with t1 and t2 the most probable based on LigPrep with Epik (Table 2).18 Monte Carlo conformational searches on models of ligands 5 and its three tautomers t1–t3 were performed followed by Jaguar 8.0 density functional theory (DFT) with M06-2X19 and the 6-31+G* basis set20,21(M06-2X/6-31+G*), and yielded the conformations shown in Table 2. Higher level M06-2X/cc-pVTZ++22 single point energy (SPE) calculations in gas and solution phase at these geometries revealed that while t1 is the most favored in the gas phase by ∼6 kcal mol−1, t2 is clearly preferred in solution (by ∼9 kcal mol−1). This is in contrast to the tautomeric probabilities from LigPrep/Epik, highlighting the value of a QM approach to such analysis. Tautomer t2 also allows for approximately two equally stable conformations (ω = 0° or 180°) in the free unbound solvated state, as defined by the dihedral angle ω defined in Table 2. The molecular electrostatic potentials (MESPs) are shown in Fig. 2. For tautomer t2, the two different conformations have little effect on the ESPs of the amide. However, the ESP in the space around a gas phase molecule is a key factor in determining its ability to accept a proton; in this regard, for t2 (ω = 180°) the maximum ESP of the triazole NH nitrogen (62.55 kcal mol−1) and minimum ESP of the connected N (−50.84 kcal mol−1) is consistent with H+ migration to form the most stable t1 tautomer in the gas phase (Table 2). The intra-molecular NH (amide) and triazole N (lone pair) contacts for the tautomers can only loosely be classified as hydrogen bonds under the current IUPAC guidelines23 (donor angles are ∼103–105°, hence less than the preferred >110°), however, there is evidence of charge transfer in the case of t1 and t2 tautomers where the magnitudes of the potential at the N are less ∼−15 kcal mol−1. In any case, favourable electrostatic stabilization occurs between these atoms and the contacts are present in all free state conformations. The conformational flexibility and MESP of t2 compared to the other tautomers become important factors when we consider the binding at GPb.| Tautomer | Dihedral angle ωb (°) | LigPrep tautomer probability | Gas phasec | Solution phasec,d | |
|---|---|---|---|---|---|
| M06-2X/6-31+G* | M06-2X/cc-pVTZ++ | M06-2X/cc-pVTZ++ | |||
| a Tautomeric states were determined using LigPrep.18 MacroModel 9.9 18 conformational searches were used to locate the above favorable conformations of the compounds, which were then used in the QM optimizations. The dihedral angle ω adopted by the atoms 1, 2, 3, 4 for rotation around the 2,3 bond is key to the conformational properties. b Values for the minima from the M06-2X/6-31+G* optimizations. c Single point energy calculations at the optimized M06-2X/6-31+G* geometries. d Continuum treatment of solvation which involved accurate numerical solution of the Poisson–Boltzmann (PBF) equation.24,25 e Relative Gibbs free energies given in parentheses. | |||||
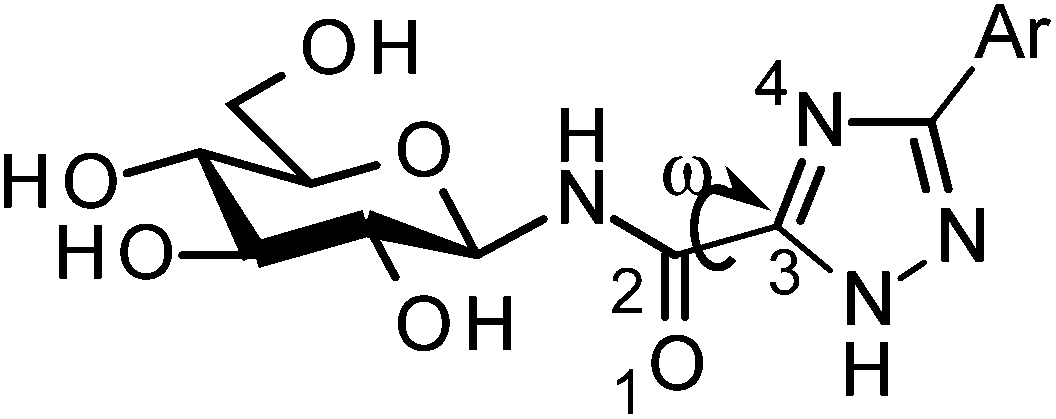 t1 t1 | 180 | 0.487 | 0.0(0.0)e | 0.0 | 8.6 |
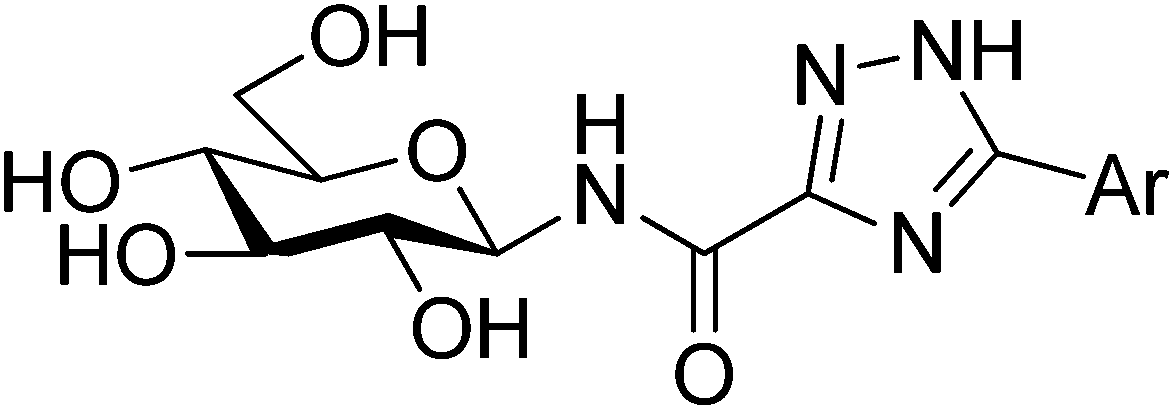 t2 t2 | 0 | 0.487 | 6.5(7.2)e | 5.9 | 0.5 |
| 180 | 5.9(6.9)e | 5.3 | 0.0 | ||
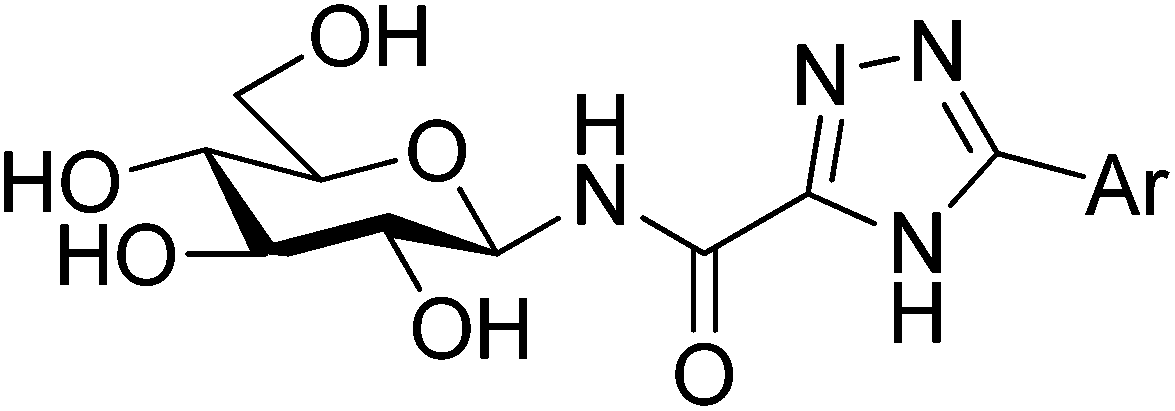 t3 t3 | 0 | 0.027 | 5.9(6.4)e | 5.5 | 9.0 |
 | ||
| Fig. 2 Molecular electrostatic potential (MESPs) can be used to provide insight into a number of hydrogen bonding phenomena. Shown are the results of MESP calculations (M06-2X/6-31+G*) on a model of the isolated 5a ligand (Me groups instead of glucopyranose) in its different tautomeric forms and most stable conformations (Table 2). The electrostatic potentials are mapped onto the electron density surface for each molecule. Only the ESP range of −50 to 50 kcal mol−1 is used for a better visual demonstration of the tautomeric effects; actual minimum–maximum ESP values were −55.4 to 63.7 kcal mol−1 obtained for the t2 tautomer. The minimum or maximum local electrostatic potential values (kcal mol−1) for the relevant atoms are also shown. | ||
Docking calculations were performed using Glide 5.8 in extra-precision (XP) mode,18,26 a docking algorithm which has proved effective in previous GPb catalytic site studies.15,27,28 The predictive capability of Glide-XP in the current study was measured by the ability of the algorithm to reproduce the trends in the kinetics results for the N-(β-D-glucopyranosyl)-oxadiazole-carboxamides with linkers 2–4. The results are shown in Table 1, with the most consistent results obtained using the Emodel scoring function which combines GlideScore, the non-bonded interaction energy and the excess internal energy of the generated ligand conformation. Non-bonded interactions in a scoring function are typically calculated as a sum of pair-wise interactions. Hence, a larger compound often receives a more favorable score than a smaller compound. In order to overcome this bias, a number of different normalization and scaling approaches have been proposed.29,30 Given that a size-dependent trend appeared to occur for our training set ligands, we used the following equation to account for the different sizes of our aryl substituents (phenyl versus naphthyl):
| Emodelnorm = Emodel/number of heavy atoms | (1) |
On initial inspection of Table 1, the predictive capability of the docking does not appear obvious. For example, the score for 3b is clearly over-estimated. However, a reasonable correlation R2 = 0.67 between predicted (Emodelnorm) and experimental (ln Ki) binding strengths was obtained. Further, the most favorable aryl group for each of the linkers 2–4 was correctly predicted: 2-naphthyl (2b) in the case of 2, 1-naphthyl (3c) in the case of 3 (1-naphthyl) and phenyl (4a) in the case of 4. In terms of our key objective, to accurately predict the potential of linker 5, we noted that the most favorable linker (from 2–4) for each aryl group was also correctly identified: linker 2 in the case of 2-naphthyl (Ki = 30 μM), linker 3 for 1-naphthyl (Ki = 33 μM), and for phenyl the linkers 3 and 4 were predicted to have similar efficiency in line with kinetics (Ki's = 104–136 μM). Encouraged by this, we then noted that for each of the phenyl, 1- and 2-naphthyl substituents, linker 5 was predicted to give rise to more potent ligands.
Despite their obvious importance, tautomeric states are still often not accurately considered in computer-aided drug design efforts.31 The most favorable tautomeric state for binding of ligands 5a–5c based on Emodel docking scores was t2 for both 5a and 5c, and t1 for 5b (Table 3). The potential of using the lowest QM/MM complex energy for protein–ligand docking pose selection has recently been highlighted, where QM/MM energies were used to re-rank docking poses and compared with their native crystallographic binding modes.32 Accordingly, to more accurately probe the binding of 5a–5c and its different tautomers at GP, QM/MM optimizations with QSite 5.8 (ref. 18) were performed on the Glide docking poses (Table 3). M06-2X/6-31+G* was used for the QM region (the ligands), while the GPb protein was described using MM with the OPLS-AA(2005) forcefield,33 with otherwise default options. Solution phase energies at the optimum geometries were then obtained, employing a self-consistent reaction field (SCRF) continuum treatment for water solvation effects and which involved accurate numerical solution of the Poisson–Boltzmann (PBF) equation (M06-2X/6-31+G* + PBF).24,25 The results of the QM/MM calculations (Table 3) revealed the most stable free state solution phase t2 tautomer of 5a–5c to also form the most favorable complexes with GPb, in both gas and solution phases. From the MESPs (Fig. 2) also, the t2 maximum and minimum atomic ESPs of the triazole NH nitrogen and amide CO oxygen (both conformations) are indicative of greater H-bond acceptor and donor capabilities,34,35 respectively, for binding at GPb compared to the other tautomers, although the value of ESP maxima on molecular surfaces for predicting hydrogen bond acidity has recently been questioned.36
| Ligand | Tautomera | Emodel (Emodelnorm)b | ω dihedral anglec (°) | QM/MM relative energies | |
|---|---|---|---|---|---|
| Gas phase | Solution phase | ||||
| a c.f. Table 2. b Normalized values as calculated using eqn (1). c Numbering scheme for dihedral angle ω as shown in Table 2. | |||||
| 5a | t1 | −99.8 (−3.99) | 34.3 | 16.6 | 18.1 |
| t1 | −93.9 (−3.76) | 178.0 | 7.7 | 19.6 | |
| t2 | −102.3 (−4.09) | 19.7 | 3.8 | 0.0 | |
| t2 | −97.1 (−3.88) | −176.7 | 0.0 | 10.7 | |
| t3 | −91.0 (−3.64) | 3.5 | 14.5 | 13.3 | |
| 5b | t1 | −116.5 (−4.02) | 33.3 | 14.5 | 15.8 |
| t2 | −115.4 (−3.98) | −10.1 | 0.0 | 0.0 | |
| t3 | −115.9 (−4.00) | 3.2 | 13.0 | 8.9 | |
| 5c | t1 | −114.3 (−3.94) | 16.2 | 18.9 | 16.5 |
| t2 | −120.4 (−4.15) | 43.3 | 6.5 | 1.3 | |
| t2 | −114.6 (−3.95) | −10.7 | 0.0 | 0.0 | |
| t3 | −112.2 (−3.87) | −0.5 | 13.8 | 13.1 | |
| t3 | −110.5 (−3.81) | −154.9 | 11.8 | 6.4 | |
For the GPb–5a complex (Fig. 3), as well as forming the standard hydrogen bonding interactions from the β-D-glucopyranosyl hydroxyl groups and a hydrogen bond from the carboxamide carbonyl O with the Leu136 backbone NH, through rotation around the dihedral angle ω the 1,2,4-triazole in its favored solution phase t2 tautomeric form has the potential to hydrogen bond with the Asp339 carboxylate on one side of the β-cavity (water-bridged), and on the other side directly with the Asp283 backbone O (ω ∼ 50°). If ω ∼ 0° or ω ∼ ±180°, although the 1,2,4-triazole NH forms no direct hydrogen bonds with GPb, there are favorable intra-molecular interactions from the carboxamide NH with the lone pair of electrons on a triazole N atom for both conformations, as mentioned earlier for the free state ligand (Fig. 2). The smaller Ar group (phenyl) of 5a also permits a flipped 1,2,4-triazole conformation (ω = −176.7°) shown in Fig. 4, where the triazole NH is a little longer than hydrogen bonding distance from the Glu88 sidechain carboxylate (distance ∼ 3.3 Å). Hence, 5a in its t2 tautomeric state exhibits considerable conformational flexibility while bound at GPb (favorable in terms of entropy). The 1,2,4-triazole has either intra- and intermolecular hydrogen bonding potential in these different conformations; while CH–π interactions from the Asp283 CH to the ligand Ar (phenyl) group can further stabilize the GPb–5a complex (Fig. 3). For the binding of ligand 5b (Fig. 5), interactions are similar to that as described for 5a in Fig. 3, except the 2-naphthyl group extends deeper into the β-cavity. However, the larger size of this Ar group limits somewhat the conformational versatility (ω dihedral angle) of the ligand, and it has much fewer poses compared to either 5a and 5c (Table 3). For 5c, the orientation of the 1-naphthyl allows for greater conformational flexibility in the GPb binding site compared to its 2-naphthyl equivalent, and indeed the t3 tautomer is able to adopt a pose (ω = −154.9°; Table 3) similar to that shown for 5a in Fig. 3, with a flip in the 1,2,4-triazole.
 | ||
| Fig. 3 The lowest solution phase energy pose of ligand 5a (tautomer t2) bound at GPb with a dihedral angle (ω = 19.7°; Table 3), as calculated using QM/MM and described in the text. | ||
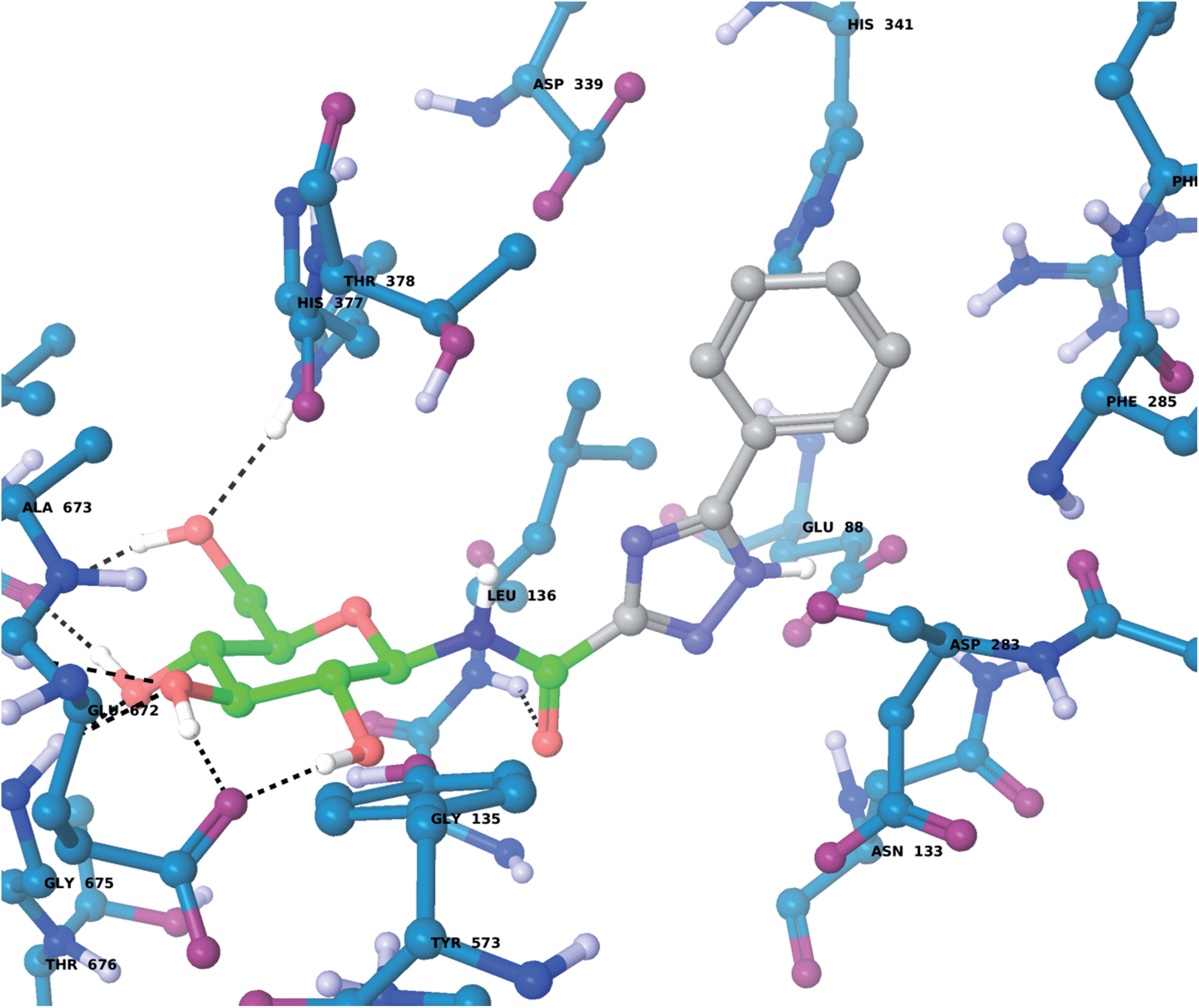 | ||
| Fig. 4 The flipped 1,2,4-triazole binding conformation of tautomer t2 of 5a at GPb (dihedral angle ω = −176.7°; Table 3), as calculated using QM/MM and described in the text. The tautomeric t2 state allows for favorable intra-molecular hydrogen bond contacts between the carboxamide NH and a 1,2,4-triazole N when ω is close to 0° (Fig. 2) or flipped (above). In the flipped conformation, the 1,2,4-triazole NH is close to hydrogen bonding distance with the Glu88 carboxylate. | ||
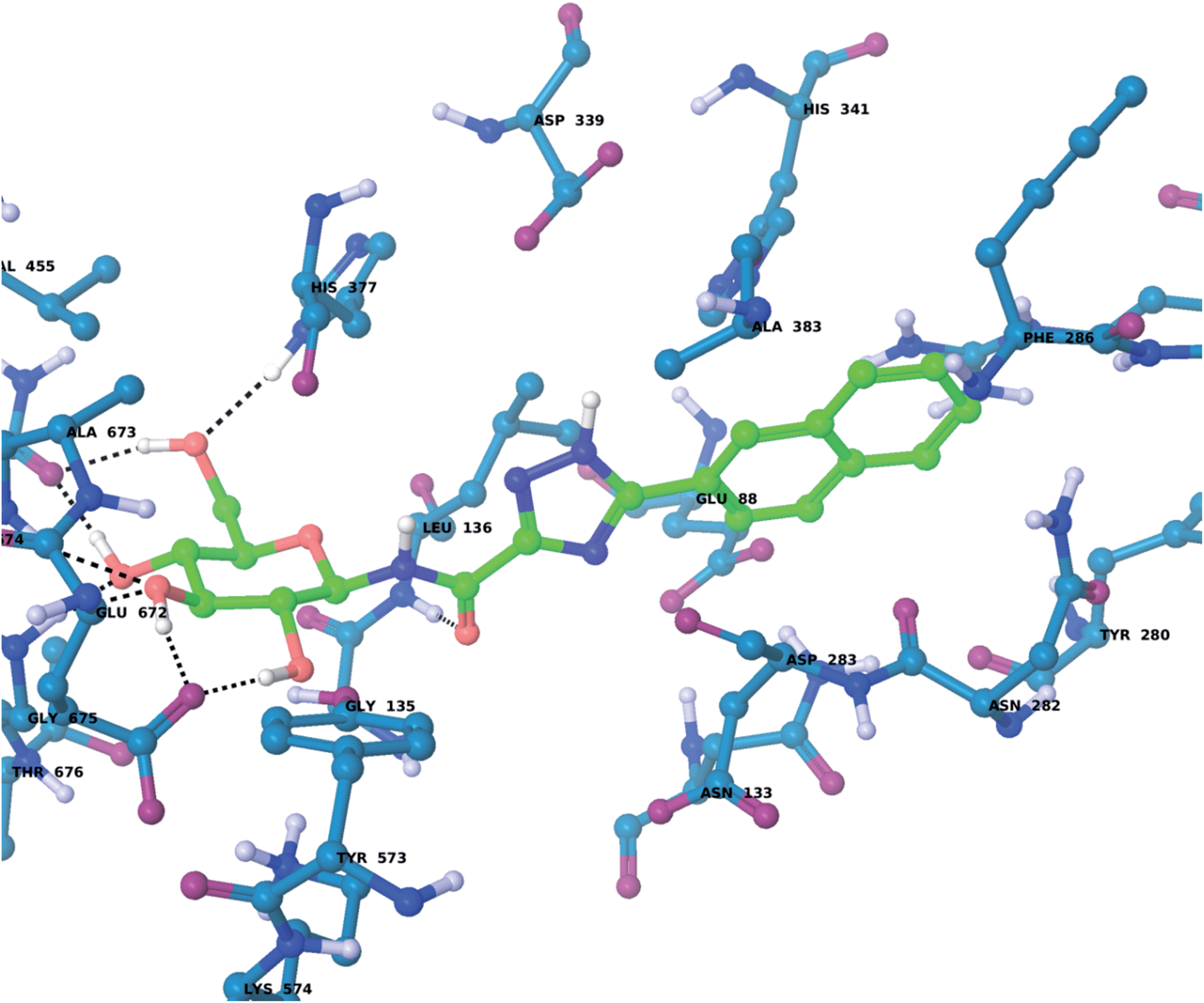 | ||
| Fig. 5 The lowest solution phase energy pose of ligand 5b (tautomer t2) bound at GPb (dihedral angle ω = −10.1°; Table 3), calculated using QM/MM as described in the text. | ||
Synthesis
Based on the computational results, synthesis of the ligands was considered worthwhile. Several methods have been published for the preparation of 3,5-disubstituted 1,2,4-triazoles.37 While acyl-amidrazones (prepared from amides, thioamides, nitriles, or amidines) could be transformed into the desired triazoles under thermal conditions,37–39 the cycloaddition of nitriles with nitrile-imines or nitrile-iminium ions gave 2,3,5-trisubstituted 1,2,4-triazoles in moderate to good yields.40–43 The reaction of 5-substituted tetrazoles with imidoyl chlorides (easily obtained from the corresponding amides by SOCl2) gave 3,4,5-trisubstituted 1,2,4-triazoles in good yields.44,45This latter method was extensively applied for the preparation of 3-(C-glycosyl)-5-substituted-1,2,4-triazoles in our group.13 The predicted target compounds of this study (5a,b) have been prepared using this methodology starting from tetrazole 6 16 as shown in Scheme 1. The imidoyl chlorides 10a,b prepared from N-benzyl-amides 9a,b as described earlier,13 were reacted with tetrazole 6 (used without any purification) to give the corresponding 4-benzyl-N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-5-aryl-1,2,4-triazole-3-carboxamides 11a,b in moderate yields. In the case of the 1-naphthyl derivative 5c, the expected formation of 11c from 10c was unsuccessful, no transformation was detected and the desired compound has not been synthesized yet using any other synthetic methods.
 | ||
| Scheme 1 Synthesis of the predicted molecules 5a and 5b. | ||
The protecting groups were cleaved in a two-steps procedure. The N-benzyl group of 11a,b was removed first by catalytic hydrogenation in excellent yields (94 and 85%). Subsequently, O-deacetylation was effected by the Zemplén method to give fully deprotected N-(β-D-glucopyranosyl)-5-aryl-1,2,4-triazole-3-carboxamides 5a,b.
Enzyme kinetics
The inhibition constants (Ki) against rabbit muscle glycogen phosphorylase b (RMGPb) of compounds 5a,b were determined according to the protocol described earlier.46 The results are summarized in Table 1 together with the Ki's of 1,2,4- and 1,3,4-oxadiazole-carboxamide type inhibitors. As the data show, the 1,2,4-triazole analogues 5a (Ki = 1 μM) and 5b (Ki = 9.2 μM) are more potent inhibitors than the oxadiazole analogues, consistent with our computational predictions for favorable binding at GPb.Pharmacokinetic predictions
The early evaluation of the pharmacokinetic properties of lead compounds in drug design efforts is desirable due to the potential for failure in late stage clinical trials. Accordingly, the absorption, distribution, metabolism and excretion (ADME) properties of 5a–5c were predicted using the QikProp 3.5 program in normal mode.18 Toxicity is the leading cause of drug attrition in clinical trials, together with lack of efficacy.47 The FAF-Drugs2 server was used to extract any potential toxicity structural warnings.48 Results are shown in Table 4.| Inhibitor/tautomer | Lipinski's rule of five and violations (V)b | V | Jorgensen's rule of three and violations (V)b | V | PSA [Å2]c (<140 Å2) | log Khsad | log BBe | TSWf | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M r [Da] (<500) | HBDg (≤5) | HBAh (≤10) | log P(o/w) (<5) | Caco-2i [nm s−1] (>22) | log S (>−5.7) | NMPj (<7) | |||||||
| a ADMET data calculated using Qikprop 3.5;18 predicted properties outside the range for 95% of known drugs indicated with an asterisk (*). b Rules as listed in the columns, with any violations of the rules highlighted in italics. c PSA represents the van der Waals (polar) surface areas of N and O atoms; recommended PSA < 140 Å2 according to Veber et al.54 d log Khsa: predicted binding to human serum albumin. e log BB: predicted blood-brain barrier coefficient. f Toxicity structural warnings from FAF-Drugs2.48 g Number of hydrogen bond donors. h Number of hydrogen bond acceptors. i Caco-2 cell permeability. j Number of primary metabolites. k Range for 95% of known drugs.18 | |||||||||||||
| 5a | |||||||||||||
| t1 | 350.3 | 6 | 10 | −1.6 | 1 | 19.2* | −2.7 | 5 | 1 | 173.3 | −1.02 | −2.8 | — |
| t2 | 350.3 | 6 | 10 | −1.1 | 1 | 19.2* | −2.8 | 5 | 1 | 173.3 | −0.93 | −2.8 | — |
| t3 | 350.3 | 6 | 10 | −1.4 | 1 | 18.9* | −2.8 | 5 | 1 | 173.4 | −0.97 | −2.8 | — |
| 5b | |||||||||||||
| t1 | 400.4 | 6 | 10 | −0.8 | 1 | 19.2* | −3.4 | 5 | 1 | 173.3 | −0.85 | −2.9 | — |
| t2 | 400.4 | 6 | 10 | −0.3 | 1 | 19.2* | −3.6 | 5 | 1 | 173.2 | −0.72 | −2.9 | — |
| t3 | 400.4 | 6 | 10 | −0.6 | 1 | 19.0* | −3.6 | 5 | 1 | 173.4 | −0.80 | −2.9 | — |
| 5c | |||||||||||||
| t1 | 400.4 | 6 | 10 | −0.8 | 1 | 18.2* | −3.4 | 5 | 1 | 174.7 | −0.85 | −2.9 | — |
| t2 | 400.4 | 6 | 10 | −0.4 | 1 | 18.8* | −3.6 | 5 | 1 | 174.2 | −0.72 | −2.9 | — |
| t3 | 400.4 | 6 | 10 | −0.7 | 1 | 18.3* | −3.5 | 5 | 1 | 174.6 | −0.80 | −2.9 | — |
| Rangek | 130–725 | 0–6 | 2–20 | −2.0–6.5 | — | <25 poor | −6.5–0.5 | 1–8 | — | 7–200 | −1.5–1.5 | −3.0–1.2 | — |
| >500 great | |||||||||||||
An orally active drug should have no more than one violation of Lipinski's ‘rule of five’,49 while for Jorgensen's ‘rule of three’50,51 more drug-like molecules have fewer violations. The N-(β-D-glucopyranosyl)-1,2,4-triazolecarboxamides 5a–5c had only one violation of each. The Lipinski violation is due to too many hydrogen bond donors (HBDs), 6 instead of ≤5 in each case, but this is still within the range for 95% of known drugs and consistent with some recent ‘beyond rule of five’ studies.52,53 In terms of Jorgensen's rules, the Caco-2 cell permeability (>22 nm s−1) is violated for the ligands (∼19 nm s−1), where previously we noted that the sensitive balance between adequate lipophilicity and solubility may need attention in lead optimization of heterocyclic derivatives conjugated to glucose.16 Meanwhile, the log Khsa's (degree of human serum albumin affecting bioavailability) is ∼−0.9 to −0.7 and within the range for 95% of known drugs (−1.5 to 1.5), while the log BB (blood-brain barrier coefficients) values (−2.9 to −2.8) are also within the desirable range (−3.0 to 1.2). Importantly, there were no toxicity structural warnings for the ligands from FAF-Drugs2. Overall, the pharmacokinetic results are similar with the results for the N-(β-D-glucopyranosyl)-oxadiazole-carboxamides previously reported.16 However, in terms of pharmacodynamics the 5a,b ligands proved via kinetics experiments to be much more potent inhibitors of GP.
Experimental
Computational details (additional)
Synthesis
The general procedures employed for the synthesis are described in the ESI,† together with the NMR data.GP inhibition assay
Glycogen phosphorylase b was prepared from rabbit skeletal muscle according to the method of Fischer and Krebs56 using 2-mercaptoethanol instead of L-cysteine, and recrystallized at least three times before use. The kinetic studies with glycogen phosphorylase were performed as described previously.46 Kinetic data for the inhibition of rabbit skeletal muscle glycogen phosphorylase by monosaccharide compounds were collected using different concentrations of α-D-glucose-1-phosphate (4, 6, 8, 10, 12 and 14 mM) and constant concentrations of glycogen (1% w/v) and AMP (1 mM). The enzymatic activities were presented in the form of double-reciprocal plots (Lineweaver–Burk) applying a nonlinear data-analysis programme. The inhibitor constants (Ki) were determined by Dixon plots, by replotting the slopes from the Lineweaver–Burk plots against the inhibitor concentrations. The means of standard errors for all calculated kinetic parameters averaged to less than 10%.3,57 IC50 values were determined in the presence of 4 mM glucose 1-phosphate, 1 mM AMP, 1% glycogen, and varying concentrations of an inhibitor.Conclusions
Molecular modeling investigations in the form of docking, QM and QM/MM studies has motivated the experimental evaluation of N-(β-D-glucopyranosyl)-1,2,4-triazolecarboxamides as GP inhibitors. The value of QM calculations to determine favorable tautomeric states is highlighted, while QM/MM optimizations were used to decipher the more likely binding interactions in the absence of time consuming X-ray crystallographic evidence. While there is often uncertainty in assigning hydrogen positions using crystallography,58 QM/MM calculations allowed us to accurately consider the binding potential of the different 5a-5c 1,2,4-triazole tautomers, with the GPb interactions formed by the most stable t2 tautomer and its conformational flexibility deemed significant. Synthesis, followed by kinetics experiments revealed 5a,b as low μM inhibitors of GP, with 5a the in the top 10 most potent catalytic site inhibitor discovered to date.4 The N-(β-D-glucopyranosyl)-1,2,4-triazolecarboxamides are predicted to have drug-like potential, but with permeability a potential issue to efficacy. While intra-molecular bonding has the potential to improve membrane permeability by reducing the polar surface areas,52 this effect is likely to be minimal in our case. However, glucose analogues have already demonstrated blood glucose lowering effects in vivo,10,11 a large number of triazole compounds are found as clinical drugs or candidates for treatment of a range of diseases,59 so that we consider the ligands studied worthy candidates for further optimization studies. Finally, the value of a computationally lead approach to GP inhibitor design has been highlighted in this work.27Acknowledgements
This work was supported by the Hungarian Scientific Research Fund (OTKA 77712, 109450), TÁMOP-4.2.2.-08/1/2008-0014, TÁMOP-4.2.2./B-10/1-2010-0024, and BAROSS REG_EA_09-1-2009-0028-LCMS_TAN projects implemented through the New Hungary Development Plan, co-financed by the European Social Fund, and Bolyai János Research Fellowships of the Hungarian Academy of Sciences (to L. J. and T. D.). T. D. credits research support from the University of Debrecen (5N5X 1IJ0 KUDT 320). J.B. and J.M.H acknowledge the University of Central Lancashire URIS scheme.References
- G. Danaei, M. M. Finucane, Y. Lu, G. M. Singh, M. J. Cowan, C. J. Paciorek, J. K. Lin, F. Farzadfar, Y.-H. Khang, G. A. Stevens, M. Rao, M. K. Ali, L. M. Riley, C. A. Robinson, M. Ezzati and C. Global Burden, Metab Risk Factors, Lancet, 2011, 378, 31–40 CrossRef CAS.
- M. Brownlee, Nature, 2001, 414, 813–820 CrossRef CAS PubMed.
- N. G. Oikonomakos, Curr. Protein Pept. Sci., 2002, 3, 561–586 CrossRef CAS.
- J. M. Hayes, A. L. Kantsadi and D. D. Leonidas, Phytochem. Rev., 2014, 13, 471–498 CrossRef CAS.
- L. Somsák, K. Czifrák, M. Tóth, É. Bokor, E. D. Chrysina, K. M. Alexacou, J. M. Hayes, C. Tiraidis, E. Lazoura, D. D. Leonidas, S. E. Zographos and N. G. Oikonomakos, Curr. Med. Chem., 2008, 15, 2933–2983 CrossRef.
- E. D. Chrysina, M. N. Kosmopolou, C. Tiraidis, R. Kardarakis, N. Bischler, D. D. Leonidas, Z. Hadady, L. Somsák, T. Docsa, P. Gergely and N. G. Oikonomakos, Protein Sci., 2005, 14, 873–888 CrossRef CAS PubMed.
- A. L. Kantsadi, A. Apostolou, S. Theofanous, G. A. Stravodimos, E. Kyriakis, V. A. Gorgogietas, D. S. M. Chatzileontiadou, K. Pegiou, V. T. Skamnaki, D. Stagos, D. Kouretas, A.-M. G. Psarra, S. A. Haroutounian and D. D. Leonidas, Food Chem. Toxicol., 2014, 67, 35–43 CrossRef CAS PubMed.
- L. Somsák, É. Bokor, K. Czifrák, L. Juhász and M. Tóth, in Topics in the Prevention, Treatment and Complications of Type 2 Diabetes, ed. M. B. Zimering, InTech Open Access Publisher, Rijeka, 2011, pp. 103–126 Search PubMed.
- L. Somsák, C. R. Chim., 2011, 14, 211–223 CrossRef PubMed.
- T. Docsa, K. Czifrák, C. Hüse, L. Somsák and P. Gergely, Mol. Med. Rep., 2011, 4, 477–481 CAS.
- L. Nagy, T. Docsa, A. Brunyánszki, M. Szántó, C. Hegedős, J. Marton, B. Kónya, L. Virág, L. Somsák, P. Gergely and P. Bai, PLoS One, 2013, 8, e69420 CAS.
- V. Nagy, K. Czifrák, A. Bényei and L. Somsák, Carbohydr. Res., 2009, 344, 921–927 CrossRef CAS PubMed.
- S. Kun, É. Bokor, G. Varga, B. Szőcs, A. Páhi, K. Czifrák, M. Tóth, L. Juhász, T. Docsa, P. Gergely and L. Somsák, Eur. J. Med. Chem., 2014, 76, 567–579 CrossRef CAS PubMed.
- É. Bokor, T. Docsa, P. Gergely and L. Somsák, ACS Med. Chem. Lett., 2013, 4, 612–615 CrossRef PubMed.
- M. Benltifa, J. M. Hayes, S. Vidal, D. Gueyrard, P. G. Goekjian, J.-P. Praly, G. Kizilis, C. Tiraidis, K.-M. Alexacou, E. D. Chrysina, S. E. Zographos, D. D. Leonidas, G. Archontis and N. G. Oikonomakos, Bioorg. Med. Chem., 2009, 17, 7368–7380 CrossRef CAS PubMed.
- M. Polyák, G. Varga, B. Szilágyi, L. Juhász, T. Docsa, P. Gergely, J. Begum, J. M. Hayes and L. Somsák, Bioorg. Med. Chem., 2013, 21, 5738–5747 CrossRef PubMed.
- Y.-C. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS.
- Schrodinger, LLC, New York, NY, 2014 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS PubMed.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- D. J. Tannor, B. Marten, R. Murphy, R. A. Friesner, D. Sitkoff, A. Nicholls, M. Ringnalda, W. A. Goddard and B. Honig, J. Am. Chem. Soc., 1994, 116, 11875–11882 CrossRef CAS.
- B. Marten, K. Kim, C. Cortis, R. A. Friesner, R. B. Murphy, M. N. Ringnalda, D. Sitkoff and B. Honig, J. Phys. Chem., 1996, 100, 11775–11788 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- J. M. Hayes and D. D. Leonidas, Mini-Rev. Med. Chem., 2010, 10, 1156–1174 CrossRef CAS.
- A. L. Kantsadi, J. M. Hayes, S. Manta, V. T. Skamnaki, C. Kiritsis, A.-M. G. Psarra, Z. Koutsogiannis, A. Dimopoulou, S. Theofanous, N. Nikoleousakos, P. Zoumpoulakis, M. Kontou, G. Papadopoulos, S. E. Zographos, D. Komiotis and D. D. Leonidas, ChemMedChem, 2012, 7, 722–732 CrossRef CAS PubMed.
- G. Carta, A. J. S. Knox and D. G. Lloyd, J. Chem. Inf. Model., 2007, 47, 1564–1571 CrossRef CAS PubMed.
- S. Zhong, Y. Zhang and Z. Xiu, Curr. Opin. Drug Discovery Dev., 2010, 13, 326–334 CAS.
- P. Pospisil, P. Ballmer, L. Scapozza and G. Folkers, J. Recept. Signal Transduction, 2003, 23, 361–371 CrossRef CAS PubMed.
- A. E. Cho, J. Y. Chung, M. Kim and K. Park, J. Chem. Phys., 2009, 131(13), 134108 CrossRef PubMed.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- J. S. Murray, S. Ranganathan and P. Politzer, J. Org. Chem., 1991, 56, 3734–3737 CrossRef CAS.
- J. S. Murray and P. Politzer, J. Org. Chem., 1991, 56, 6715–6717 CrossRef CAS.
- P. W. Kenny, J. Chem. Inf. Model., 2009, 49, 1234–1244 CrossRef CAS PubMed.
- J. B. Polya, in Comprehensive Heterocyclic Chemistry, 1984, vol. 5, pp. 733–790 Search PubMed.
- K. T. Potts, J. Chem. Soc., Dalton Trans., 1954, 3461–3464 CAS.
- K. T. Potts, Chem. Rev., 1961, 61, 87–127 CrossRef CAS.
- R. Huisgen, J. Sauer and M. Seidel, Chem. Ber., 1961, 94, 2503–2509 CrossRef CAS.
- R. Huisgen, R. Grashey, M. Seidel, G. Wallbillich, H. Knupfer and R. Schmidt, Liebigs Ann. Chem., 1962, 653, 105–113 CrossRef CAS.
- S. Condi, C. Corral and R. Madronero, Synthesis, 1974, 28–29 CrossRef.
- G. Broggini, L. Bruche, L. Garanti and G. Zecchi, J. Chem. Soc., Perkin Trans. 1, 1994, 433–438 RSC.
- R. Huisgen, J. Sauer and M. Seidel, Chem. Ber., 1960, 93, 2885–2891 CrossRef CAS.
- G. I. Koldobskii and V. A. Ostrovskii, Russ. Chem. Rev., 1994, 63, 797 CrossRef PubMed.
- E. Ősz, L. Somsák, L. Szilágyi, L. Kovács, T. Docsa, B. Tóth and P. Gergely, Bioorg. Med. Chem. Lett., 1999, 9, 1385–1390 CrossRef.
- G. F. Smith, Prog. Med. Chem., 2011, 50, 1–47 CrossRef CAS PubMed.
- D. Lagorce, J. Maupetit, J. Baell, O. Sperandio, P. Tuffery, M. A. Miteva, H. Galons and B. O. Villoutreix, Bioinformatics, 2011, 27, 2018–2020 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- W. L. Jorgensen and E. M. Duffy, Bioorg. Med. Chem. Lett., 2000, 10, 1155–1158 CrossRef CAS.
- W. L. Jorgensen and E. M. Duffy, Adv. Drug Delivery Rev., 2002, 54, 355–366 CrossRef CAS.
- A. Alex, D. S. Millan, M. Perez, F. Wakenhut and G. A. Whitlock, MedChemComm, 2011, 2, 669–674 RSC.
- G. V. Paolini, R. H. B. Shapland, W. P. van Hoorn, J. S. Mason and A. L. Hopkins, Nat. Biotechnol., 2006, 24, 805–815 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- W. C. Still, A. Tempczyk, R. C. Hawley and T. Hendrickson, J. Am. Chem. Soc., 1990, 112, 6127–6129 CrossRef CAS.
- E. H. Fischer and E. G. Krebs, Methods Enzymol., 1962, 5, 369–372 CAS.
- L. Somsák, L. Kovács, M. Tóth, E. Ősz, L. Szilágyi, Z. Györgydeák, Z. Dinya, T. Docsa, B. Tóth and P. Gergely, J. Med. Chem., 2001, 44, 2843–2848 CrossRef PubMed.
- R. Lonsdale, J. N. Harvey and A. J. Mulholland, Chem. Soc. Rev., 2012, 41, 3025–3038 RSC.
- C. H. Zhou and Y. Wang, Curr. Med. Chem., 2012, 19, 239–280 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4md00335g |
| This journal is © The Royal Society of Chemistry 2015 |