
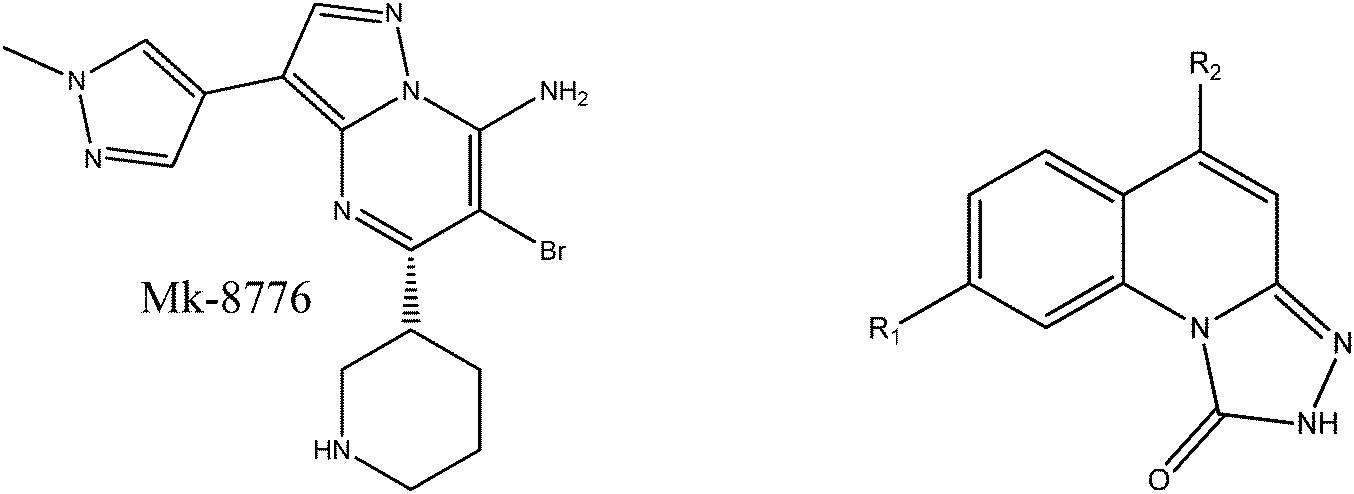
Computational studies on the binding mechanism between triazolone inhibitors and Chk1 by molecular docking and molecular dynamics
Min
Lv
,
Shuying
Ma
,
Yueli
Tian
,
Xiaoyun
Zhang
*,
Wenjuan
Lv
and
Honglin
Zhai
College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, Gansu Province, People's Republic of China. E-mail: xyzhang@lzu.edu.cn; Fax: +86 931 8912582; Tel: +86 931 8912578
First published on 28th October 2014
Abstract
Chk1, a serine/threonine protein kinase that participates in transducing DNA damage signals, is an attractive target due to its involvement in tumor initiation and progression. As a novel Chk1 inhibitor, the triazolone's bioactivity mechanism is not clear. In this study, we carried out an integrated computational study that combines molecular docking, molecular dynamics (MD) simulations, and binding free energy calculations to identify the key factors necessary for the bioactivities. With the aim of discerning the structural features that affect the inhibitory activity of triazolones, MK-8776, a Chk1 inhibitor that reached the clinical stage, was also used as a reference for simulations. A comparative analysis of the triazolone inhibitors at the molecular level offers valuable insight into the structural and energetic properties. A general feature is that all the studied inhibitors bind in the pocket characterized by residues Leu14, Val22, Ala35, Glu84, Tyr85, Cys86, and Leu136 of Chk1. Moreover, introducing hydrophobic groups into triazolone inhibitors is favorable for binding to Chk1, which is corroborated by residue Leu136 with a relatively large difference in the contribution between MK-8776 and five triazolones to the total binding free energies. A hydrogen bond between the polar hydrogen atoms at R1 and Cys86 can facilitate proper placement of the inhibitor in the binding pocket of Chk1 that favors binding. However, the introduction of hydrophilic groups into the R2 position diminishes binding affinity. The information provided by this research is of benefit for further rational design of novel promising inhibitors of Chk1.
1. Introduction
Checkpoint Kinases (Chks) are serine/threonine protein kinases which mediate the cellular response to DNA damage and coordinate cell cycle arrest. Chk1 and Chk2, as two isoforms of the Chk family, are not structurally related but functionally overlapping serine/threonine kinases subjected to multitudinous genotoxic insults.1–6 However, there is considerable variation in the biological requirements for Chk1 and Chk2 function in that Chk1 but not Chk2 is indispensable for mammalian development and viability.5Chk1 is a protein kinase that participates in the regulation of the G2/M checkpoint by preventing aberrant entry of damaged cells.7,8 A wide range of tumors exhibit defective control in the G1/S-phase checkpoint, which can be attributed to mutational events.9 Due to the dysfunction on the G1/S-phase checkpoint, cancer cells mainly rely on the G2/M checkpoint for halting the cell cycle and maintaining the genomic repair.8 In light of the involvement of Chk1 in the DNA damage response, Chk1 has now emerged as a candidate tumor suppressor in many malignancies, e.g. lung cancer, ovarian cancer, acute myeloid leukemia, breast cancer, colon cancer, pancreatic cancer and head-and-neck carcinoma.8 Despite the extensive applications in cancer therapy, chemotherapy and ionizing radiation lead to serious side effects by harming normal cells.5,10,11 Accumulating studies have proved that Chk1 inhibitors may selectively enhance the activity of genotoxic agents in tumors, whereas normal cells with competent DNA damage response will be rescued.9 Taken together, the pharmacological inhibition of Chk1 by abrogating the remaining intact checkpoint is a potential combination anticancer therapy.
In recent years, a variety of Chk1 inhibitors with different scaffolds have been designed and synthesized,9,12–29 such as ureas,12 thioquinazolinones,19 and thienopyridines.21 Because of dose-limiting toxicity and poor pharmacokinetics, many inhibitors possess modest therapeutic efficacy. To date, anticancer drugs AZD7762, Ly2603618, MK-8776, CHIR-124 and PF-477736 targeting Chk1 have entered clinical trials.30–34 Especially, a triazolone class of Chk1 inhibitors with good enzymatic and cellular activities were synthesized and evaluated by Vibha Oza and coworkers.22,28 In Vibha Oza et al.'s work, the novel scaffolds were sought using a HTS screen by a Chk1 scintillation proximity assay and more potent inhibitors were obtained through structure based drug design. However, the basis of the bioactivity variation of the triazolone inhibitors is not clear yet.22,28 As a novel class of Chk1 inhibitors, triazolones elicited our interest and attention because their scaffolds are similar to those of MK-8776 that reached the clinical stage.
In the present work, we carried out molecular modeling approaches including molecular docking, molecular dynamics and binding free energy calculations to study the triazolone inhibitors and MK-8776 systematically.22,28 The goal was to: (1) determine the binding geometries of triazolones and MK-8776; (2) highlight the key energetic and structural differences between triazolones and MK-8776; and (3) investigate the crucial structural elements responsible for the bioactivities of triazolone inhibitors. All these computational and theoretical studies can provide some valuable clues to the design of more promising inhibitors.
2. Materials and methods
2.1. Biological data
In Vibha Oza et al.'s work, the studied molecules share a similar scaffold with different substituents at different positions, covering a wide range of IC50 from 0.1 nM to 76.8 μM.22,28 In order to pinpoint features leading to improved potency, we selected five representative compounds with double substitutions to the basis scaffold and five orders of magnitude in bioactivities. The remarkable potency of inhibitor 5a to inhibit Chk1 (IC50 = 0.1 nM) suggests that this inhibitor deserves further study. The introduction of a thiazolyl moiety at the R1 position in compound 31 drastically lowers the potency (IC50 = 2.96 μM), whereas imidazolyl substitution to that position in 37 leads to a weak inhibition of Chk1 (IC50 = 0.17 μM). A hydroxymethyl group connected to the R2 position in compound 16e results in an IC50 value of 20 nM, which is comparable to 5a. A 4-pyridinyl group at the R1 position and an amino group at the R2 position in compound 14k significantly affect the potency of the IC50 value (IC50 = 1.1 μM) in comparison to 5a. On the basis of the above analysis, it can be concluded that modifications at one or two sites lead to inhibitory effects at varying degrees compared with 5a and the other four compounds, 16e, 31, 37 and 14k, were therefore included in further studies. In order to gain deep insight into the energetic and structural features of triazolones, MK-8776 was also used as a reference for simulations. The structures of these inhibitors and the corresponding biological activities (IC50 and pIC50) are summarized in Table 1. Here, half maximal inhibitory concentration, represented as IC50, was measured as a function of the ability of the compounds to inhibit Chk1 in the Chk1 SPA assay. For further investigation, IC50 values were converted into pIC50 values by representing the logarithmic values. Generally, the biological activity of a compound against a receptor is largely determined by the binding, which rests upon the geometric and energetic behavior. Therefore it is crucial to identify the binding location of a biologically active molecule on the protein in order to understand its efficacy as a therapeutic agent.|
|
||||
|---|---|---|---|---|
| No. | R1 | R2 | IC50 (nM) | pIC50 |
| MK-8776 | — | — | 3 | 8.52 |
| 5a |

|
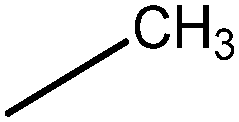
|
0.1 | 10.00 |
| 16e |

|

|
20 | 7.70 |
| 31 |
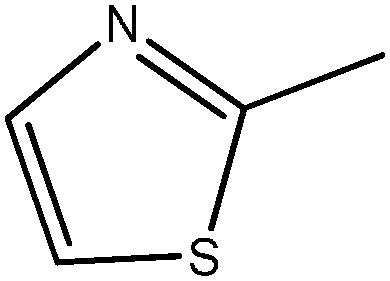
|
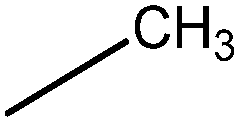
|
2960 | 5.53 |
| 37 |

|
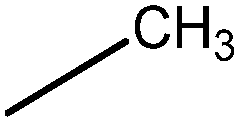
|
160 | 6.80 |
| 14k |

|

|
1100 | 6.89 |
2.2. Protein preparation
Herein, the X-ray crystal structure of Chk1 in the complex with inhibitor 5a was downloaded from the Protein Database Bank (PDB code: 2YEX) and further used as the template for molecular docking.28 Then, the structure was prepared using the protein preparation wizard, which is available in Schrödinger 9.0.35 All crystallographic water molecules were removed from the complex. Missing residues were added and refined. All hydrogen atoms were added which were subsequently minimized using optimized potentials for liquid simulations (OPLS) 2005 force field and the impact molecular mechanics engine.36 Minimization was performed to relieve steric clashes until the average root mean square deviation (RMSD) of the nonhydrogen atoms reached a maximum cutoff of 0.3 Å.2.3. Ligand preparation
The 3D molecular structures of all the studied inhibitors were constructed using Maestro and processed by using the LigPrep in Schrödinger,35 which can generate a number of structures from each input structure with various ionization states, tautomers, stereochemistries, and ring conformations, and eliminate molecules using various criteria including the molecular weight or specified number and types of functional groups present.352.4. Molecular docking
To identify the recognition between the Chk1 and inhibitors, molecular docking simulations were carried out on the 3-D structure of Chk1 since it represents the pharmacology target for the development of new drugs to treat cancer.In order to soften the potential for nonpolar parts of the receptor, the scaling factor for protein van der Waals radii was set as 0.8 in the receptor grid generation with a partial atomic charge of 0.25. The mass center of the ligand in the active site was applied as the centroid to generate the grid file for the subsequent docking process. No constraints were used for all the docking processes. The default grid size was adopted from the Grid program. Rigid receptor docking (RRD) of all the compounds to Chk1 was carried out using Glide program in the extra-precision mode.37 In the docking process, the protein was held rigid and the docked ligand was free to move. Upon completion of each docking calculation, at most 100 poses per ligand were generated. The best docked structure was chosen using a Glidescore (Gscore) function.38 The Gscore is a modified and extended version of the empirically based Chemscore function. Another scoring function used by Glide is Emodel, which itself derived from a combination of the Gscore, Coulombic, van der Waals and the strain energy of the ligand.39 The lowest-energy docked complex was found in the majority of similar docking conformations. Finally, the lowest-energy docked complex was selected for further study.
2.5. Molecular dynamic simulation
To bring back an all-atoms level of detail, the docked structures of Chk1 in complex with inhibitors 5a, 31, 37, 16e and 14k were used as the starting structures for the subsequent MD simulations.Prior to MD simulations, geometric optimization and the electrostatic potential calculations were performed on all the represented inhibitors at the HF/6-31G* level using the Gaussian 09 program.40 The general AMBER force field (gaff)41 was used for the inhibitors and the ff99SBildn force field42 was used for the protein. The parameters of the ligands were assigned by the restrained Electrostatic potential (RESP) procedure partial charges with the ANTECHAMBER module in the AMBER11.43 All missing hydrogen atoms were added by application of the LEaP module in the AMBER 11 software package.43 Then, all solutes were solvated in a rectangular periodic box of pre-equilibrated TIP3P44 water molecules with a margin distance of 12 Å. An appropriate number of sodium counter ions were placed on grids with the largest positive coulombic potentials around the complexes to maintain the electro-neutrality of all the systems.
All the molecular dynamic simulations were carried out using the AMBER11 package.43 The energy minimization and equilibration protocols of all five complexes were performed using the Sander program.45 Each system was then subjected to three consecutive minimization steps; in each step, water molecules and ions were allowed to move freely for a 1000 steps of steepest descent minimization followed by 3000 steps of conjugate gradient minimization holding protein and inhibitor atoms constrained to their original positions by a force constraint of 10 kcal mol−1 Å−1. Then each system was gradually heated in the NVT ensemble from 0 to 298.15 K for 50 ps. Subsequently, five MD equilibrations of 50 ps each with a decreased restraint weights from 10.0 to 5.0, to 2.0, to 1.0, to 0.5, and to 0.1 were carried out. These were followed by the last MD equilibration step of 150 ps without any restraint on each system. During the production simulations, PMEMD program supporting CUDA in AMBER 11 was used to improve the efficiency of the simulations. Unrestrained MD simulations were performed for 15 ns in the NPT ensemble at a temperature of 298.15 K and a pressure of 1 atm. During the simulations, periodic boundary conditions were employed and all electrostatic interactions were calculated using the particle-mesh Ewald (PME) method with a dielectric constant of unity. A 10.0 Å cutoff was used to calculate the direct space sum of PME.46 The SHAKE algorithm was used to re-strain bond lengths involving hydrogen atoms.47
2.6. Binding free energy calculations (MM-GBSA)
The binding free energy (ΔEbind) for all the studied inhibitors was calculated by the MM/GBSA methods48–53 on the basis of the obtained stable MD trajectories. A total number of 400 snapshots were extracted every 10 ps from the last 4 ns of each MD trajectory. For each snapshot, a free energy was calculated for each molecular species (complex, receptor, and ligand), and the binding free energy was calculated by the following equation:| ΔEbind = Gcomplex − Gprotein − Gligand = ΔH + ΔGsolvation − TΔS = ΔEMM + ΔGGB + ΔGSA − TΔS |
2.7. MM/GBSA binding energy decomposition analysis
In order to gain deep insight into the contribution of each residue, MM-GBSA free energy decomposition in the mm_pbsa.pl program integrated in AMBER11 was performed to decompose the interaction energies into individual residues.43,56–61 The residue-inhibitor binding interaction consists of four components: van der Waals contribution (ΔGvdw), electrostatic contribution (ΔGele), the polar part of desolvation (ΔGGB) and the non-polar part of desolvation (ΔGSA).| ΔG = ΔGvdw + ΔGele + ΔGsol = ΔGvdw + ΔGele + ΔGGB + ΔGSA |
3. Results and discussion
3.1. Validation of the molecular docking performance
The most straightforward method for evaluating the accuracy of molecular docking is to determine the discrepancy between the real and the best-scoring conformation.39,62,63 If the root mean square deviation (RMSD) value between the real and the best-scoring conformations is equal to or less than 2.0 Å, the docking process was considered to be successful, probably because the resolution in an X-ray crystal structure analysis is often about 2 Å.39,62,63 Here, the Extra Precision Glide docking procedure was validated by redocking the co-crystallized 5a to Chk1. The root mean square deviations between the predicted conformation and the observed X-ray crystallographic conformation of compound 5a equaled 1.1 Å, suggesting that Glide docking in reproducing the experimentally observed binding mode for Chk1 kinase inhibitors is reliable.3.2. Initial orientation of the inhibitors in the Chk1 active site
Molecular docking simulation was performed to determine the initial orientation of the inhibitors in the binding pocket of the protein. Fig. 1 shows the alignments of the poses of the triazolone inhibitors, and all the structures overlap well in the binding pocket. Fig. 2 shows the two dimensional inhibitor–protein diagrams of the five inhibitors. The hydrogen bond was characterized by distance (<3.5 Å) and orientation (the angle D–H⋯A > 120°).64 Obviously, all the inhibitors bind with the pocket characterized by residues Leu14, Val22, Ala35, Leu83, Glu84, Tyr85, Cys86, Gly89, Glu90, Leu136 and Ser146. For the poses of 5a and 16e, the five-member ring of triazolone binds in the deep hinge region via two hydrogen bonds with two residues Glu84 and Cys86. In addition, nitrogen of the pyrrole ring acts as a hydrogen acceptor to form a hydrogen bond with the hydroxyl group of Cys86. However, the pose of 16e shows a hydrogen bond formed between the hydroxyl moiety and Glu90. In contrast, inhibitors 31, 14k, and 37 form one hydrogen bond with Cys86, which provides evidence for their lower bioactivity. MK-8776 participates in forming two hydrogen bonds with residues Glu84 and Glu133. | ||
| Fig. 1 The alignments of five docked triazolones in the binding site of Chk1 (the inhibitors are represented as sticks and the protein is represented as a cartoon). | ||
 | ||
| Fig. 2 2D inhibitor–protein interaction diagram of (a) the crystal structure of Chk1–5a; (b) the crystal structure of Chk1–16e; (c) the best scoring docking pose of Chk1–31; (d) the best scoring docking pose of Chk1–14k; (e) the best scoring docking pose of Chk1–37; and (f) the best scoring docking pose of Chk1–MK-8776. | ||
3.3. Validation of the MD simulations
In order to bring back an all-atom level of detail, molecular dynamics simulations of five systems were performed within 12 ns. Energetic and structural properties were monitored during the entire MD simulation of each complex to evaluate the quality of the MD simulations. As shown in Fig. 3a and b, root-mean-square-deviation (RMSD) of all protein backbone atoms (Cα) and the ligand heavy atoms relative to the starting structures is monitored, respectively. Moreover, Fig. 3c plots the RMSDs of the Cα for the residues located in the binding pocket relative to their initial structures of MD simulation. According to Fig. 3a and c, the RMSD values of each system tend to converge after 8 ns, suggesting that the studied complexes reached the equilibrium state. From Fig. 3b, the RMSD values of each ligand are always stable along the entire MD trajectory of each complex, indicating that the initial structures for MD simulation are reasonable. In conclusion, the last 4 ns of the five complex simulations were taken for further analysis. | ||
| Fig. 3 Plots of the RMSD values relative to the initial structure of the Chk1–inhibitor complexes (MK-8776, 5a, 16e, 31, 37 and 14k) during the MD simulations. (a) Time evolution of the RMSD of all protein backbone atoms. (b) Time evolution of the heavy atoms for the ligand. (c) Time evolution of the RMSD of Cα atoms for the residues around 5 Å of the ligand. | ||
3.4. Binding free energy analysis
In order to gain further insight into the interactions between drugs and Chk1, the MM/GBSA method was applied to calculate the absolute binding free energy, which was divided into several contribution terms. Table 2 shows the energetic components and the total binding free energies averaged over 400 snapshots extracted evenly from the last 4 ns MD trajectories for every complex. As is shown in Table 2, the calculated binding free energies of five triazolone inhibitors in complex with Chk1 are −45.64, −35.28, −38.05, and −38.66 kcal mol−1, respectively. In addition, MK-8776 exhibits a higher binding affinity to Chk1 than all the triazolone inhibitors with the exception of 5a. Obviously, the predicted binding affinities of all the studied inhibitors are in concordance with the experimental values, which proves the reliance of the MD-simulated models and computational protocol used in this study. For all the complexes, the favorable electrostatic energies in the gas phase are opposed by the unfavorable polar solvation energies, resulting in the little polar contribution to the total free energies. In contrast, the favorable van der Waals energies and the favorable non-polar solvation energies owing to the burial of ligand's hydrophobic groups are the dominating force for inhibitor binding. In addition, to gain more information about the surrounding residues and their contribution to the whole system, the total binding free energies for five complexes were decomposed on a per-residue level using MM/PBSA.| No. | ΔEvdw | ΔEele | ΔGSA | ΔGGB | ΔGpred | pIC50 |
|---|---|---|---|---|---|---|
| MK-8776 | −46.4 ± 2.46 | −22.70 ± 3.51 | −5.44 ± 0.10 | 32.47 ± 2.67 | −42.07 ± 2.85 | 8.52 |
| 5a | −37.25 ± 2.33 | −31.38 ± 2.80 | −4.50 ± 0.09 | 27.49 ± 1.66 | −45.64 ± 2.63 | 10.00 |
| 16e | −35.22 ± 2.45 | −36.34 ± 8.38 | −4.64 ± 0.13 | 37.19 ± 6.56 | −39.02 ± 3.20 | 7.70 |
| 31 | −35.45 ± 2.67 | −20.41 ± 3.15 | −4.77 ± 0.12 | 25.35 ± 2.71 | −35.28 ± 2.89 | 5.53 |
| 37 | −33.58 ± 2.26 | −24.13 ± 3.21 | −4.40 ± 0.13 | 24.06 ± 2.42 | −38.05 ± 2.38 | 6.80 |
| 14k | −36.89 ± 2.43 | −30.51 ± 3.97 | −4.71 ± 0.12 | 33.46 ± 3.28 | −38.66 ± 3.18 | 6.89 |
The energy contributions from each residue are summarized in Fig. 4. A general feature is that the major favorable energy contributions result predominantly from the residues Leu14, Val22, Ala35, Glu84, Tyr85, Cys86, and Leu136 of Chk1, which is common in all six complexes. Prominent among these residues are Cys86 and Glu84, which form stable hydrogen bonds with each inhibitor, respectively. In addition, Leu14, Val22, Ala35, Val67, Leu83, Tyr85, Leu88, and Leu136 are hydrophobic so that they form van der Waals force with the inhibitors, leading to the dominant role of non-polar contribution in the binding free energy. Special attention should be paid to those residues with relatively large difference of the contribution between MK-8776 and five triazolones to the total binding free energies. The residue Leu136 contributes more for the binding of MK-8776 than that of the other inhibitors by about 1 kcal mol−1, which can be attributed to the stronger van der Waals interactions between the residue and the six-membered ring of MK-8776. So it is possible that introducing hydrophobic groups into triazolone inhibitors is favorable for binding to Chk1. The rest of the residues have almost the same energy contributions to the total binding free energy. It is essential for further rational design of more potent inhibitors targeting Chk1 to investigate the energetic and structural requirements of the ligand to enhance the binding affinity. Therefore, we performed structural binding mode analysis in addition to detailed energetic analysis to compare the two inhibitors in each inhibitor pair (5a–16e, 5a–31, 5a–37 or 5a–14k) in detail.
 | ||
| Fig. 4 Inhibitor–residue interaction spectra for (a) the Chk1–MK-8776 and Chk1–5a complex, (b) the Chk1–MK-8776 and Chk1–16e complex, (c) the Chk1–MK-8776 and Chk1–31 complex, (d) the Chk1–MK-8776 and Chk1–37 complex, and (e) the Chk1–MK-8776 and Chk1–14k complex. | ||
Compound 16e contains a hydroxymethyl group at R2, instead of a methyl group, as in compound 5a. One small change in structure leads to the lower activity of 16e compared to that of 5a. The calculated binding free energy for the Chk1–16e complex is lower than that for Chk1–5a, on average with the difference of 6.62 kcal mol−1, in accord with the observed activities. The polar contribution for the Chk1–16e complex (0.85 kcal mol−1) is much more unfavorable than that for the Chk1–5a complex (−3.89 kcal mol−1), and the non-polar contribution for Chk1–16e (−39.86 kcal mol−1) is slightly lower than that for the Chk1–5a complex (−41.75 kcal mol−1). As shown in Fig. 5b, many residues energetically contribute less for the binding of compound 16e than that of compound 5a. In particular, Glu90 shows obvious contribution to the difference of the binding free energy, indicating it is the key residue for better activity of 5a over that of 16e. From the Fig. 5c, the polar contribution of Glu90 is markedly promoted in 5a compared with 16e. Additionally, Ser146 forms one H-bond with inhibitor 5a while it does not form strong interactions with inhibitor 16e. This provides the reason why the contributions of Ser146 and Asp147 in the Chk1–5a complex are much lower than those in the Chk1–16e complex (−0.084 kcal mol−1). Collectively and interestingly, the hydroxymethyl group at R2 in compound 16e participates in forming an intermolecular hydrogen bond with Leu14, leading to the slight increase of the contribution of Leu14 to the binding free energy. Meanwhile, the presence of the hydrogen bond between 16e and Leu14 induced the inhibitor getting farther from the residue Glu90, thus decreasing the polar contribution of Glu90 to the binding free energy. Therefore, the addition of hydrophilic groups to the R2 position is supposed to be unfavorable to increasing the inhibitory activity of triazolone inhibitors.
 | ||
| Fig. 5 (a) The comparison of the averaged structures for the Chk1–5a and Chk1–16e complexes (carbon atoms are colored green and orange, respectively); (b) energy difference of each residue to the binding of 5a and 16e; and (c) the contributions of the individual energy terms for the key residues (ΔGnon-polar is denoted in red and olive, and ΔGpolar is denoted in blue and cyan in Chk1–5a and Chk1–16e, respectively). | ||
For inhibitors 5a and 31, the group at R1 is different. The pyrrolyl group at R1 in inhibitor 5a was replaced by a thiazolyl moiety in inhibitor 31. Despite the small differences in the substituent, compound 31 has considerable difference in its activity against Chk1 relative to compound 5a. According to Table 2, the calculated free energy for inhibitor 31 is weaker than that for inhibitor 5a by 10.32 kcal mol−1. In general, the polar interaction energy term (8.83 kcal mol−1), in particular the electrostatic interaction, plays a major role in the binding affinity of inhibitor 5a over inhibitor 31. The binding free energy decomposition reveals that two residues, Cys86 and Glu90, are responsible for the energy difference between 5a and 31. As displayed in Fig. 6b and c, Cys86 is more essential to determine the distinction in the binding affinity of these two inhibitors. The polar contribution of Cys86 in the Chk1–5a complex (−2.35 kcal mol−1) is much lower than that in the Chk1–31 complex (−0.60 kcal mol−1), though the non-polar contribution of Cys86 of the Chk1–5a complex (−0.88 kcal mol−1) is slightly higher than that of the Chk1–31 complex (−1.14 kcal mol−1). Cys86 participates in forming a hydrogen bond with the pyrrolyl moiety at R1 in inhibitor 5a whereas inhibitor 31 does not, in accord with the inhibitor–residue interaction spectrum. Formation of this hydrogen bond makes 5a well oriented and anchored in the binding site of Chk1. All in all, the presence of the thiazolyl without polar hydrogen atoms at the R1 position shows significantly reduced H-bond interactions, which suggests that maintaining groups with polar hydrogen atoms at this position favors the binding event.
 | ||
| Fig. 6 (a) The comparison of the averaged structures for the Chk1–5a and Chk1–31 complexes (carbon atoms are colored green and yellow, respectively); (b) energy difference of each residue to the binding of 5a and 31; and (c) the contributions of the individual energy terms for the key residues (ΔGnon-polar is denoted in red and olive, and ΔGpolar is denoted in blue and cyan in Chk1–5a and Chk1–31, respectively). | ||
The difference of 5a and 37 exists at R1, and the pyrrole moiety at R1 was replaced by an imidazole group in 37. Intriguingly, according to Fig. 7a, the conformations of 5a and 37 predicted by molecular dynamic simulations show one atom difference which is attributed to the docking solution that the lowest-energy docked complex is selected as the final best docked structure for further study. However, the small structural difference of the both ligands brings about considerable difference of the binding energies. The difference of the calculated binding free energies between 5a and 37 is 7.59 kcal mol−1, which is contributed by the difference of the non-polar contribution (3.77 kcal mol−1) and that of the polar contributions (3.82 kcal mol−1). As shown in Fig. 7b and c, three important residues, Glu90, Ser146, Asp147, with large difference are highlighted. Very similar behavior was observed in the case of 16e discussed above. Based on the structural alignment of the two complexes, the loss of a stabilizing hydrogen bond between 37 and Ser146 is detected, which represents the molecular origin of the decreased contributions of Ser146 and Asp147 to the total binding free energy. Without polar contacts with Ser146, 37 also stays away from Glu90 indirectly, indicating a decrease in the contribution of Glu90. Consequently, the introduction of an imidazole group at the R1 position has an appreciable effect on the ligand binding.
 | ||
| Fig. 7 (a) The comparison of the averaged structures for the Chk1–5a and Chk1–37 complexes (carbon atoms are colored green and magenta, respectively); (b) energy difference of each residue to the binding of 5a and 37; and (c) the contributions of the individual energy terms for the key residues (ΔGnon-polar is denoted in red and olive, and ΔGpolar is denoted in blue and cyan in Chk1–5a and Chk1–37, respectively). | ||
The fourth comparison was between inhibitors 14k and 5a. Compared to inhibitor 5a, modifications at two sites are found in inhibitor 14k. One is at the R1 position, where the pyrrolyl group in 5a was substituted by the 4-pyridinyl moiety. The other one is at the R2 position, where inhibitor 14k has an amino group while inhibitor 5a has a methyl group. These changes reduced the activity from 0.1 nm (inhibitor 5a) to 130 nm (inhibitor 14k). According to Table 2, the difference of the predicted binding free energy between both inhibitors is 6.98 kcal mol−1, which is consistent with the difference in the inhibitory activity. Obviously, the difference of the calculated total free energy shows a significant dependence on the difference of the polar contribution (6.84 kcal mol−1). As shown in Fig. 8b, Cys86 and Glu90 are the major contributors to the better bioactivity of 5a over 14k, whereas Glu16 and Asp147 are the key residues that provide unfavorable contributions to the better bioactivity of 5a over 14k. Although the contributions of the two pairs of the important residues are opposite, the resulting balance of the contributions is favorable for the better bioactivity of 5a over 14k. Accordingly, the amino moiety at R2 causes higher contribution of Glu16 and Asp147 to the free energy. This is obvious since the amino moiety could form polar interactions with both residues. Moreover, the contributions of the favorable residues of 14k are similar to those of 31, as shown in Fig. 8b and 6b. In addition, the loss of two hydrogen bonds between inhibitor 14k and Chk1 can be appreciably observed from the superposition of the two complexes. This is mainly due to the absence of the polar hydrogen atoms at the R1 position that renders inhibitor 14k incapable of forming hydrogen bonds with Cys86.
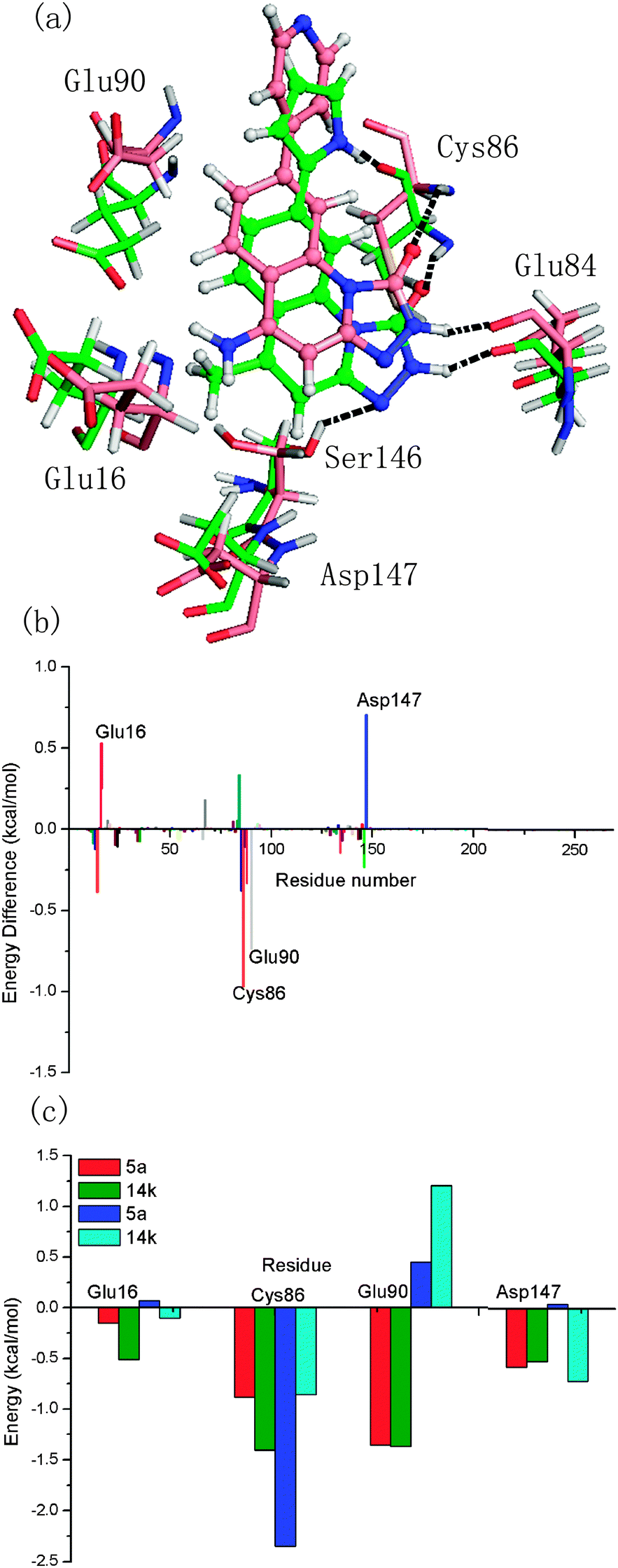 | ||
| Fig. 8 (a) The comparison of the averaged structures for the Chk1–5a and Chk1–14k complexes (carbon atoms are colored green and pink, respectively); (b) energy difference of each residue to the binding of 5a and 14k; (c) and the contributions of the individual energy terms for the key residues (ΔGnon-polar is denoted in red and olive, and ΔGpolar is denoted in blue and cyan in Chk1–5a and Chk1–14k, respectively). | ||
In summary, our binding energy data associate well with the interaction mode which quantitatively illustrates the effects of different substitutes on the bioactivity of inhibitors. Through the studies described above, the following key interactions are the primary interactions that distinguish the bioactivity from the most active inhibitor to the lower ones: (1) the hydrogen bonding interactions between R1 and two residues, Glu84 and Cys86; (2) the hydrogen bonding interactions between the inhibitor and Ser146. Based on the structural and energetic properties derived from the simulations of systems, it is rational to obtain the conclusion that different hydrogen bonds and hydrophobic interactions are mainly responsible for the different bioactivities of five compounds. In anticipation of further growth in the use of the triazolones inhibitors to target Chk1, we include results addressing the key elements necessary for the bioactivities. The polar hydrogen atoms at the R1 position could be involved in a hydrogen bond with Cys86, thus stabilizing the inhibitor in the binding site of Chk1. Moreover, the introduction of hydrophilic groups to the R2 position induces a decrease in the binding affinity compared to 5a, which can be corroborated by detailed analysis of 16e and 14k.
4. Conclusion
In the study, we used an integrated computational protocol, consisting of molecular docking, MD simulations, binding free energy calculations and decompositions to gain a deep insight into the binding mechanism of triazolone inhibitors and get to the basis of the difference in bioactivities. With the aim of discerning the structural features that affect the inhibitory activity of triazolones, Mk-8776, a Chk1 inhibitor that reached clinical stage, was also used as a reference for simulations. The reliance of computational protocol used in this study was corroborated by the result that the ranking of the calculated binding affinities of all the studied inhibitors was in concordance with the experimental values. The analysis of the components in binding free energy suggested that van der Waals contact and electrostatic interactions are the major contributions to the binding process. All the studied inhibitors, in general, present a similar pattern that reflects upon the key residues Leu14, Val22, Ala35, Glu84, Tyr85, Cys86, and Leu136 of Chk1 as shown by the decomposition of the binding free energy. However, it is significant to note that Leu136 contributes much more for the binding of MK-8776 than the other inhibitors. This can be attributed to the stronger van der Waals interactions between Leu136 and the six-membered ring of MK-8776, since this residue is hydrophobic. Thus it was indicated that introducing hydrophobic groups into triazolone inhibitors is favorable for binding to Chk1. An extensive comparison of structural and energetic properties for the triazolone inhibitors offered valuable insight into the structural elements that affect bioactivities. A hydrogen bond between the polar hydrogen atoms at R1 and Cys86 can facilitate a proper placement of the inhibitor into the binding pocket of Chk1 that favors binding. Furthermore, the introduction of hydrophilic groups to the R2 position leads to the reduction of binding affinity. The information obtained from this study may aid in a better structural understanding of inhibitors binding to Chk1 and a preliminary basis to further identify more promising inhibitors.Acknowledgements
The present work was supported by the National Natural Science Foundation of China (NSFC, No. 21275067).References
- J. Bartek and J. Lukas, Curr. Opin. Cell Biol., 2001, 13, 738–747 CrossRef CAS.
- C. H. McGowan, BioEssays, 2002, 24, 502–511 CrossRef CAS PubMed.
- Q. Liu, S. Guntuku, X.-S. Cui, S. Matsuoka, D. Cortez, K. Tamai, G. Luo, S. Carattini-Rivera, F. DeMayo and A. Bradley, Genes Dev., 2000, 14, 1448–1459 CrossRef CAS.
- H. Takai, K. Tominaga, N. Motoyama, Y. A. Minamishima, H. Nagahama, T. Tsukiyama, K. Ikeda, K. Nakayama, M. Nakanishi and K.-i. Nakayama, Genes Dev., 2000, 14, 1439–1447 CAS.
- J. Bartek and J. Lukas, Cancer Cell, 2003, 3, 421–429 CrossRef CAS.
- A. Hirao, Y.-Y. Kong, S. Matsuoka, A. Wakeham, J. Ruland, H. Yoshida, D. Liu, S. J. Elledge and T. W. Mak, Science, 2000, 287, 1824–1827 CrossRef CAS.
- C. S. Sørensen, R. G. Syljuåsen, J. Falck, T. Schroeder, L. Rönnstrand, K. K. Khanna, B.-B. Zhou, J. Bartek and J. Lukas, Cancer Cell, 2003, 3, 247–258 CrossRef.
- M. Maugeri-Saccà, M. Bartucci and R. De Maria, Cancer Treat. Rev., 2013, 39, 525–533 CrossRef PubMed.
- M. I. Walton, P. D. Eve, A. Hayes, M. R. Valenti, K. Alexis, G. Box, A. Hallsworth, E. L. Smith, K. J. Boxall and M. Lainchbury, Clin. Cancer Res., 2012, 18, 5650–5661 CrossRef CAS PubMed.
- M. H. Lam, Q. Liu, S. J. Elledge and J. M. Rosen, Cancer Cell, 2004, 6, 45–59 CrossRef CAS PubMed.
- N. T. Archie, R. Carvajal and G. K. Schwartz, Clin. Cancer Res., 2007, 13, 1955–1960 CrossRef PubMed.
- G. Li, L. A. Hasvold, Z.-F. Tao, G. T. Wang, S. L. Gwaltney II, J. Patel, P. Kovar, R. B. Credo, Z. Chen and H. Zhang, Bioorg. Med. Chem. Lett., 2006, 16, 2293–2298 CrossRef CAS PubMed.
- N.-H. Lin, P. Xia, P. Kovar, C. Park, Z. Chen, H. Zhang, S. H. Rosenberg and H. L. Sham, Bioorg. Med. Chem. Lett., 2006, 16, 421–426 CrossRef CAS PubMed.
- Z.-F. Tao, G. Li, Y. Tong, Z. Chen, P. Merta, P. Kovar, H. Zhang, S. H. Rosenberg, H. L. Sham and T. J. Sowin, Bioorg. Med. Chem. Lett., 2007, 17, 4308–4315 CrossRef CAS PubMed.
- R. M. Garbaccio, S. Huang, E. S. Tasber, M. E. Fraley, Y. Yan, S. Munshi, M. Ikuta, L. Kuo, C. Kreatsoulas and S. Stirdivant, Bioorg. Med. Chem. Lett., 2007, 17, 6280–6285 CrossRef CAS PubMed.
- Z.-F. Tao, G. Li, Y. Tong, K. D. Stewart, Z. Chen, M.-H. Bui, P. Merta, C. Park, P. Kovar and H. Zhang, Bioorg. Med. Chem. Lett., 2007, 17, 5944–5951 CrossRef CAS PubMed.
- E. J. Brnardic, R. M. Garbaccio, M. E. Fraley, E. S. Tasber, J. T. Steen, K. L. Arrington, V. Y. Dudkin, G. D. Hartman, S. M. Stirdivant and B. A. Drakas, Bioorg. Med. Chem. Lett., 2007, 17, 5989–5994 CrossRef CAS PubMed.
- J. W. Janetka, L. Almeida, S. Ashwell, P. J. Brassil, K. Daly, C. Deng, T. Gero, R. E. Glynn, C. L. Horn and S. Ioannidis, Bioorg. Med. Chem. Lett., 2008, 18, 4242–4248 CrossRef CAS PubMed.
- A. Converso, T. Hartingh, R. M. Garbaccio, E. Tasber, K. Rickert, M. E. Fraley, Y. Yan, C. Kreatsoulas, S. Stirdivant and B. Drakas, Bioorg. Med. Chem. Lett., 2009, 19, 1240–1244 CrossRef CAS PubMed.
- M. Teng, J. Zhu, M. D. Johnson, P. Chen, J. Kornmann, E. Chen, A. Blasina, J. Register, K. Anderes and C. Rogers, Bioorg. Med. Chem. Lett., 2007, 50, 5253–5256 CAS.
- L. Zhao, Y. Zhang, C. Dai, T. Guzi, D. Wiswell, W. Seghezzi, D. Parry, T. Fischmann and M. A. Siddiqui, Bioorg. Med. Chem. Lett., 2010, 20, 7216–7221 CrossRef CAS PubMed.
- V. Oza, S. Ashwell, P. Brassil, J. Breed, C. Deng, J. Ezhuthachan, H. Haye, C. Horn, J. Janetka and P. Lyne, Bioorg. Med. Chem. Lett., 2010, 20, 5133–5138 CrossRef CAS PubMed.
- J. Du, H. Sun, L. Xi, J. Li, Y. Yang, H. Liu and X. Yao, J. Comput. Chem., 2011, 32, 2800–2809 CrossRef CAS PubMed.
- Y. Li, D. J. Kim, W. Ma, R. A. Lubet, A. M. Bode and Z. Dong, J. Chem. Inf. Model., 2011, 51, 2904–2914 CrossRef CAS PubMed.
- M. P. Dwyer, K. Paruch, M. Labroli, C. Alvarez, K. M. Keertikar, C. Poker, R. Rossman, T. O. Fischmann, J. S. Duca and V. Madison, Bioorg. Med. Chem. Lett., 2011, 21, 467–470 CrossRef CAS PubMed.
- M. Labroli, K. Paruch, M. P. Dwyer, C. Alvarez, K. Keertikar, C. Poker, R. Rossman, J. S. Duca, T. O. Fischmann and V. Madison, Bioorg. Med. Chem. Lett., 2011, 21, 471–474 CrossRef CAS PubMed.
- V. Y. Dudkin, K. Rickert, C. Kreatsoulas, C. Wang, K. L. Arrington, M. E. Fraley, G. D. Hartman, Y. Yan, M. Ikuta and S. M. Stirdivant, Bioorg. Med. Chem. Lett., 2012, 22, 2609–2612 CrossRef CAS PubMed.
- V. Oza, S. Ashwell, P. Brassil, J. Breed, J. Ezhuthachan, C. Deng, M. Grondine, C. Horn, D. Liu and P. Lyne, Bioorg. Med. Chem. Lett., 2012, 22, 2330–2337 CrossRef CAS PubMed.
- X. Huang, C. C. Cheng, T. O. Fischmann, J. S. Duca, M. Richards, P. K. Tadikonda, P. A. Reddy, L. Zhao, M. Arshad Siddiqui and D. Parry, Bioorg. Med. Chem. Lett., 2013, 23, 2590–2594 CrossRef CAS PubMed.
- S. D. Zabludoff, C. Deng, M. R. Grondine, A. M. Sheehy, S. Ashwell, B. L. Caleb, S. Green, H. R. Haye, C. L. Horn and J. W. Janetka, Mol. Cancer Ther., 2008, 7, 2955–2966 CrossRef CAS PubMed.
- C. King, H. Diaz, D. Barnard, D. Barda, D. Clawson, W. Blosser, K. Cox, S. Guo and M. Marshall, Invest. New Drugs, 2014, 32, 213–226 CrossRef CAS PubMed.
- Y. Dai, S. Chen, M. Kmieciak, L. Zhou, H. Lin, X.-Y. Pei and S. Grant, Mol. Cancer Ther., 2013, 12, 878–889 CrossRef CAS PubMed.
- N. T. Archie, K. G. Rendahl, T. Sheikh, H. Cheema, K. Aardalen, M. Embry, S. Ma, E. J. Moler, Z. J. Ni and D. E. L. de Menezes, Clin. Cancer Res., 2007, 13, 591–602 CrossRef PubMed.
- P. Ferrao, E. Bukczynska, R. Johnstone and G. McArthur, Oncogene, 2011, 31, 1661–1672 CrossRef PubMed.
- Schrödinger, version 9.0, Schrödinger LLC, New York, NY, 2009 Search PubMed.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- M. D. Eldridge, C. W. Murray, T. R. Auton, G. V. Paolini and R. P. Mee, J. Comput.-Aided Mol. Des., 1997, 11, 425–445 CrossRef CAS.
- S. K. Tripathi, R. Muttineni and S. K. Singh, J. Theor. Biol., 2013, 334, 87–100 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, J. Montgomery Jr., T. Vreven, K. Kudin and J. Burant, Guassian 03, revision D. 01, Guassian, Inc, Wallingford, CT, 2004 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Bioinf., 2006, 65, 712–725 CrossRef CAS PubMed.
- D. Case, T. Darden, T. Cheatham III, C. Simmerling, J. Wang, R. Duke, R. Luo, R. Walker, W. Zhang and K. Merz, AMBER 11, University of California, San Francisco, 2010 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- J. Wang, T. Hou and X. Xu, Curr. Comput.-Aided Drug Des., 2006, 2, 287–306 CrossRef CAS.
- T. Hou and R. Yu, J. Med. Chem., 2007, 50, 1177–1188 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Comput. Chem., 2011, 32, 866–877 CrossRef CAS PubMed.
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69–82 CrossRef CAS PubMed.
- W. Xue, D. Pan, Y. Yang, H. Liu and X. Yao, Antiviral Res., 2012, 93, 126–137 CrossRef CAS PubMed.
- M. Shen, S. Zhou, Y. Li, P. Pan, L. Zhang and T. Hou, Mol. BioSyst., 2013, 9, 361–374 RSC.
- A. Onufriev, D. Bashford and D. A. Case, Proteins: Struct., Funct., Bioinf., 2004, 55, 383–394 CrossRef CAS PubMed.
- J. Weiser, P. S. Shenkin and W. C. Still, J. Comput. Chem., 1999, 20, 217–230 CrossRef CAS.
- T. Hou, Z. Xu, W. Zhang, W. A. McLaughlin, D. A. Case, Y. Xu and W. Wang, Mol. Cell. Proteomics, 2009, 8, 639–649 CAS.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan and W. Wang, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- B. Kuhn, P. Gerber, T. Schulz-Gasch and M. Stahl, J. Med. Chem., 2005, 48, 4040–4048 CrossRef CAS PubMed.
- I. Autiero, M. Saviano and E. Langella, Mol. BioSyst., 2013, 9, 2118–2124 RSC.
- L. Sacconnay, D. Smirlis, E. F. Queiroz, J. L. Wolfender, M. B. P. Soares, P.-A. Carrupt and A. Nurisso, Mol. BioSyst., 2013, 9, 2223–2230 RSC.
- R. Wang, Y. Lu and S. Wang, J. Med. Chem., 2003, 46, 2287–2303 CrossRef CAS PubMed.
- K. Onodera, K. Satou and H. Hirota, J. Chem. Inf. Model., 2007, 47, 1609–1618 CrossRef CAS PubMed.
- Y. Yang, J. Qin, H. Liu and X. Yao, J. Chem. Inf. Model., 2011, 51, 680–692 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |