
8-Hydroxyquinoline: a privileged structure with a broad-ranging pharmacological potential
Yu'ning
Song†
ac,
Hao
Xu†
b,
Wenmin
Chen
a,
Peng
Zhan
*a and
Xinyong
Liu
*a
aDepartment of Medicinal Chemistry, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, 44, West Culture Road, 250012, Jinan, Shandong, P. R. China. E-mail: zhanpeng1982@163.com; xinyongllab@163.com; Fax: +86-531-88382731; Tel: +86-531-88382005
bDepartment of Breast and Thyroid Surgery, Shandong Provincial Hospital Affiliated to Shandong University, 324 Jingwu-Weiqi Road, 250021, Jinan, Shandong, P. R. China
cDepartment of Pharmacology, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, 44, West Culture Road, 250012, Jinan, Shandong, P. R. China
First published on 15th October 2014
Abstract
Privileged structures can bind to a diverse range of targets with high affinities, thus benefiting the discovery of novel bioactive agents. 8-Hydroxyquinoline derivatives represent an important type of “privileged structure”, possessing a rich diversity of biological properties. Numerous encouraging investigations have demonstrated that this privileged structure should be further exploited for therapeutic applications in the future. In view of its predominance, and on the basis of our research interest in this scaffold, an updated and detailed account of the pharmacological properties of 8-hydroxyquinoline derivatives, as well as recent insights from structural biology, are described. Finally, some outlooks on current issues and future directions in this field of research are also provided.
1. Introduction
Privileged structures, as defined in the literature, are chemical scaffolds with versatile binding properties, which could afford potent and specific ligands for diverse biotargets via structural modifications of functional groups.1,2 Furthermore, privileged structures typically display favorable drug-like properties, which in turn provide more high-quality leads and compound libraries. No doubt, the identification of novel drug-like lead compounds is a solid base for research efforts aimed at the discovery of a new drug. Over the past two decades, the application of the privileged structure concept has emerged as a fertile strategy for overcoming shortcomings and improving the reliability of the bioactive compounds in drug discovery.1,2In the field of drug discovery and development, there is now a wealth of published papers on the identification of biologically important heterocyclic “privileged structures” as promising drugs or candidates.3–8 Quinoline has attracted considerable attention as an important heterocyclic pharmacophore, and it is amply explored for broad-ranging biological effects. Among the quinoline core compounds, the most frequently encountered in medicinal chemistry is 8-hydroxyquinoline (8-HQ), which is one of the most important strong metal ion chelator groups and represents an excellent scaffold with a wide spectrum of pharmacological applications such as iron-chelators for neuroprotection, anticancer agents, inhibitors of 2OG-dependent enzymes, chelators of metalloproteins, anti-HIV agents, antifungal agents, antileishmanial agents, antischistosomal agents, Mycobacterium tuberculosis inhibitors, botulinum neurotoxin inhibitors and many others (Fig. 1).
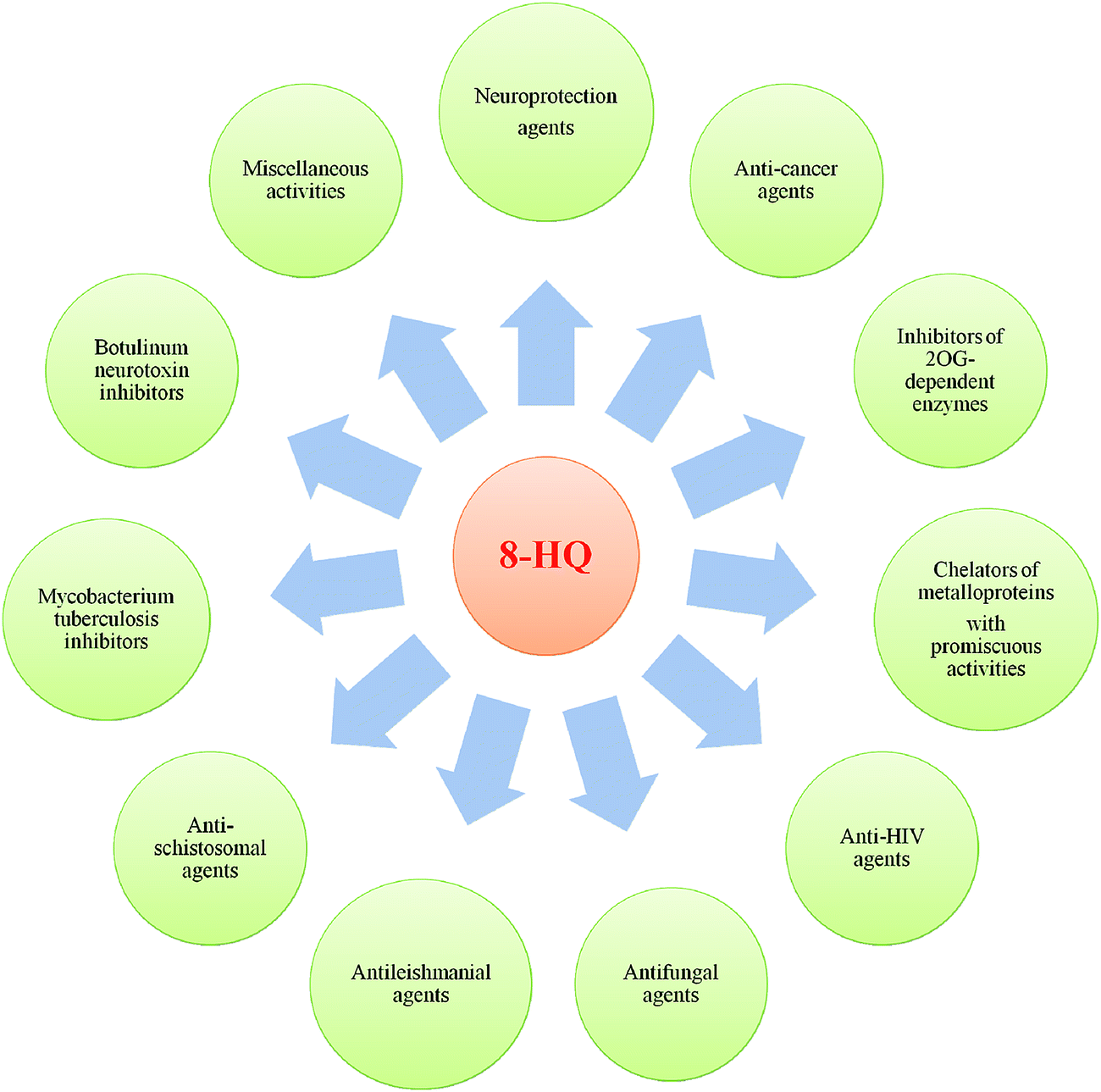 | ||
| Fig. 1 Overview of the broad-ranging pharmacological applications of 8-HQ derivatives. | ||
Hitherto, no comprehensive survey has been published to outline the wide spectrum of pharmaceutical activities of 8-HQ as a privileged heterocycle unit. Given this, and fascinated by the multifarious bioactivities of this heterocycle, we would like to comprehensively outline its pharmacological importance, which will provide insights for the further development of novel 8-HQ-based agents.
2. The broad-ranging pharmacological applications of 8-HQ derivatives
In this section, a broad range of pharmacological applications of 8-HQ derivatives are presented. It is hoped that the content and organization of this section will prove useful to medicinal chemists embarking on the exploitation of 8-hydroxyquinolines in academia and industry.2.1 Neuroprotection agents (as iron chelators)
Neurodegenerative disorder diseases, such as Alzheimer's and Parkinson's diseases (AD and PD), represent one of the principal unmet medical needs, being the third highest cause of death just behind cancer and heart disease.9 It was reported that metal-ion dysregulation and oxidative stress are involved in the progressive neurological decline (neurodegeneration) associated with neurodegenerative disorders.Several 8-HQ derivatives were reported as multifunctional metal-chelators (metal-protein attenuating compound, MPAC), acetylcholinesterase (AChE) and monoamine oxidase (MAO) inhibitors for the potential treatment of neurodegenerative disorders (Fig. 2). For instance, compound HLA20 (1), displays strong iron(III)-chelating, high antioxidant properties, favorable permeability into K562 cells and good selective MAO-B inhibition (IC50 = 110 μM), and exhibited the highest protective activity against differentiated P19 cell death induced by 6-hydroxydopamine. It was suggested that compound 1 also functions as a radical scavenger to scavenge hydroxyl radicals (OH˙) directly.10
 | ||
| Fig. 2 Structures of metal chelators with potential for the treatment of neurodegenerative diseases. | ||
VK28 (2) is a brain-permeable iron chelator with neuroprotection against 6-hydroxydopamine lesions in rats.11,12 From prototype VK28 (2), M30 (3) was identified as a novel multifunctional neuroprotective agent with strong iron-chelating and brain-selective MAO-inhibitory effects for PD.13–15
Clioquinol (4) can cross the blood–brain barrier and sequester zinc and copper ions from amyloid β (Aβ) and redistribute them into the cell. A pilot phase II clinical trial with AD patients with chelation therapy has finished, in which patients displayed lower plasma levels of Aβ42 and improved cognition. PBT2 (5) is a new generation of 8-HQ-derived MPAC with improved cognition (metal-peptide attenuating properties) and oral activity. It is in a phase II clinical trial for the treatment of AD, and further clinical testing is currently underway.16
Recently, based on the ‘multitarget-directed ligands’ (MTDLs) strategy,17a,b a set of new multifunctional compounds against neurodegenerative diseases have been designed and synthesised by combining 8-HQ and the pharmacophore elements of other bioactive fragments. For instance, DPH6 (6) was reported as an irreversible inhibitor of MAO A and B, and a mixed-type AChE inhibitor, with metal-chelating effects.18
The bis-lipoyl derivative (LA-HQ-LA, 7) and the glutathione derivative (GS(HQ)H, 8), endowed with an 8-HQ group, exhibited pronounced chelating, antioxidant, and neuroprotective effects.19,20 They could protect SHSY-5Y human neuroblastoma cells against H2O2- and 6-OHDA-induced damage.19,20
In particular, GS(HQ)H (8) showed high stability and can smoothly cross the blood–brain barrier, as assessed by in vitro assay. It is likely that this compound could selectively remove Zn(II) and Cu(II) from the Aβ peptide without leading to the depletion of copper or zinc in vivo.20
D-369 (9) and D-390 (10) were reported as highly active iron chelators, dopamine D2/D3 agonists and antioxidant agents. The in vivo activity of a mouse neuroprotection model indicated potential usage in the symptomatic and neuroprotective treatment of PD.21,22
In addition, the natural product resveratrol (11) displayed Aβ antiaggregative and cytoprotective activities in human neuroblastoma cell lines. However, the unfavorable bioavailability of resveratrol (dietary supplements) greatly limits its applications. The hybrid (E)-5-(4-hydroxystyryl)quinoline-8-ol (12), through combination of resveratrol (11) and clioquinol (4), exhibited excellent potency to inhibit self-induced (IC50 = 8.50 μM) and Cu(II)-induced Aβ aggregation and to destroy the well-structured Aβ fibrils formed by self- and Cu(II)-induced Aβ aggregation (Fig. 3). Importantly, the metal complexation of 12 could also halt copper redox cycling and then control Cu(I/II)-triggered hydroxyl radical production, as observed in a Cu–ascorbate redox system assay. Notably, 12 demonstrated extremely low acute toxicity (LD50 > 2000 mg kg−1) and could cross the blood–brain barrier.23
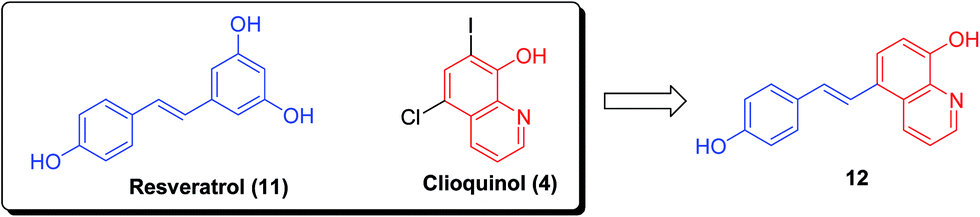 | ||
| Fig. 3 Design of the hybrid (E)-5-(4-hydroxystyryl)quinoline-8-ol (12) via the combination of resveratrol (11) and clioquinol (4). | ||
Collectively, these findings suggested that these 8-HQs could be potential neuroprotective agents for the treatment of neurodegenerative diseases, and 8-HQ will continue to offer a hugely explorable platform for the discovery of new chemical entities with neuroprotective properties for future perspectives.
2.2 Anticancer agents
In 2006, NSC3852 (13) was identified as a histone deacetylase (HDAC) inhibitor with cell differentiation and antiproliferative potency against human breast cancer cell lines and antitumor properties in mouse models containing P388 and L1210 leukemic cells.24 It was found that reactive oxygen species (ROS) formation was associated with the apoptotic and cell-differentiation responses to NSC3852 in MCF-7 cells.248-Hydroxy-2-quinolinecarbaldehyde (14) (Fig. 4) displayed strong in vitro cytotoxicity against multiple cancer cells, including T-47D, Hs578t, SaoS2, K562, MDA231, SKHep1 (MTS50: 12.5–25 μg mL−1) and Hep3B (MTS50: 6.25 ± 0.034 μg mL−1).25 The results demonstrated that the dosage of 10 mg per kg per day of 14 by intraperitoneal injection for 9 days completely suppressed the growth of the Hep3B xenograft tumor, and when compared with the control, no histological damage on vital organs was observed.25
 | ||
| Fig. 4 Structures of 8-HQ-based anticancer agents (part I). | ||
Compounds JLK 1472 (15) and JLK 1486 (16) demonstrated high potency against KB3 cell lines with EC50 values of 2.6 nM and 1.3 nM, respectively. But they were proven inactive against some cell lines including SF268 and PC3, while highly effective against other cell lines. It has tentatively been suggested that their biotarget was probably located upstream from caspase 3/7. Furthermore, their cytotoxic effect was potentiated by the pro-apoptotic effects of TRAIL (Tnf Related Apoptosis Inducing Ligand).26a Then, in 2011, it was reported that JLK 1486 (16) displayed cytostatic (not cytotoxic) effects against experimental gliomas via MyT1 and STAT1 activation and, to a lesser extent, PPARγ activation.26b
It was shown in micro-array analysis that the best performing inhibitor 17 in the 8-HQ-substituted amine series could give covalent protein thiol adducts and induce the expression of various stress-related genes involved in the cytotoxic and cytostatic activities against glioblastoma and carcinoma cells, and represents a novel promising anticancer candidate with unique mechanisms of action, targeting accessible thiols from specific proteins and inducing efficient anticancer activities.26c,d
As underlined in Fig. 5, the mechanism of action of compound JLK 1486 (16) was proposed and verified primarily. It requires two steps, a protonation process followed by a nucleophilic attack resulting in the formation of the quinone methide intermediate (16b).26c
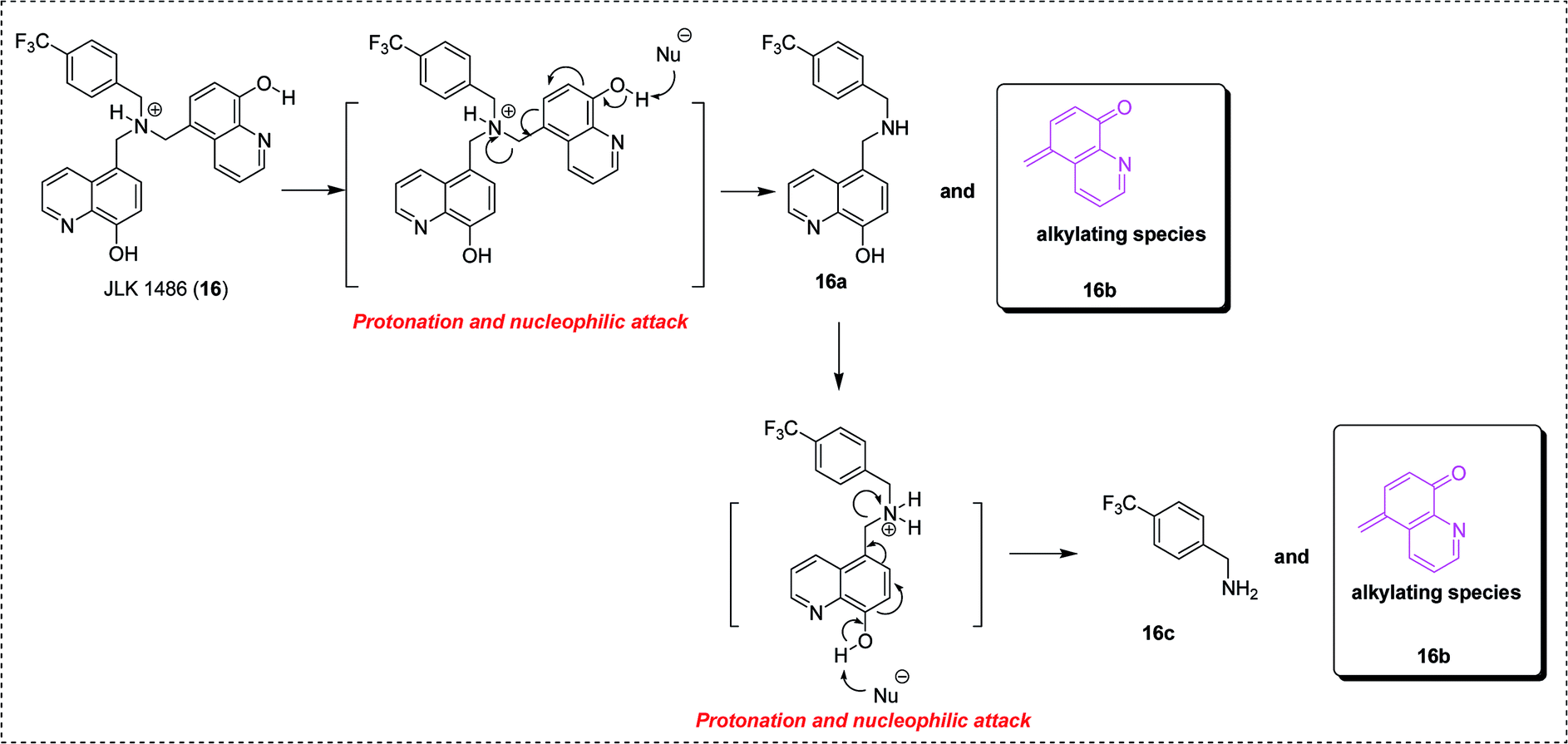 | ||
| Fig. 5 The proposed mechanism of action of compound JLK 1486 (16).26c | ||
In 2012, compound S1 (18) was reported and identified as a chelating agent binding ferric iron with a very high affinity (pFe3+ = 29.5). It also exerted a marked cytostatic effect on hepatoma cells at concentrations as low as 0.1 μM.27
Taking Mannich base 19a as a lead compound, in 2010 a series of 8-HQ-derived Mannich bases were prepared and assayed for their growth inhibition with intriguing structure–activity relationship (SAR) results in the lab of Shaw AY. All Mannich bases demonstrated moderate to low micromolar potency against four carcinoma cell lines. SAR results displayed that upon replacement of either the sulfonyl moiety with methylene or the piperazine ring with an ethylenediamine group led to an appreciable increase in activity. Besides, as 8-hydroxyquinoline was replaced with 3-hydroxypyridine, phenol and 1-naphthol, a dramatic decrease in potency was observed. The structural modifications revealed that the 8-hydroxyquinol skeleton appeared to be a crucial pharmacophore for inhibition.
Among all the Mannich bases, 19b exhibited the highest potencies against both SKHep and CE81T cells with GI50 values of 2.6 and 2.8 μM, respectively, while 19c (GI50, 0.7 and 1.9 μM against HeLa and BT483 cells, respectively) and 19d showed the highest growth-inhibitory activity against CE81T and SKHep cells (GI50, 2.8 μM against CE81T cells). These results demonstrated that the sensitivity of individual cell lines was closely related to the structures of these derivatives.28
In 2014, the 8-hydroxyquinoline derivative 20, containing a sugar and a 1,2,3-triazole moiety, was reported to show promising antiproliferative potency and high selectivity toward ovarian cancer cells (OVCAR-03, GI50 < 0.25 μg mL−1). This compound was more potent than the control drug doxorubicin (OVCAR-03, GI50 = 0.43 μg mL−1).29
Human cathepsin B, an enzyme belonging to the peptidase (or protease) families, is regarded as a promising therapeutic target for cancers. By combining structure-based design and focused library screening, the nitroxoline derivative 21 was identified as a new cathepsin B inhibitor, which was active in cell-based in vitro tumor invasion models, where it dramatically abolished invasion of MCF-10A neoT cells.30 This research will afford valuable insights for further discovery of new anticancer agents with improved potency.
The nitroxoline (22) demonstrated antiproliferative activity in endothelial cells, its action mechanisms included a dual inhibition of sirtuin 1 (SIRT1) and type 2 human methionine aminopeptidase (MetAP2). SAR investigation of nitroxoline derivatives revealed several substituted oxines (13, 23–25, Fig. 6) with potency against endothelial cell proliferation and angiogenesis. Minor modifications on the 8-HQ core resulted in oxine derivatives with entirely different mechanisms of action.31
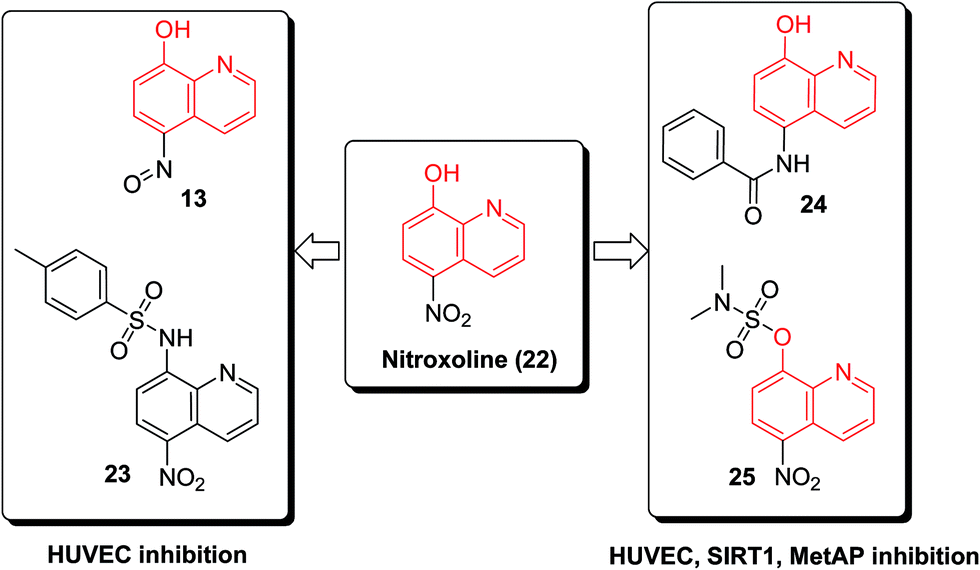 | ||
| Fig. 6 The structures of oxine derivatives with entirely different mechanisms of action. | ||
Pim-1 kinase was considered to be an important new target for anticancer drug discovery for its oncogenic potential. Compounds 2-styrylquinoline 26 and quinoline-2-carboxamide 27 were recently reported as novel and potent inhibitors of Pim-1 kinase (Fig. 7). The 8-hydroxy-quinoline-7-carboxylic acid skeleton was probably required for activity, which was preliminarily confirmed by molecular modeling.32
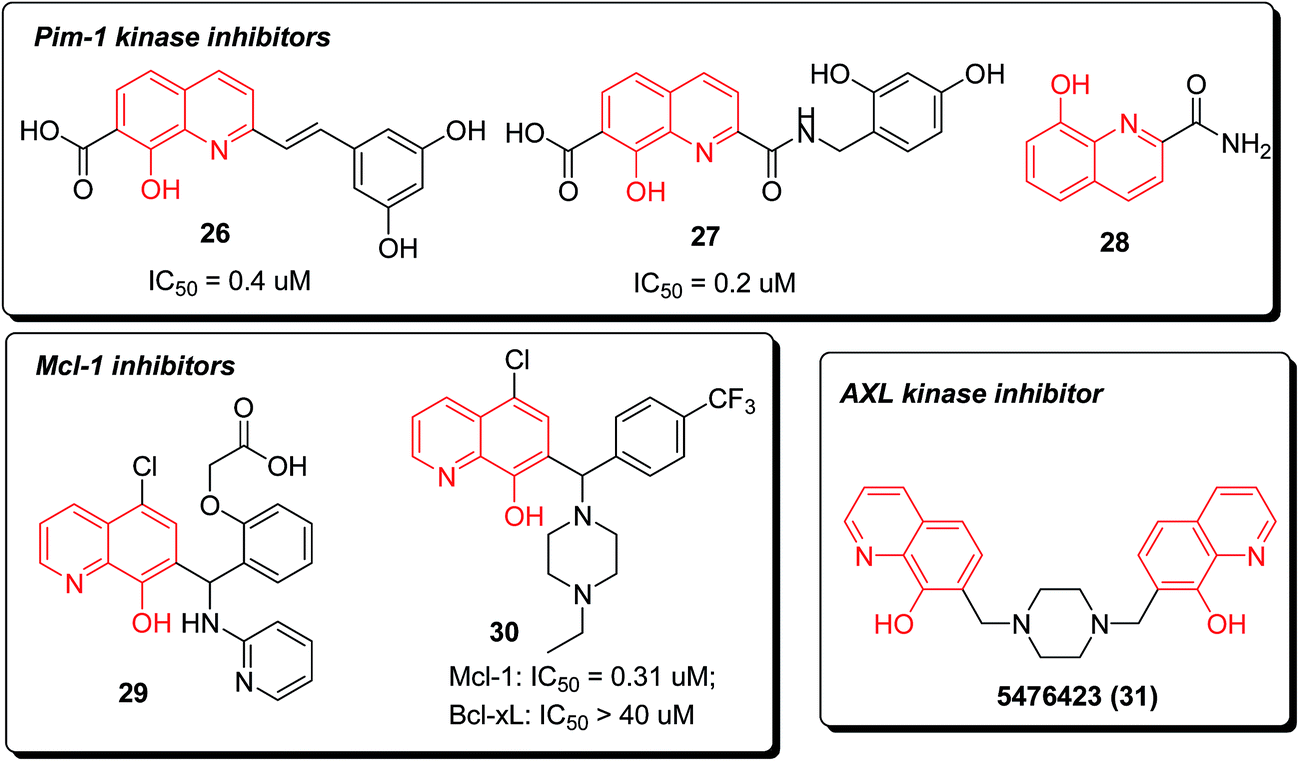 | ||
| Fig. 7 Structures of 8-HQ-based anticancer agents (part II). | ||
In addition, 8-hydroxyquinoline-2-carboxamide (28) was discovered as a fragment (IC50 = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM) bound to Pim-1 kinase via hydrogen bonding and hydrophobic interactions. The cocrystal structure investigation demonstrated the rationale of further designing potent and selective Pim-1 kinase inhibitors based on this fragment and available structural information (Fig. 8).33
000 nM) bound to Pim-1 kinase via hydrogen bonding and hydrophobic interactions. The cocrystal structure investigation demonstrated the rationale of further designing potent and selective Pim-1 kinase inhibitors based on this fragment and available structural information (Fig. 8).33
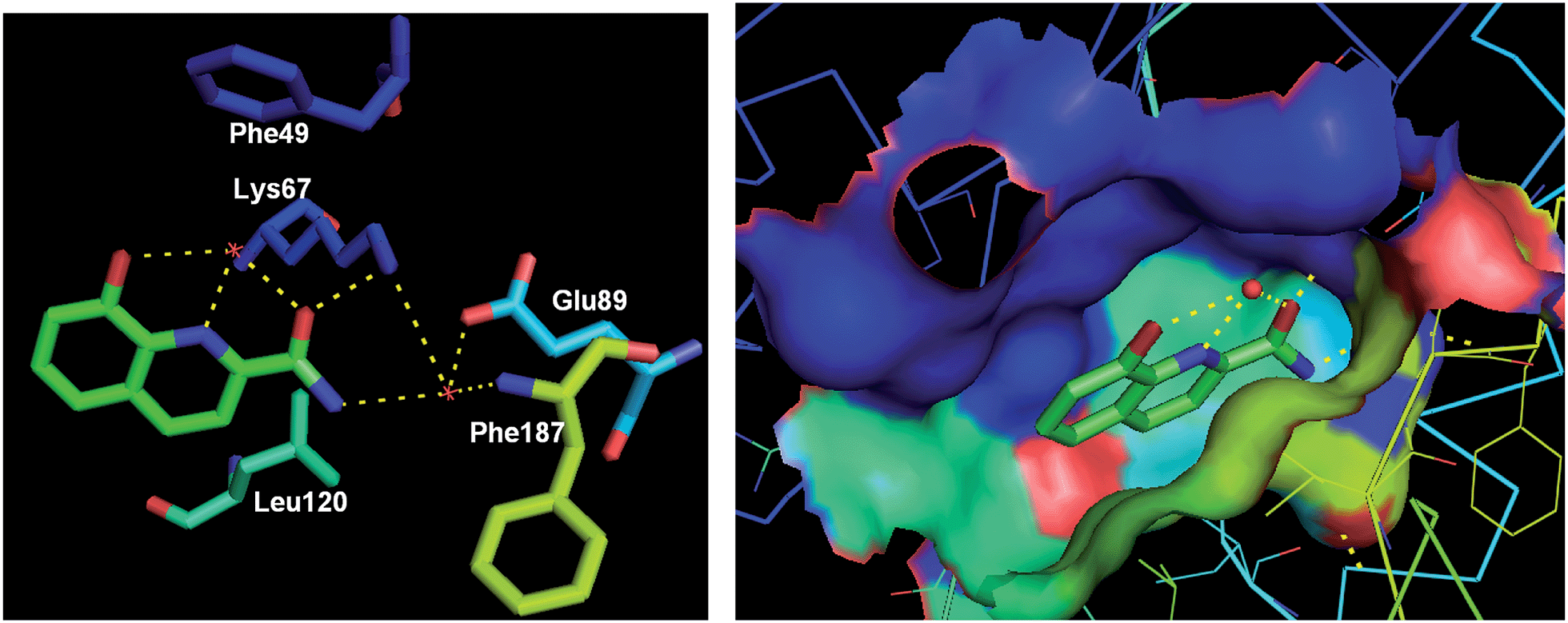 | ||
| Fig. 8 The crystal structure of a fragment 28 (IC50 = 10000 nM) bound to Pim-1 kinase (PDB code: 3VBV, resolution: 2.08 Å). Key H-bond interactions are shown as yellow dashed lines. | ||
The piperazine-substituted hydroxyquinoline 30, which is derived from a high-throughput screening (HTS) hit 29, demonstrated selective inhibition of Mcl-1 (a target central to intrinsic apoptosis that has been implicated in a cancer) relative to the related Bcl-2 family protein Bcl-xL in the fluorescence polarization assay.
This compound also displayed efficacy in promoting death in a set of cell lines derived from diverse malignancies.34
In 2014, compound 5476423 (31) was identified as a lead with potential antiproliferative effects against gallium-resistant lung cancer through an AXL kinase pathway, through virtual screening of the AXL kinase homology model. When compared with gallium acetylacetonate (GaAcAc), the IC50 values from treating gallium-resistant cells exhibited that compound 31 had 80 fold increased activity. The efficacy of GaAcAc against gallium-resistant cells was increased 2 fold combined to compound 31.35
Obviously, these studies established the proof-of-concept that 8-HQ could be employed as a favorable heterocyclic scaffold for the development of omnifarious anticancer agents.
2.3 Inhibitors of 2-oxoglutarate (2OG) and iron-dependent enzymes
2-Oxoglutarate (2OG) and iron-dependent oxygenases are considered to be promising therapeutic biotargets for omnifarious human diseases (Fig. 9).36 5-Carboxy-8-hydroxyquinoline (IOX1, 32) is the most effective broad-spectrum inhibitor of the 2OG oxygenase subfamilies (including nucleic acid demethylases and γ-butyrobetaine hydroxylase), reported to date.37 In 2010, IOX1 was validated as a cell-active inhibitor of 2OG-dependent histone lysine demethylases (KDM) (namely, the Jumonji domain-containing histone demethylases, JMJD) and an inhibitor of fat mass and obesity associated protein (FTO, a 2OG-dependent N-methyl nucleic acid demethylase that acts on substrates including 3-methylthymidine, 3-methyluracil, and 6-methyladenine) via quantitative HTS of a collection of diverse compounds.38a,b Besides, it was also reported as a ligand of AlkB, which belongs to the Fe(II)/2OG-dependent dioxygenase superfamily and oxidatively demethylates the DNA substrate.37 Though compound IOX1 suffers from low cell permeability, it has a smaller and more compact chemical structure, and represents a lead compound for further modifications and a good tool compound for studies investigating the roles of 2OG-dependent enzymes in epigenetic processes. It was envisioned that by rationally changing substituents to maximize the key contacts between the ligand and the binding site, the potency and the selectivity profiles towards the desired target will be improved. | ||
| Fig. 9 Reactions catalyzed by oxygenases and FTO. | ||
As shown in Fig. 10, cocrystal structure analysis of oxygenases complexed with IOX1 displayed the binding modes and pivotal interactions involved in target recognition, which will aid the development of more potent and selective inhibitors.38a,b
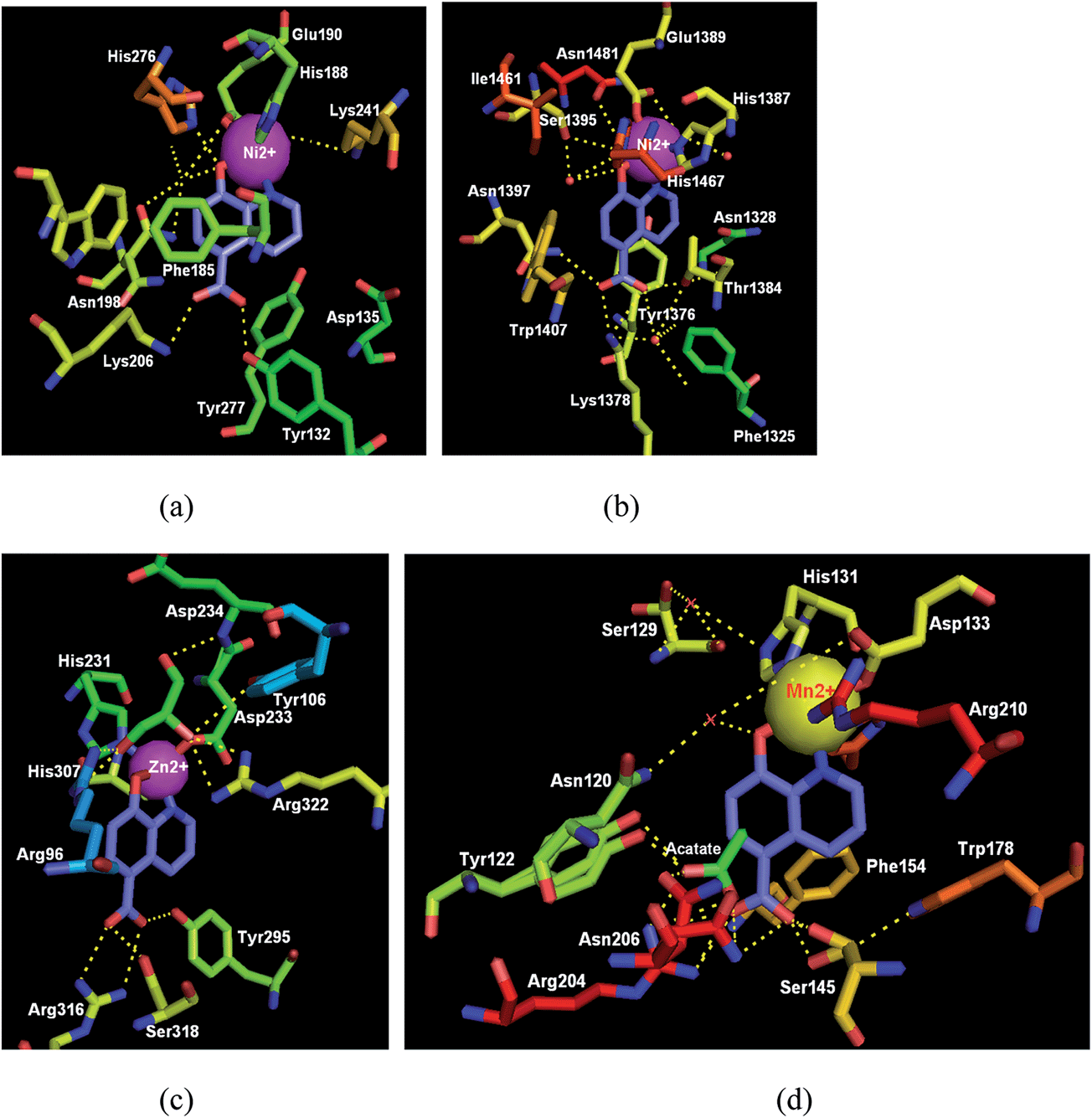 | ||
| Fig. 10 Crystal structures of oxygenases in complex with the IOX1 (32). (a) JMJD2A complexed with IOX1 (blue); (PDB code: 3NJY, resolution: 2.60 Å).38a (b) The human JMJD3 Jumonji domain with IOX1; (PDB code: 2XXZ, resolution: 1.80 Å). (c) The human FTO in complex with IOX1 (IC50 = 3.3 μM) (PDB code: 4IE4, resolution: 2.50 Å). (d) AlkB in complex with IOX1; (PDB code: 4JHT, resolution: 1.18 Å). Key H-bond interactions are shown as yellow dashed lines. | ||
As expected, the n-octyl ester (33, Fig. 11) of IOX1 demonstrated improved cellular potency (30-fold) and enhanced selectivity over the parent IOX1.39
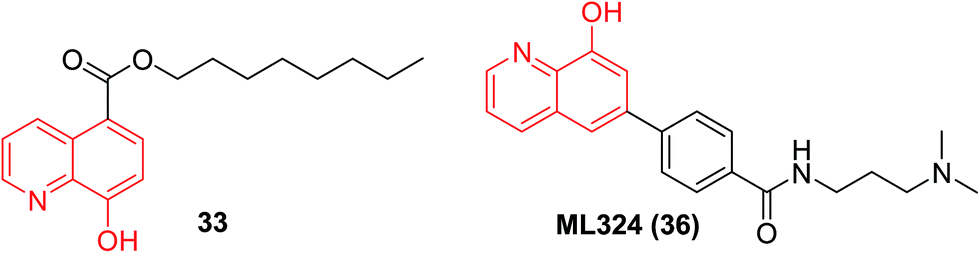 | ||
| Fig. 11 Structures of n-octyl ester (33) of IOX1 and ML324 (36). | ||
Currently, small molecule modulators of epigenetic regulation are employed as useful tools for probing biochemical mechanisms, and as lead compounds for the discovery of anticancer therapeutic agents.40 In prostate cancer, two different kinds of KDMs, KDM1 (lysine-specific demethylase 1, LSD1) and KDM4 (JMJD2), were coexpressed and colocalize with the androgen receptor. Inspired by this finding, recently, by coupling the skeleton of tranylcypromine 34, a known LSD1 inhibitor, with IOX1 (23), a 2-OG competitive JmjC inhibitor, hybrid 35 was designed as a pan-histone demethylase inhibitor simultaneously targeting Jumonji C and lysine-specific demethylases, and displayed high anticancer activities against LNCaP prostate and HCT116 colon cancer cells (Fig. 12).41
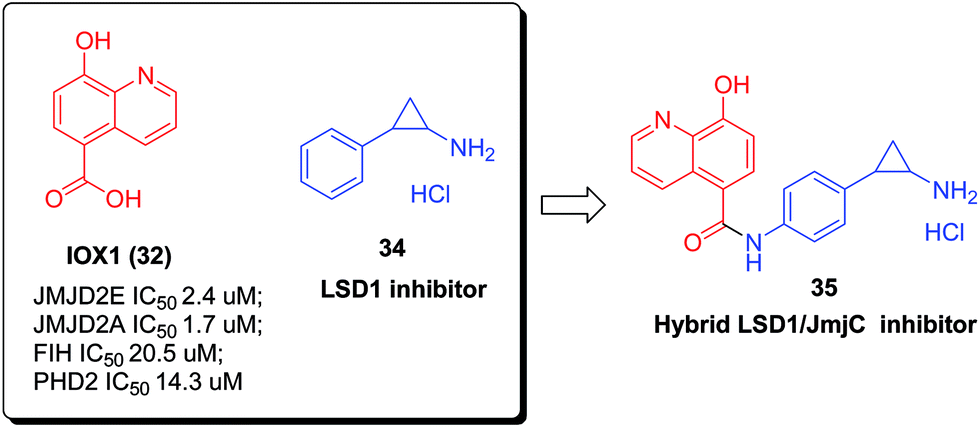 | ||
| Fig. 12 Design of a dual inhibitor of JmiC and lysine-specific demethylases. | ||
It needs to be stressed that inhibitors of the epigenetic-related targets are also showing great promise as antiviral agents. Through a quantitative HTS and subsequent structural optimization campaign, compound ML324 (36) was identified as a JMJD2E inhibitor (submicromolar inhibitory) with excellent cell permeability and in vitro absorption, distribution, metabolism and excretion (ADME) properties. In addition, ML324 showed potent antiviral effects against both human cytomegalovirus (HCMV) and herpes simplex virus (HSV) infections by inhibiting viral immediate-early (IE) gene expression. In a mouse ganglia explant model of latently infected mice, ML324 can block the formation of HSV plaques and suppress HSV-1 reactivation.42
As a transcription factor under hypoxia, hypoxia-inducible factor-1 (HIF-1) is the master regulator of the cellular hypoxia response, which can activate a variety of genes, including those involved in glucose metabolism, angiogenesis, cell proliferation and cell survival.43 Among the isoforms, HIF-1α is a vital regulator of hypoxia responses in solid tumors, and its activity is indispensable for tumors to adapt to hypoxia conditions and recover from damage caused by hypoxic insult. The HIF-1α subunit is regulated by 2-OG- and Fe(II)-dependent hydroxylases, such as Factor Inhibiting HIF-1 (FIH-1). FIH-1 can hydroxylate Asn803 of HIF-1α and block its binding with coactivating factors. Quinol derivatives, including IOX1 and clioquinol, displayed inhibition against the hydroxylation effect of FIH-1.
The cocrystal structures of FIH-1 with IOX1, clioquinol and 8-HQ (37) were determined respectively, giving an update on the molecular aspects at the basis of rational drug design. As shown in Fig. 13 and 14, these three compounds bind to the active site of FIH-1 by chelating the Fe/Zn ion, then blocking the binding of a cosubstrate, 2OG.44 Contrary to the existing FIH-1 inhibitors with negative charges, these 8-HQ derivatives are neutral in charge and can afford a scaffold for further development as FIH-1 selective inhibitors.44
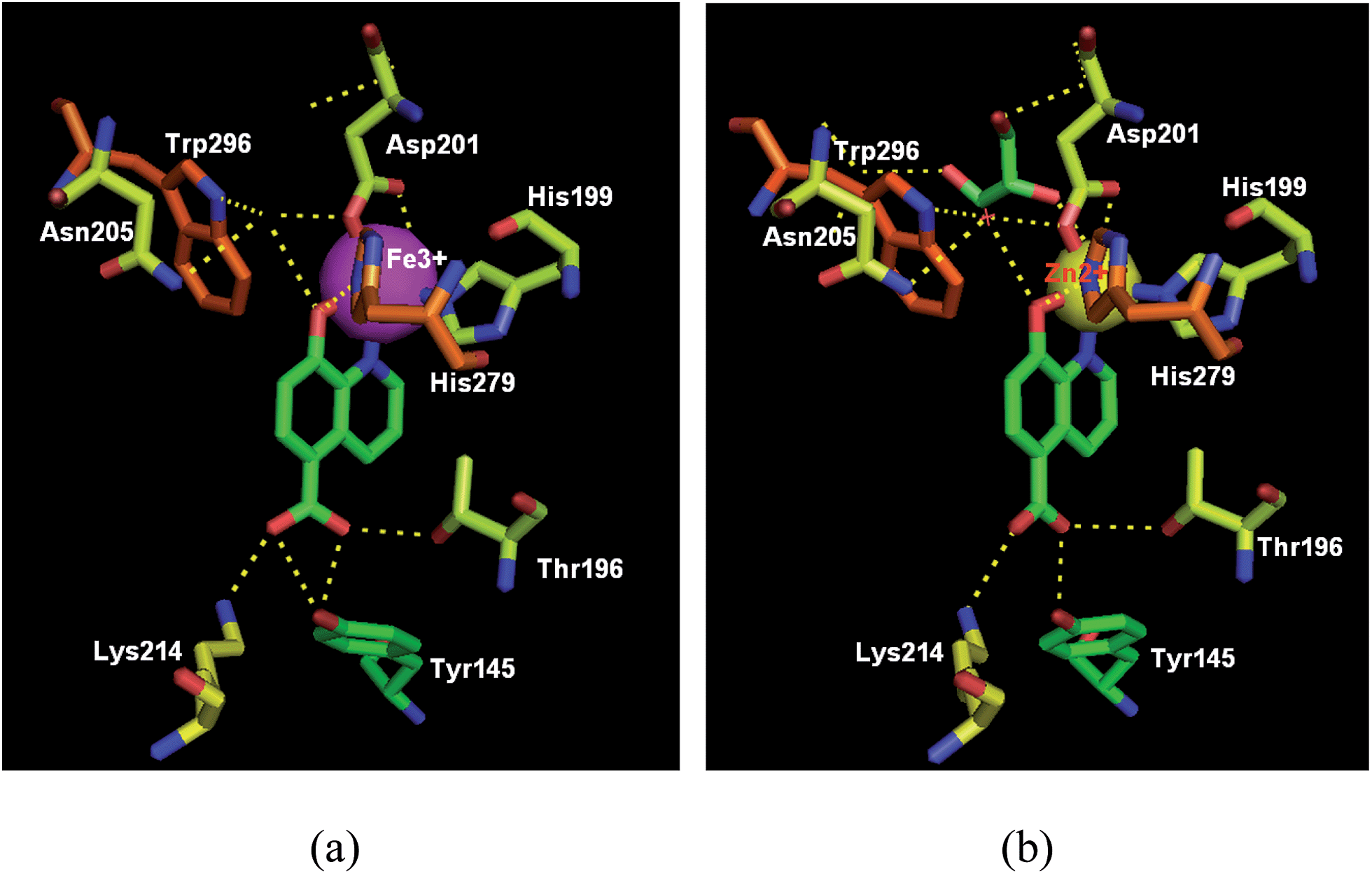 | ||
| Fig. 13 Crystal structure of FIH-1α in complex with IOX1 (green) (a) PDB code: 4BIO, resolution: 2.45 Å;37 (b) PDB code: 3OD4, resolution: 2.20 Å. | ||
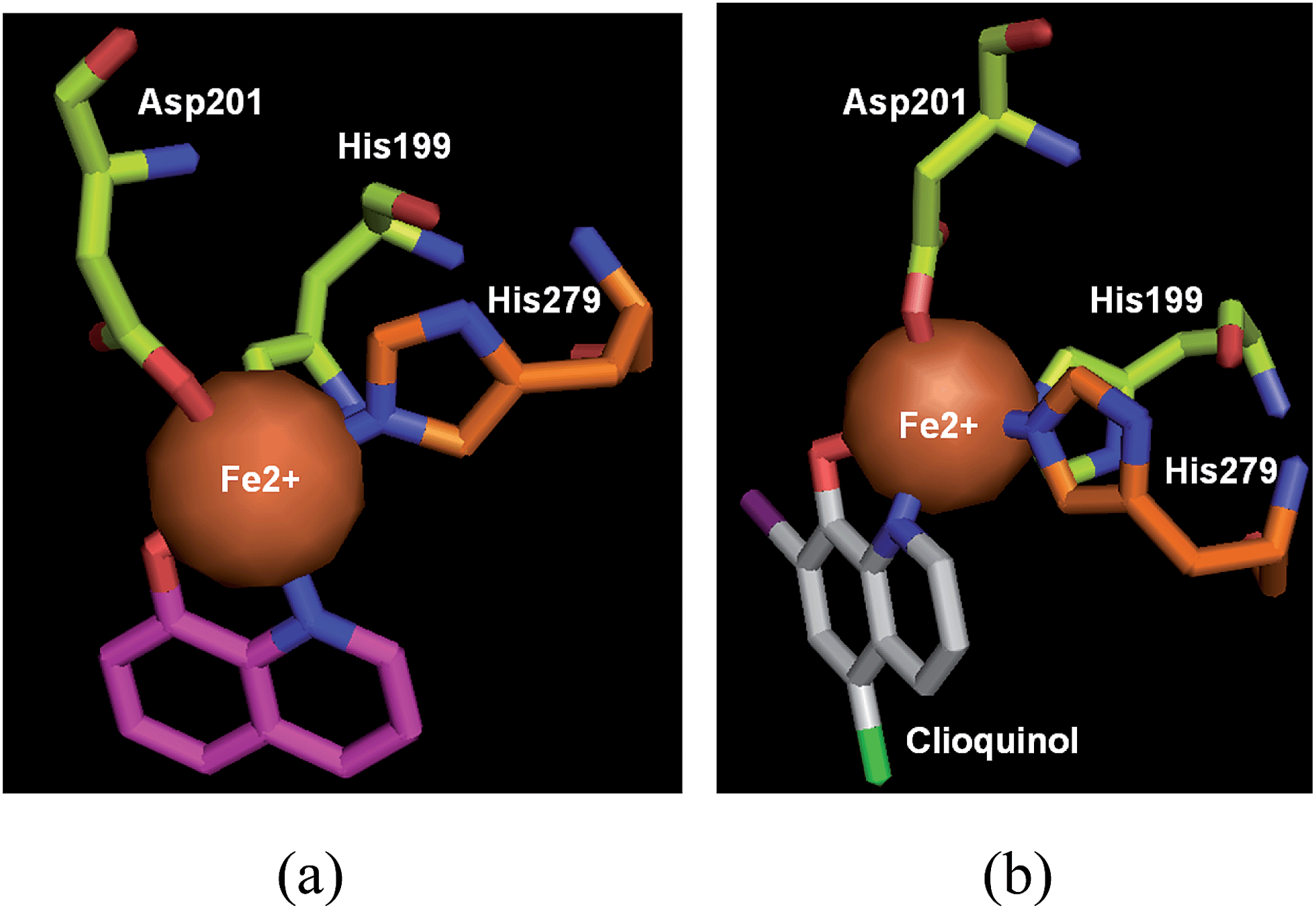 | ||
| Fig. 14 FIH-1α in complex with (a) 8-HQ (pink) (PDB code: 3KCY, resolution: 2.59 Å), (b) clioquinol (white) (PDB code: 3KCX, resolution: 2.60 Å).44 | ||
2.4 Chelators of metalloproteins (with miscellaneous therapeutic applications)
Metalloproteins are an important class of biotargets currently receiving increased attention. The ability to bind with multiple metalloproteins is a main argument for using 8-HQ as a privileged structure in drug discovery. Recently, by screening a metalloprotein-focused chelator fragment library, substituted 8-HQs at either the 5- or 7-positions were identified as active MMP-2 inhibitors (38a–d, 39a–d), with low micromolar IC50 values (Fig. 15).45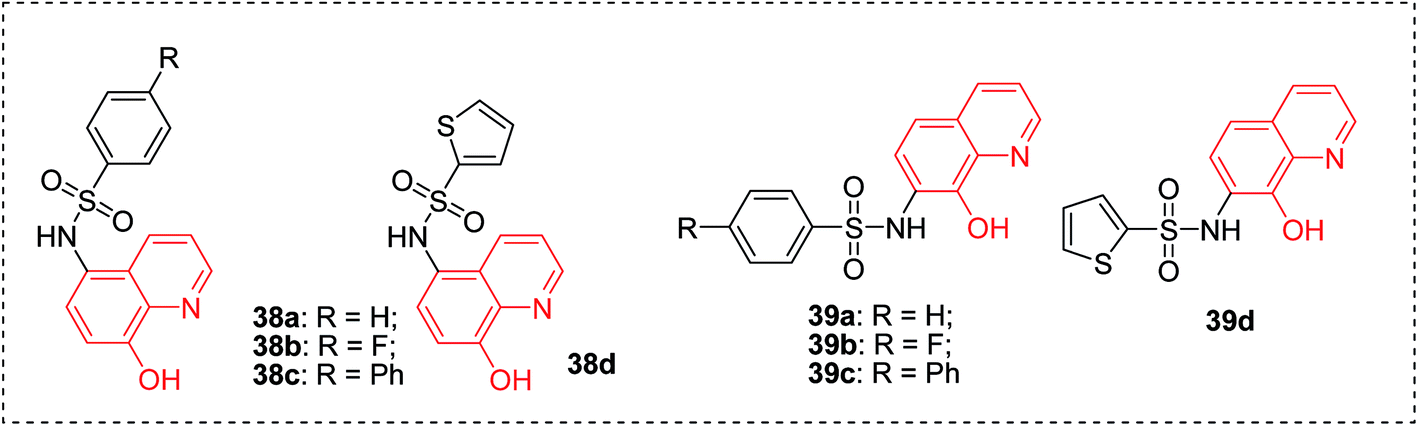 | ||
| Fig. 15 Structures of 8-HQ containing MMP-2 inhibitors. | ||
In 2012, 8-HQ (37) and its 5-substituted derivatives were reported as competitive and selective inhibitors of an aminopeptidase from Aeromonas proteolytica (AAP, a dinuclear Zn2+ hydrolase) with a Ki value of 0.16–29 μM at pH 8.0. The hydroxide at the 8-position and the nitrogen at the 1-position of 8-HQ were considered to be crucial for the inhibition of AAP. The cocrystal structure of AAP complexed with 8-HQ demonstrated that 8-HQ was able to interact with AAP in the metal-chelating mode, in which the nitrogen atom of 8-HQ coordinates to one Zn(II) ion and the hydroxide anion bridges two Zn(II) ions in the active site of AAP (Fig. 16).46
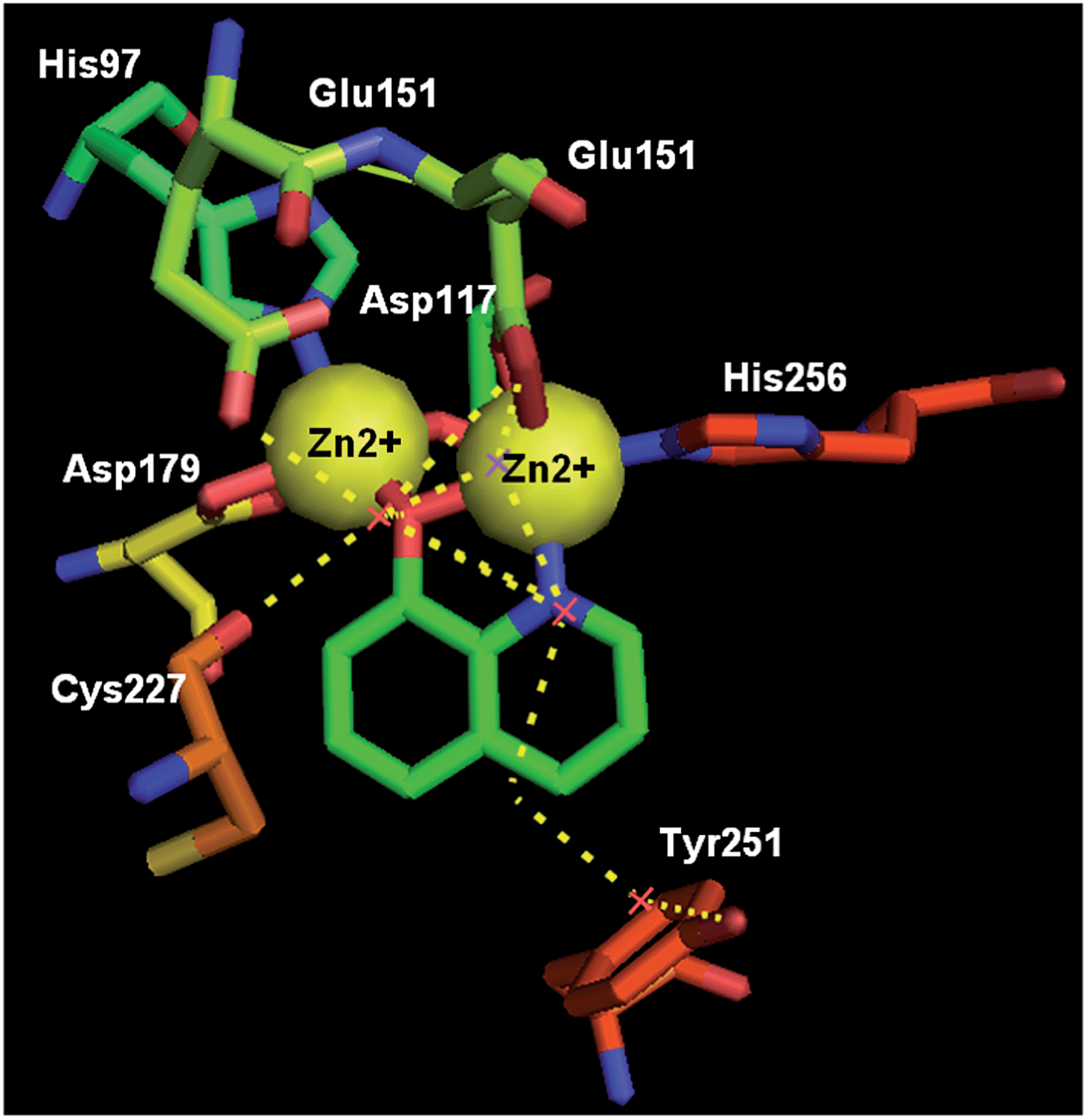 | ||
| Fig. 16 The X-ray cocrystal structure of AAP-8-HQ complex (PDB code: 3VH9, resolution: 1.29 Å). | ||
In summary, these two sections (Section 2.4 and 2.5) introduce multiple cases of the metal-chelating-related bioactivities of 8-HQs, suggesting that this heterocycle motif may be further exploited to seek novel therapeutic usages in the future. It should be pointed out that the propensity of 8-HQs to bind with multiple metalloproteins can underline their high versatility, which will result in unfavorable off-target interactions. Therefore, considerable attention in the future should be focused on the optimization of 8-HQs into highly specific agents with minimum unwanted off-target activities.
2.5 Anti-HIV agents
Improving treatments for HIV infections still remains a challenge. In particular, new chemical entities are greatly needed to overcome the emergence of drug resistant viral mutants and enrich the current paradigm. As shown in Fig. 17, several groups have already reported on the identification of 8-HQ analogues as excellent antiviral agents for targeting HIV-1 integrase (IN) (compounds 40 and 41)47,48 and IN-LEDGF/p75 interaction (compounds 42 and 43).49 We envision that with its broad-spectrum potential and flexibility of functionalization, 8-HQ will still function as an important scaffold in the discovery of HIV inhibitors in the future.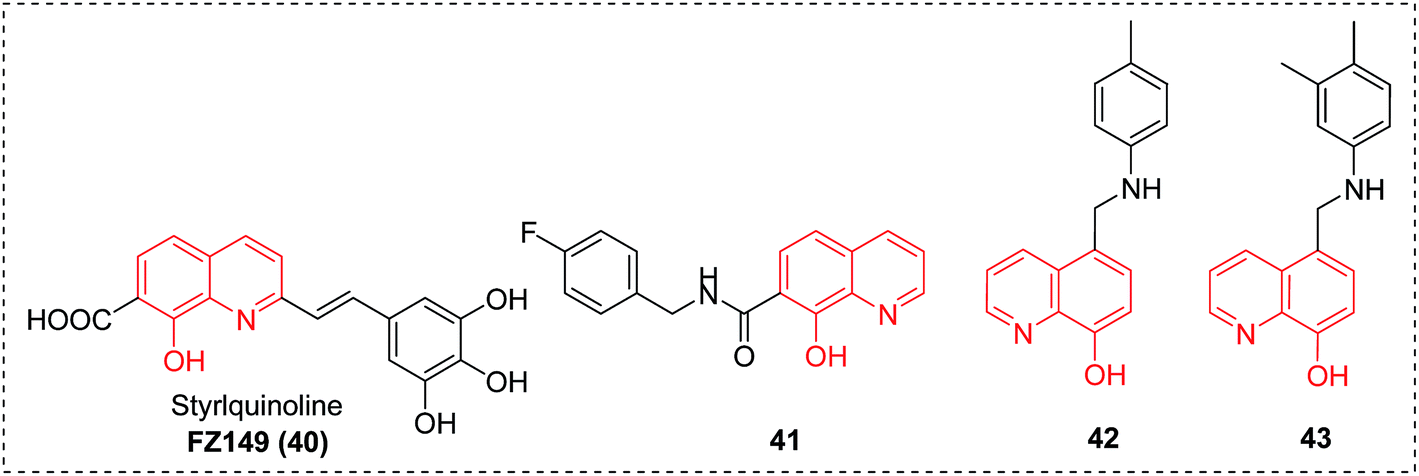 | ||
| Fig. 17 8-HQs with anti-HIV activities. | ||
2.6 Antifungal agents
8-HQ derivatives 44–46 exhibited antifungal potency in vitro comparable to or higher than that of the clinically used fluconazole (Fig. 18).50 These compounds are being carried forward as promising leads for the treatment of fungal infections.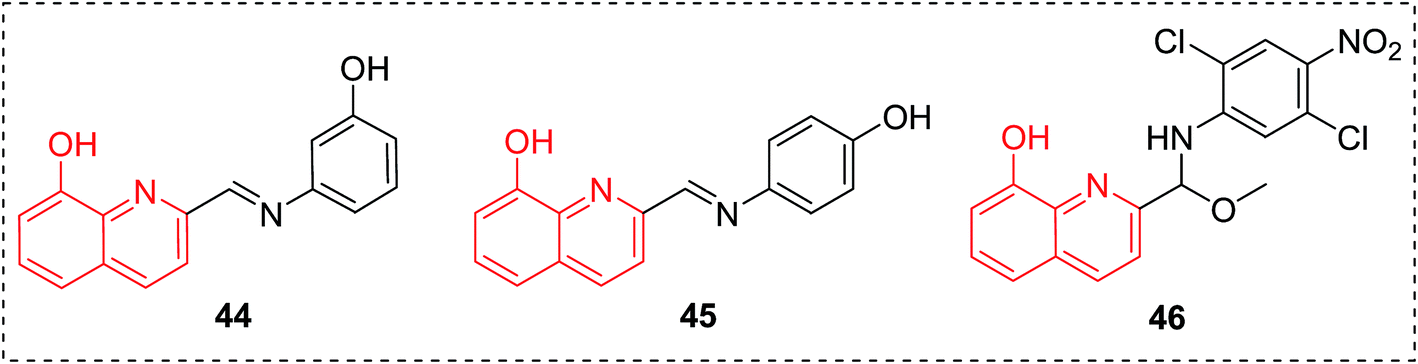 | ||
| Fig. 18 8-HQs as antifungal agents. | ||
2.7 Antileishmanial agents
In 2005, the quinoline-2-carboxylic acid derivative perspicamide A (47), which was originally isolated from the Australian ascidian Botrylloides persipicuum, has received increased attention as quinoline-2-carboxylic acid is a common moiety in numerous drug-like molecules.51 Recently, its analogue, 48 displayed potent inhibition against Leishmania donovani, with an IC50 value of 3.75 μM and a selectivity index (SI) of 25.27, which was markedly increased compared to the approved drug Miltefosine (IC50 12.4 μM; SI 4.1) (Fig. 19).52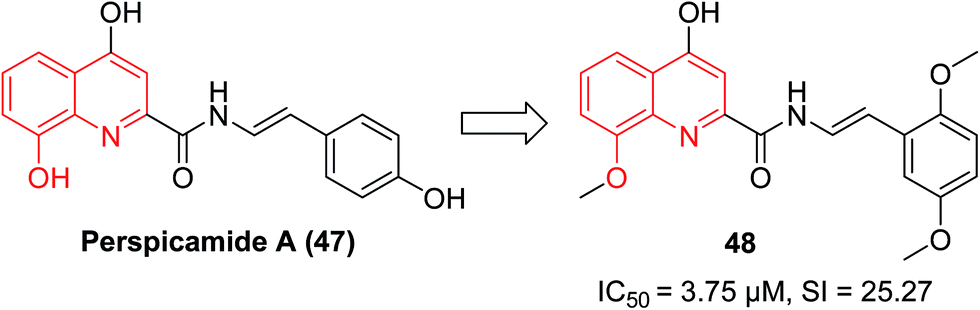 | ||
| Fig. 19 Discovery of 8-HQ derivative 48 as an antileishmanial agent. | ||
2.8 Antischistosomal agents
In 2013, the 8-hydroxyquinoline-5-sufonyl 1,4-diazepine 49 was identified as a potent antischistosomal agent (Fig. 20), which can significantly reduce oviposition of adult worms and totally diminish egg deposition in vitro. A molecular docking study showed that this compound could exert its inhibitory properties by binding with thioredoxin glutathione reductase (TGR), a vital protein for schistosome survival.53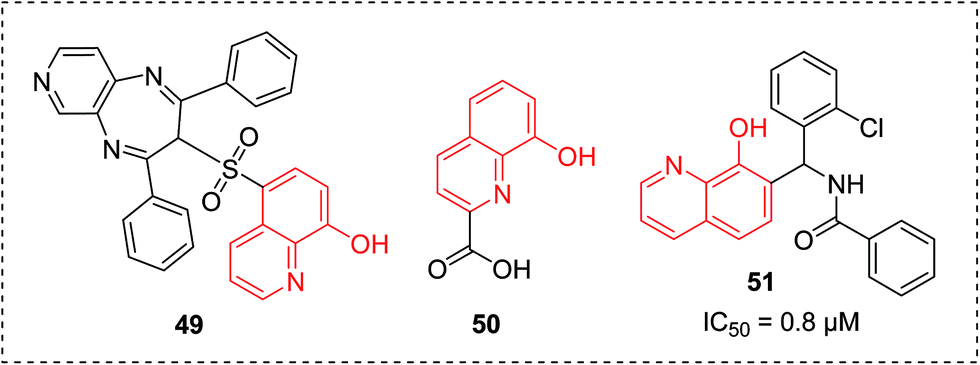 | ||
| Fig. 20 8-HQ derivatives as antischistosomal agents, Mycobacterium tuberculosis inhibitors and Botulinum neurotoxin inhibitors, respectively. | ||
2.9 Mycobacterium tuberculosis inhibitors
Class II fructose 1,6-bisphosphate aldolase (FBA) is regarded as a well-validated biotarget for the design of novel antibiotics against pathogenic bacteria including Mycobacterium tuberculosis, which is the causative agent for tuberculosis (TB). Very recently, via enzymatic and structure-guided screening, 8-HQ carboxylic acid (50) was identified as a potent inhibitor of the class II FBA present in M. tuberculosis (MtFBA), with an IC50 of 10 μM, and was shown to posses anti-TB properties (Fig. 20).Compound 50 also showed inhibitory properties for other class II FBAs, including methicillin-resistant Staphylococcus aureus. As opposed to the existing inhibitors, compound 50 functioned in a noncompetitive manner, and exhibited no inhibitory effects toward class I FBAs in humans and rabbits. Moreover, the complex crystal structure of 50 with MtFBA was determined (Fig. 21), which will allow more sophisticated, evidenced-based development of MtFBA inhibitors with improved target specificity.54
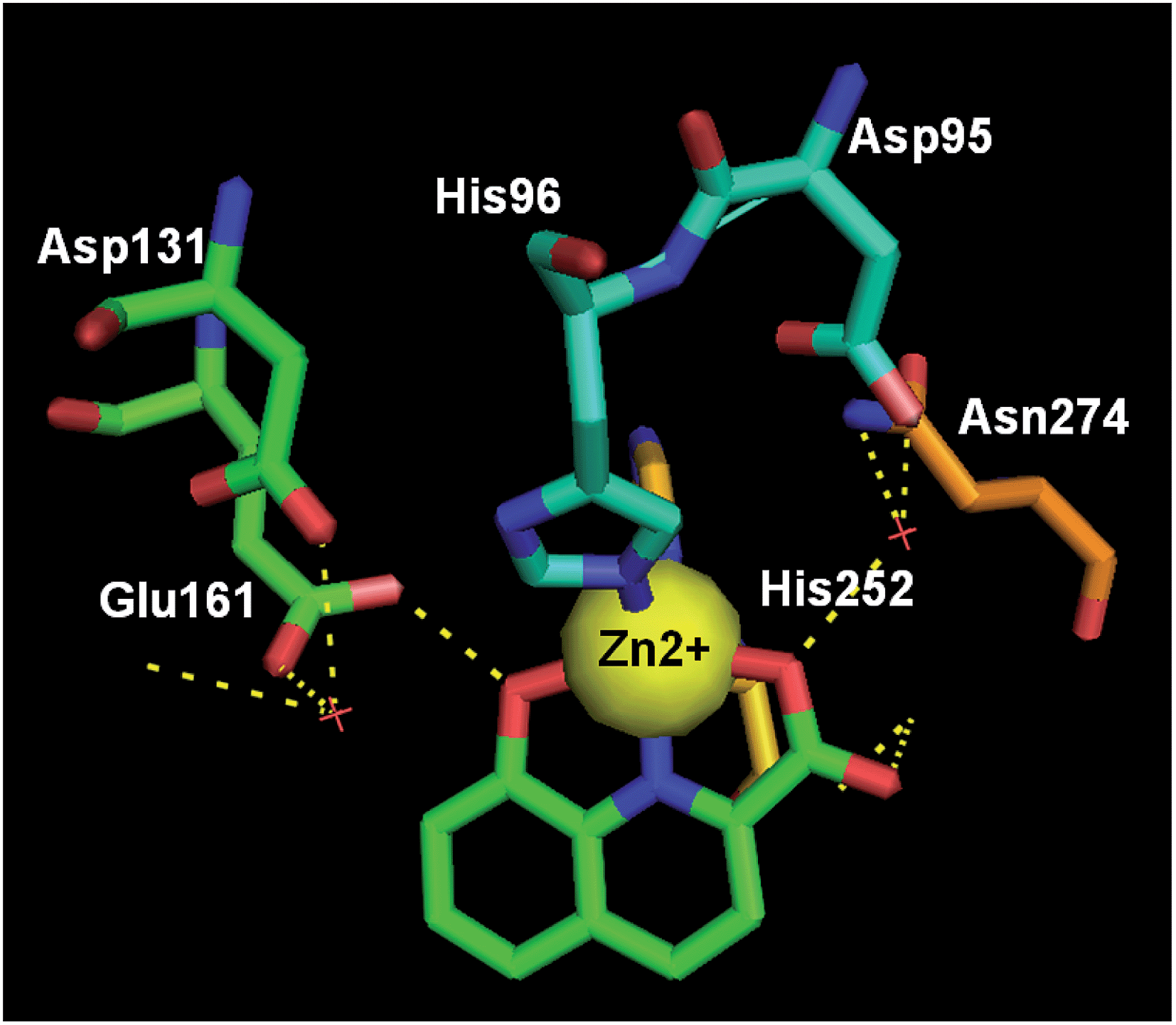 | ||
| Fig. 21 The crystal structure of 50 (green) bound to M. tuberculosis's class IIa FBA (PDB code: 4LV4, resolution: 2.08 Å). Key H-bond interactions are shown as yellow dashed lines. | ||
2.10 Botulinum neurotoxin inhibitors
Botulinum neurotoxins (BoNT) were considered to be the most active toxins known and a severe bioterrorist threat. In 2014, compound 51 (Fig. 20) was reported as a potent BoNT inhibitor with favorable in vitro potencies (IC50 = 0.8 μM) and favorable ADME properties (excellent solubility at low pH) through the screening of the commercially available and synthesized library.552.11 Miscellaneous activities
Besides the contents described above, as shown in Fig. 22, novel 8-HQ-containing 2,3-diaminopropionic acid 52 was validated as a glyoxal/methylglyoxal scavenger and as an AGE inhibitor.56 8-HQs 53–56 could induce the proliferation of rat mesenchymal stem cells (rMSCs).57 In 2014, some 5,7-bis(alkylaminosulfonyl)-8HQs (57–60) were reported as highly effective radioprotective agents with low cytotoxicity for MOLT-4 cells.58 8-HQs 61–63 were identified as chemosensors or fluorescent sensors for their interesting electro-optical properties.59–61 | ||
| Fig. 22 Other 8-HQ-containing compounds with potential applications. | ||
3. Conclusions and perspectives
Undoubtedly, the 8-HQ core structure fits the definition of a “privileged structure” in medicinal chemistry, because it forms the centerpiece of small molecule chemical entities with a wide range of pharmacological properties, which have been presented in this review. 8-HQ was introduced into drug discovery programs for different reasons. For instance, it was used as an essential part of the pharmacophore (usually as a metal-chelating moiety), favorably contributing to ligand binding. Besides, the 8-HQ moiety was shown to function as a flat, aromatic linker to orient the pharmacophore elements into the proper geometry for binding, as well as modulating the physicochemical properties by placing them in the periphery of the molecule.The prospects of 8-HQ are represented from the following aspects (Fig. 23): first, the 8-HQ structure provides a versatile platform on which multiple functional substituent groups can be placed at various sites on the core chemical skeleton, allowing medicinal chemists to utilize structure-based drug design or target-oriented synthesis (TOS) techniques to exquisitely tailor a molecule directly to its specific target, to thereby avoid or control unfavorable off-target interactions. Consequently, novel derivatives towards a given target with improved potency, selectivity and minimum off-target activities will be obtained. Second, through the design and synthesis of libraries around a given privileged structure, to exploit unchartered territories in chemical and biological space, a higher hit rate and the identification of biologically relevant hits can be expected, thus we envision that there exists a great potential in chemical approaches such as multicomponent reactions (MCRs), diversity-oriented synthesis (DOS) methodologies and more efficient, expeditious straight-forward synthetic methods,62,63 to further extend the versatility of this scaffold in various medicinal areas. Lastly, but also importantly, the “privileged structure”-guided scaffold repurposing is a useful strategy to identify structurally novel 8-HQ-related chemotypes for intellectual property purposes, by switching the central core structure with its isomeric forms, bioisosteres or structurally pertinent skeletons combined with decorating substituent groups.8 It should be firmly understood that the exploitation of the 8-HQ scaffold remains an intriguing scientific endeavour, which will lead to the discovery of more clinically-relevant agents.
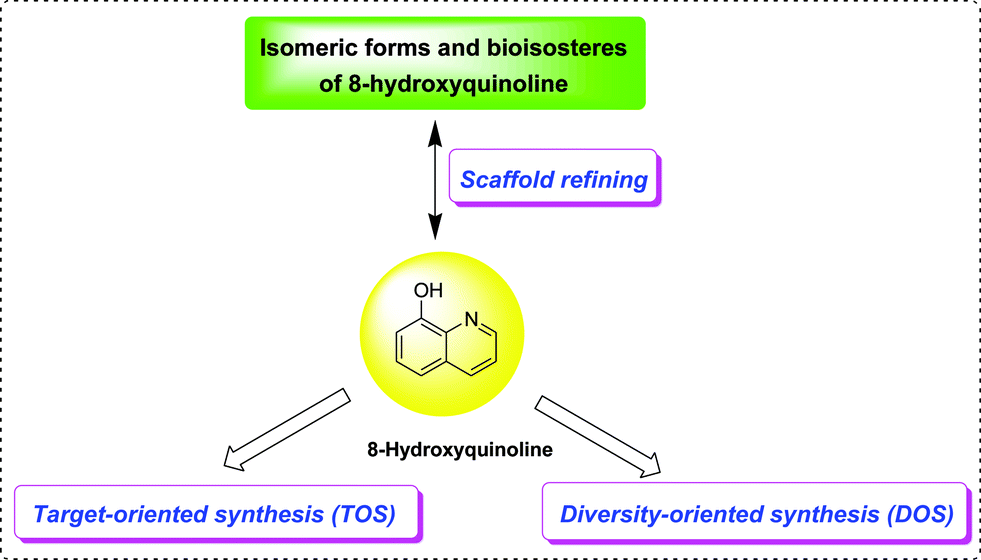 | ||
| Fig. 23 Graphical representation of the prospects for further exploitation of the 8-HQ scaffold. | ||
Conflict of interest
The authors declare no conflict of interest.Acknowledgements
The financial support from the National Natural Science Foundation of China (NSFC no. 81102320, 81273354), Key Project of NSFC for International Cooperation (no. 81420108027), Research Fund for the Doctoral Program of Higher Education of China (no. 20110131130005, 20110131120037), The Science and Technology Development Project of Shandong Province (no. 2014GGE27154), and China Postdoctoral Science Foundation funded special project (no. 2012T50584) is gratefully acknowledged.References
- R. W. DeSimone, K. S. Currie, S. A. Mitchell, J. W. Darrow and D. A. Pippin, Privileged structures: applications in drug discovery, Comb. Chem. High Throughput Screening, 2004, 7(5), 473–494 CrossRef CAS.
- C. D. Duarte, E. J. Barreiro and C. A. Fraga, Privileged structures: a useful concept for the rational design of new lead drug candidates, Mini-Rev. Med. Chem., 2007, 7(11), 1108–1119 CrossRef CAS.
- P. Zhan, X. Liu and E. De Clercq, Recent advances in antiviral activity of benzo/heterothiadiazine dioxide derivatives, Curr. Med. Chem., 2008, 15(15), 1529–1540 CrossRef CAS.
- P. Zhan, X. Liu and E. De Clercq, Functional Roles of Azoles Motif in Anti-HIV Agents, Curr. Med. Chem., 2011, 18(1), 29–46 CrossRef CAS.
- P. Zhan, D. Li, J. Li, X. Chen and X. Liu, Benzimidazole heterocycle as a privileged scaffold in antiviral agents, Mini-Rev. Org. Chem., 2012, 9(4), 397–410 CrossRef CAS.
- Y. Song, P. Zhan, Q. Zhang and X. Liu, Privileged scaffolds or promiscuous binders: a glance of pyrrolo[2,1-f][1,2,4]triazines and related bridgehead nitrogen heterocycles in medicinal chemistry, Curr. Pharm. Des., 2013, 19(8), 1528–1548 CAS.
- Y. Song, P. Zhan and X. Liu, Heterocycle-thioacetic acid motif: a privileged molecular scaffold with potent, broad-ranging pharmacological activities, Curr. Pharm. Des., 2013, 19(40), 7141–7154 CrossRef CAS.
- Y. Song, W. Chen, D. Kang, Q. Zhang, P. Zhan and X. Liu, Old friends in new guise: exploiting privileged structures for scaffold re-evolution/refining, Comb. Chem. High Throughput Screening, 2014, 17(6), 536–553 CrossRef CAS.
- R. Anand, K. D. Gill and A. A. Mahdi, Therapeutics of Alzheimer's disease: Past, present and future, Neuropharmacology, 2014, 76(Pt A), 27–50 CrossRef CAS PubMed.
- H. Zheng, L. M. Weiner, O. Bar-Am, S. Epsztejn, Z. I. Cabantchik, A. Warshawsky, M. B. Youdim and M. Fridkin, Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer's, Parkinson's, and other neurodegenerative diseases, Bioorg. Med. Chem., 2005, 13(3), 773–783 CrossRef CAS PubMed.
- M. B. Youdim, G. Stephenson and D. Ben Shachar, Ironing iron out in Parkinson's disease and other neurodegenerative diseases with iron chelators: a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28, Ann. N. Y. Acad. Sci., 2004, 1012, 306–325 CrossRef CAS PubMed.
- D. B. Shachar, N. Kahana, V. Kampel, A. Warshawsky and M. B. Youdim, Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lesion in rats, Neuropharmacology, 2004, 46(2), 254–263 CrossRef PubMed.
- Y. Avramovich-Tirosh, T. Amit, O. Bar-Am, H. Zheng, M. Fridkin and M. B. Youdim, Therapeutic targets and potential of the novel brain-permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer's disease, J. Neurochem., 2007, 100(2), 490–502 CrossRef CAS PubMed.
- O. Weinreb, S. Mandel, O. Bar-Am, M. Yogev-Falach, Y. Avramovich-Tirosh, T. Amit and M. B. Youdim, Multifunctional neuroprotective derivatives of rasagiline as anti-Alzheimer's disease drugs, Neurotherapeutics, 2009, 6(1), 163–174 CrossRef CAS PubMed.
- S. Gal, M. Fridkin, T. Amit, H. Zheng and M. B. Youdim, M30, a novel multifunctional neuroprotective drug with potent iron chelating and brain selective monoamine oxidase-ab inhibitory activity for Parkinson's disease, J. Neural Transm., Suppl., 2006, 70, 447–456 CAS.
- P. J. Crouch and K. J. Barnham, Therapeutic redistribution of metal ions to treat Alzheimer's disease, Acc. Chem. Res., 2012, 45(9), 1604–1611 CrossRef CAS PubMed.
- (a) P. Zhan and X. Liu, Rationally designed multitarget anti-HIV agents, Curr. Med. Chem., 2013, 20(13), 1743–1758 CrossRef CAS; (b) P. Zhan and X. Liu, Designed multiple ligands: an emerging anti-HIV drug discovery paradigm, Curr. Pharm. Des., 2009, 15(16), 1893–1917 CrossRef CAS.
- L. Wang, G. Esteban, M. Ojima, O. M. Bautista-Aguilera, T. Inokuchi, I. Moraleda, I. Iriepa, A. Samadi, M. B. Youdim, A. Romero, E. Soriano, R. Herrero, A. P. Fernández, R.-M. Murillo, J. Marco-Contelles and M. Unzeta, Donepezil + propargylamine + 8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer's disease, Eur. J. Med. Chem., 2014, 80, 543–561 CrossRef CAS PubMed.
- I. Cacciatore, E. Fornasari, L. Baldassarre, C. Cornacchia, S. Fulle, E. S. Di Filippo, T. Pietrangelo and F. Pinnen, A Potent (R)-alpha-bis-lipoyl derivative containing 8-hydroxyquinoline scaffold: synthesis and biological evaluation of its neuroprotective capabilities in SH-SY5Y human neuroblastoma cells, Pharmaceuticals, 2013, 6(1), 54–69 CrossRef CAS PubMed.
- I. Cacciatore, C. Cornacchia, E. Fornasari, L. Baldassarre, F. Pinnen, P. Sozio, A. Di Stefano, L. Marinelli, A. Dean, S. Fulle, E. S. Di Filippo, R. M. La Rovere, A. Patruno, A. Ferrone and V. Di Marco, A glutathione derivative with chelating and in vitro neuroprotective activities: synthesis, physicochemical properties, and biological evaluation, ChemMedChem, 2013, 8(11), 1818–1829 CrossRef CAS PubMed.
- B. Ghosh, T. Antonio, M. E. Reith and A. K. Dutta, Discovery of 4-(4-(2-((5-hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)ethyl)piperazin-1-yl)quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: in vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson's disease, J. Med. Chem., 2010, 53(5), 2114–2125 CrossRef CAS PubMed.
- S. Gogoi, T. Antonio, S. Rajagopalan, M. Reith, J. Andersen and A. K. Dutta, Dopamine D2/D3 agonists with potent iron chelation, antioxidant and neuroprotective properties: potential implication in symptomatic and neuroprotective treatment of Parkinson's disease, ChemMedChem, 2011, 6(6), 991–995 CrossRef CAS PubMed.
- F. Mao, J. Yan, J. Li, X. Jia, H. Miao, Y. Sun, L. Huang and X. Li, New Multi-Target-Directed Small Molecules Against Alzheimer's Disease: The Combination of Resveratrol and Clioquinol, Org. Biomol. Chem., 2014, 12(31), 5936–5944 CAS.
- A. Martirosyan, S. Leonard, X. Shi, B. Griffith, P. Gannett and J. Strobl, Actions of a histone deacetylase inhibitor NSC3852 (5-nitroso-8-quinolinol) link reactive oxygen species to cell differentiation and apoptosis in MCF-7 human mammary tumor cells, J. Pharmacol. Exp. Ther., 2006, 317(2), 546–552 CrossRef CAS PubMed.
- S. H. Chan, C. H. Chui, S. W. Chan, S. H. Kok, D. Chan, M. Y. Tsoi, P. H. Leung, A. K. Lam, A. S. Chan, K. H. Lam and J. C. Tang, Synthesis of 8-hydroxyquinoline derivatives as novel antitumor agents, ACS Med. Chem. Lett., 2012, 4(2), 170–174 CrossRef PubMed.
- (a) V. Moret, Y. Laras, T. Cresteil, G. Aubert, D. Q. Ping, C. Di, M. Barthélémy-Requin, C. Béclin, V. Peyrot, D. Allegro, A. Rolland, F. De Angelis, E. Gatti, P. Pierre, L. Pasquini, E. Petrucci, U. Testa and J. L. Kraus, Discovery of a new family of bis-8-hydroxyquinoline substituted benzylamines with pro-apoptotic activity in cancer cells: synthesis, structure–activity relationship, and action mechanism studies, Eur. J. Med. Chem., 2009, 44(2), 558–567 CrossRef CAS PubMed; (b) C. Bruyère, S. Madonna, G. Van Goietsenoven, V. Mathieu, J. Dessolin, J. L. Kraus, F. Lefranc and R. Kiss, JLK1486, a Bis 8-Hydroxyquinoline-Substituted Benzylamine, Displays Cytostatic Effects in Experimental Gliomas through MyT1 and STAT1 Activation and, to a Lesser Extent, PPARγ Activation, Transl. Oncol., 2011, 4(3), 126–137 CrossRef; (c) S. Madonna, C. Béclin, Y. Laras, V. Moret, A. Marcowycz, D. Lamoral-Theys, J. Dubois, M. Barthelemy-Requin, G. Lenglet, S. Depauw, T. Cresteil, G. Aubert, V. Monnier, R. Kiss, M. H. David-Cordonnier and J. L. Kraus, Structure–activity relationships and mechanism of action of antitumor bis 8-hydroxyquinoline substituted benzylamines, Eur. J. Med. Chem., 2010, 45(2), 623–638 CrossRef CAS PubMed; (d) S. Madonna, A. Marcowycz, D. Lamoral-Theys, G. Van Goietsenoven, J. Dessolin, C. Pirker, S. Spiegl-Kreinecker, C.-A. Biraboneye, W. Berger and R. Kiss, Bis-8-hydroxyquinoline and bis-8-hydroxyquinaldine N-substituted amines: a single methyl group structural difference between the two heterocycles, which modulates the antiproliferative effects, J. Heterocycl. Chem., 2010, 47(3), 719–723 CAS.
- G. Lescoat, S. Léonce, A. Pierré, L. Gouffier and F. Gaboriau, Antiproliferative and iron chelating efficiency of the new bis-8-hydroxyquinoline benzylamine chelator S1 in hepatocyte cultures, Chem.-Biol. Interact., 2012, 195(2), 165–172 CrossRef CAS PubMed.
- A. Y. Shaw, C. Y. Chang, M. Y. Hsu, P. J. Lu, C. N. Yang, H. L. Chen, C. W. Lo, C. W. Shiau and M. K. Chern, Synthesis and structure–activity relationship study of 8-hydroxyquinoline-derived Mannich bases as anticancer agents, Eur. J. Med. Chem., 2010, 45(7), 2860–2867 CrossRef CAS PubMed.
- L. B. de O Freitas, T. F. Borgati, R. P. de Freitas, A. L. Ruiz, G. M. Marchetti, J. E. de Carvalho, E. F. da Cunha, T. C. Ramalho and R. B. Alves, Synthesis and antiproliferative activity of 8-hydroxyquinoline derivatives containing a 1,2,3-triazole moiety, Eur. J. Med. Chem., 2014, 84, 595–604 CrossRef PubMed.
- I. Sosič, B. Mirković, K. Arenz, B. Stefane, J. Kos and S. Gobec, Development of new cathepsin B inhibitors: combining bioisosteric replacements and structure-based design to explore the structure–activity relationships of nitroxoline derivatives, J. Med. Chem., 2013, 56(2), 521–533 CrossRef PubMed.
- S. Bhat, J. S. Shim, F. Zhang, C. R. Chong and J. O. Liu, Substituted oxines inhibit endothelial cell proliferation and angiogenesis, Org. Biomol. Chem., 2012, 10(15), 2979–2992 CAS.
- F. Sliman, M. Blairvacq, E. Durieu, L. Meijer, J. Rodrigo and D. Desmaële, Identification and structure–activity relationship of 8-hydroxy-quinoline-7-carboxylic acid derivatives as inhibitors of Pim-1 kinase, Bioorg. Med. Chem. Lett., 2010, 20(9), 2801–2805 CrossRef CAS PubMed.
- A. C. Good, J. Liu, B. Hirth, G. Asmussen, Y. Xiang, H. P. Biemann, K. A. Bishop, T. Fremgen, M. Fitzgerald, T. Gladysheva, A. Jain, K. Jancsics, M. Metz, A. Papoulis, R. Skerlj, J. D. Stepp and R. R. Wei, Implications of promiscuous Pim-1 kinase fragment inhibitor hydrophobic interactions for fragment-based drug design, J. Med. Chem., 2012, 55(6), 2641–2648 CrossRef CAS PubMed.
- D. J. Richard, R. Lena, T. Bannister, N. Blake, W. E. Pierceall, N. E. Carlson, C. E. Keller, M. Koenig, Y. He, D. Minond, J. Mishra, M. Cameron, T. Spicer, P. Hodder and M. H. Cardone, Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker, Bioorg. Med. Chem., 2013, 21(21), 6642–6649 CrossRef CAS PubMed.
- M. O. Oyewumi, A. Alazizi, S. Liva, L. Lin and W. J. Geldenhuys, Screening and identification of novel compounds with potential anti-proliferative effects on gallium-resistant lung cancer through an AXL kinase pathway, Bioorg. Med. Chem. Lett., 2014, 24(18), 4553–4556 CrossRef CAS PubMed.
- C. Loenarz and C. J. Schofield, Expanding chemical biology of 2-oxoglutarate oxygenases, Nat. Chem. Biol., 2008, 4(3), 152–156 CrossRef CAS PubMed.
- R. J. Hopkinson, A. Tumber, C. Yapp, R. Chowdhury, W. Aik, K. H. Che, X. S. Li, J. B. L. Kristensen, O. King, M. C. Chan, K. K. Yeoh, H. Choi, L. J. Walport, C. C. Thinnes, J. T. Bush, C. Lejeune, A. M. Rydzik, N. R. Rose, E. A. L. Bagg, M. A. McDonough, T. Krojer, W. W. Yue, S. S. Ng, L. Olsen, P. Brennan, U. Oppermann, S. Muller-Knapp, R. J. Klose, P. J. Ratcliffe, C. J. Schofield and A. Kawamura, 5-Carboxy-8-hydroxyquinoline is a broad spectrum 2-oxoglutarate oxygenase inhibitor which causes iron translocation, Chem. Sci., 2013, 4, 3110–3117 RSC.
- (a) O. N. King, X. S. Li, M. Sakurai, A. Kawamura, N. R. Rose, S. S. Ng, A. M. Quinn, G. Rai, B. T. Mott, P. Beswick, R. J. Klose, U. Oppermann, A. Jadhav, T. D. Heightman, D. J. Maloney, C. J. Schofield and A. Simeonov, Quantitative high-throughput screening identifies 8-hydroxyquinolines as cell-active histone demethylase inhibitors, PLoS One, 2010, 5(11), e15535 Search PubMed; (b) W. Aik, M. Demetriades, M. K. Hamdan, E. A. Bagg, K. K. Yeoh, C. Lejeune, Z. Zhang, M. A. McDonough and C. J. Schofield, Structural basis for inhibition of the fat mass and obesity associated protein (FTO), J. Med. Chem., 2013, 56(9), 3680–3688 CrossRef CAS PubMed.
- R. Schiller, G. Scozzafava, A. Tumber, J. R. Wickens, J. T. Bush, G. Rai, C. Lejeune, H. Choi, T. L. Yeh, M. C. Chan, B. T. Mott, J. S. McCullagh, D. J. Maloney, C. J. Schofield and A. Kawamura, A cell-permeable ester derivative of the JmjC histone demethylase inhibitor IOX1, ChemMedChem, 2014, 9(3), 566–571 CrossRef CAS PubMed.
- (a) C. H. Arrowsmith, C. Bountra, P. V. Fish, K. Lee and M. Schapira, Epigenetic protein families: a new frontier for drug discovery, Nat. Rev. Drug Discovery, 2012, 11(5), 384–400 CrossRef CAS PubMed; (b) C. C. Thinnes, K. S. England, A. Kawamura, R. Chowdhury, C. J. Schofield and R. J. Hopkinson, Targeting histone lysine demethylases – Progress, challenges, and the future, Biochim. Biophys. Acta, 2014, S1874, DOI:10.1016/j.bbagrm.2014.05.009 , epub ahead of print.
- D. Rotili, S. Tomassi, M. Conte, R. Benedetti, M. Tortorici, G. Ciossani, S. Valente, B. Marrocco, D. Labella, E. Novellino, A. Mattevi, L. Altucci, A. Tumber, C. Yapp, O. N. King, R. J. Hopkinson, A. Kawamura, C. J. Schofield and A. Mai, Pan-histone demethylase inhibitors simultaneously targeting Jumonji C and lysine-specific demethylases display high anticancer activities, J. Med. Chem., 2014, 57(1), 42–55 CrossRef CAS PubMed.
- G. Rai, A. Kawamura, A. Tumber, Y. Liang, J. L. Vogel, J. H. Arbuckle, N. R. Rose, T. S. Dexheimer, T. L. Foley, O. N. King, A. Quinn, B. T. Mott, C. J. Schofield, U. Oppermann, A. Jadhav, A. Simeonov, T. M. Kristie and D. J. Maloney, Discovery of ML324, a JMJD2 demethylase inhibitor with demonstrated antiviral activity, SourceProbe Reports from the NIH Molecular Libraries Program [Internet], National Center for Biotechnology Information (US), Bethesda (MD), 2010–2012 Dec 17, updated 16 Sep 2013 Search PubMed.
- X. J. Wang and L. B. Si, Advances on hypoxia inducible factor-1, Chin. Med. J., 2013, 126(18), 3567–3571 CAS.
- H. Moon, S. Han, H. Park and J. Choe, Crystal structures of human FIH-1 in complex with quinol family inhibitors, Mol. Cells, 2010, 29(5), 471–474 CrossRef CAS PubMed.
- J. A. Jacobsen, J. L. Fullagar, M. T. Miller and S. M. Cohen, Identifying chelators for metalloprotein inhibitors using a fragment-based approach, J. Med. Chem., 2011, 54(2), 591–602 CrossRef CAS PubMed.
- K. Hanaya, M. Suetsugu, S. Saijo, I. Yamato and S. Aoki, Potent inhibition of dinuclear zinc(II) peptidase, an aminopeptidase from Aeromonas proteolytica, by 8-quinolinol derivatives: inhibitor design based on Zn2+ fluorophores, kinetic, and X-ray crystallographic study, J. Biol. Inorg. Chem., 2012, 17(4), 517–529 CrossRef CAS PubMed.
- K. Mekouar, J. F. Mouscadet, D. Desmaële, F. Subra, H. Leh, D. Savouré, C. Auclair and J. d'Angelo, Styrylquinoline derivatives: a new class of potent HIV-1 integrase inhibitors that block HIV-1 replication in CEM cells, J. Med. Chem., 1998, 41(15), 2846–2857 CrossRef CAS PubMed.
- S. M. Cohen, A. Agrawal, J. Desoto, Y. Pommier and K. Maddali, Preparation of raltegravir-chelator derivatives as HIV integrase inhibitors, PCT Int. Appl., WO 2012106534 A2, 2012, p. 20120809.
- E. Serrao, B. Debnath, H. Otake, Y. Kuang, F. Christ, Z. Debyser and N. Neamati, Fragment-Based Discovery of 8-Hydroxyquinoline Inhibitors of the HIV-1 Integrase-Lens Epithelium-Derived Growth Factor/p75 (IN-LEDGF/p75) Interaction, J. Med. Chem., 2013, 56(6), 2311–2322 CrossRef CAS PubMed.
- R. Musiol, J. Jampilek, V. Buchta, L. Silva, H. Niedbala, B. Podeszwa, A. Palka, K. Majerz-Maniecka, B. Oleksyn and J. Polanski, Antifungal properties of new series of quinoline derivatives, Bioorg. Med. Chem., 2006, 14(10), 3592–3598 CrossRef CAS PubMed.
- M. J. McKay, A. R. Carroll and R. J. Quinn, Perspicamides A and B, quinolinecarboxylic acid derivatives from the Australian ascidian Botrylloides perspicuum, J. Nat. Prod., 2005, 68(12), 1776–1778 CrossRef CAS PubMed.
- A. K. Pandey, R. Sharma, R. Shivahare, A. Arora, N. Rastogi, S. Gupta and P. M. Chauhan, Synthesis of perspicamide A and related diverse analogues: their bioevaluation as potent antileishmanial agents, J. Org. Chem., 2013, 78(4), 1534–1546 CrossRef CAS PubMed.
- A. F. Eweas, G. Allam, A. S. Abuelsaad, A. H. Alghamdi and I. A. Maghrabi, Design, synthesis, anti-schistosomal activity and molecular docking of novel 8-hydroxyquinoline-5-sufonyl 1,4-diazepine derivatives, Bioorg. Chem., 2013, 46, 17–25 CrossRef CAS PubMed.
- G. C. Capodagli, W. G. Sedhom, M. Jackson, K. A. Ahrendt and S. D. Pegan, A noncompetitive inhibitor for Mycobacterium tuberculosis's class IIa fructose 1,6-bisphosphate aldolase, Biochemistry, 2014, 53(1), 202–213 CrossRef CAS PubMed.
- D. Caglič, M. C. Krutein, K. M. Bompiani, D. J. Barlow, G. Benoni, J. C. Pelletier, A. B. Reitz, L. L. Lairson, K. L. Houseknecht, G. R. Smith and T. J. Dickerson, Identification of clinically viable quinolinol inhibitors of botulinum neurotoxin A light chain, J. Med. Chem., 2014, 57(3), 669–676 CrossRef PubMed.
- N. Audic, G. Potier and N. A. Sasaki, New 2,3-diaminopropionic acid inhibitors of AGE and ALE formation, Org. Biomol. Chem., 2013, 11(5), 773–780 CAS.
- H. P. Zeng, T. T. Wang, X. H. Ouyang, Y. D. Zhou, H. L. Jing, G. Z. Yuan, D. F. Chen, S. H. Du, H. Li and J. H. Zhou, 8-Hydroxyquinoline derivatives induce the proliferation of rat mesenchymal stem cells (rMSCs), Bioorg. Med. Chem., 2006, 14(16), 5446–5450 CrossRef CAS PubMed.
- S. Ariyasu, A. Sawa, A. Morita, K. Hanaya, M. Hoshi, I. Takahashi, B. Wang and S. Aoki, Design and synthesis of 8-hydroxyquinoline-based radioprotective agents, Bioorg. Med. Chem., 2014, 22(15), 3891–3905 CrossRef CAS PubMed.
- Y. Chen, H. Wang, L. Wan, Y. Bian and J. Jiang, 8-Hydroxyquinoline-substituted boron-dipyrromethene compounds: synthesis, structure, and OFF–ON–OFF type of pH-sensing properties, J. Org. Chem., 2011, 76(10), 3774–3781 CrossRef CAS PubMed.
- G. Farruggia, S. Iotti, M. Lombardo, C. Marraccini, D. Petruzziello, L. Prodi, M. Sgarzi, C. Trombini and N. Zaccheroni, Microwave assisted synthesis of a small library of substituted N,N′-bis((8-hydroxy-7-quinolinyl)methyl)-1,10-diaza-18-crown-6 ethers, J. Org. Chem., 2010, 75(18), 6275–6278 CrossRef CAS PubMed.
- J. A. González-Vera, E. Luković and B. Imperiali, Synthesis of red-shifted 8-hydroxyquinoline derivatives using click chemistry and their incorporation into phosphorylation chemosensors, J. Org. Chem., 2009, 74(19), 7309–7314 CrossRef PubMed.
- J. P. Heiskanen, W. A. Omar, M. K. Ylikunnari, K. M. Haavisto, M. J. Juan and O. E. Hormi, Synthesis of 4-alkoxy-8-hydroxyquinolines, J. Org. Chem., 2007, 72(3), 920–922 CrossRef CAS PubMed.
- F. Babudri, A. Cardone, C. T. Cioffi, G. M. Farinola, F. Naso and R. Ragni, A straightforward methodology for the introduction of aryl and vinyl substituents in the 5 or 7 position of 8-hydroxyquinoline, Synthesis, 2006, 8, 1325–1332 CrossRef PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |